

Summary
Inhibitory synaptic neurotransmission mediated by GABA requires a low concentration of chloride ions (Cl−) in neurons, which is established and maintained by the potassium-chloride co-transporter 2 (KCC2). While KCC2-interacting proteins are known to regulate KCC2 protein level and function, specific KCC2-interacting partners are still being identified and characterized. We asked whether SNAP25, an integral component of the SNARE-complex and a novel KCC2 interactor, regulates KCC2 protein and function in mice. We demonstrated that SNAP25 interacts with KCC2, and that this interaction is regulated by protein kinase C (PKC)-mediated phosphorylation. We also discovered that SNAP25 knockdown decreases total KCC2 in cortical neurons, and reduces the strength of synaptic inhibition, as demonstrated through a depolarization of the reversal potential for GABA (EGABA), indicating reduced KCC2 function. Our biochemical and electrophysiological data combined demonstrate that SNAP25 regulates KCC2 membrane expression and function, and in doing so, regulates inhibitory synaptic transmission.
Subject areas: Biological sciences, Neuroscience, Molecular neuroscience
Graphical abstract
Highlights
-
•
SNAP25 knockdown reduces surface and total KCC2
-
•
SNAP25 knockdown depolarizes EGABA and reduces Cl− driving force
-
•
KCC2-SNAP25 interaction is PKC-dependent
-
•
The KCC2 C-terminus plays a prominent role in KCC2-SNAP25 interaction
Biological sciences; Neuroscience; Molecular neuroscience
Introduction
Fast synaptic inhibition in the adult brain is mediated by the amino acid neurotransmitter γ-aminobutyric acid (GABA) and its corresponding ionotropic receptor, the GABAA receptor (GABAAR).1 GABAARs are largely permeable to Cl− and the efficacy of synaptic inhibition is dependent on the driving force for Cl− across the neuronal membrane.2,3 In adult neurons, the Cl− gradient is primarily maintained by KCC2.4,5 Loss of KCC2 function results in the hyperexcitability of neuronal networks and is associated with various neurological disorders,6 such as autism spectrum disorder (ASD),7 schizophrenia,8,9 Huntington’s disease,10 neuropathic pain,11 epilepsy,12,13 traumatic brain injury, and spinal cord injury.14
Given the importance of KCC2 for synaptic inhibition, and the role of KCC2 in human disease, it is critically important that we understand how this membrane transporter is regulated. Using affinity purification mass-spectrometry (AP-MS), we previously identified the KCC2-interactome, which revealed a rich set of KCC2-protein interactions.15 While KCC2-interacting proteins are known to play major roles in KCC2 trafficking,15 oligomerization,16,17 and degradation,18 many members of the KCC2-interactome have not yet been investigated, including SNAP25. This integral component of the presynaptic t-SNARE complex is responsible for synaptic vesicle fusion in regulated exocytosis.19,20 It has also been implicated in the trafficking and cell surface expression of various channels and receptors postsynaptically, including the membrane insertion and removal of NMDA receptors (NMDARs)21,22 and kainate-type receptors,23 respectively. Moreover, SNAP25 is an ASD risk gene with implications in neurological disorders.24 Single nucleotide polymorphisms and reduced expression of SNAP25 are susceptibility factors for the development of ASD.24,25,26,27 Due to the known cellular roles of SNAP25 and its link to ASD, we sought to characterize the KCC2-SNAP25 interaction biochemically and functionally.
Based on the emerging role of SNAP25 in mediating postsynaptic membrane protein expression, we hypothesized that SNAP25 regulates the cell surface expression and function of KCC2. We found that SNAP25 interacts with KCC2, and that the interaction is regulated by PKC-mediated phosphorylation. SNAP25 knockdown decreases total and membrane KCC2, and depolarizes EGABA, indicating reduced KCC2 function and weakened synaptic inhibition.
Results
SNAP25 interacts with KCC2 and regulates KCC2 protein levels
We previously observed that SNAP25 is a component of the KCC2 interactome,15 however, the KCC2-SNAP25 interaction has never been demonstrated biochemically or characterized functionally. We performed co-immunoprecipitation on cortical mouse (P50) brain lysates, following pull-down of KCC2, and showed that SNAP25 interacts with KCC2 in vivo (Figure 1A). We also validated this biochemical interaction data by looking at the spatial distribution of KCC2 and SNAP25 immunofluorescence. Consistent with the known localization of KCC2, we observed the distribution of KCC2-SNAP25 colocalization across the neuronal soma and dendrites (Figures 1B and S1A).
Figure 1.

SNAP25 interacts with KCC2 and regulates total KCC2
(A) Native KCC2 complexes from cortical fraction immunoprecipitated (IP) with non-specific IgG or KCC2 antibody, and immunoblotted (IB) with antibodies indicated on the right. Representative blot from three independent biological replicates.
(B) Representative images (maximum intensity projection) showing co-localization of SNAP25 (green) with KCC2 (red) in mouse cortical neurons. White arrow heads highlight co-localization (yellow). Scale bar, 10 μm.
(C) Immunoblot from COS-7 cells transfected with KCC2 and GFP, GFP-SNAP25 or GluK2 cDNA, and probed with antibodies indicated on the right.
(D) Summary graph (n = 4) showing total KCC2 abundance for (C), normalized to KCC2 alone. Statistical significance was determined using Brown-Forsythe and Welch ANOVA tests followed by Dunnett’s T3 multiple comparisons test. Asterisks denote significance from KCC2+GFP (0.25 μg).
(E) Immunoblot from neuro-2a cells transfected with KCC2 and scrambled shRNA (scrSNAP25), SNAP25 shRNAs (shSNAP25-1, shSNAP25-2) with or without shRNA resistant SNAP25 (Rescue SNAP25). The representative blot was probed with the antibodies indicated on the right.
(F) Summary graph (n = 5) showing total KCC2 abundance for (E), normalized to scrSNAP25. Statistical significance was determined using Brown-Forsythe and Welch ANOVA tests followed by Dunnett’s T3 multiple comparisons test. Asterisks denote significance from scrSNAP25. All graphs represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01.
Having established that KCC2 and SNAP25 interact, we next sought to identify the functional significance of this interaction. We began this functional characterization by asking if SNAP25 regulates KCC2 protein levels. To answer this question, we first transfected COS-7 cells (without endogenous SNAP25 or KCC2) with equal amounts of KCC2 cDNA and increasing amounts of SNAP25 cDNA, and analyzed the resulting total KCC2 protein levels. We found that by increasing SNAP25, we also increased KCC2 in COS-7 cells (Figures 1C and 1D). In addition, we also performed knockdown of endogenous SNAP25 in neuro-2a cells (without endogenous KCC2) and examined the levels of exogenous KCC2. Neuro-2a cells were co-transfected with equal amounts of KCC2 cDNA, and SNAP25 shRNA or scrambled shRNA followed by quantification of total KCC2 protein levels. Consistent with our previous results, reduction in SNAP25 resulted in a reduction in total KCC2 (Figures 1E and 1F).
SNAP25 knockdown reduces KCC2 and depolarizes EGABA in cultured neurons
We next asked whether the regulation of KCC2 by SNAP25 occurred in neurons, where both proteins are endogenously expressed. We quantified total KCC2 in cultured mouse cortical neurons following knockdown of SNAP25. Similar to the result from neuro-2a cells (Figures 1E and 1F), we found that SNAP25 knockdown reduced total endogenous KCC2 in neurons (Figures 2A–2C). To investigate whether this reduction in KCC2 altered intracellular Cl−, we performed gramicidin-perforated patch clamp recordings to determine the reversal potential for GABA (EGABA). EGABA is commonly used as a functional readout to assess KCC2 activity. We observed a depolarization of EGABA following SNAP25 knockdown (Figures 2D and 2E), with a significant reduction in the Cl− driving force (Figure 2F) and an increase in intracellular Cl− (Figure 2G), all indicating a reduction in KCC2-mediated Cl−-extrusion. To confirm the specificity of decreased KCC2 activity following SNAP25 reduction, we also performed gramicidin-perforated patch clamp recordings on cultured SNAP25(+/−) neurons with and without exogenous SNAP25 expression. We observed a similar depolarization of EGABA in SNAP25(+/−) neurons, which was rescued through exogenous SNAP25 expression (Figure 2H).
Figure 2.

SNAP25 regulates total KCC2 protein levels and depolarizes EGABA in neurons
(A) Representative images (maximum intensity projection) of neurons co-transfected with dTomato (to identify transfected neurons) and scrSNAP25 or shSNAP25, and immunostained with anti-SNAP25 (green) and anti-KCC2 (gray) antibodies. Scale bar, 10 μm.
(B) Summary graph corresponding to (A) showing total SNAP25 fluorescence (normalized to volume) in neurons following scrSNAP25 (n = 50) or shSNAP25-1 (n = 43) transfection. n values represent individual neurons obtained from three independent cultures. Statistical significance was determined using an unpaired t-test with Welch’s correction.
(C) Summary graph corresponding to (A) showing total KCC2 fluorescence (normalized to volume) in neurons following scrSNAP25 (n = 50) or shSNAP25 (n = 44) transfection. n values represent individual neurons obtained from three independent cultures. Statistical significance was determined using an unpaired t-test with Welch’s correction.
(D) Summary graph showing EGABA values (not corrected for liquid junction potential) in neurons transduced with scrSNAP25 (n = 11) or shSNAP25-1 (n = 15), and (E) a corresponding representative current-voltage plot and sample current traces, (F) driving force for Cl− and (G) estimated intracellular Cl− concentration. n values represent individual cells obtained from three independent cultures. Statistical significance for (D), (F) and (G) was determined using an unpaired t-test with Welch’s correction.
(H) Summary graph showing EGABA values (not corrected for liquid junction potential) in WT (n = 20), SNAP25(+/−) (n = 21) and SNAP25(+/−) neurons with SNAP25 transduction (n = 10). n values indicate cells obtained from cultures of at least five individual pups. Statistical significance for was determined using ordinary one-way ANOVA and Tukey’s multiple comparisons test, with single pooled variance. All summary graphs represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
SNAP25 reduction decreases KCC2 and depolarizes EGABA in mice
Reduced levels of SNAP25 are known to contribute to hyperexcitability in vivo,28 and hyperexcitability can also arise from a reduction in synaptic inhibition.29,30,31 Due to the critical role of KCC2 in maintaining synaptic inhibition, we hypothesized that KCC2 protein level and function are affected in mice with reduced SNAP25. We used both SNAP25(+/−) mice32 and WT mice transduced with SNAP25 shRNA as models for reduced SNAP25. SNAP25(+/−) mice have consistent and stable SNAP25 reduction,32 but both SNAP25 and KCC2 play vital roles during brain development.28,33 So, to avoid any potential developmental compensation, we also used mice with transient, shRNA-mediated SNAP25 knockdown.
SNAP25(+/−) mice (at ∼ P60) exhibited reduced KCC2 protein (Figures 3B and 3C) without significant changes in KCC2 mRNA when compared to WT littermates (Figure 3A). We also performed electrophysiology in whole-cell configuration using an intracellular solution containing high Cl− concentration (30 mM) to assess KCC2 function under a Cl− load. We observed that SNAP25(+/−) mice had depolarized EGABA and resting membrane potential compared to WT littermates (Figures 3D–3G).
Figure 3.

KCC2 protein levels and function are affected in mice following SNAP25 reduction
(A) Summary graph showing pan-KCC2, KCC2a, and KCC2b mRNA fold difference in SNAP25(+/−) (n = 4) and WT littermates (n = 5). Statistical significance was determined using an unpaired t-test with Welch’s correction.
(B) Immunoblot from SNAP25(+/−) mice (n = 5) and WT littermates (n = 7) probed with antibodies indicated on the right.
(C) Summary graph showing total KCC2 abundance for (B). Statistical significance was determined using an unpaired t-test with Welch’s correction.
(D) Summary graph showing EGABA values (not corrected for liquid junction potential) in SNAP25(+/−) mice (n = 13) and WT littermates (n = 18), and the corresponding (E) current-voltage plot, (F) sample current traces, and (G) resting membrane potential. n values represent individual cells obtained from at least four animals. Statistical significance for (D) and (G) was determined using an unpaired t-test with Welch’s correction.
