

Abstract
Background
Vasculogenic therapies explored for the treatment of peripheral artery disease (PAD) have encountered minimal success in clinical trials. Addressing this, B55α, an isoform of protein phosphatase 2A (PP2A), emerges as pivotal in vessel remodeling through activation of hypoxia-inducible factor 1α (HIF-1α). This study delves into the pharmacological profile of VCE-004.8 (Etrinabdione) and evaluates its efficacy in a preclinical model of critical limb ischemia, with a focus on its potential as a PP2A/B55α activator to induce angiogenesis and arteriogenesis.
Methods
Vascular endothelial cells were used for in vitro experiments. Aorta ring assay was performed to explore sprouting activity. Matrigel plug-in assay was used to assess the angiogenic potential. Critical limb ischemia (CLI) in mice was induced by double ligation in the femoral arteria. Endothelial vascular and fibrotic biomarkers were studied by immunohistochemistry and qPCR. Arteriogenesis was investigated by microvascular casting and micro-CT. Proteomic analysis in vascular tissues was analyzed by LC–MS/MS. Ex-vivo expression of B55α and biomarkers were investigated in artery samples from PAD patients.
Results
VCE-004.8 exhibited the ability to induce B55α expression and activate the intersecting pathways B55α/AMPK/Sirtuin 1/eNOS and B55α/PHD2/HIF-1α. VCE-004.8 prevented OxLDL and H2O2-induced cytotoxicity, senescence, and inflammation in endothelial cells. Oral VCE-004.8 increased aorta sprouting in vitro and angiogenesis in vivo. In CLI mice VCE-004.8 improved collateral vessel formation and induced endothelial cells proliferation, angiogenic gene expression and prevented fibrosis. The expression of B55α, Caveolin 1 and Sirtuin-1 is reduced in arteries from CLI mice and PAD patient, and the expression of these markers was restored in mice treated with VCE-004.8.
Conclusions
The findings presented in this study indicate that Etrinabdione holds promise in mitigating endothelial cell damage and senescence, while concurrently fostering arteriogenesis and angiogenesis. These observations position Etrinabdione as a compelling candidate for the treatment of PAD, and potentially other cardiovascular disorders.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-024-05748-w.
Keywords: Etrinabdione, VCE-004.8, PPA2/B55α, HIF-1α, Sirtuin 1, eNOS, Angiogenesis
Introduction
Peripheral arterial disease (PAD) is one of the major leading cause of atherosclerotic cardiovascular morbidity [1]. Experimental approaches to the treatment of PAD have included the use of angiogenic factors as a recombinant protein or as gene therapy as well as stem cell therapy [2] but, neither of these strategies achieved significant benefits in clinical trials. Hence, there is an urgent demand for novel pharmacological targets and therapies capable of preventing endothelial damage and promoting angiogenesis and arteriogenesis to enhance blood flow in extremities.
The protein phosphatase 2A (PP2A) belong to a family of serine-threonine phosphatases, which are formed by a catalytic C subunit, a scaffold A subunit, and a regulatory B subunit, which confers specificity of PP2A for selective proteins substrates [3]. B55α (PPP2R2A) is one of the regulatory subunits that has been shown to directly dephosphorylates prolyl hydroxylase 2 (PHD2) on Ser125, resulting in a further reduction of its activity that ultimately stabilized and accumulated the hypoxia inducing factor 1α (HIF-1α) [4]. HIF-1, a heterodimeric basic-helix-loop-helix-paste protein that responds to oxygen tension, is activated by severe and mild hypoxia. Under hypoxic conditions, HIF-1α accumulates in the nucleus and dimerizes with HIF-1β to activate a plethora of genes including angiogenic factors such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and angiopoietin, as well as an important number of genes involved in vascular and tissue remodeling [5, 6]. HIF-1α is also activated by therapeutic preconditioning hypoxia and by hypoxia mimetic small compounds [7, 8]. More recently, it has been shown that B55α stabilizes endothelial cells (ECs) in response to cell stress conditions, thereby protecting ECs from apoptosis and promoting angiogenesis. Thus, PP2A/B55α activators represent a new class of hypoxia mimetic compounds with potential in vessel remodeling, angiogenesis and endothelial cell protection [9].
The histone deacetylase Sirtuin 1 (Sirt1) belong to the family of Sirtuins, which are nicotinamide adenine dinucleotide (NAD+)-dependent enzymes with multiple metabolic functions. The expression of Sirt1 is reduced with aging both in animal models and in humans, and a reduced expression is thought to be involved in age-related cardiovascular diseases. Indeed, Sirt1, which is activated by AMPK, is a critical modulator of the vascular function [10] and endothelial senescence is regarded as a prominent precursor of cardiovascular diseases including PAD and critical limb ischemia (CLI) [11]. The key role of Sirt1 in the regulation of vascular homeostasis is mediated through complex cellular mechanisms involved in the promotion of vasodilatory and regenerative functions at ECs, endothelial progenitor cells, and smooth muscle cells (SMC) levels [12].
We have recently identified the cannabidiol aminoquinone VCE-004.8 (also known as Etrinabdione and EHP-101), as a PP2A/B55α activator that induces HIF-1α and HIF-2α stabilization [13]. In addition, VCE-004.8 is a dual activator of peroxisome proliferator-activated receptor-γ (PPARγ) and cannabinoid type 2 receptors (CB2R) [14]. VCE-004.8 showed efficacy in preclinical models of traumatic brain injury [15], multiple sclerosis [13], bleomycin- and angiotensin-induced models of dermal, lung, cardiac, and kidney fibrosis [14, 16], metabolic syndrome [17] and stroke [18].
Etrinabdione (VCE-004.8) has finalized the stage 1 of a clinical Phase IIa trial in Systemic Sclerosis patients (Clinical Trials.gov: NCT04166552), and it had acceptable safety profile and was well-tolerated. Herein we have further investigated additional pharmacological attributes of VCE-004.8 and explored its efficacy in a model of CLI.
Materials and methods
Test compound
VCE-004.8 (also called Etrinabdione and EHP-101) [(1′R,6′R)-3-(benzylamine)-6-hydroxy-3′-methyl-4-pentyl-6′-(prop-1-en-2-yl) [1,1′bi(cyclohexane)]-2′,3,6-triene-2,5-dione)] was produced under GMP conditions and provided by VivaCell Biotechnology España (Spain). For animal use VCE-004.8 was dissolved in sesame oil/Maisine CC® (50%/50%) and applied by oral gavage.
Cell cultures
EA.hy926 endothelial cells (ATCC® CRL-2922™), HEK-293T (ATCC® CRL-3216™) and NIH-3T3 (ATCC® CRL-1658™) were cultured in DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS, 2 mM l-glutamine, and 1% (v/v) penicillin/streptomycin at 37 °C in a humidified 5% CO2 incubator. Human microvascular endothelial cells (HMEC-1) (ATCC® CRL-3243™) were maintained in MCDB131 medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10 ng/mL epidermal growth factor (EGF), 1 μg/mL hydrocortisone, 10 mM glutamine and 10% FBS.
Cell viability
EA.hy926 cells were seeded in 96-well plates (104 cells/well) and incubated with increasing concentrations of VCE-004.8 in the absence or the presence of the inhibitors: YC-1 (HIF-1α inhibitor, Selleckchem, Dallas, TX, USA), Dorsomorphin (DS) (AMPK inhibitor, Selleckchem), and EX527 (Sirt1 inhibitor, Sigma, St Louis, MO, USA) for 30 min. Then, the cells were exposed either to H2O2 (200 µg/mL) or oxLDL (200 µg/mL) cell viability was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assay.
Luciferase assays and siRNA transfections
The NIH-3T3-EPO-luc cell line was previously described [19]. Cells (104/well) were seeded in 96-well plates and incubated with DS and EX527 for 30 min and treated with increasing concentrations of VCE-004.8 for 6 h. Luciferase activity in the cell lysates was quantified using Dual-Luciferase Assay (Promega, Madison, WI, USA). Scramble control oligonucleotide siRNA non-targeting pool and ON-TARGET plus SMARTpool against B55α or Sirt1 were purchased from Dharmacon (Waltham, MA, USA). B55α and Sirt1 silencing was performed using Lipofectamine RNAiMax transfection reagent (Life Technologies, Carlsbad, USA) following manufacturer’s instructions.
Immunocytochemistry analysis
EA.hy296 cells (2.5 × 103/well) were seeded onto glass coverslips in 24 well plates, pre-treated with increasing concentrations of VCE-004.8 for 1 h and then incubated either with interleukin 1 beta (IL1β) (R&D Systems, Minneapolis, MN, USA) plus tumor necrosis factor alpha (TNFα) (R&D Systems) for VCAM-1 expression, or with interleukin 6 (IL-6) (R&D Systems) plus TNFα (R&D Systems) for ZO-1, CLD1 and B55α expression. After fixation, cells were washed three times with PBS, permeabilized with 0.5% Triton X-100 at room temperature (RT) for 5 min and blocked in PBS with 3% BSA (Sigma) for 1 h. Cells were incubated overnight at 4 °C with the primary antibodies; rabbit monoclonal anti-VCAM1 (1:100, #ab134047, Abcam), rabbit polyclonal anti-ZO-1 (1:100, #40-2300, Invitrogen; Carlsbad, CA, USA), rabbit polyclonal anti-Claudin 1 (CLD1) (1:100, #ab15098, Abcam), rabbit polyclonal anti-B55α (1:100, #4953T, Cell Signaling, Danvers, MA, USA). After incubation with the appropriate secondary antibody for 1 h at RT in the dark, coverslips were mounted with Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) for nuclear staining. Images were acquired using a spectral confocal laser-scanning microscope LSM710 (Carl Zeiss, Jena, Germany) or a fluorescence microscope Leica Thunder Imager 3D assay (LEICA, Wetzlar, Germany).
