

Abstract
Background
Transcript variants and protein isoforms are central to unique tissue functions and maintenance of homeostasis, in addition to being associated with aberrant states such as cancer, where their crosstalk with the mutated tumor suppressor p53 may contribute to genomic instability and chromosomal rearrangements. We previously identified several novel splice variants in ovarian cancer RNA-sequencing datasets; herein, we aimed to elucidate the biological effects of the Integrin Subunit Beta 8 variant (termed pITGB8-205).
Methods
Resolution of the full-length sequence of pITGB8-205 through rapid amplification of cDNA ends (RACE-PCR). Cell cycle analysis and karyotype studies were performed to further explore genomic instability. RNA-seq and proteomics analyses were used to identify the differential expression of the genes.
Results
This full-length study revealed a unique 5’ sequence in pITGB8-205 that differed from the reported ITGB8-205 sequence, suggesting differential regulation of this novel transcript. Under a p53 mutant background, overexpression of pITGB8-205 triggered genetic instability reminiscent of oncogene-induced replicative stress with extensive abnormal mitoses and chromosomal and nuclear aberrations indicative of chromosomal instability, leading to near whole-genome duplication that imposes energy stress on cellular resources. Micronuclei and aneuploidy are striking features of pITGB8-205-overexpressing p53-mutant cells but are not enhanced in p53 wild-type (WT) cells. RNA-seq and proteomics analyses further suggested that p53 inactivation in ovarian cancer provides a permissive intracellular molecular niche for pITGB8-205 to mediate its effects on genomic instability. This observation is pivotal considering that most high-grade serous ovarian carcinoma (HGSC) tumors express mutant p53. The resulting aneuploid clones with enhanced self-renewal and survival capabilities disrupt clonal dominance under stress yet maintain a balance between replicative stress and prosurvival advantages.
Conclusion
pITGB8-205-overexpressing clones sustain ovarian tumor cell survival, achieve homeostasis and are formidable opponents of therapy.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13048-024-01538-6.
Keywords: ITGB8, p53, Aneuploidy, Genomic instability, Prosurvival mechanisms
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s13048-024-01538-6.
Background
Alternative splicing events in cancer often involve unappreciated cooperation, such as the use of alternative exons, exonization of introns, evasion of nonsense-mediated decay, etc [1–3]. Mutations in spliceosome components in tumors also generate novel variants and isoforms [4–6], which can be pro- or antiapoptotic and often correlate with tumor progression, metastasis, and therapeutic outcomes. These isoforms may serve as diagnostic or prognostic indicators [7, 8]. Moreover, alternative splicing variants of oncogenes and tumor suppressor genes contribute to genomic instability, a hallmark of cancer, providing survival advantages to tumor cells. Some genomic instabilities contribute to tetraploidization, whole-genome duplications and chromothripsis [9–11]. Studying splice variants associated with genomic instability offers a different perspective on gene regulation in normal cellular homeostasis and disorders such as cancer.
Our previous study identified several chimeric transcripts in ovarian cancer RNA-seq data, including novel splice variants of ITGB8, PRKAR1b, and ZFYVE28 ([12], Supplementary Fig. 1). The ITGB8 variant is particularly intriguing, as it is involved in neurovascular development, embryo implantation, cell-cell contact, and communication within complex multicellular structures [13, 14]. Elevated expression of the canonical ITGB8 protein is associated with various cancers, including lung, breast, prostate, and squamous carcinoma. Its effects stem from its role in promoting PI3K-Akt signaling and integrin surface protein interactions and altering the actin cytoskeleton to modulate tumor growth, metastasis, angiogenesis/vasculogenesis, and the regulation of tumor-associated regulatory T (Treg) cells. Its inhibition is reported to sensitize tumor cells to radiotherapy and gefitinib [15–21]. Mutated p53 underlies the etiology of several cancers. The advantages in terms of cellular growth, mesothelial cell adhesion and metastasis acquired by p53-mutant OVCAR3 cells are attributed to integrin beta-4 signaling. Mutations in EGFR and TP53 in lung cancer patients lead to poor prognosis [22–24].
The present study annotated the novel ITGB8 variant pITGB8-205 and aimed to understand its biological effects. These findings suggest that pITGB8-205 drives chromosomal instability (CIN), aneuploidy, and metabolic perturbations and has tumor-promoting functionality, but its reversal to tumor-suppressive functionality in the presence of active p53 reveals its pleiotropic behavior and ability to act as a “double-edged sword” depending on the p53 background of cell transformation.
Materials and methods
Cell culture, PCR, full-length (FL) amplification through rapid amplification of cDNA ends (RACE), transfections and splice graph generation
HGSC cell lines [(A2, A4EP, A4LP and G1M2 (RRID: CVCL_FG73)] from patient ascites, 10 human lymphoblastoid (LBL) cell lines from peripheral blood and human primary skin fibroblast cultures (SFs) were previously developed in the laboratory [25, 26]; OVCAR3 (RRID: CVCL_0465), OVCAR4 (RRID: CVCL_1627), A2780 (RRID: CVCL_0134)), MDCK (RRID: CVCL_0422) and CV1 (RRID: CVCL_0229) cells were sourced from the NCCS cell repository; IOSE364 from Dr. Ray (ACTREC, Mumbai); HEK293T (RRID: CVCL_0063) from Dr. Shiras, NCCS; OV90 (RRID: CVCL_3768), OVMZ6 (RRID: CVCL_4005), OVCA420 (RRID: CVCL_3935), PEO14 (RRID: CVCL_2687), OVCA432 (RRID: CVCL_3769), and CAOV3 (RRID: CVCL_0201) [27] The cell lines were authenticated via GeneMapper™ ID-X Software v1.5 and harvested at 80% confluency. RNA extraction was performed via TRIzolTM reagent, followed by cDNA synthesis and quantification. RT-PCR and qPCR were performed via gene-specific primers, and semiquantitative expression analysis was performed via densitometry. qPCR was performed via SYBR Green PCR Master Mix, and HPRT1 expression and the nontemplate reaction mixture were used as controls. The data were processed to determine cycle threshold values and isoform fractions.
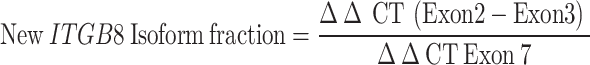 |
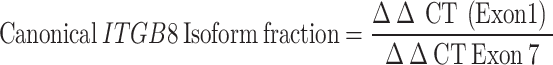 |
Full-length amplification through rapid amplification of C-DNA ends (RACE) and lentivirus generation
The PrimeScriptTM 1st strand cDNA Synthesis Kit (TaKaRa) was used for RACE cDNA synthesis from PEO14 cells. The sequence was digested via the restriction enzymes EcoRI-HF and BamHI-HF and transformed into DH5-alpha competent cells. Lentiviruses were generated in HEK293T cells, and the harvested virus was concentrated as described previously [28]. The lentiviruses were transfected into OVCAR3, A4, and PEO14 cells for overexpression and knockdown studies. Puromycin selection was used for further selection, and Sanger sequencing was used for sequence validation.