(H) Schematic showing the timeline of stereotactic surgery in C57BL/6 mice.
(I) Linear map of plasmids used for transduction (top) and representative image showing the location of RFP expression in the cortex (bottom). Scale bar, 500 μm.
(J) Representative images from C57BL/6 mice cortex transduced with scrambled shRNA (scrSNAP25) or SNAP25 shRNA (shSNAP25-1, shSNAP25-2), and immunostained for KCC2 (gray). Scale bar, 20 μm.
(K) Summary graph showing KCC2 fluorescence intensity in neurons following scrSNAP25 (n = 40), shSNAP25-1 (n = 40) or shSNAP25-2 (n = 40) transduction. n values represent individual neurons obtained from four mice. Statistical significance was determined using Brown-Forsythe and Welch ANOVA tests and Dunnett’s T3 multiple comparisons test, with individual variances computed for each comparison.
(L) Summary graph showing EGABA values (not corrected for liquid junction potential) in RFP-positive (with shRNA expression) and RFP-negative (lacking shRNA expression) cells from mice transduced with RFP alone (RFP +ve, n = 19; RFP −ve, n = 18), scrSNAP25 (RFP +ve, n = 20; RFP −ve, n = 18), shSNAP25-1 (RFP +ve, n = 16; RFP −ve, n = 15) or shSNAP25-2 (RFP +ve, n = 15; RFP −ve, n = 15), and the corresponding (M) current-voltage plot and (N) sample current traces for RFP-positive cells only. n values represent individual cells obtained from six animals each. Statistical significance for (L) was determined using ordinary two-way ANOVA and Tukey’s multiple comparisons test, with single pooled variance. All graphs represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Using previously validated SNAP25 shRNAs, which we also validated (see Figures S1B and S1C), we performed a knockdown of SNAP25 in wild type C57BL/6 mice. To extend our findings from cultured cortical neurons (Figure 2) to neurons in vivo, we delivered adeno-associated virus (AAVs) containing shRNAs into the mouse cortex by stereotactic surgery (Figures 3H and 3I). Immunofluorescence analysis of brain tissue ∼28 days post-surgery showed a reduction in KCC2 following SNAP25 knockdown compared to those infected with scrambled shRNA (Figures 3J and 3K). Again, we asked whether this reduction in KCC2 protein led to a change in KCC2 function by recording EGABA. We observed that the SNAP25 knockdown also depolarized EGABA, indicating reduced KCC2 function and increased intracellular Cl− levels (Figures 3L–3N).
SNAP25 affects KCC2 surface expression
Since the expression of KCC2 on the cell surface is essential for its function, and given that KCC2 function is reduced following SNAP25 knockdown, we hypothesized that SNAP25 affects the cell surface expression of KCC2. We performed surface biotinylation to isolate cell surface proteins in neuro-2a cells transfected with KCC2 cDNA and scrambled/SNAP25 shRNAs. Similar to the reduction in total KCC2, we also observed a reduction in cell surface KCC2 following SNAP25 knockdown (Figures 4A–4C). Because the changes in surface KCC2 may be due to the total KCC2 reduction, we also performed dual immunostaining of surface and total KCC2 by utilizing a recombinant KCC2 cDNA with an extracellular FLAG tag. This allowed us to quantify the surface/total KCC2 in the same cell to assess if there are any differential changes in surface and total KCC2. We did not observe any difference in surface/total KCC2 following SNAP25 knockdown (Figures 4D and 4E) indicating that the reduction in surface KCC2 following SNAP25 knockdown in the surface biotinylation assay results from a reduction in total KCC2 in neuro-2a cells. Overexpression of SNAP25 also reduced cell surface KCC2 in neuro-2a cells (Figures 4F and 4H) without significant changes in total KCC2 (Figure 4G), which is in line with the emerging role of SNAP25 in coupling exocytosis and endocytosis.34,35
Figure 4.

SNAP25 decreases surface KCC2 in neuro-2a cells
(A) Immunoblot showing total and surface KCC2 in neuro-2a cells co-transfected with KCC2 and scrambled (scrSNAP25) or SNAP25 shRNA (shSNAP25-1). The blot was probed with the antibodies indicated on the right. Summary graph (n = 5) showing corresponding (B) total and (C) surface KCC2 abundance normalized to cells transfected with KCC2+scrSNAP25. Statistical significance was determined using an unpaired t-test with Welch’s correction. Asterisks denote significance from KCC2+scrSNAP25.
(D) Representative images (single z-plane) showing surface (gray) and total (green) KCC2 in neuro-2a cells co-transfected with KCC2 and scrSNAP25 (n = 41), shSNAP25-1 (n = 44) or shSNAP25-2 (n = 45). Red fluorescence represents transfected cells. n values indicate individual cells from three independent coverslips. Scale bar, 10 μm.
(E) Summary graph for (D) representing surface/total KCC2 normalized to cells transfected with KCC2+scrSNAP25. Statistical significance was determined using Kruskal-Wallis test and Dunn’s multiple comparisons test.
(F) Immunoblot showing total and surface KCC2 in neuro-2a cells co-transfected with KCC2 and RFP or SNAP25-P2A-RFP. The blot was probed with the antibodies indicated on the right. Summary graph (n = 8) showing corresponding (G) total and (H) surface KCC2 abundance normalized to cells transfected with KCC2+RFP. Statistical significance was determined using Mann-Whitney test. Asterisks denote significance from KCC2+RFP.
(I) Representative images (maximum intensity projection) showing PLA puncta (KCC2-SNAP25 interaction) in neuro-2a cells transfected with full length (KCC2-FL) KCC2 cDNA, and treated with DMSO (n = 106), PMA (n = 141) or Gö6983 (n = 134) and (J) summary graph showing the corresponding PLA/total KCC2 intensity. n values indicate individual cells from four independent coverslips. Statistical significance was determined using Kruskal-Wallis test and Dunn’s multiple comparisons test. Scale bar, 10 μm.
(K) Summary graph and (L) representative images (sum projection) showing PLA puncta intensity (KCC2-SNAP25 interaction) in neuro-2a cells transfected with KCC2 and treated with DMSO (n = 72) or PMA for 24 h (n = 79), 6 h (n = 72) and 1 h (n = 79). n values indicate individual cells from three independent coverslips. Statistical significance was determined using Kruskal-Wallis test and Dunn’s multiple comparisons test. Asterisks denote significance from DMSO. Scale bar, 10 μm.
(M) Summary graph and (N) representative images (sum projection) of PLA puncta intensity (KCC2-SNAP25 interaction) in cultured neurons treated with DMSO (n = 26), Gö6983 (48h, n = 29) or PMA (1h, n = 35). n values indicate individual cells from three independent cultures. Statistical significance was determined using Kruskal-Wallis test and Dunn’s multiple comparisons test. Asterisks denote significance from DMSO. Scale bar, 10 μm. All graphs represent mean ± SEM. ns - not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
The KCC2-SNAP25 interaction is PKC dependent
Both SNAP25 and KCC2 have been widely shown as substrates of PKC-mediated phosphorylation and undergo accompanied changes in protein interactions.22,36,37,38,39 PKC acts as the downstream effector of many cellular cascades affecting neuronal excitability40,41,42 and is known to affect cell surface expression of KCC2.38 To test whether the SNAP25-KCC2 interaction is dependent on PKC-mediated phosphorylation of either or both proteins, we performed a proximity ligation assay (PLA) in neuro-2a cells (expressing endogenous SNAP25) transfected with KCC2 cDNA and treated with PKC activator (PMA) or inhibitors (Gö6983, chelerythrine chloride). We observed a significant reduction in the KCC2-SNAP25 interaction (evidenced by reduced intensity of PLA puncta) following 60 h of Gö6983 (Figures 4I and 4J) or chelerythrine chloride treatment (Figure S2D), without any significant changes in the interaction following 60 h of PMA treatment (Figures 4I and 4J). Activated PKC is more sensitive to degradation by phosphatases compared to inactive, autoinhibited PKC.43,44 Therefore, prolonged treatment with potent PKC activators such as PMA may make it more susceptible to dephosphorylation and inactivation.45,46 We confirmed this by treating the cells with PMA for 1 h, 6 h, or 24 h, followed by PLA to assess the KCC2-SNAP25 interaction. We observed a significant increase in PLA intensity in cells treated with PMA for an hour compared to DMSO treated cells (Figures 4K and 4L). Similar results were observed in neurons treated with Gö6983 (48 h) or PMA (1 h), where we saw a reduction in PLA intensity following Gö6983 treatment and an increase in PLA intensity following PMA treatment compared to DMSO treated controls (Figures 4M and 4N).
The KCC2-SNAP25 interaction requires the KCC2-C terminus
The KCC2 intracellular C-terminal domain interacts directly with several proteins,47,48 and harbors the critical serine 940 residue phosphorylated by PKC, which is important for KCC2 surface expression.38 The intracellular N-terminal domain of KCC2 is also necessary for its membrane expression.49 Since SNAP25 affects the surface expression of KCC2, we sought to identify the intracellular domain of KCC2 involved in its interaction with SNAP25. We performed PLA in neuro-2a cells transfected with full-length (FL), C-terminus deleted (ΔC) or N-terminus deleted (ΔN) KCC2 cDNA (Figure 5A). We observed a significant reduction in the intensity of PLA puncta following the deletion of the KCC2 C-terminus (Figures 5B and 5C). We also observed a reduction in PLA puncta following the deletion of the KCC2 N-terminus (Figures 5D and 5E), but the magnitude of this reduction was lower compared to the C-terminus deletion. This indicates that the C-terminus of KCC2 plays a prominent role in mediating the KCC2-SNAP25 interaction. Since the N-terminus of KCC2 is crucial for its surface expression,49 the reduction in KCC2-SNAP25 interaction following N-terminus deletion may partly be due to altered localization of KCC2.
Figure 5.

KCC2 C-terminus is necessary for KCC2-SNAP25 interaction
(A) Schematic showing the linear structure of KCC2-FL, KCC2-ΔC, ΔN-KCC2 proteins. Created with BioRender.com.
(B) Representative images (maximum intensity projection) showing total KCC2 and PLA puncta fluorescence intensity in neuro-2a cells transfected with full length (KCC2-FL) (n = 99) and C-terminus deleted (KCC2-ΔC) (n = 80) KCC2 cDNA. Scale bar, 10 μm.
(C) Summary graph showing PLA/KCC2 intensity for (B). n values represent individual cells from four independent coverslips. Statistical significance was determined using Mann-Whitney test. Asterisks denote significance from KCC2-FL cells.
(D) Representative images (maximum intensity projection) showing total KCC2 and PLA puncta fluorescence intensity in neuro-2a cells transfected with full length (KCC2-FL) (n = 69) and N-terminus deleted (ΔN-KCC2) (n = 63) KCC2 cDNA. Scale bar, 10 μm.
(E) Summary graph showing PLA/KCC2 intensity for (D). n values represent individual cells from three independent coverslips. Statistical significance was determined using Mann-Whitney test. Asterisks denote significance from KCC2-FL cells. Immunoblot from neuro-2a cells transfected with (F) KCC2-ΔC or (G) ΔN-KCC2 and scrambled (scrSNAP25) or SNAP25 shRNA (shSNAP25-1). The blots were probed with the antibodies indicated on the right.
(H) Immunoblot from neuro-2a cells transfected with KCC2-FL and scrSNAP25 or shSNAP25-1 followed by treatment with DMSO, lactacystin or bafilomycin A1. The blots were probed with antibodies indicated on the right.
(I) Summary graph (n = 11) showing total KCC2-ΔC abundance from (F), normalized to cells transfected with KCC2-ΔC + scrSNAP25. Statistical significance was determined using an unpaired t-test with Welch’s correction. Asterisks denote significance from KCC2-ΔC + scrSNAP25.