Western blots
Western blot was performed as previously described [16]. Briefly, 40 µg of cellular proteins were electrophoresed in 10% SDS/PAGE gels. Immunodetection of specific proteins was carried out by incubation with primary antibody against HIF-1α (1:500 dilution, # ab179483, Abcam), B55α (1:1000 dilution, #5689S, Cell Signaling), VCAM1 (1:1000, #ab134047, Abcam), ZO-1 (1:1000, #40-2300, Invitrogen), CLD1 (1:1000, #ab15098, Abcam, phospho-AMPK (1:1000, #2535S, Cell Signaling), total-AMPK (1:1000, #ab80039, Abcam), Sirtuin 1 (1:1000, #8469, Cell Signaling), Visfatin (1:1000, #ab236874, Abcam), p21 Waf1/Cip1 (p21) (1:1000, #2947, Cell Signaling), HSP 90 (1:1000, #4874, Cell Signaling), eNOS (1:1000, #32027S, Cell Signaling), phospho-eNOS (1:1000, # ab215717, Abcam), α-tubulin (1:5000, #T9026, Sigma) and β-actin (1:10,000 dilution, #ab49900, Abcam), overnight at 4 °C. After washing membranes, secondary antibody was added for 1 h and detected using the immunofluorescence ChemiDoc MP Imaging System (Bio-rad, Hercules, CA, USA). All the experiments were repeated at least three times.
Nitric oxide (NO) detection
NO production was detected using the fluorescent dye DAF-2DA (Sigma). EA.hy296 cells were cultured in 96 well plates until 90% confluence and then serum starved overnight. The cells were pre-incubated with 5 μM DAF-2 DA for 30 min at 37 °C in darkness and rinsed with fresh suspension media to remove excess fluorophore and then treated with either sulforaphane (50 µM), as a positive control, or VCE-004.8 (10 µM). After 6 h, fluorescence signals were measured using an IncuCyte Zoom apparatus (Sartorius, Gotinga, Germany).
Senescence assay
HMEC-1 cells were treated with increasing concentrations of VCE-004.8 for 1 h and exposed to H2O2 for 4 h on days 2 and 5. At day 7 cellular Senescence-Associated β-Galactosidase (SA-β-gal) staining was performed according to manufacturer instructions (Cell Signaling).
SIRT1 activity assay
SIRT1 activity was determined with the SIRT1 Activity Assay Kit (Fluorometric) according to the manufacturer’s instructions (Abcam).
NAD+/NADH assay
EA.hy926 (2 × 106/well) were treated with VCE-004.8 during 4 h and the levels of NAD+ and NADH were measured with a commercially available NAD+/NADH assay kit according to the manufacturer’s instructions (Abcam).
THP-1 cell differentiation and foam cell formation
THP-1 cells were cultured in 24 well plates (1.5 × 106 cells/well) and treated with PMA (160 nM) for 24 h to induce macrophage differentiation. Then, the undifferentiated macrophages were removed, and foam cell formation was induced with oxLDL (80 μg/mL) in the absence or the presence of VCE-004.8 for another 48 h. Nile red staining was used to identify foam cells.
Mouse aortic ring assay
Male C57BL/6 mice were obtained from Charles River (Barcelona, Spain) and housed with free access to standard food and water under controlled conditions (temperature 20 °C (± 2 °C), 40–50% relative humidity and 12 h light/dark cycle). The thoracic aorta was sectioned into 1 mm long aortic rings and cultured in Opti-MEM (Thermo Scientific) containing penicillin (100 U/mL) and streptomycin (100 μg/mL) overnight. Aortic rings were encapsulated in growth factor reduced Matrigel (Corning, New York, NY, USA) in 24-well plates and cultured in Opti-MEM supplemented with 2.5% FBS. The samples were treated with VCE-004.8 (2.5 and 5 µM) in a humidified 37 °C, 5% CO2 incubator for 10 days. The rings were fed with fresh growth medium every 2 days. Human VEGF-A165 (R&D Systems) (30 ng/mL) and dimethyloxalylglycine (DMOG) (0.2 mM) were used as positive controls. Images were acquired and the sprouting area analyzed with ImageJ.
Angiogenesis in vivo
C57BL/6 male mice (n = 3 animals per group) were subcutaneously injected into flanks of mice with 500 μL containing Matrigel Gel (Thermo Fischer Scientific) mixed with heparin (0.1 mg/mL). For positive control recombinant human VEGF-A165 (200 ng/mL) (R&D Systems) and fibroblast growth factor 2 (1 μg/mL) (Inmunotools) were added to the gel mix. Oral VCE-004.8 (10 or 20 mg/kg) was administered daily and the plugs were explanted on day 10 fixed in 10% formaldehyde and paraffin embedded for histological analysis. The mice were used at the age of 8–10 weeks.
Critical limb ischemia (CLI)
Male C57BL/6 mice aged 10–12 weeks were divided into 2 groups (CLI group and sham group). In the CLI group the femoral artery and its side branches were double ligated with 6–0 silk sutures immediately distal to the inguinal ligament and proximal to the popliteal bifurcation. Femoral nerves were carefully preserved. The wound was irrigated with sterile saline and then the overlying skin was closed using 4–0 vicryl sutures. Post-operative pain was reduced using Lignocaine. A similar surgery without ligation of the femoral artery was performed on the sham controls. The CLI group was administered VCE-004.8 (20 mg/kg) by oral gavage every day until the end of the study and the sham control group was treated with vehicle. The whole limb and muscles were collected for studies 10 and 28 days after ligation.
Mice were anesthetized and injected with 1000 UI of heparin (Sigma), then perfused in the left ventricle with a vasodilator solution [100 μM adenosine (Sigma)], 10 μM sodium nitroprusside (Sigma) and 0.05% BSA Dulbecco’s phosphate buffered saline containing Ca2+ and Mg2+ to avoid vasoconstriction. The limb muscles were collected and progressively frozen in isopentane suspended liquid nitrogen to preserve optimal skeletal muscle morphology. Muscle samples were mounted in optimal cutting temperature compound (ProSciTech, Kirwan QLD, Australia) and they were cut at − 21 °C into 5 μm-thick sections of muscle fibers oriented in the transverse direction. All histological assessments were performed on sections that were examined in a blinded fashion.
Vascular casting
Mice were anesthetized and injected 1000 UI of heparin (i.p.). After 10 min, the mice were euthanized, the thorax was opened, and the aorta exposed. A catheter was inserted into the descending aorta and fixed with sutures. Then mice were perfused with PBS (37 °C, 80 mL) containing heparin, nitroglycerin, adenosine and sodium nitroprusside to remove the blood and enhanced the vasodilatation of the vessels. Microfil [MV-112 (white); Flow Tech Inc, South Windsor, CT, USA] was perfused and then filled the entire vascular bed. The Microfil polymerized overnight at 4 °C, and the samples were harvested and clarified in graded glycerol solutions (40%–100% glycerol in water; glycerol increased by 20% at 24-h intervals). The clarified specimens were viewed on a dissecting microscope and Microfil retained in the vasculature was investigated by µ-CT (X-ray tomography).
Image acquisition µ-CT
After Microfilm polymerization, the animals were hold inside a Bruker SkyScan 1172 high resolution microtomography machine (Bruker micro-CT, Kontich, Belgium). The X-ray source was set to a voltage of 50 kV and a current of 498 μA with a 0.5-mm Al filter in the beam path with an angular increment of 0.3°. Data were transferred to a computer with NRecon, CTAn, CT Vol software, (Bruker), ImageJ (http://rsb.info.nih.gov/ij/) and 3DSlicer v.3.4.0 open-source software (https://pubmed.ncbi.nlm.nih.gov/22770690/) was using to analyze vessel numbers, diameter, area, and volume, as well as arterial density.
Laser capture microdissection (LCM) targeted mass spectroscopy
LCM and subsequent molecular analysis were carried out on slides immuno-stained using anti-α-Smooth Muscle Actin (α-SMA) antibody of gastrocnemius muscles sections. α-SMA+ vessels were micro dissected with a ZEISS PALM Microbeam (Carl Zeiss) and collected for mass spectroscopy analysis.
Immunofluorescence
Sections form tissues were prepared as described previously [15, 16] and incubated overnight at 4 °C with the following primary antibodies diluted in PBS with 3% BSA: anti-α-Smooth Muscle Actin (α-SMA) (1:500, #53-9760-80; Invitrogen), anti-CD31, (1:100, #ab28364, Abcam or 1:100, #14-0311-85, Invitrogen), anti-Ki67 (1:100, #ab15580, Abcam), anti-PP2A/B55α (1:100, #4953T, Cell Signaling), anti-CD34 (1:100, #553731 BD Biosciences, San Jose, CA, USA) anti-Tenascin (TNC) (1:100 dilution, #MAB2138, R&D system), anti-Sirtuin-1 (Sirt1) (1:50, #8469, Cell Signaling), and anti-Caveolin 1 (CAV1) (1:1000, #3267, Cell Signaling), phopho-eNOS (1:100, #ab215717, Abcam). The next day sections were washed and incubated in darkness at RT for 1 h with the appropriated secondary antibody. The slides were then mounted using Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). All images were acquired using a spectral confocal laser-scanning microscope LSM710 (Carl Zeiss) with a 20×/0.8, 25, 40, or 63×/0.8 Plan-Apochromat oil immersion lens and quantified in randomly chosen fields using ImageJ software (http://rsbweb.nih.gov/ij/).
Quantitative reverse transcriptase PCR analysis
Total RNA was isolated from gastrocnemius muscle using QIAzol lysis reagent and the RNeasy Lipid mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For quantitative reverse transcriptase-PCR assays, total RNA (1 μg) was retrotranscribed using the iScript cDNA Synthesis Kit (Bio-Rad) and the cDNA was analyzed by real-time PCR using the iQTM SYBR Green Supermix (Bio-Rad) and a CFX96 Real-time PCR Detection System (Bio-Rad). Gene expression of several markers indicated in the table: VegfA, Epo, Hgf, Hif1α, B55α, Sirt1 and Cav1 were quantified using the 2−ΔΔCt method and the percentage of relative expression against Sham group were represented. Glyceraldehyde-3-Phosphate Dehydrogenase (Gapdh) gene were used to standardize mRNA expression in each sample. The primers used in this study are listed in Additional file 1: Table S1.