Cell cycle analysis, label-chase, cell doubling, colony formation, gamma-irradiation, glucose consumption, lactate production, ROS, and apoptosis assays
Cell cycle analysis was conducted via propidium iodide and RNase A treatment. The cell suspension was washed with PBS, fixed with 70% ethanol, and subjected to FACS acquisition. Data analysis was performed via BD FACS Diva software 6.0. PKH26GL was used for label-chase [29]. The cells were seeded in 12-well plates for cell doubling and colony formation assays and were counted manually. Postirradiation survival was determined. Glucose consumption and lactate production assays were performed via the GLUCOSE-LQ and LACTATE kits, respectively. An Annexin V-FITC apoptosis assay was performed by suspending 105 cells in binding buffer, and data were acquired via the FITC and PE channels. Cellular reactive oxygen species (ROS) levels were examined by incubating 0.5 × 106 cells with 5 µM 2-7-dichlorodihydrofluorescein dye at 37 °C in the dark for 30 min.
Estimation of nuclear abnormalities and karyotype studies
The cells were fixed with 2% PFA at 4 °C for 10 min, permeabilized with Triton X-100, washed 3 times with 1X PBS, blocked with 10% goat serum, and incubated with primary antibodies for 1–1.5 h at room temperature. Coverslips were washed 3 times with 1x PBS three times and then incubated with secondary antibodies. Nuclei and micronuclei positive for γ-H2AX were examined and scored via ImageJ software [30]. The nuclear area was calculated as a function of the mean correlated total cell fluorescence (CTCF).
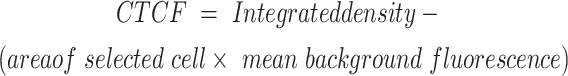 |
Nuclei and micronuclei positive for γ-H2AX were examined and scored via ImageJ software. Nuclear anomalies such as lagging chromosomes were examined in OVCAR3, OVCAR3-OE A4, and A4-OE cells. CBMC assays were performed with unirradiated and g-irradiated cells treated with 0.2 µM/ML cytochalasin D for 40 h. A total of 10,000 harvested cells were deposited on poly-L-lysine-coated slides, fixed and stained with Hoechst. Nuclei and micronuclei were captured at 40X and 60X, and their areas were quantified via QuPath 0.4.0. Metaphase spreads and G-bandings were examined for marker chromosomes via Ikaros software.
RNA sequencing, proteomics, and xenograft generation
RNA sequencing, proteomics, differential expression, and pathway analysis were conducted on the cell pellets. The cells were subjected to lysis and centrifuged, and the protein precipitates were redissolved in dissolution buffer [31]. The protein concentration was estimated via a 2D quant kit, and the proteins were reduced with DTT, alkylated with IAA, and digested with trypsin. Proteins and transcripts were subjected to REACTOME and gene set enrichment analysis (GSEA) for pathway analysis. Protein interaction networks were generated via STRING online network analysis. Xenograft generation involved injecting 2 × 106 A4-OE or A4 cells into subcutaneous NOD/SCID female mice. The tumor volume was measured each week after the 15th day of tumor injection, and the tumors were harvested, weighed, and captured after the 36th day. All animal experiments were carried out at the NCCS animal experiment facility and approved by the institutional animal ethics committee.
Graphical representations and statistics
The study used data from Ensembl for the ITGB8, PRKAR1B, and ZFYVE28 genomes and splice variants. Splice graphs and dual Y bar plots were generated via Adobe Illustrator CS5.1, SigmaPlot 10.0, SRplot, and Prism 8.4.2. Statistical significance was determined via SigmaStat 3.5. The samples’ statistical significance is as follows. p < 0.001=***, p < 0.01=**, p < 0.05=*.
Results
A new transcript variant of ITGB8 (pITGB8-205) closely resembling an uncharacterized variant was discovered, with observed clinical implications
A unique characteristic of the overlapping chimeric reads of ITGB8, PRKAR1B and ZFYVE28 identified in our previous study was the inclusion of hitherto unreported sequences of exons in the continuum (Fig. 1a; Supplementary Fig. 1a-i, a-ii, a-iii, 1b). We consolidated reads of each of these across all tumors to identify extended reads and mapped them to the specific exons in their genomic sequences. The extended ITGB8 sequence is localized to exons 2 and 4 (47 bp and 86 bp, respectively). Exon 2 is present in the noncoding variants ITGB8-205: ENST00000537992 and ITGB8-202: ENST00000477859, whereas Exon 4 is included in two protein coding variants (canonical ITGB8-201: ENST00000222573 and ITGB8-205: ENST00000537992) and a noncoding variant, ITGB8-204: ENST00000478974. Owing to these features and the reported association of ITGB8 with cancer, we focused on the elucidation of this novel variant, which we termed pITGB8-205. Splice site analyses predicted a consensus sequence (CAG/GT) at the donor sites of all three novel sequences that affirmed alternative exon usage [Spliceator [32]; Supplementary Fig. 1c]; further mapping of their isoform fractions revealed enrichment of the rare exon at the 5’ location of ITGB8 (Fig. 1b; Supplementary Fig. 1d). The expression of pITGB8-205 was validated in five HGSC cell lines through PCR followed by Sanger sequencing (Fig. 1d; Supplementary Fig. 1e). Alternative splicing PCR revealed the coexpression of canonical and pITGB8-205 transcripts across various cell types. A lower isoform fraction of the new variant was observed in human cells (lymphoblastoid, HGSC, and primary fibroblasts) but was comparable in other species, including NMDF, MDCK, and mouse ovary cells. (Supplementary Fig. 1f). Such differential expression suggests subtle yet distinct cellular functions. As a first step, we derived its full-length sequence (FL_pITGB8-205, 2316 bp) through rapid amplification of the 5’ and 3’ cDNA ends (RACE; Supplementary Data 1; Methods). This revealed a unique 88 bp 5’ UTR sequence (localizing within Exon 1 in the Ensembl Genome Browser), which is not expressed in other ITGB8 variants), while its 3’ UTR was conserved with the canonical sequence (Fig. 1e; Supplementary Fig. 2a). We also predicted four high-confidence Pol-II transcription start sites in the 5.597 kb upstream sequence (Promoter 2.0; [33]; Supplementary Fig. 2b-i, 2b-ii), in addition to major U2-mediated spliceosome binding sites at the 5’ and 3’ borders of Exon 1/Intron 1 and Intron 1/Exon 2 with high likelihoods and scores [ESEfinder 3.0, ([34, 35]; Fig. 1f)]. In an earlier study to predict the translation potential of chimeric transcripts, we did not identify any peptides associated with pITGB8-205 at a significantly high frequency or coverage (Dixit and Bapat, manuscript under review; data not shown), suggesting that it functions as a long noncoding RNA (lncRNA).
