

Abstract
Background
RNA secondary structural alignment serves as a foundational procedure in identifying conserved structural motifs among RNA sequences, crucially advancing our understanding of novel RNAs via comparative genomic analysis. While various computational strategies for RNA structural alignment exist, they often come with high computational complexity. Specifically, when addressing a set of RNAs with unknown structures, the task of simultaneously predicting their consensus secondary structure and determining the optimal sequence alignment requires an overwhelming computational effort of for each RNA pair. Such an extremely high computational complexity makes these methods impractical for large-scale analysis despite their accurate alignment capabilities.
Results
In this paper, we introduce REDalign, an innovative approach based on deep learning for RNA secondary structural alignment. By utilizing a residual encoder-decoder network, REDalign can efficiently capture consensus structures and optimize structural alignments. In this learning model, the encoder network leverages a hierarchical pyramid to assimilate high-level structural features. Concurrently, the decoder network, enhanced with residual skip connections, integrates multi-level encoded features to learn detailed feature hierarchies with fewer parameter sets. REDalign significantly reduces computational complexity compared to Sankoff-style algorithms and effectively handles non-nested structures, including pseudoknots, which are challenging for traditional alignment methods. Extensive evaluations demonstrate that REDalign provides superior accuracy and substantial computational efficiency.
Conclusion
REDalign presents a significant advancement in RNA secondary structural alignment, balancing high alignment accuracy with lower computational demands. Its ability to handle complex RNA structures, including pseudoknots, makes it an effective tool for large-scale RNA analysis, with potential implications for accelerating discoveries in RNA research and comparative genomics.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12859-024-05956-7.
Keywords: RNA secondary structure, Structural alignment, Pseudoknot structure, Deep learning, Residual encoder decoder network
Introduction
RNA is a single-stranded sequence composed of four nucleotide bases (A, C, G, and U), exhibiting a wide range of structural motifs due to local hydrogen bonding interactions [1]. The interactions between complementary nucleotides lead to the formation of base pairs, which contribute to the stability of RNA structures. Additionally, the structural motifs and specific folding patterns of RNA enable it to interact with other biomolecules, such as proteins and other RNAs, where it can play pivotal roles in biological processes. RNA structural motifs can be classified into three different categories: i) primary structure, ii) secondary structure, and iii) tertiary structure. Although the primary structure of RNA is a linear sequence of nucleotides connected by phosphodiester bonds, the secondary and tertiary structures form specific motifs that can be represented in two-dimensional and three-dimensional spaces, respectively. While analyzing the native three-dimensional structure of RNA is challenging due to its intricate interactions, its secondary structure, characterized by base pairing between nucleotides, is relatively more stable and predictable [2–4]. Hence, RNA secondary structure is more accessible for computational analysis and prediction.
The secondary structure of RNA can be decomposed into stem and loop structures, wherein base pairs are formed by canonical (Watson-Crick and GU) base pairs, as illustrated in Fig. 2a. These base pairs tend to form in a nested arrangement, where the base positions and follow the order or . In addition to the nested arrangement, RNA can also form pseudoknots, which are non-nested crossing base pairs. Pseudoknots are known to play roles in structural stability and regulatory function [5, 6]. However, the presence of pseudoknots complicates computational structure analysis, including structure prediction and structural alignment.
Fig. 2.

Illustration of the input conformations of the dinucleotides for aligning RNA Sequence 1 and Sequence 2. a The illustration of RNA Sequence 1 with a stem-loop structure motif. The stem consists of consecutive stacked base pairs, while the loop represents unpaired segments enclosed by the base pairs. b The illustration of RNA Sequence 2 with a stem-loop structure motif. c The corresponding contact matrix for the AU dinucleotide. d The corresponding contact matrix for the GU dinucleotide. e The corresponding contact matrix for the AA dinucleotide. f The corresponding contact matrix for the GG dinucleotide
RNA secondary structures form the core framework for RNA folding and are crucial for its function. RNA secondary structural alignment emerges as a critical procedure in bioinformatics, playing a pivotal role in comparative genomic analysis. The alignment aids in identifying homologous RNA families, thereby accelerating the functional studies and annotation of newly discovered genes [7–9]. Though sequence alignment based on similarity is suitable for highly similar sequences, it becomes less effective for sequences with low similarity due to accumulated mutations [10]. Conversely, comparative structural analyses have revealed that RNA secondary structures tend to be more conserved than their primary sequences [11, 12]. Hence, RNA sequence alignment should integrate the underlying RNA folding structures to accurately identify homologous RNA sequences and uncover their functional characteristics along with evolutionary relationships.