Proteomic sample preparation
Samples were digested with advanced iST (in-Stage-Tip) double digestion using Trypsin and LysC as enzymes. Then, the samples were suspended by the IMSMI staff in a phase A solution and measured using microfluorimetry (Qubit™ Protein Assay). Then, they were analyzed by LC–MS/MS using DDA-PASEF mode on the EvosepOne-TIMSTOF-Flex Clinical Proteomics Platform IMSMI. During acquisition, the samples were identified in real time by PaSER software (https://cutt.ly/L0hMYde) to ensure the correct data acquisition (quality control). All samples were loaded in triplicate. Later, the samples were analyzed using LC run (60SPD) and standard DDA-PASEF method. Fragpipe software and the MSstats shiny application were utilized. In total, 36 runs were performed, including 12 HeLaQC analysis (external QC) to system conditioning and system monitoring, 4 pool QCs, 20 DDA-PASEF analyses containing the triplicates from each sample.
LC–MS analysis
Clinical Proteomics Platform of IMSMI was used. It consists of an EvosepOne nanoLC (Evosep, Odense, Denmark, 2021) coupled to a TIMSTOF-Flex (Bruker Daltonics, Bremen, Germany, 2022). For the DDA-PASEF method, each top N acquisition cycle consisted in ten PASEF MS/MS. The collision energy was linearly decreased from 59 eV at 1/K0 = 1.60 Vs cm−2 to 20 eV at 1/K0 = 0.60 Vs cm−2. The accumulation and ramp times were set to 100 ms. Singly charged precursors were excluded from fragmentation using a polygon filter in the (m/z, 1/K0) plane. Additionally, all precursors that reached the target value of 20,000 were excluded for 0.4 min. Precursors were isolated using a Q window of 2 m/z for m/z < 700 and 3 m/z for m/z > 800.
Proteomics data analysis
To generate Fig. 11B we employed the log transformed intensities for proteins annotated with the Gene Ontology term ‘Angiogenesis’ (GO: 0001525). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050360.
Fig. 11.

Oral VCE-004.8 upregulates CAV1 expression in vivo. A Overview of laser capture microdissection. Representative images vessel marked with αSMA in gastrocnemius before (left) and after (right) laser dissection indicated by red asterisk. Scale bar equivalent to 50 μm. B Heatmap showing the expression levels for the resulting list of 11 proteins. Color indicates the mean of scaled regularized log transformed expression values. Red square indicates CAV1. C Representatives images of double immunostaining of CAV1(red) and CD31 (green) in gastrocnemius 10 days after ischemia (left) and the CAV1 staining quantification (right). Scale bar equivalent to 50 μm. The significance was determined non-parametric followed by a Kruskal–Wallis test. In both quantification, values are mean ± SEM, (n = 3). P values indicated in panels, significant as *p < 0.05. D Expression of Cav1 mRNA. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups test (n = 3 animals per group). The results are shown as mean ± SEM. Values indicated in panels, significant as *p < 0.05; **p < 0.01. E Representative immunofluorescence staining of CAV1 in cross-section of healthy and disease human arteries. Scale bar equivalent to 50 μm
Data analysis
Acquired DDA-PASEF runs of all samples present in the study were used to perform the entire analysis including identification and quantification of each replicate of each sample. To achieve this, we used Fragpipe v19.1 (https://fragpipe.nesvilab.org) with default parameters was used. MSStats shiny package software was used to perform the analysis between user’s defined groups. In summary, the entire sample dataset (with exception of samples QC replicates) with all conditions/groups (4 different groups) were used for the comparisons. A log2 data transformation was done and the normalization of transformed data was done by medians equalization. All features were used during the analysis considering a model-based imputation (internally calculated by MSstats) and all NaN values were censored. T-test analyses were performed and a table with potential candidate results was generated. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050360.
Statistical analysis
Data were expressed as the mean ± SEM or SD. The data were evaluated by normality test performance by Kolmogorov–Smirnov, Shapiro–Wilk or D’Agostino and Pearson test. Then, one-way analysis of variance (ANOVA) followed by the Tukey’s or Dunnett’s post-hoc test or for parametric analysis or Kruskal–Wallis post-hoc test in the case of non-parametric analysis tests were used to determine the statistical significance. The level of significance was set at p ≤ 0.05. Statistical analyses were performed using GraphPad Prism version 9 (GraphPad, San Diego, CA, USA).
Results
VCE-004.8 induces B55α/PP2A expression in endothelial vascular cells
Ehling and colleagues have demonstrated that in adult mice, the expression of B55α is relatively low in CD31+ blood vessels, but it increases significantly in sites of active angiogenesis [9]. Additionally, it has been observed that B55α is induced in human umbilical vein endothelial cells (HUVECs) when seeded at high density compared to those seeded at low density, and it prevents the induction of apoptosis [20]. Herein, we show that B55α expression in EA-hy926 cells is primarily localized perinuclearly and is significantly upregulated following exposure to VCE-004.8 (1 and 5 µM) (Fig. 1A, B). Furthermore, VCE-004.8 also maintained its capacity to induce the expression of B55α even under inflammatory conditions (Fig. 2A, lower panel and Fig. 2B).
Fig. 1.

VCE-004.8 induces B55α/PP2A expression and activates HIF-1α in endothelial EA.hy926 cells. A Representative confocal image of B55α expression in EA.hy926 cells untreated and treated with VCE-004.8 for 24 h. Magnified region (marked by the white rectangle) shows preferential perinuclear expression of B55α as indicated by white arrows. Scale bars equivalent to 25 μm. B Effect of VCE-004.8 on EA.hy926 cells treated for 24 h. Representative western blots of three independent experiments are shown. B55α protein expression is normalized relative to actin protein expression and results were expressed relative to control defined as 1. C Effect of VCE-004.8 on cell viability. EA.hy926 cells were pre-incubated with increasing concentrations of VCE-004.8 during 1 h and then exposed to H2O2 for 24 h. Cell viability was investigated by MTT assay. The data passed the normality test performed by the Kolmogorov–Smirnov test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n > 4 independent experiments per group), the results are shown as mean ± SEM. D Cytoprotective activity of VCE-004.8 is partially dependent on HIF-1α activation. EA.hy926 cells were pretreated with VCE-004.8 in the absence and the presence of the HIF-1α inhibitor YC-1 and then exposed to H2O2. Cell viability was investigated by MTT assay. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n > 3 independent experiments per group); the results are shown as means ± SEM. The results are shown as means ± SEM. P values indicated in panels, significant as **p < 0.01, ***p < 0.001, ****p < 0.0001
Fig. 2.

VCE-004.8 alleviates inflammation of vascular endothelial cells. A Representative image of VCAM, ZO-1, CLD1 and B55α expression in EA.hy926 cells stimulated for 24 h with proinflammatory cytokines and their quantifications. Scale bars (25 μm). The data passed the normality test performed by the Shapiro Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n = 3 independent experiments per group). B EA.hy926 cells preincubated with VCE-004.8 for 1 h and then treated with TNFα and IL6 for 24 h. ZO-1, CLD1 and B55α expression was analyzed by immunoblotting. Representative western blots of three independent analyses are shown. B55α and CLD1 protein expressions are normalized to actin. ZO-1 protein expression is normalized relative to vinculin. Results were expressed relative to control defined as 1. C VCAM-1 expression was analyzed by immunoblotting. Protein expression is normalized relative to actin and results were expressed relative to control defined as 1. The results are shown as mean ± SEM. P values indicated in panels, significant as **p < 0.01, ***p < 0.001 ****p < 0.0001
To investigate the functional impact of VCE-004.8 and HIF-1α on cell viability, we subjected the cells to H2O2 treatment in the presence or absence of the compound. VCE-004.8 greatly prevented H2O2-cytotoxicity in a concentration-dependent manner (Fig. 1C), which was significantly counteracted by YC-1, a HIF-1α inhibitor (Fig. 1D). Moreover, the cytoprotective effect of VCE-004.8 was evident in cells exposed to both high glucose and OxLDL (Additional file 2: Fig. S1). Subsequently, to explore the anti-inflammatory properties of VCE-004.8, EA-hy926 cells were incubated with proinflammatory cytokines, and the expression of various markers was analyzed via confocal microscopy and immunoblotting. VCE-004.8 attenuated IL-1β+TNFα-induced VCAM-1 expression and restored the expression of tight-junction proteins ZO-1 and CLD1 in IL-6/TNFα-treated cells (Fig. 2). Notably, tight-junction proteins play a crucial role in maintaining the integrity of vascular endothelial cells [20].
VCE-004.8 activates AMPK/Sirtuin 1 and prevents senescence in endothelial vascular cells
We have demonstrated that VCE-004.8, dephosphorylates PHD2 at ser-125 via a PP2A/B55α-dependent pathway, leading to the induction of HIF-1α expression [15]. Knockdown of B55α mRNA in various cell types, including endothelial cells, significantly reduced the activation of HIF-1α induced by VCE-004.8 (Fig. 3A) [15]. Nonetheless, it is likely that VCE-004.8 also engages other convergent pathways to activate HIF.
Fig. 3.