(J) Summary graph (n = 6) showing total ΔN-KCC2 abundance from (G), normalized to cells transfected with ΔN-KCC2+scrSNAP25. Statistical significance was determined using an unpaired t-test with Welch’s correction. Asterisks denote significance from ΔN-KCC2+scrSNAP25. Summary graph showing KCC2 abundance in neuro-2a cells transfected with KCC2-FL and scrSNAP25 or shSNAP25 followed by treatment with (K) DMSO (n = 5), (L) lactacystin (n = 9) or (M) bafilomycin A1 (n = 9). Statistical significance was determined using an unpaired t-test with Welch’s correction for (K) and (L), and Mann-Whitney test for (M). Asterisks denote significance from KCC2+scrSNAP25. All graphs represent mean ± SEM. ns - not significant, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
To validate whether either of these intracellular domains is necessary for SNAP25-mediated regulation of KCC2, we asked whether SNAP25 knockdown affects KCC2-ΔC or ΔN-KCC2 protein abundance. Both KCC2-ΔC and ΔN-KCC2 monomers underwent reduction following knockdown of SNAP25 (Figures 5F, 5G, 5I, and 5J), indicating that neither of these domains are necessary for the regulation of KCC2 by SNAP25 and that this regulation may involve the interplay of multiple domains. Following this, we sought to identify the degradation pathway involved in the reduction of KCC2 following SNAP25 knockdown. Both lysosomal and proteasomal degradation pathways are involved in KCC2 degradation.50,51 So, we transfected neuro-2a cells with KCC2-FL cDNA and scrambled or SNAP25 shRNA followed by treatment with a lysosomal (bafilomycin A1, leupeptin) or proteasomal (lactacystin, MG-132) inhibitor. Treatment with both lysosomal and proteasomal inhibitors rescued SNAP25 knockdown induced reduction in KCC2 (Figures 5H, 5K–5M, and S3E–S3H) indicating that the reduction is KCC2 is mediated by the proteasomal and lysosomal degradation pathways.
SNARE complex proteins regulate KCC2 in neuro-2a cells
SNAP25 is an integral component of the SNARE complex which includes the VAMP and Syntaxin1 proteins.52 To investigate the involvement of other SNARE complex proteins in regulating KCC2, we performed a knockdown of common SNAP25 SNARE partners, VAMP1, VAMP2, Syntaxin1A (STX1A) or Syntaxin1B (STX1B) in neuro-2a cells and quantified the amount of exogenous KCC2. Neuro-2a cells were co-transfected with equal amounts of KCC2 cDNA, and scrambled shRNA or shRNAs targeting either SNAP25, VAMP1, VAMP2, STX1A or STX1B, followed by quantification of total KCC2 protein levels. We observed that a knockdown of these proteins also reduced KCC2 protein levels indicating the involvement of the SNARE complex in mediating KCC2 regulation (Figure 6).
Figure 6.

KCC2 is regulated by SNARE complex proteins
Immunoblot from neuro-2a cells transfected with KCC2 and scrambled shRNA (shScrambled), SNAP25 shRNA (shSNAP25-1), (A) VAMP1 shRNAs (shVAMP1-1, shVAMP1-2), or (B) VAMP2 shRNAs (shVAMP2-1, shVAMP2-2), or (C) STX1A shRNAs (shSTX1A-1, shSTX1A-2), or (D) STX1B shRNAs (shSTX1B-1, shSTX1B-2). The representative blot was probed with the antibodies indicated on the right. Summary graph (n = 3) showing total KCC2 abundance following (E) VAMP1 knockdown as shown in (A), (F) VAMP2 knockdown as shown in (B), (G) STX1A knockdown as shown in (C), (H) STX1B knockdown as shown in (D), normalized to shScrambled. Statistical significance was determined using ordinary one-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. Asterisks denote significance from shScrambled. All graphs represent mean ± SEM. ns - not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
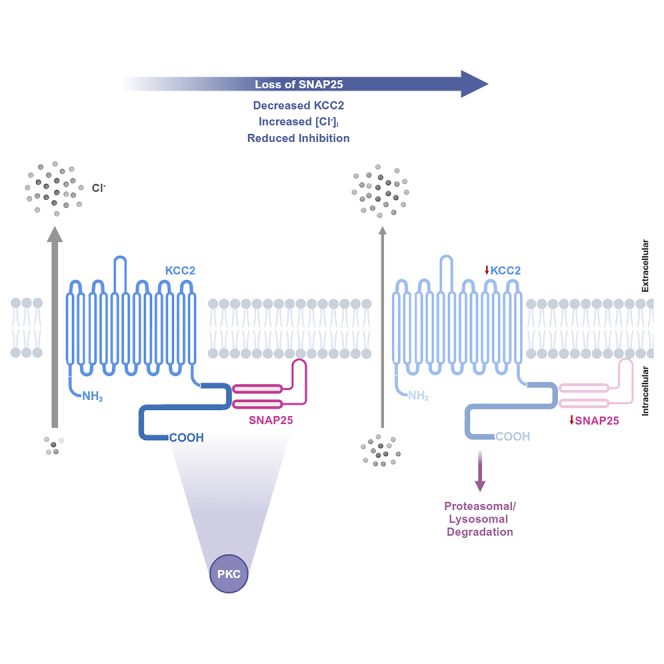
In this study we used co-immunoprecipitation and immunofluorescence to provide the first experimental demonstration that SNAP25 and KCC2 interact in neurons of the mouse brain. Moreover, we discovered that the SNAP25-KCC2 interaction regulates both KCC2 protein level and KCC2 function. By knocking down SNAP25 we found a reduction in both total and surface KCC2, and we recorded a depolarization of EGABA, indicative of a reduction in KCC2 function. The SNAP25-KCC2 interaction regulates KCC2 via PKC-mediated phosphorylation, and proteasomal and lysosomal degradation. This newly discovered mechanistic insight into KCC2 regulation is important for our understanding of both KCC2 biology and inhibitory synaptic transmission.
SNAP25 interacts with and regulates KCC2
We previously reported the KCC2-interactome in mouse brain using functional proteomics and performed an ingenuity pathway analysis to examine the interactome based on cellular functions.15 We found that KCC2 partners segregated into multiple nodes and that the node for receptor trafficking had the densest network. This suggested a tight link between KCC2 and the proteins in this node, which included SNAP25. In the present study, we provide the first biochemical characterization of this predicted KCC2-SNAP25 interaction, and moreover, demonstrate that this interaction regulates the protein level of KCC2 in both cell lines and neurons. In neurons, the knockdown of SNAP25 resulted in a reduction of KCC2, while SNAP25 overexpression did not significantly alter total endogenous KCC2 (S1 D, E). Since SNAP25 is highly expressed in neurons, increasing expression of this protein further would not necessarily have a functional impact, due to the ceiling effect (explained further).
SNAP25 affects KCC2 surface expression
Since the homozygous SNAP25 knockout is neonatally lethal,32 we used either SNAP25(+/−) mice or performed a knockdown of SNAP25 in wild-type C57BL/6 mice; in both models we observed a reduction in KCC2 and a corresponding increase in intracellular Cl− levels. In vertebrates, the SNAP25 protein sub-family consists of SNAP25, SNAP23, SNAP29, and SNAP47.20 Both SNAP25 and its ubiquitous homolog SNAP23 are involved in the insertion of postsynaptic membrane proteins.20 Rescue experiments have previously shown that SNAP23 can partly substitute for SNAP25.53,54 It was also recently shown that SNAP23 regulates KCC2 membrane insertion and activity,55 but it was unknown if SNAP25, which is expressed at relatively higher levels in cortical and hippocampal neurons32,56 was involved in KCC2 regulation. We used a previously validated SNAP25 specific shRNA (shSNAP25-1) that doesn’t affect SNAP23,57 which suggests that SNAP23 in neurons can’t completely compensate for the lack of SNAP25 in maintaining KCC2 protein level or function.
SNAP25 knockdown reduced both surface and total KCC2, but SNAP25 overexpression in neuro-2a cells also reduced surface KCC2 without an accompanied change in total KCC2. In neuro-2a cells, knockdown of SNAP25 interfered with KCC2 protein levels via increased lysosomal and proteasomal degradation, which led to an overall reduction in both total and surface KCC2. Therefore, we could not separate the effect of SNAP25 knockdown on KCC2 surface expression. However, following SNAP25 overexpression in neuro-2a cells, total KCC2 protein levels remained unaltered, whereas a significant reduction in surface KCC2 expression was observed. This SNAP25-mediated robust reduction in KCC2 surface expression likely results from the enhanced trafficking of KCC2 away from the cell surface, which further substantiates the emerging role of SNAP25 in coupling exocytosis and endocytosis.34,35 Additionally, SNAP25 is involved in the regulation and membrane expression of NMDA receptors,21,22 kainate-type receptors,23 and voltage-gated calcium channels.58,59,60,61 Thus, while seemingly paradoxical, both overexpression and knockdown of SNAP25 in neuro-2a cells results in KCC2 loss of function. Taken together with our findings, this also indicates that SNAP25 plays a global role in regulating the excitability of neurons postsynaptically.
Botulinum neurotoxins can effectively cleave SNARE complex proteins62,63 and treatment with botulinum toxins that specifically cleave SNAP25 cause neuronal death in vitro.64 However, cleavage of SNAP25, or its SNARE partners using botulinum toxins also revealed a major role played by these proteins during inflammation and injury, specifically by altering the membrane trafficking of transient receptor potential (TRP) ion channels in sensory neurons.65 These channels relay sensory information and are implicated in the transduction of pain signals.65 KCC2 also plays a major role in the pathology of traumatic injury, and attenuation of KCC2 expression is observed in the spinal cord motorneurons following injury.14,66 This has made KCC2 an important target for the recovery of motor function following spinal cord injury.14 Our results indicating the regulation of KCC2 by SNAP25, and possibly by the SNARE complex, provide additional insights into the known therapeutic role of botulinum toxins following injury.
PKC mediated KCC2-SNAP25 interaction requires C- terminus
Both KCC2 and SNAP25 are direct phosphorylation targets of PKC,22,38,61 and PKC-mediated phosphorylation increases SNAP25-mediated exocytosis21,22 and KCC2 membrane insertion,38 independently. Serine-940 (S940), which is located on the KCC2 intracellular C-terminus, is directly phosphorylated by PKC and mediates the activity-dependent regulation of this transporter.38 In hippocampal neurons, PKC-mediated phosphorylation of S940 increases the cell surface stability and activity of KCC2.38 Conversely, the use of PKC inhibitors prevents oxytocin-mediated upregulation of KCC2 activity.67
We observed that treatment with PKC activator (PMA) and inhibitors (Gö6983, chelerythrine chloride) increased and reduced the KCC2-SNAP25 interaction, respectively. This indicates that the KCC2-SNAP25 interaction is phosphorylation dependent. We also saw a reduction in the KCC2-SNAP25 interaction following the deletion of KCC2 N-/C-terminus. It has been previously shown that KCC2 N- and C-terminus are necessary for the membrane insertion and membrane stability of this transporter, respectively.49 Additionally, the C-terminal domain of KCC2 is involved in its oligomerization68 and contains PKC-mediated phosphorylation sites that affect its surface expression.38 Therefore, the direct lack of these domains along with indirect changes in KCC2 localization, protein structure,69 and activity may regulate the level of the KCC2-SNAP25 interaction.
KCC2 degradation pathway following SNAP25 knockdown
KCC2 is degraded by lysosomal or proteasomal pathways, depending on the cellular context.50,51 Similarly, we observed that both lysosomal and proteasomal inhibitors could rescue SNAP25 knockdown-induced KCC2 degradation.
SNAP25 affects the function of membrane proteins through both SNARE-dependent, and presumably SNARE-independent mechanisms.21,23,28 The core SNARE complex is formed by the association of SNAP25-Syntaxin-VAMP proteins.70,71 Even though there are multiple isoforms of VAMP and Syntaxin proteins, VAMP1 and VAMP2 are specifically expressed in the brain where they are known to mediate vesicular fusion,70,72,73 and syntaxin1A and 1B are shown to have high affinity to SNAP25.56,74 We show that other components of the neuronal SNARE complex, including VAMP1/2 and Sytaxin1A/B can also regulate KCC2 in neuro-2a cells, indicating the possibility that KCC2 regulation is mediated by the SNARE complex.