Fig. 1.

(a) ITGB8 splice graph positioning known variants and novel reads (denoted as dashed lines above and below exons, respectively); (b) Box and whisker plot representing isoform fractions of the alternative continuum of exons in the new variants; (c) pie chart showing the distribution of the number of dead and living patients expressing pITGB8-205; (d) Sanger sequencing-based validation of 133 bp ITGB8 (Exons 2, 4); (e) Novel 5’ pITGB8-205 sequences; (f) Predicted major U2-mediated spliceosome sites in pITGB8-205; (g.i) Overexpression of pITGB8-205 affects cell growth, cycling and metabolism. (a) Profiling of pITGB8-205 full length (2.4 kb) in 10 HGSC cell lines (X-axis indicates the HGSC cell lines, Y-Axis indicates the relative pITGB8-205 FL expression compared with the endogenous control; (g.ii) Screening of pITGB8-205 overexpressing (OVCAR3-OE, A4-OE) and knockdown (PEO14-KD) clones; (h) Bar graph representation of glucose consumption and lactate production (mg/dL; normalized with cell number) of OVCAR3-OE and control cells at 24, 48, 72, 96 and 120 h in culture; (i) Bar graph representing the cell cycle phases in OVCAR3-OE, A4-OE and PEO14-KD vs. control cells; (j) Histogram representing differential label quenching (cell cycling) dynamics of i-OVCAR3-OE, ii-A4-OE and iii-PEO14-KD vs. control cells (Solid lines- OVCAR3-OE/A4-OE/PEO14-KD, Dotted lines - respective controls at 0 (Green) and 96 h (black); (k) Representative doubling times of OVCAR3-OE, A4-OE and PEO14
Increased expression of pITGB8-205 perturbs cell growth, cycling and metabolism in p53 mutant cells
FL_pITGB8-205 was expressed at varying levels across several HGSC cell lines, with high expression in PEO14 and OVCA420 and low expression in G1M2 and OVCAR3 (Fig. 1g-i). Owing to its high mutation rate in HGSC, p53 has been identified as a key contributor to genomic instability and chromosomal and amplification instability [36] and is directly associated with the upregulation of canonical ITGB8 [50]. We explored the association of pITGB8-205 with the mutational status of p53 toward cellular functions and transformation. We thus ectopically expressed pITGB8-205 in the OVCAR3 (p53 mutant) and A4 (wild-type) HGSC cell lines and depleted it in PEO14 (p53 mutant) cells to generate OVCAR3-overexpressing (OVCAR3-OE), A4-overexpressing (A4-OE) and PEO14-knockdown clones (PEO14-KD; Fig. 1g-ii). pITGB8-205 overexpression was highly lethal and resulted in very few viable OVCAR3-OE cells (∼ 5%), whereas its knockdown did not significantly affect cell viability. The most striking feature of OVCAR3-OE was its distinctly altered metabolism (rapidly lowering the pH of the culture media), which we experimentally attributed to increased glucose consumption and lactate production (Fig. 1h). This otherwise reflected a highly proliferative state, whereas the OVCAR3-OE cells were slow cycling compared with the control cells (higher G0/G1, reduced S and G2M; Fig. 1i). Label-chase confirmed the decreased rate of cellular turnover following pITGB8-205 overexpression, which was further reflected by an increased cell doubling time (Fig. 1j-i and k; Supplementary Fig. 2j-i). OVCAR3-OE cells presented decreased colony dynamics (colony size/area and density of foci), yet surprisingly, an unaltered colony formation frequency correlated with upregulation of the expression of the self-renewal marker Nanog (Fig. 1f, Supplementary Fig. 2e-i, e-ii). Interestingly, the effect of pITGB8-205 overexpression was very subtle in wild-type p53 cells. Although A4-OE was indicated to be slow cycling, as evidenced by the greater G0/G1 ratio, reduced S population, increased doubling time and significantly slower quenching of the label chase (PKH dye) than those of OVCAR3-OE, they did not show any metabolic perturbations (Fig. 2d and e-iii, 2f, 2 g-i, 2 g-ii). Depletion of pITGB8-205, on the other hand, did not significantly alter the phenotype, cell cycling dynamics or metabolism of PEO14-KD cells; however, these cells presented a lower colony formation frequency and dynamics with lower Nanog and Oct4 expression (Fig. 1h and j-ii, 1k; Supplementary Fig. 2d, 2e-i, e-ii; 2 h). Primistically, pITGB8-205 expression perturbs cell growth and cycling independent of p53 mutational status but alters metabolism in a way that is reliant on p53 mutational status and may contribute to self-renewal. These findings suggest that the ectopic expression of pITGB8-205 in p53-mutant cells may affect cell survival.
Fig. 2.

pITGB8-205 overexpression in OVACR3-OE cells triggers genomic and chromosomal instability, altered ploidy, extensive DNA damage, aneuploidy and near-whole-genome duplication. A FACS dot plot comparing cell size and granularity in OVCAR3-OE (right) with control (left) cells; (b) Bar graph comparing the cell area (mm2) of OVCAR3-OE with the respective control cells; (c) Representative FACS histogram comparing the DNA content of OVCAR3-OE with control cells (upper and lower panels, respectively); (d) Bar graph representation of quantified nuclear and chromosomal anomalies (1-lagging chromosomes, 2-anaphase bridges, 3-multinucleated cells, 4-micronuclei); (e) Bar graph representation of increased micronuclei (1), mononucleated (2), binucleate (3), trinucleate (4), and tetranucleated (5) cells in OVACR3-OE over control cells; (f-i) Graphical representation of the percentage of γ-H2AX-positive nuclei and micronuclei in OVCAR3-OE and control cell nuclei, (f-ii) Representative immunofluorescence images of phosphorylated H2AX expression in OVCAR3-OE and control cell nuclei, γ-H2AX (cyan) and nuclei (DAPI); (g-i) Graph indicating the number of γ-H2AX foci per nucleus in OVCAR3-OE vs. control cells, (g-ii) Representative immunofluorescence images of phosphorylated H2AX foci in PEO14-KD vs. control; (h) Comparison of the range of chromosome numbers in OVCAR3-OE and control cells (100 metaphases screened for each); (i) Bar graph representation of the fold change in individual chromosome copy numbers; (j) Representative karyotypes of (i) OVCAR3-OE cells and (ii) Controls; (k) Bar graph representation of translocation events; (l) Dot plot comparing several marker chromosomes in control and OVCAR3-OE cells; (m) Frequency (number of metaphases) harboring specific chromosomes in the marker fraction resolved by Ikaros 6.3
Overexpression of pITGB8-205 in a mutant p53 background is associated with replicative stress through increased genomic and mitotic instabilities
Cell cycle profiling and confocal image quantification revealed that enhanced expression of pITGB8-205 in OVCAR3 cells led to altered cellular granularity and size, along with an increase in the range of DNA content (from 2n–4n to 4n–8n compared with the control), unlike in A4-OE cells, suggesting that overexpression may perturb the dynamics of cell division and genomic stability only under the mutant P53 background. (Figure 1a, b and c; Supplementary Fig. 3a, 3b, 3c). Mitotic defects as well as nonmitotic nuclear aberrations were indeed highly pronounced in OVCAR3-OE cells (Fig. 2d, Supplementary Fig. 3d, 4a-i, ii;). The most significant of these were chromosomal segregation errors, including lagging chromosomes, anaphase bridges and mitotic cortical blebbing associated with cytokinesis failure, and cleavage furrow regression, which progresses to mitotic catastrophe. Other emerging nuclear aberrations included a significantly increased frequency of micronuclei along with the emergence of notched, ring, blebbed and lobed nuclei. Some of these mitotic errors in OVCAR3-OE cells culminated in enhanced generation of binucleate (tetraploid) cells, a fraction of which persevere and divide to generate trinucleate and tetra-nucleate cells ([37]; Supplementary Fig. 4a-i). This extensive instability, along with increased ROS levels in OVCAR3-OE cells, likely led to apoptosis, whereas necrosis was more pronounced in PEO14-KD cells (Supplementary Fig. 3e, 3f). Strikingly, no significant increase in these nuclear abnormalities, ROS levels or apoptosis was detected in p53 WT A4-OE cells (Supplementary Fig. 3 h, 4a-ii).