To accurately predict RNA secondary structure alignments, Sankoff initially proposed a dynamic programming algorithm for RNA structural alignment, which simultaneously solves the RNA sequence alignment and consensus folding structure problem [13]. Subsequently, various implementations of Sankoff-style algorithms have been developed. Among these methods, Dynalign and Foldalign utilize the nearest-neighbor thermodynamic model to assess the potential structures and find the consensus structure with the lowest free energy for the structural alignment [14–16]. Similarly, PARTS employs a pseudo-free energy model based on base pairing and alignment probabilities to identify the structural alignment with the maximum joint probability [17]. However, the computational complexity of the Sankoff algorithm for RNA sequences of length L is considerable, with time complexity of and space complexity of . The extreme time complexity of the Sankoff algorithm makes it impractical for large-scale genome analysis, leading to the development of several simplified algorithms for structural alignment [18, 19]. By applying the base pairing probability as a lightweight energy model, PMcomp simplifies the dynamic programming to reduce the computational complexity to in time [20]. Building upon this lightweight energy model, LocARNA and SPARSE further simplify the alignment approach by exploiting the sparse property of the base pairing, ultimately achieving the quadratic time complexity [21, 22]. LinearTurboFold utilizes a linearized partition function to iteratively refine base pairing and alignment probabilities, enabling efficient structural alignments [23]. In contrast to the Sankoff-style algorithms, TOPAS integrates RNA sequence and structure information through a topological network for RNA structural alignment [24]. While the network-based structural alignment method is efficient, the accuracy relies on the precise prior estimation of RNA base pairing and alignment probability.
Deep learning has emerged as a powerful tool in various domains, including biomedicine and bioinformatics, owing to its ability to handle high-dimensional data and learn hierarchical representations [25–27]. Taking advantage of this, RNABERT employs deep learning to acquire informative base embedding for RNA structural alignment, resulting in reduced time complexity [28]. In this study, we introduce REDalign, a novel method that utilizes the Residual Encoder-Decoder network for RNA structural alignment. Inspired by the success of REDfold [29], an accurate deep learning-based RNA secondary structure prediction algorithm, we adapt the encoder-decoder network to efficiently learn RNA structure and directly align RNA sequences. By incorporating the ResNet network, REDalign can effectively learn residual information and mitigate the vanishing gradient problem. In comparative tests with several well-known RNA structure alignment algorithms, REDalign demonstrates superior performance in terms of speed and accuracy. The contributions of this work include a novel methodology that utilizes an efficient learning network to capture complex RNA secondary structural features in RNA sequence pairs with low computational complexity, enabling direct and highly accurate RNA secondary structural alignment. Additionally, we propose an effective data representation for RNA sequence pairs that enables the use of sophisticated neural networks, such as residual encoder-decoder networks. Furthermore, we have developed a user-friendly web server that allows for the convenient utilization of REDalign with customizable parameters for RNA structural alignment. Users can submit their RNA sequences in FASTA format to the server and obtain the corresponding RNA alignment results, even if they lack sufficient software background and computing resources.
Materials and methods
The objective of RNA secondary structural alignment is to align the consensus folding structure across a given set of RNA sequences. To achieve this goal, we developed a rapid and accurate structural alignment method using deep neural networks. REDalign adopts a residual encoder-decoder network similar to REDfold, leveraging its high computational efficiency and ability to accurately model RNA structures. This approach ensures that REDalign can effectively transform RNA sequences into a high-dimensional data representation tailored to a deep learning framework, allowing it to accurately capture and analyze the complex structural features underlying RNA sequences and drive the accurate alignment of RNA secondary structures. Figure 1 provides a graphical overview of the proposed RNA structural alignment algorithm. The process begins with the transformation of the RNA sequences into an input conformation that integrates contact matrices for the dinucleotides. In the following stage, an encoder-decoder network is employed to extract essential features and formulate a score map intended to assess dinucleotide alignment. In the subsequent postprocessing phase, dynamic programming is applied to the score map to derive the optimal alignment. After postprocessing, REDalign outputs the forecasted contact map corresponding to the resulting structural alignment. Further details of the procedure are discussed in subsequent subsections.
Fig. 1.