The high expression of SNAP25 in neurons and/or the involvement of other SNARE complex proteins in regulating KCC2, may explain the ceiling effect observed with SNAP25 overexpression. If regulation of KCC2 by SNAP25 is mediated by the SNARE complex, overexpression of one of its components (i.e., SNAP25) may not be sufficient to increase the function of this complex. For example, membrane insertion of NMDARs and AMPARs require the SNAP25-syntaxin4-VAMP1 and SNAP25-syntaxin1A/B-VAMP2 SNARE complex, respectively.21,57 Therefore, overexpression of other SNARE components including syntaxin1 and VAMP proteins may be required in combination with SNAP25 to increase KCC2 protein levels.
The complex interplay of other KCC2 interacting proteins may also be responsible for this regulation. For example, 14-3-3 proteins, which are known for binding to phosphorylated targets, were also enriched in the receptor trafficking node of the KCC2 interactome.15 Additionally, both SNAP25 and KCC2 are critical for the formation and maintenance of dendritic spines.47,75,76 It is established that SNAP25 affects dendritic spine formation through its interaction with the p140cap/SRCIN1 protein,76 which also showed up as a high confidence interactor of KCC2.15 Therefore, it is possible that the KCC2-SRCIN1-SNAP25 complex may be involved in the formation and maintenance of dendritic spines. This complex may also recruit additional molecular adaptors such as 14-3-3 proteins for the regulation/stabilization of this complex and therefore mediate the non-canonical functions of KCC2. This would also explain the ceiling effect observed with SNAP25 overexpression, as enhanced delivery of KCC2 to the cell surface requires additional adaptor proteins and cytoskeletal elements to stabilize KCC2 at the surface and reduce the protein turnover.
The existence of higher order KCC2 oligomers16,77 further opens the possibility that these proteins may be a part of a larger macromolecular complex containing other proteins responsible for the membrane insertion, anchorage, or removal of KCC2 depending on the cellular context. Real-time assays investigating the role of SNAP25 in dynamic trafficking of KCC2 is necessary to further untangle the specifics of this regulation. To conclude, we identified SNAP25 as a major component in regulating KCC2 protein and function, and our findings have identified a potential therapeutic target that could improve the efficacy of synaptic inhibition in disease states.
Limitations of the study
Our current study demonstrates the role of SNARE-complex protein SNAP25 in regulating KCC2 protein level and function. Even though we have shown that other components of the SNARE-complex, Syntaxin1, and VAMP proteins are involved in the regulation of KCC2 in neuro-2a cells, detailed investigation of the mechanisms of this regulation in neurons is necessary. This includes investigating the interaction of KCC2 with other SNARE-complex proteins and using dynamic assays to track the insertion and/or removal of KCC2 at the cell membrane. We have extensively investigated the effects of SNAP25 knockdown on KCC2 protein level and function, but changes in surface expression and function of KCC2 following SNAP25 overexpression needs to be further explored. This is also important as we saw a significant reduction in surface KCC2 following SNAP25 overexpression.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Melanie A. Woodin (m.woodin@utoronto.ca).
Materials availability
Plasmids generated in this study (pAAV-Syn-RFP, pAAV-Syn-SNAP25-P2A-RFP, pAAV-U6-scrambledshRNA-Syn-RFP, pAAV-U6-shSNAP25-1-Syn-RFP, pAAV-U6-shSNAP25-2-Syn-RFP) are available from the lead contact with a completed materials transfer agreement.
Data and code availability
-
•
All data reported in this study will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Acknowledgments
We thank Dr. Pavel Uvarov (University of Helsinki) for the KCC2-Myc and KCC2-ΔC constructs, Dr. Keith Murai (McGill University) for the GFP construct, Dr. Michela Matteoli (University of Milan) for the GFP-SNAP25 construct, Dr. Christophe Mulle (University of Bordeaux) for the GluK2(GluR6a)-Myc construct, Dr. Richard Huganir (Johns Hopkins University School of Medicine) for the SNAP25 (shRNA-resistant), scrambled shRNA (scrSNAP25) and shSNAP25-1 constructs, Dr. Jean Christophe Poncer (INSERM) for the KCC2-FLAG (extracellular) construct, and the CNP Viral Vector Core at the CERVO Research Center for preparing the Syn-SNAP25-P2A-RFP, U6-scrSNAP25-Syn-RFP, U6-shSNAP25-1-Syn-RFP, and U6-shSNAP25-2-Syn-RFP plasmids and packaging them into respective AAVs. We would also like to acknowledge the technical support provided by Kelly Chen, Vivarium Research Technician, University of Toronto for stereotactic surgeries. This research was funded by a research grant from the Canadian Institutes of Health Research (CIHR FRN 190000) to M.A.W.
Author contributions
Conceptualization, V.A.R., J.C.P., and M.A.W.; methodology, V.A.R., A.A., J.C.P., J.A.M., and M.A.W.; investigation, V.A.R., A.A., M.S., M.C., S.M., and M.S.R.; writing—original draft, V.A.R., A.A., J.C.P., and M.A.W.; writing—review and editing, M.A.W.; visualization, V.A.R. and A.A.; supervision, M.A.W.; funding acquisition, M.A.W.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-KCC2 | Sigma-Aldrich | Cat#07–432; RRID: AB_310611 |
| Mouse monoclonal anti-KCC2 (N1/12) | NeuroMab | Cat#75-013; RRID: AB_10672851 |
| Rabbit polyclonal anti-KCC2 | Abcam | Cat#ab97502; RRID: AB_10866258 |
| Mouse monoclonal anti-SNAP25 | Synaptic Systems | Cat#111011; RRID: AB_887794 |
| Rabbit polyclonal anti-SNAP25 | Abcam | Cat#ab5666; RRID: AB_305033 |
| Rabbit polyclonal anti-β-actin | Cell Signaling Technology | Cat#4967S; RRID: AB_330288 |
| Mouse monoclonal anti-Transferrin Receptor | Invitrogen | Cat#13–6800; RRID: AB_2533029 |
| Chicken polyclonal anti-MAP2 | EMD Millipore | Cat#AB5543; RRID: AB_571049 |
| Mouse monoclonal anti-TUJ1 | BioLegend | Cat#801202; RRID: AB_2728521 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Purified Mouse IgG | Antibodies Inc | Cat#43-637-0010; RRID: AB_2797169 |
| Goat anti-Mouse IgG, Alexa Flour 488 | Thermo Fisher | Cat#A-11029; RRID: AB_2534088 |
| Goat anti-Mouse IgG, Alexa Flour 555 | Thermo Fisher | Cat#A-21422; RRID: AB_2535844 |
| Goat anti-Mouse IgG, Alexa Flour 647 | Thermo Fisher | Cat#A-21235; RRID: AB_2535804 |
| Goat anti-Rabbit IgG, Alexa Flour 488 | Thermo Fisher | Cat#A-11008; RRID: AB_143165 |
| Goat anti-Rabbit IgG, Alexa Flour 555 | Thermo Fisher | Cat#A-21428; RRID: AB_2535849 |
| Goat anti-Rabbit IgG, Alexa Flour 647 | Thermo Fisher | Cat#A-21244; RRID: AB_2535812 |
| Goat anti-Chicken IgY, Alexa Flour 647 | Thermo Fisher | Cat#A-21449; RRID: AB_2535866 |
| Goat anti-Mouse IgG, HRP-linked | Cell Signaling Technology | Cat#7076S; RRID: AB_330924 |
| Goat anti-Rabbit IgG, HRP-linked | Cell Signaling Technology | Cat#7074S; RRID: AB_2099233 |
| Bacterial and virus strains | ||
| AAV2/DJ-Syn-RFP | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| AAV2/DJ-U6-scrambledshRNA-Syn-RFP (also called scrSNAP25) | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| AAV2/DJ-U6-shSNAP25-1-Syn-RFP (also called shSNAP25-1) | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| AAV2/DJ-U6-shSNAP25-2-Syn-RFP (also called shNSAP25-2) | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| AAV2/DJ-Syn-SNAP25-P2A -RFP | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Phorbol 12-Myrisate 13-Acetate (PMA) | BioShop | Cat#PMA168.1 |
| Gö 6983 | Sigma-Aldrich | Cat#G1918-500UG |
| B-27 Supplement (50×), serum free | Gibco | Cat#17504044 |
| EZ-Link Sulfo-NHS-SS-Biotin | Thermo Fisher | Cat#21331 |
| Lactacystin | HelloBio | Cat#HB3953 |
| Bafilomycin A1 | Cell Signaling Technology | Cat#54645S |
| Chelerythrine chloride | HelloBio | Cat#HB0190 |
| MG-132 | HelloBio | Cat#HB4135 |
| Leupeptin hemisulfate | Cell Signaling Technology | Cat#73618S |
| Critical commercial assays | ||
| Duolink In Situ PLA Probe Anti-Mouse PLUS | Sigma-Aldrich | Cat#DUO92001-100RXN |
| Duolink In Situ PLA Probe Anti-Rabbit MINUS | Sigma-Aldrich | Cat#DUO92005-100RXN |
| Duolink In Situ Detection Reagents FarRed | Sigma-Aldrich | Cat#DUO92013-100RXN |
| Experimental models: Cell lines | ||
| COS-7 cells | ATCC | CRL-1651; RRID: CVCL_0224 |
| Neuro-2a cells | ATCC | CCL-131; RRID: CVCL_0470 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: B6.129X1-Snap25tm1Mcw/J | The Jackson Laboratory | RRID: IMSR_JAX:004863 |
| Oligonucleotides | ||
| Primer: KCC2a, Forward: GCCGGCTCCCGAGGGAAG, Reverse: TGAAGGGACTGCTCTCTTTGG | Uvarov et al.78 | N/A |
| Primer: KCC2b, Forward: GCCACCATGCTCAACAACCT, Reverse: TGAAGGGACTGCTCTCTTTGG | Uvarov et al.78 | N/A |
| Primer: pan-KCC2-1, Forward: GGCATTCTTCCAGGCAGTAG, Reverse: CCCTAATTGGTGTCGATGCT | Cheung et al.66 | N/A |
| Primer: pan-KCC2-2, Forward: TCAGTCACAGGGATCATGG, Reverse: GGATAGTTCCAGTAGGGATAGAC | Bhandage et al.79 | N/A |
| Primer: GAPDH, Forward: GCAAAGTGGAGATTGTTGCCAT, Reverse: CCTTGACTGTGCCGTTGAATTT | Shulga et al.80 | N/A |
| Recombinant DNA | ||
| KCC2-Myc | Dr. Pavel Uvarov, University of Helsinki | N/A |
| GFP | Dr. Keith Murai, McGill University | N/A |
| GFP-SNAP25 | Dr. Michela Matteoli, University of Milan | N/A |
| GluK2-Myc | Dr. Christophe Mulle (Bordeaux University) | N/A |
| SNAP25 (shRNA-resistant) | Dr. Richard Huganir, Johns Hopkins University School of Medicine | N/A |
| scrambled shRNA (also called scrSNAP25) | Dr. Richard Huganir, Johns Hopkins University School of Medicine | N/A |
| shSNAP25-1 | Dr. Richard Huganir, Johns Hopkins University School of Medicine | N/A |
| TRCN0000334139 (sh#139) | Sigma-Aldrich | TRCN0000334139 |
| TRCN0000348198 (sh#198) | Sigma-Aldrich | TRCN0000348198 |
| TRCN0000348199 (sh#199, also called shSNAP25-2) | Sigma-Aldrich | TRCN0000348199 |
| TRCN0000348200 (sh#200) | Sigma-Aldrich | TRCN0000348200 |
| TRCN0000110592 (sh#592) | Sigma-Aldrich | TRCN0000110592 |
| TRCN0000110586 (shVAMP1-1) | Sigma-Aldrich | TRCN0000110586 |
| TRCN0000110587 (shVAMP1-2) | Sigma-Aldrich | TRCN0000110587 |
| TRCN0000306004 (shVAMP2-1) | Sigma-Aldrich | TRCN0000306004 |
| TRCN0000325526 (shVAMP2-2) | Sigma-Aldrich | TRCN0000325526 |
| TRCN0000380322 (shSTX1A-1) | Sigma-Aldrich | TRCN0000380322 |
| TRCN0000236762 (shSTX1A-2) | Sigma-Aldrich | TRCN0000236762 |
| TRCN0000110496 (shSTX1B-1) | Sigma-Aldrich | TRCN0000110496 |
| TRCN0000110497 (shSTX1B-2) | Sigma-Aldrich | TRCN0000110497 |
| pAAV-Syn-RFP | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| pAAV-U6-scrambledshRNA-Syn-RFP (also called scrSNAP25) | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| pAAV-U6-shSNAP25-1-Syn-RFP (also called shSNAP25-1) | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| pAAV-U6-shSNAP25-2-Syn-RFP (also called shSNAP25-2) | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| pAAV-Syn-SNAP25-P2A-RFP | CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477) | N/A |
| KCC2-FLAG (extracellular) | Dr. Jean Christophe Poncer, INSERM | N/A |
| KCC2-ΔC | Dr. Pavel Uvarov, University of Helsinki | N/A |
| ΔN-KCC2 | GenScript | N/A |
| Software and algorithms | ||
| ImageJ (FIJI) | Schindelin et al.81 | https://fiji.sc/ |
| Imaris 7.6 | Oxford Instruments | https://imaris.oxinst.com/ |
| BioRender | BioRender | https://biorender.com |
| GraphPad Prism 10 | GraphPad Software | https://www.graphpad.com/ |
| Image Lab 6.1 | Bio-Rad | https://www.bio-rad.com/ |
| Adobe Photoshop CC 2023 | Adobe | https://www.adobe.com/ca/creativecloud.html |
| Adobe Illustrator CC 2023 | Adobe | https://www.adobe.com/ca/creativecloud.html |
Experimental model and study participant details
Animals and approvals
All procedures were performed with the approval of, and in accordance with, the guidelines from the University of Toronto Animal Care Committee and the Canadian Council on Animal Care. Wildtype C57BL/6J (JAX: 000664) and SNAP25(+/−) (B6.129X1-Snap25tm1Mcw/J, JAX: 004863) mice were originally purchased from the Jackson Laboratory. Mice were kept in individually ventilated cages (Techniplast GM500) in a temperature-controlled room. Maximum of 4 mice were housed in a cage with a floor area of 501 cm2 with corncob bedding, and mixed enrichment of nestlet, crinkle paper and hut. The cages were maintained on a 12 h light/dark cycle with ad libitum access to food and water. Genotyping protocol provided by the Jackson Laboratory was used for the maintenance of the SNAP25(+/−) mice. Animals of both sexes were used to prepare the cortical cultures (P0-2) and male mice (∼P21) were used for stereotactic surgeries. Littermates were randomly assigned to experimental groups.