The high rates of ROS in the OVCAR3-OE and PEO14-KD cells suggested that these cells may experience high levels of DNA damage. To test this hypothesis, we quantified the levels of γ-H2AX (an early marker of double-strand DNA breaks (DSBs), indicating impending DNA repair and cell survival) in the nuclei and micronuclei of these cells and their corresponding controls and found that the levels of γ-H2AX in both micronuclei and main nuclei were significantly greater in the OVCAR3-OE cells than in the controls (Fig. 2f, Supplementary Fig. 3g), whereas the difference was not as striking for PEO14-KD (Fig. 2g). Finally, we assessed whether any changes in the cytoskeleton occurred in response to multinucleation and increased cell size in OVCAR3-OE cells. We found that the levels of actin were increased in OVCAR3-OE vs. OVCAR3 cells, whereas no differences were observed in the levels of tubulin (Supplementary Fig. 3i-j). Overall, pITGB8-205 overexpression led to chromosome missegregation and other phenotypes suggestive of genomic instability in the context of a p53 mutant background (OVCAR3-OE) but not in the presence of functional p53 (A4-OE).
pITGB8-205 overexpression leads to aneuploidy associated with diverse structural and numerical chromosomal aberrations
To confirm the chromosomal instabilities (CINs) suggested above, we analyzed numerical and structural differences in chromosomes (100 metaphases and 10 G-banded karyotypes, respectively). This revealed that, as reported earlier, the OVCAR3 cells were hyperdiploid (modal number = 62; [38]), whereas the OVCAR3-OE cells were strikingly hyperploid (> 100 chromosomes; modal number = 114), with all except chromosome 21 displaying increased copy numbers and a striking twofold increase in the number of marker (Mar) chromosomes (Fig. 2h and j; Supplementary Fig. 5a, 5b, 5c). The latter were classified as such since they did not appear to match known chromosomes vis-à-vis their length, appearance or G-banding as perceived in manual analysis. Structural alterations included the emergence of several unbalanced translocations (involving chromosomes 8, 9, 11, 12, 13, X; Fig. 2i-i and i-ii, 2 h), isochromosomes (i5q, i15q) and a rare triple translocation t(1:11:14; Supplementary Fig. 5d). Further use of an AI-based classifier (Ikaros 6.3, MetaSystems Hard & Software GmbH) for automated categorization and resolution of the identity of marker chromosomes revealed an additional, almost complete haploid set of chromosomes camouflaged in the mar fraction of OVCAR3-OE cells (average ∼ 20 chromosomes per cell), which were absent in parental cells (Fig. 2j and k; Supplementary Fig. 6). Taken together, pITGB8-205 overexpression conclusively drives a multitude of errors in the cell cycle, causing CIN and replicative stress that ultimately culminate in complex aneuploidy approaching whole-genome duplication (WGD).
pITGB8-205 overexpression-associated replicative stress enhances resistance to apoptosis but is unsustainable and reverted during tumor homeostasis
Since WGD is known to confer a survival advantage to tumor cells, we profiled the recovery of OVCAR3-OE and PEO14-KD cells and their respective controls following exposure to 8.5 Gy gamma radiation (γ-IR) to ascertain the same. A higher rate of cell survival was evident in irradiated OVCAR3-OE cells than in control cells, while control PEO14 cells (expressing endogenous pITGB8-205) also survived better than PEO14-KD cells under irradiation (Fig. 3a-i). An immediate response of all the cell derivatives was an initial reduction in pITGB8-205 expression, which was subsequently restored only in OVCAR3-OE cells (Fig. 3a-ii). We speculate that while cells expressing moderate levels of pITGB8-205 do not survive irradiation, those with high levels of pITGB8-205 are initially arrested in the cell cycle but resume cycling later, causing a spike in pITGB8-205 expression (Fig. 3a-ii, 3b-iii). This resulted in improved recovery over other cells, along with an increased number of micronuclei, increased nuclear size and altered cell cycle dynamics following initial G2M arrest in both OVCAR3-OE and control cells (Fig. 3b-i and b-ii, 3b-iii). OVCAR3-OE cells progress into further phases of the cell cycle, as evidenced by the increased number of cells in the G0G1 and S phases, consequently generating superaneuploid fractions (> 8 C; Fig. 3b-iv), which contribute positively to postirradiation recovery.
Fig. 3.

