

Abstract
Background
The copy number status (CNS) of the survival motor neuron (SMN) gene may influence the risk and prognosis of amyotrophic lateral sclerosis (ALS) and lower motor neuron diseases (LMND) other than spinal muscular atrophy (SMA). However, previous studies of this association, mainly from Europe, have yielded controversial results, suggesting possible regional differences. Here, we investigated the effect of the SMN gene in Japanese patients with ALS and LMND.
Methods
We examined the SMN copy numbers and clinical histories of 487 Japanese patients with sporadic ALS (281 men; mean age at onset 61.5 years), 50 with adult LMND (50 men; mean age at onset 58.4 years) and 399 Japanese controls (171 men; mean age 62.2 years). Patients with pathogenic mutations in ALS-causing genes were excluded. SMN1 and SMN2 copy numbers were determined using the droplet digital polymerase chain reaction.
Results
The frequency of a copy number of one for the SMN2 gene was higher in patients with ALS (38.0%) than in healthy controls (30.8%) (odds ratio (OR) = 1.37, 95% confidence interval (CI) = 1.04–1.82, p < 0.05). The SMN2 copy number affected the survival time of patients with ALS (median time: 0 copies, 34 months; 1 copy, 39 months; 2 copies, 44 months; 3 copies, 54 months; log-rank test, p < 0.05). Cox regression analysis revealed that the SMN2 copy number was associated with increased mortality (hazard ratio = 0.84, 95% CI = 0.72–0.98, p < 0.05). Also, null SMN2 cases were significantly more frequent in the LMND group (12.0%) than in the control group (4.8%) (OR = 2.73, 95% CI = 1.06–6.98, p < 0.05).
Conclusions
Our findings suggest that SMN2 copy number reduction may adversely affect the onset and prognosis of MND, including ALS and LMND, in Japanese.
Keywords: ALS, SMA, LMND, SMN, Copy number status
Background
Amyotrophic lateral sclerosis (ALS) is a fatal adult-onset upper and lower motor neuron disease (MND) with a diverse genetic background; 5–10% of ALS case are familial, and more than 30 genes are involved, including TARDBP, FUS, TBK1, c9orf72, and SOD1 [1–3]. In addition, several related genes have been reported to influence disease development, one being SMN, whose copy number status (CNS) appears to be of importance in this context [3, 4].
SMN has two homologs, SMN1 and SMN2, which are the causative and disease-modifying genes of spinal muscular atrophy (SMA), a lower motor neuron disease that develops in early childhood [5, 6]. SMA is caused by a deficiency of SMN1, and the severity is reduced as the copy number of SMN2 increases [6]. Differences in SMN1 and SMN2 messenger RNA (mRNA) splicing are the major factor in the pathomechanism of SMA. SMN1 mRNA produces normal SMN protein, whereas mRNA derived from SMN2 produces mostly unstable SMN protein, although a small amount undergoes normal splicing [7]. The amount of normal SMN protein produced plays a major role in determining the severity of SMA [6].
ALS and SMN are also associated with dysregulation of nuclear function. Nucleolar gemini bodies (GEM), in which the SMN protein is a major component, are involved in the maturation of functional small nuclear RNAs (snRNAs) and play an important role in mRNA splicing [8]. In SMA-affected tissues, the levels of SMN protein are markedly reduced, resulting in GEM depletion and impairment of mRNA splicing function [9]. Interestingly, in ALS, GEM are decreased in affected tissues, and snRNA expression is also altered [10, 11], suggesting that SMN CNS could be a noteworthy factor in ALS.
A number of studies have examined the association between SMN CNS and ALS [12–18], and a recent large-scale investigation found no link between SMN CNS and the development and prognosis of the disease [19]. That study, however, and most of the previous ones, were conducted in Europe, suggesting the need for a wider survey of regional differences in genetic background [4].
The frequency of genetic mutations often varies widely by region and can influence the diagnosis of ALS and formulation of treatment strategies. For example, the most frequent ALS-causing variant in Caucasians is the hexanucleotide repeat expansion of C9orf72 [20, 21]; however, this mutation is very rare in Asians [22]. Furthermore, SMN CNS in controls varies widely in Europe [13, 14], Asia [23, 24], and Africa [25] (Table 1). Regarding the association between SMN CNS and MND in East Asia, two studies from Korea have reported that deletion of the SMN2 gene is involved in the development of ALS [16] and lower motor neuron disease (LMND) [26], although each of those studies involved only a small number of cases.