VCE-004.8 activates AMPK/Sirt1 pathway and prevents cellular senescence. A EA.hy926 cells were transfected with siB55α or scrambled siRNAs (siCTR) and 48 h later and treated with VCE-004.8 for 3 h. B55α and HIF-1α protein expression were analyzed by immunoblotting, normalized relative to actin protein and the results expressed relative to control defined as 1. B HEK-293T cells preincubated with or without DS for 30 min and stimulated for 3 h with increasing concentrations of VCE-004.8 and the steady state levels of pAMPK total AMPK and actin detected by western blot. pAMPK protein expression is normalized relative to total AMPK protein and results were expressed relative to control defined as 1. C EA.hy296 cells were stimulated with VCE-004.8 for 3 h and the expression of Sirt1 and Visfatin (NAMPT) was analyzed by western blot. Sirt1 and Visfatin (NAMPT) protein expressions are normalized to actin and results were expressed relative to control defined as 1. D VCE-004.8 induces Sirt1 activity measured by a fluorometric assay. SRT1720 was used as a positive control. Data represent the mean ± SD (n = 3). E NAD+/NADH ratio was determined in EA.hy296 cells. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n = 3 independent experiments per group). The results are shown as means ± SEM. P value indicated in panel, significant as **p < 0.01. F VCE-004.8 prevents H2O2-induced senescence measured by SA-β-gal staining in HMEC-1 cells. The data passed the normality test performed by the D’Agostino and Pearson test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n > 18 represent areas of cell surface from 3 independent experiments). The results are shown as means ± SD. P values indicated in panel, significant as ****p < 0.0001. G HMEC-1 cells were incubated with VCE-004.8 and exposed to H2O2 and the expression of Sirt1, p21 and tubulin detected by western blot. Western blots are representative of 3 independent experiments. Sirt1 and p21 protein expressions are normalized relative to tubulin protein expression and results were expressed relative to control defined as 1
The protective role of the AMP-activated Protein Kinase (AMPK) pathway, a pivotal cellular energy sensor, extends beyond metabolic homeostasis to encompass cardiovascular protection. Vascular AMPK plays a crucial role in regulating endothelial function, redox homeostasis, and inflammation [21]. Moreover, emerging evidence indicates a functional interplay between AMPK and HIF pathways, contributing to cellular survival adaptation [22]. Thus, we interrogated the participation of AMPK on the pathways activated by VCE-004.8 in endothelial cells. VCE-004.8 induced AMPK phosphorylation and upregulated the expression of Sirt1 and nicotinamide phosphoribosyl transferase (NAMPT), also known as Visfatin, in EA.hy926 cells (Fig. 3B, C). A connection between AMPK and Sirt1 has been proposed since AMPK activates Sirt1 through an increase in cellular NAD+ level [23], while Sirt1 deacetylates the AMPK kinase LKB1, leading to phosphorylation and activation of AMPK [24]. We found that VCE-004.8 increases the enzymatic activity of Sirt1 and the NAD+/NADPH ratio in EA-hy926 cells (Fig. 3D, E). To further explore the effect of VCE-004.8 on cellular senescence, EA.hy926 cells were treated with H2O2 and the expression of both SA-β-gal+ cells, Sirt1 and p21 proteins analyzed. VCE-004.8 significantly inhibited H2O2-induced senescence and p21 expression and restored the expression of Sirt1 that was downregulated with the H2O2 treatment (Fig. 3F, G). Moreover, knockdown of Sirt1 mRNA prevented the induction of HIF-1α expression by VCE-004.8 (Fig. 4A). In addition, pretreatment with dorsomorphin (DS), an AMPK inhibitor, or EX527, a Sirt1 inhibitor, separately or in combination, also inhibited both VCE-004.8-induced EPO-Luc, serving as a surrogated marker of HIF activation (Fig. 4B), and H2O2-induced cytotoxicity (Fig. 4C). We also investigated eNOS phosphorylation at ser1177, another target of AMPK, and observed that VCE-004.8 induced eNOS phosphorylation (Fig. 5A) as well as NO production (Fig. 5B) in EA.hy926 cells. Interestingly, eNOS phosphorylation was significantly diminished in B55α siRNA cells (Fig. 5C), providing further evidence for the functional interaction between B55α and AMPK in response to VCE-004.8. Taken together, these findings suggest that in addition to PHD2/HIF-1α activation, the AMPK/Sirt1/eNOS signaling pathways play a significant role in the mechanism of action of VCE-004.8.
Fig. 4.
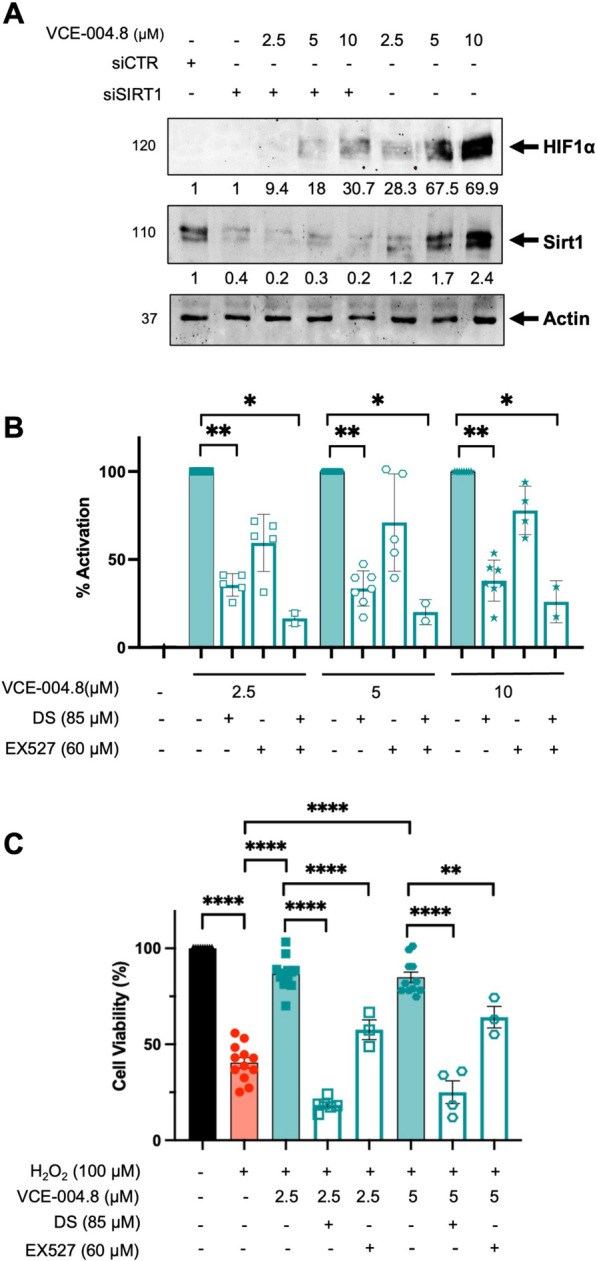
VCE-004.8 activates HIF-1α and prevents H2O2-induced cytotoxicity through AMPK/Sirt1. A HEK-293T cells were transfected with siSIRT1 or scrambled siRNA and treated with VCE-004.8 for 3 h Western blots are representative of 3 independent experiments. HIF-1α and Sirt1 protein expressions are normalized relative to actin protein and results were expressed relative to control defined as 1. B NIH-3T3-EPO-luc cells were treated with VCE-004.8 in the absence and the presence of AMPK and Sirt1 inhibitors for 6 h and luciferase activity measured and expressed as percentage of activation considering VCE-004.8 as 100% activation. Data represent the mean ± SD (n = 2–7). The significance was determined non-parametric followed by a Kruskal–Wallis test. P values indicated in panels, significant as *p < 0.05, **p < 0.01. C EA.hy926 cells were preincubated either with DS or Ex527 and treated with VCE-004.8 during 1 h before exposure to H2O2 for 24 h. Cytotoxicity was measured by the MTT method. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Dunnett’s post hoc multiple comparisons to compare between the groups. Data represent the mean ± SD (n = 3–12). P values indicated in panels, significant as **p < 0.01, ****p < 0.0001
Fig. 5.

VCE-004.8 induces eNOS phosphorylation and NO synthesis. A EA.hy926 cells were treated with VCE-004.8 for 3 h. peNOS protein expression is normalized to eNOS and results were expressed relative to control defined as 1. B VCE-004.8 induces NO production. EA.hy296 cells were seeded, preincubated with DAF-2DA and treated with sulforaphane (SFN) (50 µM) or VCE-004.8 (10 µM). The figure indicated the percentage of NO induction over untreated cells. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n = 3 independent experiments per group). The results are shown as means ± SD. P value indicated in panel, significant as ****p < 0.0001. C VCE-004.8 induces phosphorylation in vascular endothelial cells in a B55α dependent manner. EA.hy296 cells were transfected with siB55α or scrambled siRNAs (siCTR) and 48 h later treated with VCE-004.8 for 3 h. Representative western blots of three independent experiments is shown. peNOS and B55α protein expressions are normalized to eNOS and actin respectively and the results were expressed relative to control defined as 1
Angiogenic and arteriogenic effects of VCE-004.8
Vascular regeneration, angiogenesis, arteriogenesis and vasculogenesis are the main objectives for potential vasculogenic therapies for PAD [25]. To evaluate the effect of VCE-004.8 in vascularization and sprouting, we performed an ex vivo aortic ring assay. We found that VCE-004.8, as well as VEGF and dimethyloxallyl glycine (DMOG), an enzymatic Pan-PHD inhibitor, induced vascularization in vitro, indicated by the formation of new sprouts as well as sprout area Fig. 6A. Next, to investigate the angiogenic potential of VCE-004.8 in vivo, we conducted a Matrigel plug in situ assay. After 10 days following the subcutaneous injection of Matrigel into the flank, no signs of angiogenesis were observed in the negative control group (without treatment), in contrast to Matrigel embedded with VEGF (positive control). Oral treatment with VCE-004.8 (10 and 20 mg/kg) clearly increased the formation of functional vessel identified by CD31+/αSMA+ staining and the proliferative activity of vascular endothelial cells were identified by expression of CD31+/Ki67+ cells. Moreover, we also confirmed that B55α is also expressed in the new formed vessels (Fig. 6B).
Fig. 6.