Effects of g-irradiation on pITGB8-205 derivative cells and their controls. (a-i) Ratios of surviving cells (irradiated: unirradiated OVCAR3 controls), (irradiated: unirradiated OVCAR3-OE), cells (irradiated: unirradiated PEO14 controls), (irradiated: unirradiated PEO14-KD); (a-ii) Relative expression of pITGB8-205 (vis-à-vis 133 bp of Exons 2 and 4) in postirradiation surviving cells at specific time points; (b-i) Box and whisker plot representation of micronuclei-containing cells in unirradiated and irradiated OVCAR3-OE and control cells as revealed through a cytokinesis-block micronucleus cytome assay; (b-ii) Bar graph showing increased nuclear size in the same cell groups; (b-iii) Line graph indicating the cell cycle perturbation in OVCAR3-OE vs. control cells after irradiation; (b-iv) Histogram representing the altered ploidy in OVCAR3-OE vs. control cells after 0 h, 20 h and 40 h (from top to bottom) after irradiation (blue– OVCAR3 control, green– OVCAR3-OE); (c) Graphical representation of reduced (i) relative mRNA expression of pITGB8-205, (ii) nuclear and chromosomal instabilities in early- and late-passage OVCAR3-OE cells; misaligned metaphases (1), lagging chromosomes (2), anaphase bridges (3), binucleated (4), trinucleated (5), tetranucleated (6), hexanucleated (7), micronuclei (8), lobed nuclei (9), blebbed nuclei (10), notched nuclei (11), ring nuclei (12), cytokinesis defects (13); (d) Bar graph representation γ-H2AX foci in OVCAR3-OE early and late passages (p < 15 and p > 30, respectively) vs. controls; e. Ratios of surviving cells (irradiated: unirradiated OVCAR3 controls) (irradiated: unirradiated OVCAR3-OE-EP) (irradiated: unirradiated OVCAR3-OE-LP)
Further observations and quantification of the same features revealed a sharp reduction in pITGB-205 expression, aberrant mitoses and nuclear and cell cycle anomalies in later passages (∼ 30) of OVCAR3-OE cells maintained at steady-state conditions in culture, which were associated with reduced γ-H2AX foci per nucleus, confirming the restoration of homeostasis (Fig. 3c and d). Despite a restored near-normal ploidy level, these cells, however, continued to maintain a survival advantage over controls. Taken together, these data suggest that although replicative stress and exaggerated genomic content provide survival advantages, they may be utilized when homeostasis necessitates a balance between energy utilization and cell cycling in response to competitive forces within populations that deplete aneuploid cells lacking a growth advantage. This ultimately reflects the long-term effects of pITGB8-205, which can be activated following irradiation and possibly other types of stress.
pITGB8-205 perturbs several molecular pathways, including metabolism-, cell cycle- and self-renewal-associated modules, and its ectopic expression may have a tumor suppressive effect on WT-p53 cells
We then performed differential transcriptomics and proteomics of the OVCAR3OE, A4-OE and PEO14-KD cell derivatives and their respective controls to elucidate the molecular pathways underlying the effects mediated by pITGB8-205. The top 25 enriched gene sets (derived from exclusive and upregulated transcripts) in OVCAR3-OE reflected an involvement in chromosome maintenance, cytoskeleton-actin filament organization, etc., whereas a similar analysis revealed enrichment of gene sets associated with metabolism-associated changes (glycosylation, lipid and organic acid metabolism, small molecule catabolism, etc.) along with epithelial cell differentiation and gland formation that may emerge from repression of self-renewal pathways in PEO14-KD (Fig. 4a-i and a-ii; Supplementary Fig. 7a, 7b). Compared with OVCAR3-OE, A4-OE was enriched in several immune-related pathways (tumor necrosis factor superfamily cytokine production, adaptive immune response, immunoglobulin superfamily domains, cytokine activity, etc.), indicating distinctive ways in which pITGB8-205 affects a cell depending on the p53 mutational status (Supplementary Fig. 5c, 5d). Differential proteomics strikingly revealed greater protein enrichment in both OVCAR3-OE and PEO14-KD cell derivatives than in their controls (Supplementary Fig. 8a-i, 8a-ii, 8b-i, 8b-ii). Compared with the control, A4-OE failed to show such protein enrichment (Supplementary Fig. 8c-i, 8c-ii). Corroborating their transcriptional profiles, proteins related to actin filament organization, cell cycle regulation, oxidative phosphorylation, and organelle assembly were enriched in OVCAR3-OE cells, whereas the knockdown of pITGB8-205 in PEO14-KD cells led to the derepression of proteins associated with focal adhesion and actin binding, anatomical structure and epithelial tube morphogenesis, the regulation of microtubules, the G1-S transition and the cell cycle (Supplementary Fig. 8a-iii, 8b-iii). On the other hand, A4-OE-enriched proteins largely included components of lipid metabolism and mitochondrial ribosomes (Supplementary Fig. 8c-iii). STRING-based network analyses of the most significant protein hubs (exclusive proteins at the core with a minimum of 10 interactors) in each of these derivatives (A4 and OVCAR3 OE/KD) not only supported the correlations between transcriptomics and proteomics but also revealed different modules of the DNA damage response and repair in the two derivatives, OVCAR3-OE and PEO14-KD (Fig. 4b). String analysis of A4-OE transcripts (yellow cluster, supported immune-modulatory functions with collagen synthesis enrichment; the former could indicate the tumor suppressive effect of pITGB-205 in response to WT p53) was performed (Supplementary Fig. 6d).
Fig. 4.

Molecular pathway perturbations driven by pITGB8-205; (a) Bar graph representing GSEA-enriched gene sets in the transcriptomics data of a-i. OVCAR3-OE and a-ii. PEO14-KD cells; (b) STRING protein interaction network of exclusive proteins in b-i. OVCAR3-OE, b-ii. PEO14KD and b-iii A4-OE; (c) Dot plot of enriched proteins for the cellular response to DNA damage stimulus and DNA damage repair in OVCAR3-OE and PEO14-KD; (d) Venn diagram representing the overlap between enriched transcripts and proteins (n = 14), in OVCAR3OE over control, from transcriptomics and proteomics data; (e) The table represents the overlap between OVCAR3-OE enriched proteins with the gene targets of different transcription factors (TFs); (f) Venn diagram representing the overlapping proteins enriched in OE and KD derivatives and their respective controls. (g-i) A4 control vs. A4-OE tumor images harvested on the 36th day after cell injection; (g-ii) Line plot indicating the A4-OE and A4 control tumor volumes at different time intervals (1st week after tumor palpation); (g-ii) Line plot representing the body weights of the mice at the indicated time intervals; (g-iv) Violin plot representing the weight of the harvested tumors
Enriched proteins in OVCAR3-OE cells contribute to genomic/chromosomal perturbation, chromatin condensation and spindle disassembly (ATR, PAXIP1, NPLOC4, USB7, USB10 and UBE2L6), whereas proteins upregulated in PEO14-KD cells are associated with DNA repair and maintenance of homeostasis (APTX, ERCC2, GINS4, POLD1, and XRCC4) (Fig. 4c) [39, 40]. Moreover, compared with the control, OVCAR-OE resulted in the downregulation of several pathways, which are pivotal for cytokinesis regulation, further validating the aneuploidy observed in OVCAR3-OE derivatives. Pathways such as Rho GTPase, AURKA activation, the PLK1 pathway, and the AURORA B pathway were among the top downregulated pathways in OVCAR3-OE cells (Supplementary Fig. 8d). Overlapping (common) enriched transcripts and proteins of OVCAR3-OE supported the biochemical association of glycolysis and glucose metabolism, yet others were revealed to be targets of E2F4 (potential regulators of cell cycle arrest) or Nanog (regulating self-renewal; Fig. 6d and e). To further explore the suggested functionalities with cellular networks positively regulated by endogenous pITGB8-205 expression, we identified a subset of proteins common to OVCAR3-OE and PEO14 cells (high endogenous pITGB8-205 levels) that mediate increased mRNA splicing, RNA metabolism and SUMOylation for transcript and protein processing, along with IFN-g signaling (Fig. 4f; Supplementary Table 1). A similar comparison to probe derepression from depleted/low endogenous pITGB8-205 expression (OVCAR3 and PEO14-KD cells) indicated the activation of NFE2L2 targets (TXNRD1, GSTA1, GCLC, G6PD, PGD, which crucially prevent cellular damage from reactive oxygen species and products of peroxidation by providing substrates such as glutathione and NADPH), Rho GTPase signaling (involved in cytoskeleton regulation) and protein ubiquitination and posttranslational modifications (Fig. 4f, Supplemental Table 1). The latter is most likely to protect the survival of PEO14 cells after pITGB8-205 knockdown and contribute to the poor initial survival of OVCAR3 cells after transfection with pITGB8-205.