Table 1.
Differences in SMN1 and SMN2 copy number in control cases by country/region
| SMN1 CNS | China [23] | Taiwan [24] | France [13] | Netherlands [14] | Mali [25] |
|---|---|---|---|---|---|
| 0 (%) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 1 (%) | 2.4 | 2.1 | 2.1 | 2.3 | 0.5 |
| 2 (%) | 89.7 | 90.3 | 95.5 | 94.1 | 47.0 |
| 3 (%) | 7.0 | 7.5 | 2.4 | 3.6 | 39.6 |
| 4 (%) | 1.0 | 0.2 | 0.0 | 0.0 | 12.9 |
| Total n= | 1712 | 107,611 | 621 | 984 | 628 |
| SMN2 CNS | China [23] | Taiwan [24] | France [13] | Netherlands [14] | Mali [25] |
| 0 (%) | 5.2 | 4.6 | 8.4 | 7.9 | 23.9 |
| 1 (%) | 29.0 | 31.9 | 38.5 | 37.8 | 43.9 |
| 2 (%) | 61.1 | 60.5 | 51.7 | 49.4 | 27.4 |
| 3 (%) | 3.9 | 2.8 | 1.4 | 4.7 | 1.8 |
| 4 (%) | 0.9 | 0.2 | 0.0 | 0.2 | 0.6 |
| Total n= | 1712 | 107,611 | 621 | 984 | 613 |
Country names followed the notation in the referenced papers
As the effects of SMN CNS on ALS and LMND may differ between Europe and Asia, a larger study is warranted. Here, we examined the association between SMN CNS and the onset and outcome of ALS or LMND in Japanese patients.
Methods
This study included 487 Japanese patients with sporadic ALS (SALS) (281 men, 206 women; mean age at onset 61.5 years; bulbar onset 121 patients), 50 Japanese patients with adult lower motor neuron disease (LMND) (50 men; mean age at onset 58.4 years) and 399 Japanese controls (171 men, 228 women; mean age at sampling 62.2 years) (Table 2). Of the 487 ALS cases, 440 were registered in the Japanese Consortium for Amyotrophic Lateral Sclerosis (JaCALS) data bank, and 47 were autopsy cases at Niigata University. The LMND patients were 50 adults who were negative for spinal and bulbar muscular atrophy (SBMA) and SMA by genetic testing among 100 consecutive patients who had requested genetic testing for SBMA at Niigata University. For this reason, all of the LMND patients were male. As controls, we also included 299 spouses of patients with ALS registered in the JaCALS. Another 100 of the controls were patients with other diseases, including 41 with spinocerebellar ataxia(SCA) (SCA3: 3 cases, SCA6: 10 cases, SCA31: 7 cases, DRPLA: 1 case, undetermined: 20 cases), 5 with early onset SCA (EAOH: 3 cases, undetermined: 2 cases), 4 suspected of Huntington’s disease (HD: 1 case, undetermined: 3 cases), 42 with leukoencephalopathy or cerebral small vessel disease (CADASIL: 2 cases, HDLS: 1 case, undetermined: 39 cases), 5 with parkinsonism (undetermined: 5 cases), 2 with dementia (undetermined: 2 cases), and 1 normal control seen at Niigata University. The age at onset and the prognosis of the ALS cases were investigated on the basis of the JaCALS registry information and autopsy summaries. The LMND patients were not followed up and only age at onset was investigated based on the order sheets for genetic testing. The date when individual patients had first noticed symptoms was denoted as the onset of ALS or LMND. The date of death from any cause, or the introduction of invasive tracheostomy ventilation was set as the endpoint of ALS.
Table 2.