VCE-004.8 induces angiogenesis in vitro and in vivo A Representative images of aortic ring assay. Red arrows indicate sprouts, and red lines delimit sprouting area. The scales are indicated in the images. B Macroscopic (top) (scale bar equivalent to 1 mm) and confocal images of immunofluorescence staining of Matrigel sections of double immunofluorescence staining of α-SMA (green)/CD31 (red), CD31 (green)/Ki67(red), B55α (green)/CD31(red). Scale bars equivalent to 50 μm for confocal images. For α-SMA staining quantification, the data passed the normality test performed by the Kolmogorov–Smirnov test. P value is calculated using one-way ANOVA followed by Tukey test compare between the groups (n = 3). For CD31 staining quantification, the significance was determined non-parametric followed by a Kruskal–Wallis test (n = 3). Double staining CD31/Ki67 was used to quantify vascular endothelial cells proliferation per area (CD31+/Ki67+ cells). The significance was determined non-parametric followed by a Kruskal–Wallis test (n = 3). The results are shown as mean ± SEM. P values indicated in panels, significant as *p < 0.05, **p < 0.01, ***p < 0.001
To further explore the effect of VCE-004.8 in a model of critical limb ischemia (CLI) a double ligation in the femoral arteria was performed in mice and 24 h later animals were treated daily with oral VCE-004.8. After 10 or 28 days of treatment the effect of VCE-004.8 on collateral vessels was investigated by vascular casting and microcomputed tomography (µ-CT) imaging. It is shown in Fig. 7A that treatment with VCE-004.8 clearly induced collateral artery formation in the ligated limb compared to the non-ligated control limb, which was not affected by the treatment. These results are in good correlation with the mRNA expression of Hif1α and the HIF-regulated proangiogenic genes: Vegfa, Epo and Hgf in the gastrocnemius muscle in the affected limb but not in control limb Fig. 7B suggesting that the effect of VCE-004.8 is specific to damaged tissues.
Fig. 7.

Effects of oral VCE-004.8 in a Critical Limb Ischemia mouse model. A Representative whole-mount images of Microfil vascular cast limbs 10 days after ischemia (upper panel) (Scale bar 2 mm), µ-CT reconstruction, and their segmentation of arterial vasculature 28 days (middle and lower panel) (Scale bar 1 mm) after femoral artery ligation. Collateral artery growth is indicated by black arrows. B Expression of Vegf-a, Epo, Hgf, and Hif1α mRNA in the gastrocnemius after 10 days of ischemia. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n = 3 animals per group). The results are shown as means ± SEM. P value indicated in panel, significant as *p < 0.05, **p < 0.01
Angiogenic and antifibrotic effects of VCE-004.8 in CLI mice
Reperfusion of ischemic tissues depends on angiogenesis that is required for a proper tissue blood flow [26, 27]. Thus, we explored the effect of VCE-004.8 on angiogenesis and vascular density in the gastrocnemius tissue. VCE-004.8 induced angiogenesis measured by the number of vessels (CD31+/αSMA+) and lumen perimeter vessel per area in the ligated limb without affecting the contralateral limb Fig. 8A. We also investigated endothelial cells proliferation, a key event in angiogenesis, and found that VCE-004.8 significantly induced cell proliferation as measured by CD31/Ki67 co-staining Fig. 8B. eNOS phosphorylation has been shown to contribute to vascular repair by rescuing impaired angiogenesis [28] and we found that in CLI mice the treatment with VCE-004.8 enhanced the expression of peNOS in vascular endothelium and myocytes compared with untreated mice (Fig. 8C). CD34 is one of the surface markers for endothelial progenitor cells, which plays a major role in angiogenic and vasculogenic activities during neovascularization [29]. Indeed, CD34+ cells isolated from peripheral blood have been widely used in experimental therapies including cardiovascular diseases and critical leg ischemia [30, 31]. VCE-004.8 treatment significantly increased the average the of number of CD34+/CD31+ as well as CD34+/CD31− cells in the ligated limb compared with the contralateral limb Fig. 9A.
Fig. 8.

Oral VCE-004.8 enhances ischemia-induced vessel formation and vascular endothelial cells proliferation in vivo. A Double immunofluorescence confocal staining of α-SMA (green)/CD31 (red) in gastrocnemius muscles 28 days after ischemia. Quantification of the number of vessel peer area and the perimeter of the lumen vessel. As the data cannot passed the normality test. P values are calculated using a nonparametric Kruskal–Wallis test followed by Dunn multiple. The results are shown as mean ± SEM. P values indicated in panels, significant as **p < 0.01, ***p < 0.001, ****p < 0.0001. B The number of CD31+/Ki67+ positive cells per area and number of CD31+/Ki67+ cells per total cells were quantified using double immunofluorescence confocal staining of CD31 (green) and Ki67 (red) in gastrocnemius muscles 28 day after ischemia. Significance was determined by one-way ANOVA followed by Tukey’s test. The results are shown as means ± SEM (n = 3). P values indicated in panels, significant as ****p < 0.0001. C Double immunofluorescence confocal staining of peNOS (green) and CD31 (red) in gastrocnemius muscles 10 days after ligation and quantifications. Magnified region (marked by the white rectangle) shows detailed colocalization of peNOS and CD31 expression. Scale bars equivalent to 50 μm. The data do not pass the normality test performed, and the significance was determined non-parametric followed by a Kruskal–Wallis test. Values are mean ± SEM (n = 3–4). P values indicated in panels, significant as *p < 0.1, ***p < 0.001
Fig. 9.

Oral VCE-004.8 enhanced ischemia-induced de novo vessel and alleviated fibrosis in vivo. A Double immunofluorescence confocal staining of CD34 (green)/CD31 (red) in gastrocnemius muscles 28 days was used to calculate the number of CD34+/CD31− cells (immature endothelial vascular cells) and CD34+/CD31+ (mature endothelial vascular cells). Scale bars equivalent to 50 μm. Values are mean ± SEM (n = 3–4). In both quantifications, the significance was determined by one-way ANOVA non-parametric followed by a Kruskal–Wallis test. P values indicated in panels, significant as **p < 0.01; ***p < 0.001; ****p < 0.0001. B Immunofluorescence staining of TNC (green) in gastrocnemius muscles 28 days after ischemia and quantifications. Scale bars equivalent to 50 μm. Values are mean ± SEM (n = 3–4), and significance was determined by one-way ANOVA non-parametric followed by Kruskal–Wallis test. P values indicated in panels, significant as ***p < 0.001; ****p < 0.0001. C Representative immunofluorescence staining of TNC in cross-section of healthy and disease human arteries. Scale bars equivalent to 50 μm
Evidence supports the involvement of fibrosis in the pathophysiology of PAD [32]. Fibrosis is associated with various pathological processes in vascular tissues and may contribute to the progression of PAD [33]. Herein, we investigated the expression of the profibrotic marker Tenascin C (TNC) in both CLI mice and in ex-vivo human samples from PAD patients [34]. TNC expression was detected in the ligated limb of CLI mice (Fig. 9B) and human aorta from PAD patients (Fig. 9C) and treatment with VCE-004.8 significantly alleviated TNC deposition in the ischemic tissue relative to the contralateral leg (Fig. 9B).
B55α and Sirtuin 1 expression in CLI mice and human arteries
As demonstrated above, our data revealed a mechanistic link between B55α and Sirt1 expression and activity in vitro. Consequently, we investigated the tissular expression of both proteins. B55α and Sirt1 expression were found to be downregulated in the ligated limb from CLI mice compared to the contralateral limb (Fig. 10A, B), and similar results were observed in the aorta from PAD patients (disease) compared to healthy tissues (Fig. 10D). VCE-004.8 treatment completely restored the expression of both proteins in the ligated limb of CLI mice. The mRNA expression levels of B55α and Sirt1 confirmed the findings obtained from confocal analysis. Additionally, VCE-004.8 exhibited a tendency to increase the mRNA expression of Sirt1 in the non-ligated limb of CLI mice (Fig. 10C).
Fig. 10.

Effects of VCE-004.8 on B55α and Sirt1 expression in vivo. A Representative double immunofluorescence confocal staining of B55α (red)/CD31 (green) (left) (scale bar equivalent to 50 µm) and immunofluorescence staining of Sirt1(green) (right) (scale bar equivalent to 20 µm) in gastrocnemius muscles 10 days after ischemia. B Quantifications of B55α (left) and Sirt1 (right). For B55α and Sirt1 quantification, the data do not pass the normality test performed, and the significance was determined non-parametric followed by a Kruskal–Wallis test. Values are mean ± SEM (n = 3–4). P values indicated in panels, significant as *p < 0.05 as **p < 0.01; ***p < 0.001. C Expression of B55α and Sirt1 mRNA in mice gastrocnemius at 10 days after femoral ligation. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups (n = 3 animals per group). The results are shown as mean ± SEM. P value indicated in panel, significant as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. D Representative immunofluorescence image of B55α (red) in cross-section of healthy and disease human arteries (left) (scale bar equivalent to 50 µm) and representative double immunofluorescence staining of Sirt1 (green) and CD31 (red) in cross-section of healthy and disease human arteries (right) (scale bar equivalent to 50 µm). Magnified region (white rectangle) showing details of Sirt1 expression (yellow arrow) (scale bar equivalent to 10 µm)
Caveolin 1 is downregulated in CLI mice and human arteries
To investigate the effects of VCE-004.8 on the expression of other markers, we labeled the tissues with αSMA to identify mature vessels. These vessels were subsequently isolated and studied using Laser Capture Microdissection coupled to Mass Spectrometry (Fig. 11A). From the total bulk of proteins (210), eleven proteins involved in the angiogenesis process were identified according to Gene Ontology Annotations from Mice Genomic Identification [35]. Among them, caveolin 1 (CAV1) is of special interest in our study since its gene is regulated by HIF-1 [36] and it is required for collateral development in CLI mice [37] (Fig. 11B). The expression of CAV1 was detected in both the control limb of CLI mice and in healthy human arteries (Fig. 11C, E), while it was downregulated in the ligated limb of CLI mice and in arteries affected by PAD (Fig. 11C, E). Treatment with VCE-004.8 normalized the expression of CAV1 protein and upregulated its mRNA expression (Fig. 11C, D).