To understand the effects of TP53 mutations in vivo, we generated xenograft models using A4-OE and A4 cells. A drastic difference in tumor formation was observed between the two groups, with control tumors growing rapidly in association with weight loss in the mice, whereas A4-OE-injected tumors were extremely small to negligible in size and had no effect on the weight of the animals (Fig. 4g-i and g-ii). Compared with those of the control A4 tumors, the volume and weight of the A4-OE tumors 36 days after injection were markedly lower (Fig. 4g-iii, 4 g-iv). These findings strongly suggest that in the presence of active p53, pITGB8-205 does not have tumor-promoting effects but rather achieves tumor-suppressive effects.
Discussion
HGSC is a prevalent and deadly subtype of epithelial ovarian cancer, primarily due to mutations in the p53 tumor suppressor gene, which maintains stable genomic and nuclear contents via its regulatory effects on cell cycle checkpoints; in corollary, inactive p53 can contribute to genomic instability, a hallmark of cancer [22, 41]. Ectopic overexpression of pITGB8-205 in p53 mutant cells triggers genetic instability reminiscent of oncogene-induced replicative stress, which results in extensive chromosomal instability (CIN) in association with the downstream outcomes of micronuclei generation, aneuploidy, and larger nuclei with distinctive architectural abnormalities [13, 42–44]. The enhanced glycolysis in OVCAR3-OE cells can deliver enough nucleotides and intermediates for the synthesis of other biomolecules, which are necessary for superaneuploid daughter cells. The generation of these tetraploid cells or multinucleated cells in the presence of mutant p53 is a result of cytokinesis failure, as evidenced by the deregulation of the Rho GTPase and PLK1 pathways in OVCAR3-OE cells due to the presence of lagging chromosomes, which results in cleavage furrow regression [45]. The presence of micronuclei that contain one or very few chromosomes is considered a WGD, and the increased complexity of chromosomes within a cell changes dynamically with its niche prior to chromothripsis [46, 47], which in turn mediates various types of gross structural–numerical chromosomal changes [13–15]. A majority of the duplicated chromosomes following overexpression of pITGB8-205 in OVCAR3 cells were difficult to classify manually because of atypical features and hence were considered marker chromosomes. However, a machine learning-based comparison with reference ideograms mapped their identities revealed the possibility of pITGB8-205 triggering WGD in p53-mutant OVCAR3 cells. This finding is further supported by the fact that WGD is one of the main evolutionary trajectories, other than homologous recombination repair (HRD), in HGSCs, 95% of which are p53 mutants, affecting patient survival and altering genomic content [46]. Following the ectopic expression of pITGB8, similar to the p53 mutant derivative, p53 WT A4 cells exhibited delayed cell cycling. However, ploidy, granularity, cell dimensions and metabolism were completely unaltered in A4-OE cells compared with control cells. This observation, together with the remarkable in vivo tumor suppressive effect of pITGB8-205 overexpression in A4 cells, reinforces the notion that pITGB8-205 overexpression requires a p53 mutant background to contribute to genomic instability and aggravate HGSC.
Replicative stress involving abrogation of cell cycle regulation, mitotic slippage or bypass and cytokinesis defects as a driver of WGD is a consequence of p53 mutations and/or CCNE1, both of which are frequently associated with HGSC [45]. These two features may be involved in the development of a permissive intracellular milieu that is a likely prerequisite for the effects mediated by pITGB8-205. USP10-, UBE2L6- and USP7-mediated ubiquitination, ISGylation and degradation (respectively) of p53 in cells overexpressing pITGB8-205 are also suggested to provide additional mechanisms of inactivation. Complementing this is the possible repression of the NFE2L2 network and its target genes that protect against the cytotoxic effects of reactive oxygen species [46, 47]. The molecular heterogeneity of HGSC, however, suggests that several other features are involved in the generation of a permissive niche in tumors toward the mediation of genomic instability by pITGB8-205.
An increased number of chromosomes arising from abnormal replication in normal cells often degenerate and are lost because of their inconsequence [44]. However, the expansion of genomic content in tumor cells provides a survival advantage in unfavorable micro/macro environments during metastasis or following therapy. However, it is difficult to envision a scenario in which cells with decreased turnover kinetics, increased glycolysis and energy demands that drain available cellular resources are retained in a tumor population [48, 49]. This conundrum was resolved through our observation that p53-mutant tumor cells overexpressing pITGB8-205 soon adapt to the microenvironment and progress to a stress-free steady state in which fewer abnormal mitoses, perturbed nuclear phenotypes and overall reduced genetic instability maximize the efficiency of energy utilization, as evidenced by the late passages of the OVCAR3-OE. In addition to generating intratumor heterogeneity, self-renewal and reversible quiescence by aneuploid clones within a population are essential drivers of tumor evolution since they can promote long-term survival and potentially lead to recurrent disease [30]. This observation is highly relevant for further study, as most HGSCs are p53 mutants, and under this mutational background, aneuploid clones are easily generated and evolve. Taken together, the results of our study reveal the pleiotropic behavior of pITGB8-205 in that it either cooperates with mutant p53 in a manner similar to oncogene-induced replicative stress to increase genetic instability or complements WT p53 activities and mediates tumor suppressor-like effects. Further mechanistic elucidation of these pathways in ovarian cancer is essential for a deeper understanding of their involvement.