Characteristics and SMN1 or SMN2 copy number groups in this study
| Control (n = 399) |
ALS (n = 487) |
LMND (n = 50) |
Control vs. ALS OR (95% CI) |
Control vs. LMND OR (95% CI) |
|
|---|---|---|---|---|---|
| Characteristics | |||||
|
Age (years) Mean (SD) |
62.2 (11.0) | 61.5 (11.1) | 58.4 (12.6) | - | - |
| M/F | 171/228 | 281/206 | 50/0 | - | - |
| SMN1 copies | |||||
| 1 copy | 5 (1.3%) | 4 (0.8%) | 0 (0%) | 0.65 (0.20–2.17) | - |
| 2 copies | 364 (91.2%) | 457 (93.4%) | 47 (94.0%) | 1.46 (0.88–2.42) | 1.51 (0.46–4.82) |
| 3 copies | 28 (7.0%) | 23 (4.7%) | 2 (4.0%) | 0.66 (0.37–1.13) | 0.55 (0.13–2.10) |
| 4 copies | 2 (0.5%) | 3 (0.6%) | 1 (2.0%) | 1.23 (0.25–6.96) | 4.05 (0.27–35.2) |
| SMN2 copies | |||||
| 0 copy | 19 (4.8%) | 35 (7.2%) | 6 (12.0%) | 1.55 (0.88–2.75) | 2.73 (1.06–6.98)* |
| 1 copy | 123 (30.8%) | 185 (38.0%) | 14 (28.0%) | 1.38 (1.04–1.81)* | 0.87 (0.46–1.68) |
| 2 copies | 251 (62.9%) | 260 (53.4%) | 29 (58.0%) | 0.68 (0.52–0.89)* | 0.81 (0.45–1.47) |
| 3 copies | 6 (1.5%) | 7 (1.4%) | 1 (2.0%) | 0.96 (0.35–2.87) | 1.34 (0.11–8.43) |
* P< 0.05
The JaCALS method for extraction of DNA has been reported previously [27]. DNA samples from autopsy cases were also collected from central nervous system (occipital lobe, motor cortex, and cerebellum) tissue using a DNA extraction kit (QIAamp® DNA Mini Kit Cat. No. 56304; Qiagen, Venlo, Netherlands). DNA of LMND patients was extracted from blood samples.
We examined the causative genes of ALS and excluded those with pathological mutations to assess the effects of SMN copy number more accurately. For 344 of the JaCALS-registered patients, a comprehensive analysis had been conducted previously [27], and the remaining 96 had undergone analysis of repeat expansions in C9orf72 [28] and high-frequency causative gene mutations (SOD1, TDP-43 and FUS) using high-resolution melting (HRM) [29]. For the 47 autopsy cases, we performed comprehensive Illumina NovaSeq 6000 exome analysis by outsourcing (Takara Bio, Shiga. Japan) and excluded patients with non-synonymous or truncated variants of specific genes (TARDBP, OPTN, FUS, SOD1, TBK1, SQSTM1, MATR3, TUBA4A, NEK1, HNRNPA2B1, VCP, ELP3, SETX, HNRNPA1, CCNF, VAPB, C21orf2, CHCHD10, NEFH, ANG, DCTN1, CHMP2B, UBQLN2, Fig. 4, PFN1, ARHGEF28, EWSR1, TAF15, ANXA11, DAO, ERBB4, MAPT, TIA1, GLE1, PRPH, C9orf72, ALS2, SPG11, SIGMAR1 and DNAJC7) and C9orf72 repeat expansions. ALS causative gene mutations were observed in five JaCALS cases and three Niigata University cases. These 8 cases are not included in the 487 cases analyzed in this paper. In the LMND group, the number of CAG repeats in the AR gene was determined in order to exclude SBMA, and cases with 0 copies of the SMN1 gene, i.e., SMA cases, were also excluded.
SMN1 and SMN2 copy numbers were determined using TaqMan® droplet digital PCR (ddPCR) on a QX200 system (Bio-Rad Laboratories, Hercules, CA, USA) and ddPCR™ Supermix for Probes (No dUTP) (Cat. No. 1863024; Bio-Rad Laboratories). BCKDHA was used as a reference gene [30]. Primer and TaqMan probe sequences were designed for distinguishing between single base differences in exon 7 of the SMN1 and SMN2 genes [30]. The thermal cycler settings were as follows: (1) 94ºC for 10 min, (2) 94ºC for 30 s, (3) 50ºC for 2 min, (4) return to step 2) 49 times, (5) 90ºC for 10 min, and (6) maintain at 4ºC. The copy number of the target gene was determined as the ratio of the number of droplets positive for the target genes to those positive for BCKDHA.
To test for differences in the frequency of each SMN CNS between ALS or LMND patients and controls, the Fisher exact test was applied. Kaplan-Meier survival curves for the SMN1 or SMN2 copy number groups in ALS were compared, and a log-rank test was performed. Cox regression analysis was used to determine independent prognostic factors for survival, after adjusting for SMN1 or SMN2 copy number, sex, type of onset (bulbar or other), and age at onset. Each Cox regression analysis was performed independently, since the copy numbers of SMN1 and SMN2 are related. GraphPAD Prism 10 was used for all statistical analyses.