Discussion
Effective induction and promotion of angiogenesis have been so far very challenging. A better knowledge of the basic pro-angiogenic players will be crucial to achieve a clinical benefit for PAD patients and other cardiovascular diseases. Therapeutic angiogenesis or vascular regeneration remains an attractive treatment modality for PAD and chronic tissue ischemia. Despite decades of research into angiogenic and cell-based therapies for PAD, their translation to clinical use has been limited. Therefore, there is an urgent need for novel therapeutic angiogenic approaches.
Vascular regeneration strategies include restoration of vascular function and structure that can be partially achieved through HIF activation. While severe hypoxia can be detrimental, leading to cell death and organ failure, mild hypoxia, also known as hypoxia preconditioning, has shown promising therapeutic potential, particularly in the treatment of cardiovascular diseases [38]. Mild hypoxia can be achieved by hypoxia mimetic enzymatic inhibitors of PHDs such are JNJ-42041935, FG-4497, FG-4592 (Roxadustat), TRC160334 (Desidustat), and AKB-4924 [39] or by novel PPA2/B55α activators such are Etrinabdione (VCE-004.8) and VCE-005.1 [15, 40]. Indeed, it has been observed that the enzymatic inhibitor of PHDs, FG-4497, confers protection against the development of atherosclerosis, a pivotal factor in PAD pathogenesis [41]. Herein, we show the sprouting and angiogenic activity of VCE-004.8 in vitro and in vivo and its efficacy in CLI mice induced by femoral ligation. Etrinabdione induced endothelial cells proliferation, angiogenic gene expression and prevented fibrosis. One striking and interesting observation is that the effects of Etrinabdione occur only in the affected limbs compared to non-ischemic limbs. A similar observation was found with sodium nitrite therapy and metformin in CLI mice [42, 43] and these results agree with our previous finding showing that VCE-004.8 induced angiogenesis and restored damaged brain blood barrier in the affected brain hemisphere of mice with traumatic brain injury but in the contralateral hemisphere [15]. Thus, we could speculate that the therapeutic benefit of Etrinabdione will be akin to converting severe hypoxia into therapeutic mild hypoxia in the affected limb.
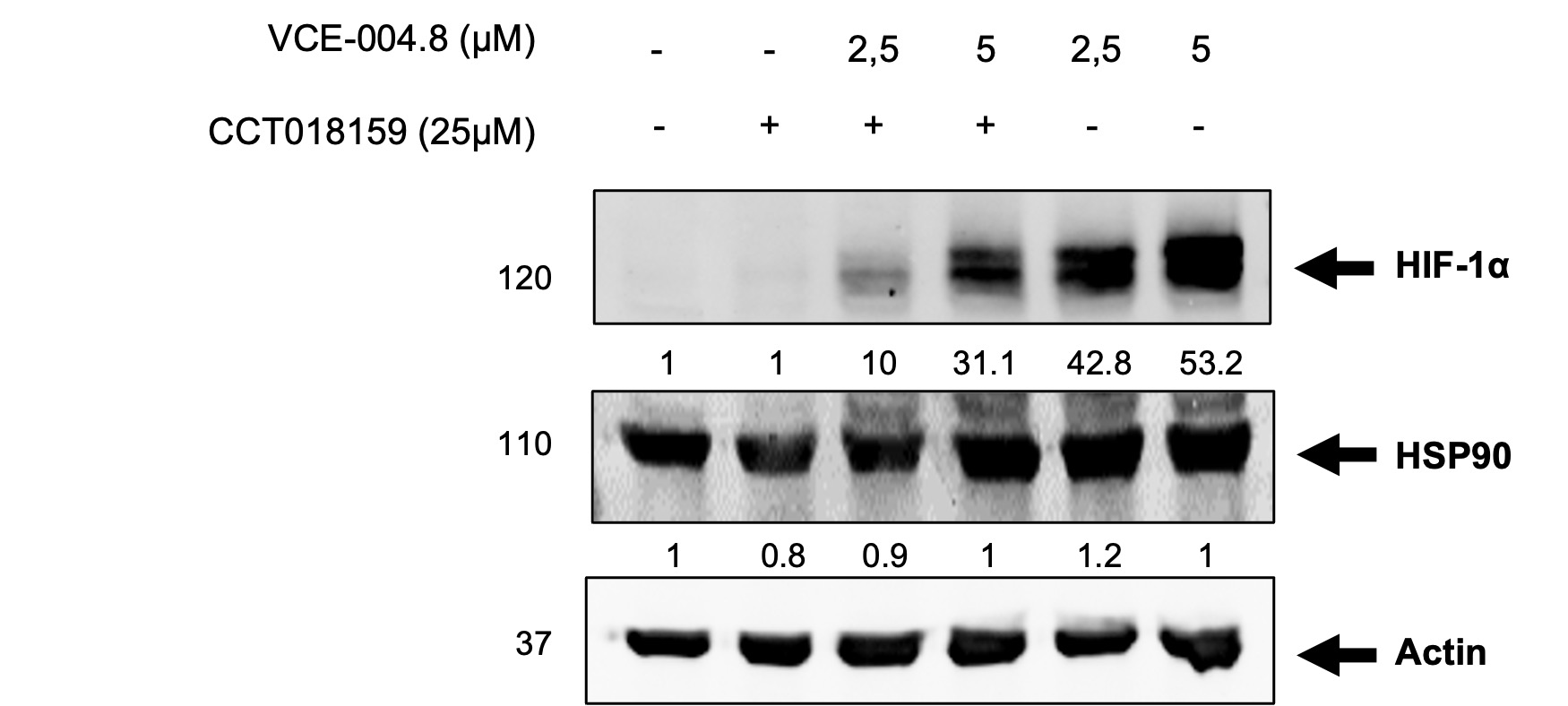
The pathophysiology of atherothrombosis and PAD is complex, and involves many cells, proteins and pathways, giving the possibility to explore multitarget therapeutic strategies, which by covering different angles of the disease may be beneficial for the outcome of patients. Herein we show that Etrinabdione, a multitarget drug candidate in clinical phases, induces HIF-1α expression in endothelial cells through a novel pathway that potentially involves two axes: B55α/PHD2 and B55α/AMPK/Sirt1 signaling that may converge on HIF stabilization. In addition, VCE-004.8 induces the phosphorylation of eNOS at ser1177, which is a target for AMPK. The exact molecular mechanisms by which B55α regulates AMPK are still to be determined. Previous coimmunoprecipitation experiments have indicated that B55α does not physically interact with AMPK [9], implying that their regulation might occur through an indirect functional interaction. AMPK signaling regulates the nuclear accumulation of HIF-1α through a complex process involving HDAC5 phosphorylation. This process leads to the export of HDAC5 from the nuclei to the cytosol, where it dissociates the complex formed by the HSP70 chaperone and nascent HIF-1α [44]. Subsequently, HIF-1α associates with HSP90, facilitating its maturation and relocation into the nucleus. Notably, treatment with CCT018159, an HSP90 inhibitor, partially inhibits VCE-004.8-induced HIF-1α stabilization (Additional file 3: Fig. S2). Moreover, Sirt1 interacts with AMPK and HIF-1α, and pharmacological inhibition of Sirt1 impairs HIF-1α nuclear accumulation and the expression of HIF-1α-dependent genes [45]. Therefore, it is probable that VCE-004.8 requires AMPK to enhance the synthesis of nascent HIF-1α, and B55α/PHD2 to stabilize and translocate this nuclear factor into nuclei. This is supported by our results showing that metformin, an AMPK activator that phosphorylates eNOS [41] but did not activate HIF-1α in vascular endothelial cells [46]. However, it is noteworthy that metformin has been shown to promote revascularization in CLI mice independently of HIF-1α [43].
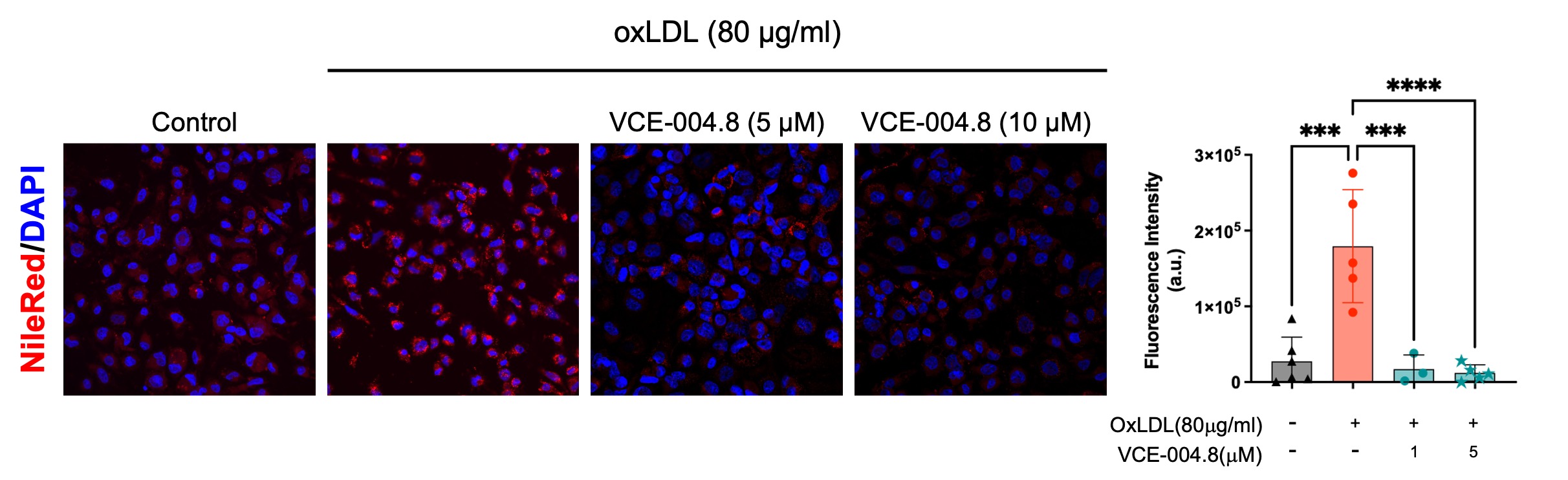
In addition, VCE-004.8 is a dual agonist of PPARγ and CB2R [14]. PPARγ and CB2R are preclinically validated therapeutic targets for vascular inflammation and atherosclerosis [47–49]. For instance, foam cells play a vital role in the initiation and development of atherosclerosis since they are the major sources of necrotic core in atherosclerotic plaques. Interestingly, it has been shown that PPARγ agonists inhibit the formation of macrophage foam cells in vivo [50]. Moreover, JHW-015, a CB2R agonist, significantly prevented oxLDL accumulation in foam cells and reduced the production of proinflammatory cytokines and the expression of CD36, a scavenger receptor that plays a critical role in the formation of foam cells. In agreement with finding CB2R activation by WIN55,212-2 also reduces an OxLDL-induced inflammatory response in rat macrophages [51]. In line with this, we have found that VCE-004.8 inhibits the induction of foam cells in OxLDL-treated macrophages (Additional file 4: Fig. S3).