Primary neuronal culture
Cortical region from P0-2 C57BL/6 pups (tissue from littermates of both sexes were pooled together) or SNAP25(+/−) pups (each pup and their wildtype littermates of both sexes were cultured individually) were dissected out in ice-cold dissection medium [D(+)glucose, 25 μM; sucrose, 5 μM; HEPES, 15 μM in HBSS (without Ca2+ or Mg2+) at pH 7.4] and triturated into single cell suspension following 30 min incubation in 0.05% trypsin-EDTA (Gibco, 25300062) at 37°C. Cells were plated onto poly-D-Lysine (Gibco, A3890401) coated coverslips placed in plating medium [sodium pyruvate, 1 mM; Glutamax (Gibco, 35050061), 1×; fetal bovine serum (Gibco, 12484028), 10% in DMEM medium (Gibco, 11995065)] and replaced with Neurobasal-A growth medium (Gibco, 10888022) supplemented with 1× Glutamax, 2% B27 (Gibco, 17504044), 2% penicillin-streptamycin (Sigma-Aldrich, P4333) after 3–4 h at 37°C. The neurons were maintained at 37°C and 5% CO2, and 30% growth medium was replaced every 3 days. Cells were transfected using Lipofectamine-2000 (Invitrogen, 11668019) or Lipofectamine-3000 (Invitrogen, L3000015) as per manufacturer supplied protocol, and experiments were carried out between DIV 12–15.
COS-7 and neuro-2a culture
COS-7 (ATCC, CRL-1651) and neuro-2a (ATCC, CCL-131) cells were maintained at 37°C and 5% CO2 in DMEM medium (Gibco, 11995065) supplemented with 10% fetal bovine serum (Gibco, 12484028) and 1% penicillin-streptomycin (Sigma-Aldrich, P4333). Cells were plated in 6-well (Sarstedt, 83.3920) or 12-well (Sarstedt, 83.3921) plates for experiments and were transfected at 50–70% confluency using Lipofectamine-2000 (Invitrogen, 11668019) or Lipofectamine-3000 (Invitrogen, L3000015) as per manufacturer supplied protocol. All experiments performed on neuro-2a cells were done in an undifferentiated state.
Method details
Stereotactic surgery
AAV2/DJ-Syn-RFP, AAV2/DJ-U6-scrambledshRNA-Syn-RFP, AAV2/DJ-U6-shSNAP25-1-Syn-RFP and AAV2/DJ-U6-shSNAP25-2-Syn-RFP viral vectors were packaged by the Molecular Tools Platform, CERVO Brain Research Center at Laval University. Mice were maintained on isoflurane anesthesia and viral vectors were bilaterally infused into the cortex (AP: +2.15, DV: −1.65, ML: ±1.9) in a total volume of 1 μL (titer 1012 GC/mL). The infusion was done at a rate of 50 nL/min by pressure ejection using a cannula connected by Tygon tubing to a 10 μL Hamilton syringe mounted on an infusion pump. Following surgery, animals were group-housed, and experiments began following ∼28 days.
Brain slice preparation
Mice were deeply anesthetized with a mix of ketamine (150 mg/kg) and xylazine (15 mg/kg), and transcardially perfused with 20 mL of ice-cold carbogenated (95% O2, 5% CO2) N-methyl-D-glucamine (NMDG)-based slicing solution containing (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-glucose, 2 thiourea, 5 sodium ascorbate, 3 sodium pyruvate, 0.5 CaCl2, and 10 MgSO4 (pH 7.4, 290–300 mOsm). After decapitation, the brain was placed into ice-cold NMDG-HEPES-aCSF and coronal cortical slices (300 μm) were obtained using a compresstome VF-300-0Z (Precisionary Instruments LLC, MA). Slices were transferred into a chamber (30°C–32°C) filled with NMDG-HEPES-aCSF for 12 min, followed by incubation in carbogenated (95% O2, 5% CO2) HEPES-holding aCSF (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-glucose, 2 thiourea, 5 sodium ascorbate, 3 sodium pyruvate, 2 CaCl2·2H2O, and 2 MgSO4·7H2O (pH 7.3, 300–305 mOsm) for an hour at room temperature. Slices were then transferred to the recording chamber (Warner Instrument) and perfused (2–3 mL/min) with carbogenated (95% O2, 5% CO2) standard aCSF containing (in mM): 125 NaCl, 25 D-glucose, 26 NaHCO3, 2 CaCl2, 1 MgSO4, 2.5 KCl and 1.25 NaH2PO4 (pH 7.4, 290–300 mOsm) and maintained at room temperature.
Patch clamp recording
For gramicidin-perforated patch clamp recordings, cultured cortical neurons were transduced at DIV 7 and recordings were made between DIV 12–15. Healthy neurons with oval/pyramidal cell bodies and positive for RFP fluorescence (where applicable) were used for recordings. Glass pipettes (4–6 MΩ) were filled with an intracellular solution containing (in mM): 150 KCl, 10 HEPES (pH 7.4, 310 mOsm) and gramicidin (50 μg/mL). Cells were continuously perfused (at ∼1 mL/min) with an extracellular solution at room temperature containing (in mM): 120 NaCl, 2 KCl, 2 CaCl2.2H2O, 3 MgCl2.6H2O, 10 HEPES, and 20 glucose (pH 7.4 with NaOH, 290 mOsm). Recordings were amplified with Multiclamp 700B (Molecular Devices) and digitized using an Axon Digidata 1440 (Molecular Devices). EGABA values were determined from the reversal potential of currents activated by the puffing of muscimol (10 μM) onto the neuronal soma and holding the membrane potential in 10 mV increments from −110 to −60 mV. EGABA values have not been corrected for liquid junction potential of ∼4.3 mV. Intracellular Cl− concentration was calculated using the Nernst equation: ECl- = RT/zF.ln [Cl−]out/[Cl−]in.
For slice-electrophysiology with Cl− loading, whole-cell voltage-clamp recordings were obtained from layer 5 neurons using borosilicate glass pipettes (5–6 MΩ) filled with an intracellular solution containing (in mM): 110 K+-Gluconate, 30 KCl, 0.2 EGTA, 10 HEPES, 4 MgATP, 0.3 NaGTP and 10 Na2-phosphocreatine (pH 7.3, 290–295 mOsm). Recordings were obtained in the presence of DL-2-amino-5-phosphonopentanoic acid (DL-AP5; 50 μM) and 6,7-dinitroquinoxaline-2, 3-dione (DNQX; 20 μM) to block NMDA and AMPA receptors, respectively. To record EGABA from shRNA transduced slices, neurons were held at −80 mV under whole-cell voltage clamp, and the membrane potential was increased in +10 mV increments from −110 to −10 mV. To record EGABA from SNAP25(+/−) slices, neurons were held at −70 mV under whole-cell voltage clamp, and the membrane potential was increased in +10 mV increments from −100 to 0 mV. Local electrical stimulation was used during each membrane potential step, to activate inhibitory postsynaptic currents (IPSCs). Signals were amplified using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) and digitized at 10 kHz using Digidata 1550b and pClamp 11.1 software (Molecular Devices, Sunnyvale, CA, USA). Recordings were low pass filtered at 2 kHz. Access resistance was monitored throughout experiments and data were included only if the holding current and series resistance were stable (changes <25% of initial value). Values have not been corrected for whole cell liquid junction potential of 3.9 mV. All chemicals were purchased from Sigma (Oakville, ON) unless otherwise specified.
DNA constructs
KCC2-Myc and KCC2-ΔC constructs were a gift from Dr. Pavel Uvarov, University of Helsinki. GFP construct was a gift from Dr. Keith Murai, McGill University. GFP-SNAP25 was a gift from Dr. Michela Matteoli, University of Milan. GluK2 (GluR6a)-Myc was a gift from Dr. Christophe Mulle, University of Bordeaux, France. SNAP25 (shRNA-resistant), scrambled shRNA or scrSNAP25 (sense, 5′-GAT CCC CGC GCG CTT TGT AGG ATT CGT TCA AGA GAC GAA TCC TAC AAA GCG CGC TTT TTA-3′) in pSuper vector and shSNAP25-1 (sense, 5′-GAT CCC CGG CTT CAT CCG CAG GGT AAT TCA AGA GAT TAC CCT GCG GAT GAA GCC TTT TTC-3′) in pSuper vector were a gift from Dr. Richard Huganir, Johns Hopkins University School of Medicine. Additional SNAP25 shRNAs, TRCN0000334139 (sh#139), TRCN0000348198 (sh#198), TRCN0000348199 (sh#199, also called shSNAP25-2), TRCN0000348200 (sh#200), TRCN0000110592 (sh#592) were obtained from Sigma-Aldrich. The SNAP25, scrSNAP25, shSNAP25-1, shSNAP25-2 sequences were further subcloned as Syn-SNAP25-P2A-RFP (also called SNAP25-P2A-RFP), U6-scrSNAP25-Syn-RFP (also called scrSNAP25), U6-shSNAP25-1-Syn-RFP (also called shSNAP25-1), U6-shSNAP25-2-Syn-RFP (also called shSNAP25-2) respectively and packaged into AAVs (AAV2/DJ) by the CNP Viral Vector Core at the CERVO Research Center (RRID:SCR_016477). KCC2-FLAG (extracellular) was a gift from Dr. Jean Christophe Poncer, INSERM, France. ΔN-KCC2 was created from KCC2-Myc via deletion of 100 aa from the N-terminus of KCC2 by GenScript. VAMP1 shRNAs (shVAMP1-1, TRCN0000110586; shVAMP1-2, TRCN0000110587), VAMP2 shRNAs (shVAMP2-1, TRCN0000306004; shVAMP2-2, TRCN0000325526), STX1A shRNAs (shSTX1A-1, TRCN0000380322; shSTX1A-2, TRCN0000236762) and STX1B shRNAs (shSTX1B-1, TRCN0000110496; shSTX1B-2, TRCN0000110497) were obtained from Sigma-Aldrich.