Results
The distribution of SMN1 and SMN2 copy numbers is summarized in Tables 2 and 3. Most control cases had two copies of SMN1: more than 90% of individuals had two copies, 7.0% had three copies, and only 1.3% had one copy of SMN1. The frequency of SMN1 copy numbers did not differ significantly between ALS patients and the control group. In contrast, genetic variations were observed in the copy numbers of SMN2 in both groups. In the control group, 4.8% had null alleles, 30.8% had one copy, 62.9% had two copies, and 1.5% had three copies. The presence of one SMN2 copy was more common in patients with ALS (38.0%) than in the controls (OR = 1.38, 95% CI = 1.04–1.81, p < 0.05).
Table 3.
Details of SMN1 or SMN2 copy number groups
| SMN1:SMN2 copies |
Control (n = 399) |
ALS (n = 487) |
LMND (n = 50) |
Control vs. ALS OR (95% CI) |
Control vs. LMND OR (95% CI) |
|---|---|---|---|---|---|
| 1:1 | 3 (0.8%) | 1 (0.2%) | 0 | 0.27 (0.02–1.83) | - |
| 1:2 | 1 (0.3%) | 3 (0.6%) | 0 | 2.46 (0.37-32.0) | - |
| 1:3 | 1 (0.3%) | 0 | 0 | - | - |
| 2:0 | 14 (3.5%) | 29 (6.0%) | 4 (8.0%) | 1.74 (0.90–3.28) | 2.39 (0.83–7.33) |
| 2:1 | 103 (25.8%) | 168 (34.5%) | 13 (26.0%) | 1.51 (1.13–2.03)* | 1.01 (0.51-2.00) |
| 2:2 | 242 (60.7%) | 254 (52.2%) | 29 (58.0%) | 0.70 (0.54–0.93)* | 0.90 (0.50–1.62) |
| 2:3 | 5 (1.3%) | 7 (1.4%) | 1 (2.0%) | 1.15 (0.40–3.22) | 1.61 (0.13–12.1) |
| 3:0 | 4 (1.0%) | 3 (0.6%) | 1 (2.0%) | 0.61 (0.15–2.29) | 2.02 (0.16–12.5) |
| 3:1 | 17 (4.3%) | 16 (3.3%) | 1 (2.0%) | 0.76 (0.40–1.57) | 0.46 (0.04–2.76) |
| 3:2 | 7 (1.8%) | 3 (0.6%) | 0 | 0.35 (0.10–1.24) | - |
| 4:0 | 1 (0.3%) | 3 (0.6%) | 1 (2.0%) | 2.47 (0.37–32.1) | 8.12 (0.42–154.5) |
| 4:2 | 1 (0.3%) | 0 | 0 | - | - |
* P<0.05
We classified ALS patients based on their SMN1 and SMN2 copy number and investigated their survival time (Fig. 1a, b). This revealed no significant differences among the groups classified according to SMN1 copy number (Fig. 1a). However, classification by SMN2 copy number revealed significant differences in survival time. Patients with the SMN2 null allele showed shorter survival than the other patients [median/mean survival: null SMN2 (n = 35) = 34/38.6 months; one copy (n = 185) = 39/62.0 months; two copies (n = 260) = 44/61.0 months; three copies (n = 7) = 54/66.6 months; log-rank test, p < 0.05] (Fig. 1b). Cox regression analysis revealed that age at onset (p < 0.001) and SMN2 copy number (hazard ratio = 0.84, 95% CI = 0.72–0.98, p < 0.05) were independently associated with the mortality rate.
Fig. 1.