Conclusion
Our results suggest that pITGB8-205 dynamic overexpression in pITGB8-205 mutant HGSC cells aggravates CIN in a way similar to oncogene-induced replicative stress with significant genomic alterations (e.g., micronuclei generation and aneuploidy). This instability is supported by altered metabolism, which provides the necessary biomolecules for abnormal cell division. In contrast, in p53 wild-type cells, pITGB8-205 has a minimal effect on ploidy and metabolism, suggesting that its oncogenic role is p53 mutation dependent. We also show that p53-mutant cells are adaptable and evolve to reach a new normal status of decreased instability while maintaining high energy usage to support continued tumor progression.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Lymphoblastoid cell lines and primary skin fibroblasts were established under the human genetic mutation repository program that was approved by the NCCS Institutional Ethics Committee. OVCAR3 cells were procured from the NCCS Repository; other cell lines used in the present study were kindly gifted by Prof. Judith Clements (Translational Research Institute, Australia; OVCA420, PEO14, OVCA432 CAOV3), Dr. S. Mok (MD Anderson Cancer Center, TX; OV90) Prof. V. Magdalen (Klinische Forschergruppe der Frauenklinik der TU München; OVMZ6), Dr. Pritha Ray (Advanced Centre for Treatment Research & Education in Cancer, Tata Memorial Centre; IOSE364), Dr. Ramray Bhat (Indian Institute of Science; FTE). We also extend our gratitude to Dr. Shiras (late), Dr. Sahu, and Dr. Majumdar (NCCS) for providing NMDF, mouse iPSC, and HEK293T, CV1 and S2 cells for RNA isolation. We thank Dr Joseph, NCCS, Pune for providing the cytochalasin D drug and Dr. V. Tripathi, NCCS, Pune for anti-γ-H2AX (phospho), CENP-E and anti-tubulin antibodies. The data used in this publication were generated by the Clinical Proteomic Tumor Analysis Consortium [NCI/NIH] and are acknowledged. We extend our gratitude to Prof. David Fenyö and Dr. McKerrow, NYU Langone Medical Center, USA, for sharing workflows of the FenyoLab tandem pipeline on the SBGenomics Cloud. The collaborative grant with Seven Bridges is deeply appreciated. The Seven Bridges Cancer Research Data Commons Cloud Resource has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, Contract No. HHSN261201400008C and ID/IQ Agreement No. 17 × 146 under Contract No. HHSN261201500003I and 75N91019D00024. The authors acknowledge the contribution of Shreya Junnarkar to the generation of the pITGB8-205 full-length sequence. We are grateful to Dr. Moghe, Department of Genetics, Deenanath Mangeshkar Hospital, Pune, for the karyotyping of OVCAR3 and OVCAR3-OE. We also thank the flow cytometry and bioimaging facilities at the NCCS for their assistance in data acquisition.
Abbreviations
- HGSC
High-grade serous ovarian carcinoma
- WT
Wild-type
- RACE
Rapid amplification of cDNA ends
- CIN
Chromosomal instability
- WGD
Whole-genome duplication
- FL
Full-length
- ROS
Reactive oxygen species
- CBMN
Cytokinesis-block micronucleus
- LFQ
Label-free quantification
- GSEA
Gene set enrichment analysis
- OE
Overexpression
- KD
Knockdown
- Mar
Marker chromosome
Author contributions
SAB: Conceptualized and planned the study design, developed the methodology, analyzed and interpreted the data, supervised the project, project administration, reviewed and finalized the original manuscript draft; AN: Experimentation, data curation, data analysis, data interpretation, editing, writing – original draft; ASM: Experimentation, data curation, data analysis, data interpretation, editing, writing – original draft; MT: Experimentation, data curation, data analysis, data interpretation; AM: Experimentation; SBV: Experimentation.
Funding
and disclosure of conflicts of interest.
None of the authors have any competing financial interests to declare in this work. This work was supported by funding to SAB from the Science and Engineering Research Board (SERB), the Government of India, New Delhi (extramural grants CRG/2019/001157) and intramural grants from the NCCS, Pune, India. Research fellowships were as follows: AN from the NCCS, Pune, India; ASM from the Council of Scientific and Industrial Research, New Delhi, India; and SBV from SERB extramural grants (CRG/2019/001157).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Conflicting of interests
The authors state that none of their known conflicting financial interests or personal connections are likely to influence the work that was published in this paper.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Aravindan Narayanan and Ankita S. More contributed equally to this work.
References
- 1.Singh P, Fragoza R, Blengini CS, et al. Human MLH1/3 variants causing aneuploidy, pregnancy loss, and premature reproductive aging. Nat Commun. 2021. 10.1038/s41467-021-25028-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol. 2019;20:406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Da Costa PJ, Menezes J, Romão L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int J Biochem Cell Biol. 2017;91:168–75. [DOI] [PubMed] [Google Scholar]
- 4.Marzese DM, Huynh JL, Kawas NP, Hoon DSB. Multiplatform genome-wide analysis of melanoma progression to brain metastasis. Genomics Data. 2014;2:150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Marabti E, Younis I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front Mol Biosci. 2018. 10.3389/fmolb.2018.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finci LI L, Zhang X, Huang X, Zhou Q, Tsai J, Teng T. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action, Genes and Development (2018). [DOI] [PMC free article] [PubMed]
- 7.Climente-Gonzlez H, Porta-Pardo E, Godzik A, Eyras E. The functional impact of alternative splicing in Cancer. Cell Rep. 2017;20:2215–26. [DOI] [PubMed] [Google Scholar]
- 8.Kim Y-J, Kim H-S. Alternative splicing and its impact as a Cancer Diagnostic marker. Genomics Inf. 2012;10:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon M, Leibowitz ML, Lee J-H. Small but mighty: the causes and consequences of micronucleus rupture. Exp Mol Med. 2020;52:1777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y-F, Hu Q, Mazzagatti A, et al. Mitotic clustering of pulverized chromosomes from micronuclei. Nature. 2023;618:1041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shorokhova M, Nikolsky N, Grinchuk T. Chromothripsis—Explosion in Genetic Science. Cells. 2021;10:1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sridhar A, More AS, Jadhav AR, Patil K, Mavlankar A, Dixit VM, Bapat SA. Pattern recognition in the landscape of seemingly random chimeric transcripts. Comput Struct Biotechnol J. 2023;21:5153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De A, Morales JE, Chen Z, Sebastian S, McCarty JH. The β8 integrin cytoplasmic domain activates extracellular matrix adhesion to promote brain neurovascular development. Development. 2022. 10.1242/dev.200472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar V, Maurya VK, Joshi A, Meeran SM, Jha RK. Integrin beta 8 (ITGB8) regulates embryo implantation potentially by controlling the activity of TGF-B1 in mice, Biology of reproduction (2015). [DOI] [PubMed]