On the other hand, VCE-004.8 has also shown efficacy in a murine model of metabolic syndrome induced by high-fat diet. VCE-004.8 showed anti-obesity and anti-inflammatory activity, prevented liver damage and significantly improved glycolipid metabolism [17]. If this activity had translation in humans, we could expect an added benefit in PAD patients with type II diabetes as co-morbidity. The same hold true for coexisting cerebrovascular or coronary diseases since VCE-004.8 showed efficacy in a preclinical stroke model with an excellent therapeutic window and inhibited angiotensin-II signaling and cardiac inflammation and fibrosis [16, 18].
In many cases, diseases with complex physiopathology are very difficult to be treated by monotargeted therapies, and usually requires concomitant therapies (polypharmacy) that have limitations such as therapy adherence and pharmacological interactions. In contrast, multipronged therapies (polypharmacology) may represent potential pharmacological therapies with better clinical benefits after providing a safe profile in humans and an appropriated target engagement as it is the case of reprofiling therapies with small molecules [52].
Conclusions
In conclusion, our results collectively demonstrate that VCE-004.8 is a first-in-class activator of PP2A/B55α, a phosphatase critical for vascular endothelial cell homeostasis through HIF-1α stabilization and activation. Furthermore, VCE-004.8 activates the AMPK/Sirt1/eNOS pathway in a PP2A/B55α-dependent manner, showing potential in preventing endothelial cell damage and senescence, while also promoting arteriogenesis and angiogenesis in CLI mice (Fig. 12). Moreover, the supplementary anti-inflammatory properties of Etrinabdione position it as a potential disease-modifying drug, offering clinical benefits to PAD patients with diverse comorbidities.
Fig. 12.

Potential mechanism of action of Etrinabdione (VCE-004.8)
Supplementary Information
Additional file 1: Table S1. Primers used in this study.
{kind=link}
Additional file 2: Fig. S1. VCE-004.8 protects endothelial vascular cell. EA.hy926 cells were pre-incubated with different concentrations of VCE-004.8 during 1 h and then exposed to either Glucoseor OxLDL. Cell viability was calculated by MTT assay. The data passed the normality test performed by the Kolmogorov–Smirnov test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups, the results are shown as mean ± SEM. P value indicated in panel, significant as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
{kind=link}
Additional file 3: Fig. S2. The SP90 inhibitor CCT018159 impaired VCE-004.8-induced HIF-1α expression. EA.hy926 cells were preincubated with CCT018159 and treated with VCE-004.8 during 3 h. Western blots are representative of 3 independent experiments. HIF-1α and HSP90 protein expression are normalized to actin and results were expressed relative to control defined as 1.
{kind=link}
Additional file 4: Fig. S3. VCE-004.8 prevented oxLDL-induced foam cells Nile Red staining of lipid droplets. Representative images of Nile red staining in THP-1 cells stimulated for 48 h with oxLDL and their quantifications. Scale bars equivalent to 25 μm. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups tests. Data represent the mean ± SD. P values indicated in panels, significant as ***p < 0.001, ****p < 0.0001.
Acknowledgements
We acknowledge the Microscopy Advanced Optical Microscopy and the Proteomic Units of IMIBIC for its support. Portions of the Figure 12 utilized from Servier Medical Art, licensed under Creative Commons Attribution 3.0 Unported License.
Abbreviations
- AMPK
AMP-activated protein kinase
- B55α PP2A
Protein phosphatase 2A-B55α subunit (PPP2R2A)
- CAV1
Caveolin 1
- CB2R
Cannabinoid type 2 receptors
- CLI
Critical limb ischemia
- µ-CT
Microcomputed tomography
- DMOG
Dimethyloxallyl glycine
- DS
Dorsomorphin
- EC
Endothelial cells
- eNOS
Endothelial nitric oxide synthase
- Epo
Erythropoietin
- FGF
Fibroblast growth factor
- Gapdh
Glyceraldehyde-3-phosphate dehydrogenase
- HG
High glucose
- Hgf
Hepatocyte growth factor
- HIF
Hypoxia inducing factor
- LCM
Laser capture microdissection
- IL1β
Interleukin 1 beta
- IL-6
Interleukin 6
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAD+
Nicotinamide adenine dinucleotide
- NADPH
Nicotinamide adenine dinucleotide phosphate hydrogen
- NAMPT
Nicotinamide phosphoribosyl transferase or Visfatin
- NO
Nitric oxide
- Ox-LDL
Oxidized low-density lipoprotein
- PAD
Peripheral arterial disease
- PHD2
Prolyl hydroxylase 2
- PPARγ
Peroxisome proliferator-activated receptor-γ
- SD
Standard deviation
- SEM
Standard error of the mean
- SFN
Sulforaphane
- α-SMA
Smooth muscle actin
- Sirt1
Sirtuin 1
- TNC
Tenascin
- TNFα
Tumor necrosis factor alpha
- VCAM
Vascular cell adhesion molecule 1
- VCE-004.8
Etrinabdione or EHP-101, [(1′R,6′R)-3-(benzylamine)-6-hydroxy-3′-methyl-4-pentyl-6′-(prop-1-en-2-yl) [1,1′bi(cyclohexane)]-2′,3,6-triene-2,5-dione)
- VEGF
Vascular endothelial growth factor
- ZO-1
Zonula occludens 1
Author contributions
AGM., MEP., ILC., and ABR: performed the in vivo experiments. MEP., ILC., FJPD., CN and LC. performed the in vitro experiments. MGR performed the bioinformatic analysis. RP advised the µCT analysis. IM., JM., AM., JAM., AC., JGR., collected the human samples and performed data analysis. AGM and EM designed the overall study. AGM and EM managed, supervised the study, and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by PID2020-114753RB-I00, PID2023-148340OB-I00 (Agencia Estatal de Investigación, Spain; co-funded with EU funds from FEDER Program), CPP2021-008557/AEI/10.13039/501100011033/Unión Europea NextGenerationEU/PRTR and CNS2022-135922/Unión Europea NextGenerationEU/Plan de Recuperación, Tranformación y Resilencia, Agencia Estatal de Investigación.
Availability of data and materials
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050360.
Declarations
Ethics approval and consent to participate
All experimental protocols followed the guidelines of animal care set by the EU guidelines 86/609/EEC, the Ethic Committee on Animal Experimentation at the University of Córdoba (Spain) and the Andalusian Regional Committee for Animal Experimentation approved all the procedures described in this study (31/03/2022/057). Human biopsies from patients were obtained by the Cardiovascular Surgery Unit of Hospital Universitario Reina Sofia of Cordoba, Spain (RSUH) and the protocol approved from the Ethics Committee of RSUH (reference: HIP-CI:1.0-01/06/2023).
Consent for publication
Not applicable.
Competing interests
AGM, ILC, and EM have applied for a European Patent based on some of the data presented herein.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Adela García-Martín, Email: bc2gamam@uco.es.
Eduardo Muñoz, Email: fi1muble@uco.es.