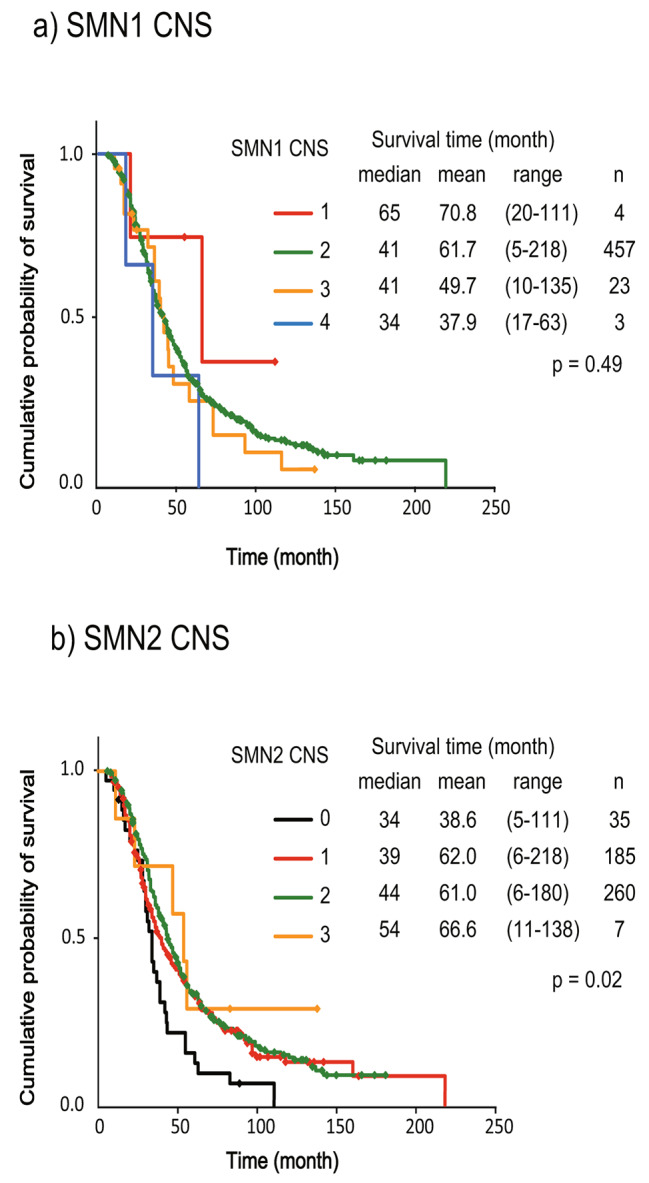
Kaplan-Meier analysis of survival time in the (a) SMN1 and (b) SMN2 copy number groups. Survival time differed between the SMN2 copy number groups (Cochran Mentel Haenszel test; p = 0.02)
SMN2 copy number had no effect on the age at onset of ALS (mean 62.9 years for null cases, 61.7 years for one copy, 61.2 years for two copies, and 63.9 years for three copies; one-way ANOVA, p = 0.77). There was no significant difference in SMN2 copy number between patients with bulbar onset and those with non-bulbar onset (43.0% and 36.3%, respectively, for one SMN2 copy; chi-squared test, p = 0.19). There was also no significant difference in SMN2 copy number between JaCALS-registered control patients and the control patients from Niigata University (30.4% versus 32.0%, respectively, for one SMN2 copy).
In addition, null SMN2 cases were significantly more frequent in the LMND group than in the control group (12.0% versus 4.8%, respectively; OR = 2.73, 95% CI = 1.06–6.98, p < 0.05) (Table 2). There were no statistically significant differences in SMN1 copy number. As no prognostic data were available for the LMND group, prognostic evaluation was not performed.
Discussion
Our present study of 487 Japanese ALS patients revealed that the CNS of SMN2 differed significantly from that in the controls: ALS patients with one SMN2 copy were significantly more frequent, and SMN2-null ALS patients had a significantly worse outcome. Furthermore, in the LMND group (excluding SBMA), SMN2-null patients were significantly more frequent than in the control group. These findings suggest that, at least in Japanese patients with adult-onset MND, including ALS and LMND, SMN2 copy number reduction adversely affects the onset and prognosis of MND.
However, a recent large European study showed that SMN copy number did not affect the risk for development of ALS or its prognosis [19]. This discrepancy may be attributable to differences in genetic background. SMN CNS in healthy subjects varies by region (Table 1). For example, normal individuals with only one copy of SMN2 are reportedly more frequent among Caucasians (37.9 − 42.2% ) [13–15] than among Asians (29.0 − 31.9%) [16, 23, 24]. A study from South Korea, located in the East Asian region as Japan, yielded results similar to those of the present study, in which SMN2 gene deficiency was associated with the development of ALS [16] and LMND [26], although the results were based on only a small number of cases. These results suggest that SMN2 copy number reduction may adversely affect the incidence and prognosis of adult-onset MND in East Asia, including Japan and South Korea.
The main issue raised by this study is why the copy number of SMN2, and not that of SMN1, affects the development and prognosis of ALS and LMND, even though most of the SMN protein is expressed from the SMN1 gene. A simple decrease in the SMN protein level due to SMN2 gene reduction cannot explain the development of MND and its prognostic impact.