- 15.Zhu T, Chen R, Wang J, Yue H, Lu X, Li J. The prognostic value of ITGA and ITGB superfamily members in patients with high grade serous ovarian cancer. Cancer Cell Int. 2020. 10.1186/s12935-020-01344-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He J, Liu Y, Zhang L, Zhang H. Integrin subunit beta 8 (ITGB8) upregulation is an independent predictor of unfavorable survival of high-Grade Serous Ovarian Carcinoma patients. Med Sci Monit. 2018;24:8933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial–mesenchymal transition. Cell Adhes Migr. 2015;9:317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Downs-Canner S, Berkey S, Delgoffe GM, Edwards RP, Curiel T, Odunsi K, Bartlett DL, Obermajer N. Suppressive IL-17A + Foxp3 + and ex-Th17 IL-17AnegFoxp3 + Treg cells are a source of tumor-associated Treg cells. Nat Commun. 2017. 10.1038/ncomms14649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malric L, Monferran L, Delmas S C, et al. Inhibiting integrin Œ≤8 to Differentiate and Radiosensitize Glioblastoma-initiating cells. Mol Cancer Res. 2019;17:384–97. [DOI] [PubMed] [Google Scholar]
- 20.SUN W, MA Y, CHEN P, WANG D. MicroRNA-10a silencing reverses cisplatin resistance in the A549/cisplatin human lung cancer cell line via the transforming growth factor-Œ≤/Smad2/STAT3/STAT5 pathway. Mol Med Rep. 2015;11:3854–9. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Zhu T, Weng Y, Cheng F, Sun Q, Yang K, Su Z, Ma H. Exosomal circDNER enhances paclitaxel resistance and tumorigenicity of lung cancer by targeting miR‚Äê139‚Äê5p/ITGB8. Thorac Cancer. 2022;13:1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Z, Wu R. Alteration in Metastasis potential and gene expression in Human Lung Cancer Cell lines by ITGB8 silencing. Anat Rec. 2012;295:1446–54. [DOI] [PubMed] [Google Scholar]
- 23.Marques JF, Kops GJPL. Permission to pass: on the role of p53 as a gatekeeper for aneuploidy. Chromosome Res. 2023. 10.1007/s10577-023-09741-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng C, Li X, Ren Y, Yin Z, Zhou B. Coexisting EGFR and TP53 mutations in lung adenocarcinoma patients are Associated with COMP and ITGB8 upregulation and poor prognosis. Front Mol Biosci. 2020. 10.3389/fmolb.2020.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurrey NK, Bapat KA SA. Snail and slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol. 2005;97:155–65. [DOI] [PubMed] [Google Scholar]
- 26.Sharma N, Mali AM, Bapat SA. Spectrum of CREBBP mutations in Indian patients with Rubinstein-Taybi syndrome. J Biosci. 2010;35:187–202. [DOI] [PubMed] [Google Scholar]
- 27.Varankar SS, More M, Abraham A, Pansare K, Kumar B, Narayanan NJ, Jolly MK, Mali AM, Bapat SA. Functional balance between Tcf21-Slug defines cellular plasticity and migratory modalities in high grade serous ovarian cancer cell lines. Carcinogenesis. 2019;41:515–26. [DOI] [PubMed] [Google Scholar]
- 28.Shivalingappa PKM, Singh DK, Sharma V, Arora V, Shiras A, Bapat SA. RBM47 is a critical Regulator of Mouse Embryonic Stem Cell differentiation. Stem Cell Reviews Rep. 2022;19:475–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusumbe AP, Bapat SA. Cancer Stem cells and aneuploid populations within developing tumors are the major determinants of Tumor Dormancy. Cancer Res. 2009;69:9245–53. [DOI] [PubMed] [Google Scholar]
- 30.Guzmán C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ Plugin to automatically quantify colony formation in Clonogenic assays. PLoS ONE. 2014;9:e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalra R, Bapat SA. Enhanced levels of double-strand DNA break repair proteins protect ovarian cancer cells against genotoxic stress-induced apoptosis. J Ovarian Res. 2013;6:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scalzitti N, Kress A, Orhand R, Weber T, Moulinier L, Jeannin-Girardon A, Collet P, Poch O, Thompson JD. Spliceator: multispecies splice site prediction using convolutional neural networks. BMC Bioinformatics. 2021. 10.1186/s12859-021-04471-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen S. Promoter2.0: for the recognition of PolII promoter sequences. Bioinformatics. 1999;15:356–61. [DOI] [PubMed] [Google Scholar]
- 34.Cartegni L. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–508. [DOI] [PubMed] [Google Scholar]
- 36.Zhang M, Zhuang G, Sun X, Shen Y, Wang W, Li Q, Di W. TP53 mutation-mediated genomic instability induces the evolution of chemoresistance and recurrence in epithelial ovarian cancer. Diagn Pathol. 2017. 10.1186/s13000-017-0605-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lens SMA, Medema RH. Cytokinesis defects and cancer. Nat Rev Cancer. 2018;19:32–45. [DOI] [PubMed] [Google Scholar]
- 38.Du M, Zhang S, Liu X, Xu C, Zhang X. Ploidy Status of Ovarian Cancer Cell lines and their association with gene expression profiles. Biomolecules. 2023;13:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsegay PS, Lai Y, Liu Y. Replication stress and consequential instability of the genome and Epigenome. Molecules. 2019;24:3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2013;16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saleh A, Perets R. Mutated p53 in HGSC—From a common mutation to a target for Therapy. Cancers. 2021;13:3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276–89. [DOI] [PubMed] [Google Scholar]
- 43.Newcomb R, Dean E, McKinney BJ, Alvarez JV. Context-dependent effects of whole-genome duplication during mammary tumor recurrence. Sci Rep. 2021. 10.1038/s41598-021-94332-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinton RJ, DiDomizio A, Vittoria MA, et al. Whole-genome doubling confers unique genetic vulnerabilities on tumor cells. Nature. 2021;590:492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng J, Hills SA, Ozono E, Diffley JFX. Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell. 2023;186:528–e54214. [DOI] [PubMed] [Google Scholar]
- 46.Ewing A, Meynert A, Silk R, Aitken S, Bendixsen DP, Churchman M, Brown SL et al. Divergent trajectories to structural diversity impact patient survival in high grade serous ovarian cancer. bioRxiv (2024): 2024–01.
- 47.Taguchi K, Yamamoto M. The KEAP1–NRF2 system in Cancer. Front Oncol. 2017. 10.3389/fonc.2017.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baird L, Yamamoto M. The Molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol. 2020. 10.1128/mcb.00099-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pober BR. Overview of epidemiology, genetics, birth defects, and chromosome abnormalities associated with CDH. Am J Med Genet Part C: Seminars Med Genet. 2007;145 C:158–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shiratori R, Furuichi K, Yamaguchi M, Miyazaki N, Aoki H, Chibana H, Ito K, Aoki S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci Rep. 2019. 10.1038/s41598-019-55296- [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.