RT-qPCR
RNA was isolated from the cortex of SNAP25(+/−) or WT littermate mice (P60). Briefly, 50 mg frozen brain tissue was powdered and resuspended in 500 μL TRIzol reagent (Ambion, 15596018), incubated at room temperature for 5 min and centrifuged at 12000 × g, 4°C for 10 min. The supernatant was mixed with 100 μL chloroform (Sigma-Aldrich, 472476) and centrifuged at 12,000 × g, 4°C for 15 min. The RNA was precipitated using 250 μL isopropanol through overnight incubation at −20°C, and pelleted by centrifugation at 12,000 × g, 4°C for 10 min. The RNA pellet was washed with 500 μL 75% ethanol and air-dried for 20 min. The pellet was dissolved in 30 μL RNAse-free water (Qiagen, 56601542) and incubated at 55°C for 10 min. RNA sample at 200 ng/μL was mixed with 0.1 volume 10× TURBO DNase Buffer (TURBO DNA-free Kit, ThermoFisher Scientific, AM1907) and incubated at 37°C for 30 min with 3 μL TURBO DNase, followed by additional incubation for 30 min with 2 μL TURBO DNase. The samples were incubated with 0.2 volume TURBO DNase inactivator for 5 min at room temperature, and centrifuged at 10,000 × g, room temperature for 1.5 min.
High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814) and 2000 ng DNase-treated RNA sample was used for cDNA preparation. For each sample, negative control was prepared by substituting reverse transcriptase with water. The samples were incubated in a thermocycler under the following conditions: 25°C, 10 min; 37°C, 2 h; 85°C, 5 min; 4°C, ∞. For qPCR, SYBR Select Master Mix (Applied Biosystems, 4472908) and the following amplification conditions (Thermocycler: Bio-Rad CFX Opus 384) were used: 95°C, 3 min; 40 cycles of 95°C, 10 s and 60°C, 30 s (includes plate read); 95°C, 10 s; melt curve analysis with 0.5°C increment for 5 s (includes plate read) from 65°C to 95°C. See key resources table for detailed list of primers used. The resulting Cq values were imported using the Bio-Rad CFX Maestro Software. ΔΔCt method was used to determine relative fold change of mRNA.
Co-immunoprecipitation
In vivo co-immunoprecipitation was performed as per previously published protocol.15 All biochemical preparations and centrifugations were done at 4°C. C57BL/6 mouse brain cortex (P50) was homogenized in ice-cold lysis buffer [Tris-HCl, 50 mM, pH 7.4; EDTA, 1 mM; protease (Roche, 11836170001) and phosphatase inhibitor tablets (Roche, 4906837001)] using pellet pestle and centrifuged for 30 min at 21,000 × g. The membrane pellet was re-suspended in ice-cold solubilization buffer (4Xw/v) [Tris·HCl, 50 mM, pH 7.4; NaCl, 150 mM; EDTA, 0.05 mM; C12E9, 1.5%; protease (Roche, 11836170001) and phosphatase inhibitor tablets (Roche, 4906837001)] and solubilized by rotating (10 RPM) for 3 h at 4°C, and centrifuged for 1 h at 21,000 × g. Total protein estimation was performed on protein supernatant using DC Protein Assay Kit (BioRad, 5000112). For immuno-precipitation, 1000 μg total protein was incubated with 5 μg anti-KCC2 antibody (Mouse, NeuroMab, 75-013) or normal IgG (Mouse, Antibodies Inc, 43-637-0010) for overnight at 4°C with rotation (10 RPM). The samples were then incubated in 30 μL pre-washed GammaBind G Sepharose beads (GE Healthcare Life Sciences, 17-0885-01) for an hour at 4°C. The beads were then washed twice with normal solubilization buffer, and twice with solubilization buffer without 1.5% C12E9 detergent. The bound proteins were eluted using 30 μL 6× SDS buffer (Tris, pH 6.8, 375 mM; sodium dodecyl sulfate, 12% w/v; glycerol, 60% v/v; bromophenol blue, 0.3% w/v) containing 50 mM DTT. Similarly, 5% input and unbound fractions were prepared. The samples were heated at 37°C for an hour and resolved via SDS-poly-acrylamide gel electrophoresis (PAGE) using 10% acrylamide gels followed by western blot analysis.
Immunofluorescence
Cultured neurons were fixed 48 h post-transfection using 4% PFA in PBS for 15 min, followed by permeabilization for 5 min with 0.5% Triton X-100. The cells were blocked with 10% normal goat serum in PBS for an hour and incubated in appropriate concentration of primary antibody (see Table S1) diluted in blocking solution for 2 h. The cells were then incubated in Alexa Flour conjugated secondary antibodies (see key resources table) for an hour, followed by a quick rinse in DAPI solution and mounted using ProLong Diamond (Invitrogen, P36970) or ProLong Gold (Invitrogen, P10144) Antifade Mountant.
Mice were transcardially perfused with ice-cold PBS ∼28 days post-surgery. Brain tissue was extracted and postfixed in 4% PFA in PBS for 24 h at 4°C, and cryoprotected by sequential addition of 15% (24 h) and 30% (72 h) sucrose in PBS. Cryoprotected brains were embedded in Tissue-Tek O.C.T. Compound (Sakura, 4583) and 20 μm thick coronal slices were obtained using Leica cryostat (CM3050 S). Free-floating sections were rinsed thrice in PBS for 5 min each, followed by 30 min incubation in 0.5% Triton X-100 in PBS. Permeabilized sections were blocked using 0.25% Triton X-100 and 10% goat serum in PBS (blocking solution) for 3 h. Sections were incubated in appropriate dilution of primary antibody (see Table S1) in blocking solution for 48 h at 4°C. After rinsing thrice in PBS for 5 min each, sections were incubated in appropriate dilution of Alexa Flour conjugated secondary antibodies (see key resources table) for 3 h at room temperature. Sections were finally rinsed thrice in PBS for 10 min each, containing DAPI in the final wash and mounted using Prolong Diamond Antifade Mountant (Invitrogen, P36970).
Surface biotinylation assay
Neuro-2a cells were incubated in 1 mg/mL EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific, 21331) in PBS (containing Ca2+ and Mg2+, pH 8) for 30 min at 4°C with gentle agitation. The reaction was quenched by incubating the cells twice for 10 min each in TBS solution at 4°C with gentle agitation. The cells were then lysed in 1.5% C12E9 buffer [Tris·HCl, 50 mM, pH 7.4; NaCl, 150 mM; EDTA, 0.05 mM; C12E9, 1.5%; protease (Roche, 11836170001) and phosphatase inhibitor tablets (Roche, 4906837001)], by rotating (25 RPM) the samples at 4°C for 30 min with intermittent vortexing. After lysis, the samples were centrifuged at 21000 × g for 15 min at 4°C and total protein concentration of the supernatant was estimated using DC Protein Assay Kit (BioRad, 5000112). 50 μg total protein at 0.5 μg/μL in 1.5% C12E9 buffer was incubated with 25 μL of 50% slurry of Pierce High Capacity NeutrAvidin agarose beads (Thermo Scientific, 29202) and rotated (10 RPM) at 4°C for 3 h. 10 μg total protein was saved as total fraction. The beads (surface fraction) were subsequently washed thrice with 1.5% C12E9 buffer for 15 min each. Total and surface fractions were denatured in 6× SDS sample buffer (Tris, pH 6.8, 375 mM; sodium dodecyl sulfate, 12% w/v; glycerol, 60% v/v; bromophenol blue, 0.3% w/v) containing 50 mM DTT and heated at 37°C for and hour and resolved through SDS-PAGE using 8% acrylamide gel. Western blot analysis was performed as described below.
Surface labeling assay
Neuro-2a cells were incubated at 4°C for 10 min followed by a quick rinse with cold HBSS solution (containing Ca2+ and Mg2+). The cells were incubated with anti-Flag antibody (Sigma-Aldrich, F1804, 1:500) in HBSS solution (with Ca2+ and Mg2+) containing 10% normal goat serum (Gibco, 16210064), at 4°C for 30 min. After rinsing with cold HBSS solution (with Ca2+ and Mg2+), the cells were fixed with 4% (w/v) paraformaldehyde in PBS containing 4% (w/v) sucrose, for 5 min at room temperature (all subsequent steps performed at room temperature). The cells were permeabilized with 0.25% (v/v) Triton X-100 in PBS for 5 min and incubated for 30 min in blocking buffer (10% normal goat serum, 0.01% Triton X-100 in PBS). Following this, the cells were sequentially incubated in anti-KCC2 antibody (EMD Millipore, 07–432), and Goat anti-Mouse IgG, Alexa Fluor 647 (Invitrogen, A-21235) and Goat anti-Rabbit IgG, Alexa Fluor 488 (Invitrogen, A-11008) in blocking buffer for an hour each. The cells were rinsed twice with PBS (containing Ca2+ and Mg2+), containing DAPI in the first wash. Finally, the coverslips were mounted using ProLong Diamond Antifade Mountant (Invitrogen, P36970).
Proximity ligation assay
For identification of the KCC2 intracellular domain mediating its interaction with SNAP25, neuro-2a cells were collected 60 h post-transfection. To test whether PKC is involved in mediating SNAP25-KCC2 interaction, transfected neuro-2a cells were grown in complete growth medium and collected 60 h post-transfection. These cells were treated with DMSO, PMA (BioShop, PMA168.1, 200 nM), Gö6983 (Sigma-Aldrich, G1918-500UG, 100 nM) or chelerythrine chloride (HelloBio, HB0190, 5 μM) for 60 h, 24 h, 6 h or an hour toward collection depending on the experiment. Similarly, neurons were treated with PMA (200 nM, 1 h) or Gö6983 (100 nM, 48 h) and collected at DIV 14. Cells were fixed using 4% PFA in PBS for 5 min, followed by permeabilization for 5 min with 0.25% Triton X-100. We used Duolink In Situ PLA Probe Anti-Mouse PLUS (Sigma-Aldrich, DUO92001-100RXN), Duolink In Situ PLA Probe Anti-Rabbit MINUS (Sigma-Aldrich, DUO92005-100RXN), Duolink In Situ Detection Reagents FarRed (Sigma-Aldrich, DUO92013-100RXN) as per manufacturer supplied protocol for PLA. During PLA, the coverslips were incubated in appropriate dilutions of primary antibody (see Table S1). Finally, the coverslips were incubated in Duolink In Situ Wash Buffer B (DUO820409) containing an appropriate dilution of Alexa Flour conjugated secondary antibodies (see key resources table) and DAPI for 30 min, and were mounted using ProLong Diamond Antifade Mountant (Invitrogen, P36970).
Western blot
For total KCC2 quantification, cortex was dissected out from SNAP25(+/−) mice or WT littermates and lysed in C12E9 buffer (4× w/v) [Tris·HCl, 50 mM, pH 7.4; NaCl, 150 mM; EDTA, 0.05 mM; C12E9, 1.5%, protease (Roche, 11836170001) and phosphatase inhibitor tablets (Roche, 4906837001)], solubilized for 3 h on a rotating platform at 4°C, and centrifuged at 21,000 × g for an hour. For total KCC2 quantification without any treatments, COS-7 and neuro-2a cell samples were collected 48 h post-transfection. For identifying KCC2 degradation pathway, transfected neuro-2a cells were treated with 500 nM lactacystin (HelloBio, HB3953), 50 nM MG-132 (HelloBio), 500 μM leupeptin hemisulfate (Cell Signaling Technology, 73618S) or 500 nM bafilomycin A1 (Cell Signaling Technology, 54645S) 24 h post-transfection and the samples were collected 36 h post-treatment. All biochemical preparations and centrifugations were done at 4°C. For sample collection, cells were washed with ice-cold 1× PBS and lysed in RIPA buffer [Tris·HCl, 50 mM, pH 7.4; NaCl, 150 mM; EDTA, 1 mM; Nonidet P-40, 1%; SDS, 0.1%; DOC, 0.5%, protease (Roche, 11836170001) and phosphatase inhibitor tablets (Roche, 4906837001)]. Lysed cells were rotated (20 RPM) at 4°C for 30 min, vortexed every 10 min and were centrifuged at 21,000 × g for 15 min at 4°C. Total protein estimation was performed on supernatant using DC Protein Assay Kit (BioRad, 5000112). Protein samples (20–30 μg) were prepared using SDS sample buffer (Tris, pH 6.8, 50 mM; sodium dodecyl sulfate, 2% w/v; glycerol, 10% v/v; bromophenol blue, 0.05% w/v) containing 50 mM DTT. The samples were heated at 37°C for an hour and resolved via SDS-poly-acrylamide gel electrophoresis (PAGE) using 8–10% acrylamide gels.