There are several hypotheses that could explain this situation: the first is that the SMN2-derived defective SMN protein has physiological importance. There are several rare isoforms of SMN in addition to the normal type, and axonal SMN with intron 3 retention plays an important role in mammalian brain [31]. However, the Δ7 SMN protein, which is produced mainly from SMN2, is less stable because it lacks the exon 7-derived amino acid [31] and no important physiological function has been reported for it to date.
A second possibility is that SMN2 may have tissue-specific importance, as the splicing patterns of SMN2 mRNA vary from tissue to tissue. In the testes of mice experimentally expressing SMN2, the splicing pattern of SMN2-derived mRNA is greatly altered and full-length SMN protein is produced in large amounts [32, 33]. SMN2 CNS changes would be important if SMN2 mRNA splicing is altered and involved in normal SMN protein expression in the human nervous system, muscle, and cardiopulmonary tissues, and would influence ALS pathogenesis or prognosis, as in the mouse testis model. Currently, however, there is no such evidence, and therefore this possibility remains merely speculative.
A third possibility is simultaneous loss of genes near SMN2. NAIP, located close to SMN1, is reportedly associated with ALS [34], whereas SERF1B, located close to SMN2, has not yet been linked to any neurological disease. Thus, the mechanism by which the CNS of SMN2 influences the onset and prognosis of adult MND remains unclear and requires further investigation.
One of the limitations of this study was the small number of cases examined in comparison to recent large-scale studies. The proportion of SMA carriers in our reported control cohort does not differ significantly from previously reported data in Japan [35], suggesting that the analysis group in this study reflects the SMN CNS status in Japan to a certain extent. However, there are limitations to what can be concluded from an analysis based on several hundred cases. Additional studies of East Asian populations are therefore warranted. In addition, as the LMND group comprised only male patients, and prognosis evaluation has not been conducted. Future studies should also include female patients and investigate the relationship between the prognosis of LMND cases and SMN CNS. Additionally, several mutations in the SMN1 and SMN2 genes are known, and these may also impact the disease prognosis of SMA [36]. The influence of these genetic mutations on ALS and LMND is an important subject for future investigation.
This study has shown that the CNS of SMN2 affects the incidence and prognosis of ALS and LMND in Japanese patients, contrary to the results of previous studies in Europe, suggesting regional differences in genetic background.
Acknowledgements
Thanks to all researchers and editors who contributed to this study.
Abbreviations
- CNS
Copy number status
- SMN
Survival motor neuron
- ALS
Amyotrophic lateral sclerosis
- LMND
Lower motor neuron diseases
- SMA
Spinal muscular atrophy
- OR
Odds ratio
- CI
Confidence interval
- MND
Motor neuron disease
- mRNA
Messenger RNA
- snRNAs
Small nuclear RNA
- SALS
Sporadic ALS
- JaCALS
Japanese Consortium for Amyotrophic Lateral Sclerosis
- SBMA
Spinal and bulbar muscular atrophy
- SCA
Spinocerebellar ataxia
- DRPLA
Dentatorubral-Pallidoluysian Atrophy
- EAOH
Early onset ataxia associated with hypoalubuminemia and oculomotor apraxia
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- HDLS
Hereditary diffuse leukoencephalopathy with axonal spheroids
- HRM
High-resolution melting
- ddPCR
Droplet digital PCR
Author contributions
TI, AKo, AY, and OO contributed to the conception and design of the study. TI wrote the main manuscript text and prepared all figures and tables. TI, AKo, ST, and KK analyzed the genomic data for SMN CNS. TI, NA, SH and YH performed ALS-related gene mutation analysis of the target case genes. NA, RN, GT, YI, RK, MM, AT, OK, MA, SK and GS contributed to the data collection and management of cases from JaCALS. MT and AKa contributed to the data collection and management of autopsy cases. TI, SH and OO contributed to the data collection and management of LMND cases. Statistical review was performed by TI, AY, YH and NA. All authors reviewed the manuscript.
Funding
Study Funding: This research was supported by a grant-in-aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control; 26117006) from MEXT; grants-in-aid for Scientific Research (C) (17K09750, 21K07272, 24K10506) from the Japan Society for the Promotion of Science; grants-in-aid (17ek0109284h0001 and 181k1601002h0001) from the Japan Agency for Medical Research and Development; a grant-in-aid from the Mitsubishi Tanabe Pharma; and a grant-in-aid from the Japan ALS Association; MHLW Research on rare and intractable diseases Program Grant Number JPMH23FC1008.