References
- 1.Annex BH, Cooke JP. New directions in therapeutic angiogenesis and arteriogenesis in peripheral arterial disease. Circ Res. 2021;128:1944–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckman JA, Schneider PA, Conte MS. Advances in revascularization for peripheral artery disease: revascularization in PAD. Circ Res. 2021;128:1885–912. [DOI] [PubMed] [Google Scholar]
- 3.Clark AR, Ohlmeyer M. Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol Ther. 2019;201:181–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Conza G, Trusso Cafarello S, Loroch S, Mennerich D, Deschoemaeker S, Di Matteo M, et al. The mTOR and pp2a pathways regulate PHD2 phosphorylation to fine-tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Rep. 2017;18:1699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appelhoff RJ, Tian Y-M, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–65. [DOI] [PubMed] [Google Scholar]
- 6.Watts ER, Walmsley SR. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med. 2019;25:33–46. [DOI] [PubMed] [Google Scholar]
- 7.Olenchock BA, Moslehi J, Baik AH, Davidson SM, Williams J, Gibson WJ, et al. EGLN1 inhibition and rerouting of α-ketoglutarate suffice for remote ischemic protection. Cell. 2016;164:884–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang Y, Ruan W, Jiang Y, Smalling R, Yuan X, Eltzschig HK. Interplay of hypoxia-inducible factors and oxygen therapy in cardiovascular medicine. Nat Rev Cardiol. 2023;20:723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehling M, Celus W, Martín-Pérez R, Alba-Rovira R, Willox S, Ponti D, et al. B55α/PP2A limits endothelial cell apoptosis during vascular remodeling: a complementary approach to disrupt pathological vessels? Circ Res. 2020;127:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ministrini S, Puspitasari YM, Beer G, Liberale L, Montecucco F, Camici GG. Sirtuin 1 in endothelial dysfunction and cardiovascular aging. Front Physiol. 2021;12: 733696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Y, Kim SY. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med. 2023;55:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Onofrio N, Vitiello M, Casale R, Servillo L, Giovane A, Balestrieri ML. Sirtuins in vascular diseases: emerging roles and therapeutic potential. Biochim Biophys Acta BBA Mol Basis Dis. 2015;1852:1311–22. [DOI] [PubMed] [Google Scholar]
- 13.Navarrete C, Carrillo-Salinas F, Palomares B, Mecha M, Jiménez-Jiménez C, Mestre L, et al. Hypoxia mimetic activity of VCE-004.8, a cannabidiol quinone derivative: implications for multiple sclerosis therapy. J Neuroinflamm. 2018;15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Del Río C, Navarrete C, Collado JA, Bellido ML, Gómez-Cañas M, Pazos MR, et al. The cannabinoid quinol VCE-004.8 alleviates bleomycin-induced scleroderma and exerts potent antifibrotic effects through peroxisome proliferator-activated receptor-γ and CB2 pathways. Sci Rep. 2016;6:21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navarrete C, García-Martín A, Correa-Sáez A, Prados ME, Fernández F, Pineda R, et al. A cannabidiol aminoquinone derivative activates the PP2A/B55α/HIF pathway and shows protective effects in a murine model of traumatic brain injury. J Neuroinflamm. 2022;19:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.García-Martín A, Navarrete C, Garrido-Rodríguez M, Prados ME, Caprioglio D, Appendino G, et al. EHP-101 alleviates angiotensin II-induced fibrosis and inflammation in mice. Biomed Pharmacother. 2021;142: 112007. [DOI] [PubMed] [Google Scholar]
- 17.Palomares B, Ruiz-Pino F, Navarrete C, Velasco I, Sánchez-Garrido MA, Jimenez-Jimenez C, et al. VCE-004.8, a multitarget cannabinoquinone, attenuates adipogenesis and prevents diet-induced obesity. Sci Rep. 2018;8:16092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lavayen BP, Yang C, Larochelle J, Liu L, Tishko RJ, De Oliveira ACP, et al. Neuroprotection by the cannabidiol aminoquinone VCE-004.8 in experimental ischemic stroke in mice. Neurochem Int. 2023;165: 105508. [DOI] [PubMed] [Google Scholar]
- 19.Minassi A, Rogati F, Cruz C, Prados ME, Galera N, Jinénez C, et al. Triterpenoid hydroxamates as HIF prolyl hydrolase inhibitors. J Nat Prod. 2018;81:2235–43. [DOI] [PubMed] [Google Scholar]
- 20.Claesson-Welsh L, Dejana E, McDonald DM. Permeability of the endothelial barrier: identifying and reconciling controversies. Trends Mol Med. 2021;27:314–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodríguez C, Muñoz M, Contreras C, Prieto D. AMPK, metabolism, and vascular function. FEBS J. 2021;288:3746–71. [DOI] [PubMed] [Google Scholar]
- 22.Dengler F. Activation of AMPK under hypoxia: many roads leading to Rome. Int J Mol Sci. 2020;21:2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. J Biol Chem. 2008;283:27628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooke JP, Meng S. Vascular regeneration in peripheral artery disease. Arterioscler Thromb Vasc Biol. 2020;40:1627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wahlberg E. Angiogenesis and arteriogenesis in limb ischemia. J Vasc Surg. 2003;38:198–203. [DOI] [PubMed] [Google Scholar]
- 27.Han J, Luo L, Marcelina O, Kasim V, Wu S. Therapeutic angiogenesis-based strategy for peripheral artery disease. Theranostics. 2022;12:5015–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen TD, Rahman N-T, Sessa WC, Lee MY. Endothelial nitric oxide synthase (eNOS) S1176 phosphorylation status governs atherosclerotic lesion formation. Front Cardiovasc Med. 2023;10:1279868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Ii M, Kamei N, Alev C, Kwon S-M, Kawamoto A, et al. CD34+ cells represent highly functional endothelial progenitor cells in murine bone marrow. PLoS ONE. 2011;6: e20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Huyen J-PD, Smadja DM, Bruneval P, Gaussem P, Dal-Cortivo L, Julia P, et al. Bone marrow-derived mononuclear cell therapy induces distal angiogenesis after local injection in critical leg ischemia. Mod Pathol. 2008;21:837–46. [DOI] [PubMed] [Google Scholar]
- 31.Dong Z, Pan T, Fang Y, Wei Z, Gu S, Fang G, et al. Purified CD34+ cells versus peripheral blood mononuclear cells in the treatment of angiitis-induced no-option critical limb ischaemia: 12-month results of a prospective randomised single-blinded non-inferiority trial. EBioMedicine. 2018;35:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calvier L, Miana M, Reboul P, Cachofeiro V, Martinez-Martinez E, De Boer RA, et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol. 2013;33:67–75. [DOI] [PubMed] [Google Scholar]
- 33.Ding N, Yang C, Ballew SH, Kalbaugh CA, McEvoy JW, Salameh M, et al. Fibrosis and inflammatory markers and long-term risk of peripheral artery disease: the ARIC study. Arterioscler Thromb Vasc Biol. 2020;40:2322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7:11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eppig JT. Mouse genome informatics (MGI) resource: genetic, genomic, and biological knowledgebase for the laboratory mouse. ILAR J. 2017;58:17–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor–mediated upregulation of caveolin-1. Proc Natl Acad Sci. 2012;109:4892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonveaux P, Martinive P, DeWever J, Batova Z, Daneau G, Pelat M, et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95:154–61. [DOI] [PubMed] [Google Scholar]
- 38.Bagali S, Das KK. Hypoxia and its preconditioning on cardiac and vascular remodelling in experimental animals. Respir Physiol Neurobiol. 2021;285: 103588. [DOI] [PubMed] [Google Scholar]
- 39.Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR. Prolyl hydroxylase inhibitors: a breakthrough in the therapy of anemia associated with chronic diseases. J Med Chem. 2018;61:6964–82. [DOI] [PubMed] [Google Scholar]
- 40.Prados ME, Correa-Sáez A, Unciti-Broceta JD, Garrido-Rodríguez M, Jimenez-Jimenez C, Mazzone M, et al. Betulinic acid hydroxamate is neuroprotective and induces protein phosphatase 2A-dependent HIF-1α stabilization and post-transcriptional dephosphorylation of prolyl hydrolase 2. Neurotherapeutics. 2021;18:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahtu-Korpela L, Määttä J, Dimova EY, Hörkkö S, Gylling H, Walkinshaw G, et al. Hypoxia-inducible factor prolyl 4-hydroxylase-2 inhibition protects against development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:608–17. [DOI] [PubMed] [Google Scholar]
- 42.Kumar D, Branch BG, Pattillo CB, Hood J, Thoma S, Simpson S, et al. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proc Natl Acad Sci. 2008;105:7540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi N, Shibata R, Ouchi N, Sugimoto M, Murohara T, Komori K. Metformin stimulates ischemia-induced revascularization through an eNOS dependent pathway in the ischemic hindlimb mice model. J Vasc Surg. 2015;61:489–96. [DOI] [PubMed] [Google Scholar]
- 44.Chen S, Yin C, Lao T, Liang D, He D, Wang C, et al. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle. 2015;14:2520–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laemmle A, Lechleiter A, Roh V, Schwarz C, Portmann S, Furer C, et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE. 2012;7: e33433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prados ME, Navarrete C, García-Martín A, Lastres-Cubillo I, Ponce-Díaz F, Martínez-Orgado J, et al. VCE-005.1, an hypoxia mimetic betulinic acid derivative, induces angiogenesis and shows efficacy in a murine model of traumatic brain injury. Biomed Pharmacother. 2023;162: 114715. [DOI] [PubMed] [Google Scholar]
- 47.Yang X, Shang D. The role of peroxisome proliferator-activated receptor γ in lipid metabolism and inflammation in atherosclerosis. Cell Biol Int. 2023;47:1469–87. [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Guo H, Jing Z, Yang L, Chen C, Peng L, et al. N-Oleoylethanolamine reduces inflammatory cytokines and adhesion molecules in TNF-α-induced human umbilical vein endothelial cells by activating CB2 and PPAR-α. J Cardiovasc Pharmacol. 2016;68:280–91. [DOI] [PubMed] [Google Scholar]
- 49.Steffens S, Pacher P. Targeting cannabinoid receptor CB2 in cardiovascular disorders: promises and controversies. Br J Pharmacol. 2012;167:313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J Clin Invest. 2004;114:1564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turcotte C, Blanchet M-R, Laviolette M, Flamand N. The CB2 receptor and its role as a regulator of inflammation. Cell Mol Life Sci. 2016;73:4449–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryszkiewicz P, Malinowska B, Schlicker E. Polypharmacology: promises and new drugs in 2022. Pharmacol Rep. 2023;75:755–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Primers used in this study.
Additional file 2: Fig. S1. VCE-004.8 protects endothelial vascular cell. EA.hy926 cells were pre-incubated with different concentrations of VCE-004.8 during 1 h and then exposed to either Glucoseor OxLDL. Cell viability was calculated by MTT assay. The data passed the normality test performed by the Kolmogorov–Smirnov test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups, the results are shown as mean ± SEM. P value indicated in panel, significant as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Additional file 3: Fig. S2. The SP90 inhibitor CCT018159 impaired VCE-004.8-induced HIF-1α expression. EA.hy926 cells were preincubated with CCT018159 and treated with VCE-004.8 during 3 h. Western blots are representative of 3 independent experiments. HIF-1α and HSP90 protein expression are normalized to actin and results were expressed relative to control defined as 1.
Additional file 4: Fig. S3. VCE-004.8 prevented oxLDL-induced foam cells Nile Red staining of lipid droplets. Representative images of Nile red staining in THP-1 cells stimulated for 48 h with oxLDL and their quantifications. Scale bars equivalent to 25 μm. The data passed the normality test performed by the Shapiro–Wilk test. P value is calculated using 1-way ANOVA followed by Tukey post hoc multiple comparisons to compare between the groups tests. Data represent the mean ± SD. P values indicated in panels, significant as ***p < 0.001, ****p < 0.0001.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050360.