The acrylamide gels were transferred to nitrocellulose membrane using Trans-Blot Turbo Transfer Kit (BioRad, 1704270) and blocked with 5% skimmed milk or BSA in TBS-T (NaCl 150 mM; KCl 2.5 mM; Tris.HCl 50 mM; Tween 20 0.05%; pH 7.4), followed by incubation in appropriate dilution of primary antibody (see Table S1) at 4°C for overnight. The blot was incubated in appropriate concentration of HRP-linked secondary antibody (see key resources table), developed using Immobilon Crescendo Western HRP substrate (Millipore, WBLUR0500) and imaged using BioRad ChemiDoc imager. Densitometric quantification of protein bands were done using FIJI81 (ImageJ 1.54f). Representative immunoblots have been cropped (denoted by white space within the panel) to show one or two replicates for each condition.
Antibodies
Complete details and dilutions of all antibodies used in this study are listed in Table S1.
Image acquisition and analysis
Images of immunolabeled cells were acquired on Eclipse Ti2-E inverted microscope (Nikon Instruments) equipped with an Apo λ 60× oil objective, CSU-X1 confocal scanner (Yokogawa Corporation), iChrome MLE laser combiner (Toptica Photonics), CoolSnap Myo CCD camera (Roper Scientific), and a dual galvanometer laser scanner (Bruker Corporation). Images were acquired at a z-step of 0.2–0.3 μm. Alternatively, images of immunolabelled brain cryosections were obtained using Leica TCS SP8 (standard) Confocal system with HCX PL APO 10×/0.40 oil or HC PL APO 63×/1.40 oil objective and Leica DMI 6000 inverted microscope (Quorum Technologies). Imaging experiments were performed and analyzed in a blinded manner. Colocalization analysis and total KCC2 quantification from neuronal cultures was performed on selected z-stacks using Imaris 7.6 software. Quantification of KCC2 fluorescence intensity from mouse brain cryo-sections (see Figure S2B), surface/total KCC2, PLA puncta/KCC2 intensity was done using FIJI81 (ImageJ 1.54f). Adobe Photoshop CC 2023 and Adobe Illustrator CC 2023 were used to create figures.
Quantification and statistical analysis
Statistical analysis
Statistical analysis was done using GraphPad Prism 10. All graphs represent mean ± SEM. For immunostaining, PLA and electrophysiology experiments, n value represents data from individual cells, with a minimum of three separate coverslips or four individual animals for each dataset (see figure legend for details), respectively. For immunoblotting, n value represents data from independent replicates. Data points that were outliers as determined by the ROUT method with Q = 1% were excluded from further analysis. Normality of data were tested using D'Agostino-Pearson omnibus test or Shapiro-Wilk test (only if the n value is too small). Unpaired t-test (with Welch’s correction, two-tailed), Brown-Forsythe and Welch ANOVA tests and Dunnett’s T3 multiple comparisons test, with individual variances computed for each comparison or Ordinary two-way ANOVA and Tukey’s multiple comparisons test, with single pooled variance was performed on normal data. When data did not pass the normality test, we used Mann-Whitney test or Kruskal-Wallis test and Dunn’s multiple comparisons test. Statistical significance was determined as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Details of corresponding statistical analysis can be found in the figure legend.
Published: October 11, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111156.
Supplemental information
References
- 1.Moss S.J., Smart T.G. Constructing inhibitory synapses. Nat. Rev. Neurosci. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- 2.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog. Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 3.Raimondo J.V., Richards B.A., Woodin M.A. Neuronal chloride and excitability - the big impact of small changes. Curr. Opin. Neurobiol. 2017;43:35–42. doi: 10.1016/j.conb.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Kaila K., Price T.J., Payne J.A., Puskarjov M., Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivera C., Voipio J., Payne J.A., Ruusuvuori E., Lahtinen H., Lamsa K., Pirvola U., Saarma M., Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 6.Pressey J.C., de Saint-Rome M., Raveendran V.A., Woodin M.A. Chloride transporters controlling neuronal excitability. Physiol. Rev. 2023;103:1095–1135. doi: 10.1152/physrev.00025.2021. [DOI] [PubMed] [Google Scholar]
- 7.Cellot G., Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2014;2:70. doi: 10.3389/fped.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao R., Li C., Newburn E.N., Ye T., Lipska B.K., Herman M.M., Weinberger D.R., Kleinman J.E., Hyde T.M. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J. Neurosci. 2012;32:5216–5222. doi: 10.1523/JNEUROSCI.4626-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hyde T.M., Lipska B.K., Ali T., Mathew S.V., Law A.J., Metitiri O.E., Straub R.E., Ye T., Colantuoni C., Herman M.M., et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews K., Josiah S.S., Zhang J. The Therapeutic Potential of Neuronal K-Cl Co-Transporter KCC2 in Huntington's Disease and Its Comorbidities. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21239142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahle K.T., Khanna A., Clapham D.E., Woolf C.J. Therapeutic restoration of spinal inhibition via druggable enhancement of potassium-chloride cotransporter KCC2-mediated chloride extrusion in peripheral neuropathic pain. JAMA Neurol. 2014;71:640–645. doi: 10.1001/jamaneurol.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu R., Wang J., Liang S., Zhang G., Yang X. Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front. Neurol. 2019;10:1407. doi: 10.3389/fneur.2019.01407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silayeva L., Deeb T.Z., Hines R.M., Kelley M.R., Munoz M.B., Lee H.H.C., Brandon N.J., Dunlop J., Maguire J., Davies P.A., Moss S.J. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. USA. 2015;112:3523–3528. doi: 10.1073/pnas.1415126112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talifu Z., Pan Y., Gong H., Xu X., Zhang C., Yang D., Gao F., Yu Y., Du L., Li J. The role of KCC2 and NKCC1 in spinal cord injury: From physiology to pathology. Front. Physiol. 2022;13 doi: 10.3389/fphys.2022.1045520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahadevan V., Khademullah C.S., Dargaei Z., Chevrier J., Uvarov P., Kwan J., Bagshaw R.D., Pawson T., Emili A., De Koninck Y., et al. Native KCC2 interactome reveals PACSIN1 as a critical regulator of synaptic inhibition. Elife. 2017;6 doi: 10.7554/eLife.28270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahadevan V., Pressey J.C., Acton B.A., Uvarov P., Huang M.Y., Chevrier J., Puchalski A., Li C.M., Ivakine E.A., Airaksinen M.S., et al. Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 2014;7:1762–1770. doi: 10.1016/j.celrep.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pressey J.C., Mahadevan V., Khademullah C.S., Dargaei Z., Chevrier J., Ye W., Huang M., Chauhan A.K., Meas S.J., Uvarov P., et al. A kainate receptor subunit promotes the recycling of the neuron-specific K(+)-Cl(-) co-transporter KCC2 in hippocampal neurons. J. Biol. Chem. 2017;292:6190–6201. doi: 10.1074/jbc.M116.767236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puskarjov M., Ahmad F., Kaila K., Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J. Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söllner T., Whiteheart S.W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J.E. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 20.Kádková A., Radecke J., Sorensen J.B. The SNAP-25 Protein Family. Neuroscience. 2019;420:50–71. doi: 10.1016/j.neuroscience.2018.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Gu Y., Huganir R.L. Identification of the SNARE complex mediating the exocytosis of NMDA receptors. Proc. Natl. Acad. Sci. USA. 2016;113:12280–12285. doi: 10.1073/pnas.1614042113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau C.G., Takayasu Y., Rodenas-Ruano A., Paternain A.V., Lerma J., Bennett M.V.L., Zukin R.S. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J. Neurosci. 2010;30:242–254. doi: 10.1523/JNEUROSCI.4933-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selak S., Paternain A.V., Aller M.I., Picó E., Rivera R., Lerma J. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron. 2009;63:357–371. doi: 10.1016/j.neuron.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Guerini F.R., Bolognesi E., Chiappedi M., Manca S., Ghezzo A., Agliardi C., Sotgiu S., Usai S., Matteoli M., Clerici M. SNAP-25 single nucleotide polymorphisms are associated with hyperactivity in autism spectrum disorders. Pharmacol. Res. 2011;64:283–288. doi: 10.1016/j.phrs.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Safari M.R., Omrani M.D., Noroozi R., Sayad A., Sarrafzadeh S., Komaki A., Manjili F.A., Mazdeh M., Ghaleiha A., Taheri M. Synaptosome-Associated Protein 25 (SNAP25) Gene Association Analysis Revealed Risk Variants for ASD, in Iranian Population. J. Mol. Neurosci. 2017;61:305–311. doi: 10.1007/s12031-016-0860-2. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z., Li J., Zhang T., Lu T., Wang H., Jia M., Liu J., Xiong J., Zhang D., Wang L. Family-based association study identifies SNAP25 as a susceptibility gene for autism in the Han Chinese population. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2021;105 doi: 10.1016/j.pnpbp.2020.109985. [DOI] [PubMed] [Google Scholar]
- 27.Braida D., Guerini F.R., Ponzoni L., Corradini I., De Astis S., Pattini L., Bolognesi E., Benfante R., Fornasari D., Chiappedi M., et al. Association between SNAP-25 gene polymorphisms and cognition in autism: functional consequences and potential therapeutic strategies. Transl. Psychiatry. 2015;5:e500. doi: 10.1038/tp.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonucci F., Corradini I., Fossati G., Tomasoni R., Menna E., Matteoli M. SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions. Front. Synaptic Neurosci. 2016;8:7. doi: 10.3389/fnsyn.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raol Y.H., Joksimovic S.M., Sampath D., Matter B.A., Lam P.M., Kompella U.B., Todorovic S.M., González M.I. The role of KCC2 in hyperexcitability of the neonatal brain. Neurosci. Lett. 2020;738 doi: 10.1016/j.neulet.2020.135324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sivakumaran S., Cardarelli R.A., Maguire J., Kelley M.R., Silayeva L., Morrow D.H., Mukherjee J., Moore Y.E., Mather R.J., Duggan M.E., et al. Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J. Neurosci. 2015;35:8291–8296. doi: 10.1523/JNEUROSCI.5205-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMoneagle E., Zhou J., Zhang S., Huang W., Josiah S.S., Ding K., Wang Y., Zhang J. Neuronal K(+)-Cl(-) cotransporter KCC2 as a promising drug target for epilepsy treatment. Acta Pharmacol. Sin. 2024;45:1–22. doi: 10.1038/s41401-023-01149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Washbourne P., Thompson P.M., Carta M., Costa E.T., Mathews J.R., Lopez-Benditó G., Molnár Z., Becher M.W., Valenzuela C.F., Partridge L.D., Wilson M.C. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat. Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- 33.Virtanen M.A., Uvarov P., Mavrovic M., Poncer J.C., Kaila K. The Multifaceted Roles of KCC2 in Cortical Development. Trends Neurosci. 2021;44:378–392. doi: 10.1016/j.tins.2021.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Xu J., Luo F., Zhang Z., Xue L., Wu X.S., Chiang H.C., Shin W., Wu L.G. SNARE proteins synaptobrevin, SNAP-25, and syntaxin are involved in rapid and slow endocytosis at synapses. Cell Rep. 2013;3:1414–1421. doi: 10.1016/j.celrep.