Data availability
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Ethical approval was obtained from the research ethics committees of Niigata University (G2015-0781, G2020-0031) in accordance with the Declaration of Helsinki. ALS cases and their spouses participating in JaCALS have given written consent for genetic samples and clinical data to be provided for JaCALS and its collaborations. In the patients of genetic testing at Niigata University, written consent was obtained to use genetic information for research purposes. Written informed consent for autopsy cases, including the use of tissues for research purposes, was obtained from the families of the patients.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.de Boer EMJ, Orie VK, Williams T, Baker MR, De Oliveira HM, Polvikoski T, Silsby M, Menon P, van den Bos M, Halliday GM. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2021;92(1):86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dols-Icardo O, Garcia-Redondo A, Rojas-Garcia R, Borrego-Hernandez D, Illan-Gala I, Munoz-Blanco JL, Rabano A, Cervera-Carles L, Juarez-Rufian A, Spataro N, et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J Neurol Neurosurg Psychiatry. 2018;89(2):162–8. [DOI] [PubMed] [Google Scholar]
- 3.Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Reviews Neurol. 2017;13(2):96–104. [DOI] [PubMed] [Google Scholar]
- 4.Nagy ZF, Pal M, Engelhardt JI, Molnar MJ, Klivenyi P, Szell M. Beyond C9orf72: repeat expansions and copy number variations as risk factors of amyotrophic lateral sclerosis across various populations. BMC Med Genomics. 2024;17(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65. [DOI] [PubMed] [Google Scholar]
- 6.Wadman RI, Stam M, Jansen MD, van der Weegen Y, Wijngaarde CA, Harschnitz O, Sodaar P, Braun KP, Dooijes D, Lemmink HH, et al. A comparative study of SMN protein and mRNA in blood and fibroblasts in patients with spinal muscular atrophy and healthy controls. PLoS ONE. 2016;11(11):e0167087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96(11):6307–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8(4):340–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133(4):585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishihara T, Ariizumi Y, Shiga A, Kato T, Tan CF, Sato T, Miki Y, Yokoo M, Fujino T, Koyama A, et al. Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22(20):4136–47. [DOI] [PubMed] [Google Scholar]
- 11.Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med. 2013;5(2):221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veldink JH, Kalmijn S, Van der Hout AH, Lemmink HH, Groeneveld GJ, Lummen C, Scheffer H, Wokke JH, Van den Berg LH. SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology. 2005;65(6):820–5. [DOI] [PubMed] [Google Scholar]
- 13.Corcia P, Camu W, Halimi JM, Vourc’h P, Antar C, Vedrine S, Giraudeau B, de Toffol B, Andres CR, French ALSSG. SMN1 gene, but not SMN2, is a risk factor for sporadic ALS. Neurology. 2006;67(7):1147–50. [DOI] [PubMed] [Google Scholar]
- 14.Blauw HM, Barnes CP, van Vught PW, van Rheenen W, Verheul M, Cuppen E, Veldink JH, van den Berg LH. SMN1 gene duplications are associated with sporadic ALS. Neurology. 2012;78(11):776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corcia P, Ingre C, Blasco H, Press R, Praline J, Antar C, Veyrat-Durebex C, Guettard YO, Camu W, Andersen PM, et al. Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur J Hum Genet. 2012;20(5):588–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J-B, Lee K-A, Hong J-M, Suh G-I, Choi Y-C. Homozygous SMN2 deletion is a major risk factor among twenty-five Korean sporadic amyotrophic lateral sclerosis patients. Yonsei Med J. 2012;53(1):53–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XB, Cui NH, Gao JJ, Qiu XP, Zheng F. SMN1 duplications contribute to sporadic amyotrophic lateral sclerosis susceptibility: evidence from a meta-analysis. J Neurol Sci. 2014;340(1–2):63–8. [DOI] [PubMed] [Google Scholar]
- 18.Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci. 2016;3(7):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moisse M, Zwamborn RA, van Vugt J, van Der Spek R, van Rheenen W, Kenna B, Van Eijk K, Kenna K, Corcia P, Couratier P. The effect of SMN gene dosage on ALS risk and disease severity. Ann Neurol. 2021;89(4):686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Rheenen W, van Blitterswijk M, Huisman MH, Vlam L, van Doormaal PT, Seelen M, Medic J, Dooijes D, de Visser M, van der Kooi AJ, et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology. 2012;79(9):878–82. [DOI] [PubMed] [Google Scholar]
- 22.Konno T, Shiga A, Tsujino A, Sugai A, Kato T, Kanai K, Yokoseki A, Eguchi H, Kuwabara S, Nishizawa M, et al. Japanese amyotrophic lateral sclerosis patients with GGGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry. 2013;84(4):398–401. [DOI] [PubMed] [Google Scholar]
- 23.Sheng-Yuan Z, Xiong F, Chen Y-J, Yan T-Z, Zeng J, Li L, Zhang Y-N, Chen W-Q, Bao X-H, Zhang C. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur J Hum Genet. 2010;18(9):978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su Y-N, Hung C-C, Lin S-Y, Chen F-Y, Chern JP, Tsai C, Chang T-S, Yang C-C, Li H, Ho H-N. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005–2009: a prospective population-based cohort study. PLoS ONE. 2011;6(2):e17067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sangare M, Hendrickson B, Sango HA, Chen K, Nofziger J, Amara A, Dutra A, Schindler AB, Guindo A, Traore M, et al. Genetics of low spinal muscular atrophy carrier frequency in sub-saharan Africa. Ann Neurol. 2014;75(4):525–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Lee S-G, Choi Y-C, Kang S-W, Lee J-B, Choi JR, Lee KA. Association between survivor motor neuron 2 (SMN2) gene homozygous deletion and sporadic lower motor neuron disease in a Korean population. Ann Clin Lab Sci. 2010;40(4):368–74. [PubMed] [Google Scholar]
- 27.Nakamura R, Sone J, Atsuta N, Tohnai G, Watanabe H, Yokoi D, Nakatochi M, Watanabe H, Ito M, Senda J, et al. Next-generation sequencing of 28 ALS-related genes in a Japanese ALS cohort. Neurobiol Aging. 2016;39:e219211–218. [DOI] [PubMed] [Google Scholar]
- 28.Ogaki K, Li Y, Atsuta N, Tomiyama H, Funayama M, Watanabe H, Nakamura R, Yoshino H, Yato S, Tamura A. Analysis of C9orf72 repeat expansion in 563 Japanese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(10):e11–2527. e2516. [DOI] [PubMed] [Google Scholar]
- 29.Akimoto C, Morita M, Atsuta N, Sobue G, Nakano I. High-resolution melting (HRM) analysis of the Cu/Zn superoxide dismutase (SOD1) gene in Japanese sporadic amyotrophic lateral sclerosis (SALS) patients. Neurol Res Int. 2011; 165415. [DOI] [PMC free article] [PubMed]
- 30.Zhong Q, Bhattacharya S, Kotsopoulos S, Olson J, Taly V, Griffiths AD, Link DR, Larson JW. Multiplex digital PCR: breaking the one target per color barrier of quantitative PCR. Lab Chip. 2011;11(13):2167–74. [DOI] [PubMed] [Google Scholar]
- 31.Singh RN, Howell MD, Ottesen EW, Singh NN. Diverse role of survival motor neuron protein. Biochim Biophys Acta. 2017;1860(3):299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ottesen EW, Howell MD, Singh NN, Seo J, Whitley EM, Singh RN. Severe impairment of male reproductive organ development in a low SMN expressing mouse model of spinal muscular atrophy. Sci Rep. 2016;6:20193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seo J, Singh NN, Ottesen EW, Lee BM, Singh RN. A novel human-specific splice isoform alters the critical C-terminus of survival motor neuron protein. Sci Rep. 2016;6:30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kano O, Tanaka K, Kanno T, Iwasaki Y, Ikeda J-E. Neuronal apoptosis inhibitory protein is implicated in amyotrophic lateral sclerosis symptoms. Sci Rep. 2018;8(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sawada T, Kido J, Sugawara K, Yoshida S, Ozasa S, Nomura K, Okada K, Fujiyama N, Nakamura K. Newborn screening for spinal muscular atrophy in Japan: one year of experience. Mol Genet Metab Rep. 2022;32:100908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruhno C, McGovern VL, Avenarius MR, Snyder PJ, Prior TW, Nery FC, Muhtaseb A, Roggenbuck JS, Kissel JT, Sansone VA, et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum Genet. 2019;138(3):241–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.