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z., Wang D., Sun T., Xu J., Chiang H.C., Shin W., Wu L.G. The SNARE proteins SNAP25 and synaptobrevin are involved in endocytosis at hippocampal synapses. J. Neurosci. 2013;33:9169–9175. doi: 10.1523/JNEUROSCI.0301-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao J., Hirata M., Mizokami A., Zhao J., Takahashi I., Takeuchi H., Hirata M. Differential role of SNAP-25 phosphorylation by protein kinases A and C in the regulation of SNARE complex formation and exocytosis in PC12 cells. Cell. Signal. 2016;28:425–437. doi: 10.1016/j.cellsig.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 37.Katayama N., Yamamori S., Fukaya M., Kobayashi S., Watanabe M., Takahashi M., Manabe T. SNAP-25 phosphorylation at Ser187 regulates synaptic facilitation and short-term plasticity in an age-dependent manner. Sci. Rep. 2017;7:7996. doi: 10.1038/s41598-017-08237-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H.H., Walker J.A., Williams J.R., Goodier R.J., Payne J.A., Moss S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- 39.Kfir A., Awasthi R., Ghosh S., Kundu S., Paul B., Lamprecht R., Barkai E. Cellular Mechanism of Learning-Induced Enhancement of Synaptic Inhibition: PKC-Dependent Upregulation of KCC2 Activation. Sci. Rep. 2020;10:962. doi: 10.1038/s41598-020-57626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franceschetti S., Taverna S., Sancini G., Panzica F., Lombardi R., Avanzini G. Protein kinase C-dependent modulation of Na+ currents increases the excitability of rat neocortical pyramidal neurones. J. Physiol. 2000;528:291–304. doi: 10.1111/j.1469-7793.2000.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Callender J.A., Newton A.C. Conventional protein kinase C in the brain: 40 years later. Neuronal Signal. 2017;1 doi: 10.1042/NS20160005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Astman N., Gutnick M.J., Fleidervish I.A. Activation of protein kinase C increases neuronal excitability by regulating persistent Na+ current in mouse neocortical slices. J. Neurophysiol. 1998;80:1547–1551. doi: 10.1152/jn.1998.80.3.1547. [DOI] [PubMed] [Google Scholar]
- 43.Dutil E.M., Keranen L.M., DePaoli-Roach A.A., Newton A.C. In vivo regulation of protein kinase C by trans-phosphorylation followed by autophosphorylation. J. Biol. Chem. 1994;269:29359–29362. [PubMed] [Google Scholar]
- 44.Hansra G., Garcia-Paramio P., Prevostel C., Whelan R.D.H., Bornancin F., Parker P.J. Multisite dephosphorylation and desensitization of conventional protein kinase C isotypes. Biochem. J. 1999;342:337–344. [PMC free article] [PubMed] [Google Scholar]
- 45.Kraft A.S., Anderson W.B., Cooper H.L., Sando J.J. Decrease in cytosolic calcium/phospholipid-dependent protein kinase activity following phorbol ester treatment of EL4 thymoma cells. J. Biol. Chem. 1982;257:13193–13196. [PubMed] [Google Scholar]
- 46.Szallasi Z., Smith C.B., Pettit G.R., Blumberg P.M. Differential regulation of protein kinase C isozymes by bryostatin 1 and phorbol 12-myristate 13-acetate in NIH 3T3 fibroblasts. J. Biol. Chem. 1994;269:2118–2124. [PubMed] [Google Scholar]
- 47.Li H., Khirug S., Cai C., Ludwig A., Blaesse P., Kolikova J., Afzalov R., Coleman S.K., Lauri S., Airaksinen M.S., et al. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 48.Chevy Q., Heubl M., Goutierre M., Backer S., Moutkine I., Eugène E., Bloch-Gallego E., Lévi S., Poncer J.C. KCC2 Gates Activity-Driven AMPA Receptor Traffic through Cofilin Phosphorylation. J. Neurosci. 2015;35:15772–15786. doi: 10.1523/JNEUROSCI.1735-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedel P., Ludwig A., Pellegrino C., Agez M., Jawhari A., Rivera C., Medina I. A Novel View on the Role of Intracellular Tails in Surface Delivery of the Potassium-Chloride Cotransporter KCC2. eNeuro. 2017;4 doi: 10.1523/ENEURO.0055-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee H.H.C., Jurd R., Moss S.J. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 2010;45:173–179. doi: 10.1016/j.mcn.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu J.J., Liu Y., Yao H., Cao B., Liao H., Yang R., Chen P., Song X.J. Emergence of consciousness from anesthesia through ubiquitin degradation of KCC2 in the ventral posteromedial nucleus of the thalamus. Nat. Neurosci. 2023;26:751–764. doi: 10.1038/s41593-023-01290-y. [DOI] [PubMed] [Google Scholar]
- 52.Han J., Pluhackova K., Böckmann R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017;8:5. doi: 10.3389/fphys.2017.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arora S., Saarloos I., Kooistra R., van de Bospoort R., Verhage M., Toonen R.F. SNAP-25 gene family members differentially support secretory vesicle fusion. J. Cell Sci. 2017;130:1877–1889. doi: 10.1242/jcs.201889. [DOI] [PubMed] [Google Scholar]
- 54.Delgado-Martínez I., Nehring R.B., Sorensen J.B. Differential abilities of SNAP-25 homologs to support neuronal function. J. Neurosci. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asraf H., Bogdanovic M., Gottesman N., Sekler I., Aizenman E., Hershfinkel M. SNAP23 regulates KCC2 membrane insertion and activity following mZnR/GPR39 activation in hippocampalneurons. iScience. 2022;25 doi: 10.1016/j.isci.2022.103751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen D., Minger S.L., Honer W.G., Whiteheart S.W. Organization of the secretory machinery in the rodent brain: distribution of the t-SNAREs, SNAP-25 and SNAP-23. Brain Res. 1999;831:11–24. doi: 10.1016/s0006-8993(99)01371-2. [DOI] [PubMed] [Google Scholar]
- 57.Gu Y., Chiu S.L., Liu B., Wu P.H., Delannoy M., Lin D.T., Wirtz D., Huganir R.L. Differential vesicular sorting of AMPA and GABAA receptors. Proc. Natl. Acad. Sci. USA. 2016;113:E922–E931. doi: 10.1073/pnas.1525726113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Condliffe S.B., Matteoli M. Inactivation kinetics of voltage-gated calcium channels in glutamatergic neurons are influenced by SNAP-25. Channels. 2011;5:304–307. doi: 10.4161/chan.5.4.16228. [DOI] [PubMed] [Google Scholar]
- 59.Condliffe S.B., Corradini I., Pozzi D., Verderio C., Matteoli M. Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. J. Biol. Chem. 2010;285:24968–24976. doi: 10.1074/jbc.M110.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verderio C., Pozzi D., Pravettoni E., Inverardi F., Schenk U., Coco S., Proux-Gillardeaux V., Galli T., Rossetto O., Frassoni C., Matteoli M. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron. 2004;41:599–610. doi: 10.1016/s0896-6273(04)00077-7. [DOI] [PubMed] [Google Scholar]
- 61.Pozzi D., Condliffe S., Bozzi Y., Chikhladze M., Grumelli C., Proux-Gillardeaux V., Takahashi M., Franceschetti S., Verderio C., Matteoli M. Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc. Natl. Acad. Sci. USA. 2008;105:323–328. doi: 10.1073/pnas.0706211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davletov B., Bajohrs M., Binz T. Beyond BOTOX: advantages and limitations of individual botulinum neurotoxins. Trends Neurosci. 2005;28:446–452. doi: 10.1016/j.tins.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 63.Rasetti-Escargueil C., Lemichez E., Popoff M.R. Variability of Botulinum Toxins: Challenges and Opportunities for the Future. Toxins. 2018;10 doi: 10.3390/toxins10090374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peng L., Liu H., Ruan H., Tepp W.H., Stoothoff W.H., Brown R.H., Johnson E.A., Yao W.D., Zhang S.C., Dong M. Cytotoxicity of botulinum neurotoxins reveals a direct role of syntaxin 1 and SNAP-25 in neuron survival. Nat. Commun. 2013;4:1472. doi: 10.1038/ncomms2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meng J., Wang J., Steinhoff M., Dolly J.O. TNFalpha induces co-trafficking of TRPV1/TRPA1 in VAMP1-containing vesicles to the plasmalemma via Munc18-1/syntaxin1/SNAP-25 mediated fusion. Sci. Rep. 2016;6 doi: 10.1038/srep21226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheung D.L., Toda T., Narushima M., Eto K., Takayama C., Ooba T., Wake H., Moorhouse A.J., Nabekura J. KCC2 downregulation after sciatic nerve injury enhances motor function recovery. Sci. Rep. 2023;13:7871. doi: 10.1038/s41598-023-34701-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leonzino M., Busnelli M., Antonucci F., Verderio C., Mazzanti M., Chini B. The Timing of the Excitatory-to-Inhibitory GABA Switch Is Regulated by the Oxytocin Receptor via KCC2. Cell Rep. 2016;15:96–103. doi: 10.1016/j.celrep.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie Y., Chang S., Zhao C., Wang F., Liu S., Wang J., Delpire E., Ye S., Guo J. Structures and an activation mechanism of human potassium-chloride cotransporters. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abc5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hartmann A.M., Nothwang H.G. NKCC1 and KCC2: Structural insights into phospho-regulation. Front. Mol. Neurosci. 2022;15 doi: 10.3389/fnmol.2022.964488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong W. SNAREs and traffic. Biochim. Biophys. Acta. 2005;1744:493–517. [PubMed] [Google Scholar]
- 71.Ungar D., Hughson F.M. SNARE protein structure and function. Annu. Rev. Cell Dev. Biol. 2003;19:493–517. doi: 10.1146/annurev.cellbio.19.110701.155609. [DOI] [PubMed] [Google Scholar]
- 72.Bennett M.K., García-Arrarás J.E., Elferink L.A., Peterson K., Fleming A.M., Hazuka C.D., Scheller R.H. The syntaxin family of vesicular transport receptors. Cell. 1993;74:863–873. doi: 10.1016/0092-8674(93)90466-4. [DOI] [PubMed] [Google Scholar]
- 73.Lin R.C., Scheller R.H. Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 2000;16:19–49. doi: 10.1146/annurev.cellbio.16.1.19. [DOI] [PubMed] [Google Scholar]
- 74.Pevsner J., Hsu S.C., Braun J.E., Calakos N., Ting A.E., Bennett M.K., Scheller R.H. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 75.Awad P.N., Amegandjin C.A., Szczurkowska J., Carriço J.N., Fernandes do Nascimento A.S., Baho E., Chattopadhyaya B., Cancedda L., Carmant L., Di Cristo G. KCC2 Regulates Dendritic Spine Formation in a Brain-Region Specific and BDNF Dependent Manner. Cereb. Cortex. 2018;28:4049–4062. doi: 10.1093/cercor/bhy198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomasoni R., Repetto D., Morini R., Elia C., Gardoni F., Di Luca M., Turco E., Defilippi P., Matteoli M. SNAP-25 regulates spine formation through postsynaptic binding to p140Cap. Nat. Commun. 2013;4:2136. doi: 10.1038/ncomms3136. [DOI] [PubMed] [Google Scholar]
- 77.Smalley J.L., Kontou G., Choi C., Ren Q., Albrecht D., Abiraman K., Santos M.A.R., Bope C.E., Deeb T.Z., Davies P.A., et al. Isolation and Characterization of Multi-Protein Complexes Enriched in the K-Cl Co-transporter 2 From Brain Plasma Membranes. Front. Mol. Neurosci. 2020;13 doi: 10.3389/fnmol.2020.563091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uvarov P., Ludwig A., Markkanen M., Pruunsild P., Kaila K., Delpire E., Timmusk T., Rivera C., Airaksinen M.S. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 2007;282:30570–30576. doi: 10.1074/jbc.M705095200. [DOI] [PubMed] [Google Scholar]
- 79.Bhandage A.K., Olivera G.C., Kanatani S., Thompson E., Loré K., Varas-Godoy M., Barragan A. A motogenic GABAergic system of mononuclear phagocytes facilitates dissemination of coccidian parasites. Elife. 2020;9 doi: 10.7554/eLife.60528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shulga A., Thomas-Crusells J., Sigl T., Blaesse A., Mestres P., Meyer M., Yan Q., Kaila K., Saarma M., Rivera C., Giehl K.M. Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci. 2008;28:6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this study will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.