Graphical overview of REDalign architecture. REDalign includes three main steps to derive a precise RNA structural alignment. First, REDalign transforms the pair of input RNA sequences into two-dimensional binary contact matrices in order to represent the relative position of dinucleotides in the RNA sequences. Next, through the residual encoder and decoder network architecture, it learns the conserved structures of RNA sequences and can yield the alignment probability of dinucleotides within RNA sequences. Finally, REDalign derives the accurate structural alignment of RNA sequences using dynamic programming
Input conformation
In the initial step, REDalign transforms the input RNA sequences into two-dimensional binary contact matrices, which serve as the primary input conformation. These matrices represent the relative positions of dinucleotides within the RNA sequences processed by REDalign. Consider RNA sequences and with bases and from the set . Here, and denote the lengths of the respective sequences. The contact matrices for the dinucleotide, denoted by , where the dinucleotide , are used to trace all 16 possible combinations of the dinucleotide within the sequences. Take Fig. 2 as an example, the element of the contact matrix M(AU) is set to one if the dinucleotide corresponds to the dinucleotide set ; otherwise it is zero. The contact matrices provide information not only about the alignment of identical bases between sequences but also offer clues regarding the consensus secondary structure. As shown in Fig. 2, the contact matrices M(AA) and M(GG) indicate the potential alignments for bases A and G, respectively. Furthermore, the sum of matrices encompasses the consensus stem structure between RNA Sequence 1 and RNA Sequence 2. Hence, the neural network has the potential to simultaneously learn the structural alignment of bases and the consensus structure, enabling a more comprehensive understanding of RNA sequence relationships. The input conformation consists of contact matrices with overall size for the input RNA sequences. Based on the input conformation, the following neural network can effectively extract feature maps and output a score map for structural alignment.
Network architecture
The deep learning network (DNN) of REDalign is organized into three phases: feature extraction, residual encoder-decoder network, and normalization. Given both input sequences of length L, the input conformation has a size of . As the input conformation consists of contact matrices with high sparsity, REDalign employs three-layer basic convolution modules (BCMs) to extract essential features for consensus structure and alignment. Here, the BCM serves as the fundamental processing unit, incorporating 2-dimensional convolution, batch normalization, and rectified linear unit (ReLU) activation functions. After feature extraction, the condensed feature map retains the size of , which is then fed into the subsequent residual encoder-decoder network, as illustrated in Fig. 3.
Fig. 3.

The REDalign architecture. a The learning network schematic encompasses feature extraction, a residual encoder-decoder network, and normalization. The RNA sequences are first transformed into an input conformation, and then fed into the deep neural network. Based on the extracted feature map, the encoder-decoder network outputs a score map for the structural alignment. b Dense Connected Module (DCM). The DCM consists of a series of BCM layers that are densely interconnected. The output feature map is formed by concatenating all feature maps from the BCM layers, including the input feature map and the output map of the encoder network. This design ensures that each layer receives all feature maps from the preceding layers, thereby improving the network’s parameter efficiency
The residual encoder-decoder network is designed based on a fusion of the FC-DenseNet and ResNet architectures, capitalizing on their strengths to enhance performance. Since the input feature map contains low-level structure and pattern details, the encoder network in the DNN employs a hierarchical pyramid structure to capture more generalized high-level structural features. Hence, it can ultimately integrate low-level local features and high-level structural features in a balanced manner. This enables the learning network to recognize larger patterns and structures with a decreased computational complexity. To avoid forming learning bottlenecks in the encoding pathway, the dense connected module (DCM) is employed. The DCM, as shown in Fig. 3b, consists of a series of BCM layers with densely connected links between layers and serves to increase the depth of the feature map. Within each BCM layer of the DCM, new feature maps are generated and combined with feature maps from all preceding layers, including the input feature map. As a result, the output feature map of the DCM integrates all feature maps, effectively reusing preceding features. This approach allows the DCM to accommodate a more diverse set of features, enhancing the efficiency of the network parameters and contributing to overall network performance [30, 31].
In the decoder network, DCMs and transition up modules are utilized to reform high-level encoded features for the structural alignment. The transition up module utilizes up-sampling and BCM operations to expand the size of the feature map while reducing its depth. This ensures that spatial information is efficiently integrated during the decoding process. Furthermore, multi-level encoded features are incorporated into the decoding pathway through skip connections and additions, as illustrated in Fig. 3a. This approach resembles the residual learning connection found in ResNet [32], enabling effective information flow across different levels and facilitating the capture of intricate feature hierarchies. In contrast to FC-DenseNet [33], which makes use of skip and concatenation, the residual learning connection can efficiently learn the detailed information with fewer parameters. Ultimately, the decoder network leverages all multi-level features and produces a feature map of size .
Afterward, batch normalization is applied to regulate covariate shift and cap the maximum value at one, resulting in the raw score map . Although the raw score map can effectively capture the structural alignment probability due to the effective learning capabilities of the deep neural networks, we may need to further integrate the contact matrix in a balanced manner depending on the different levels of sequence identity in order to enhance the flexibility of the method. Using this raw score map, specific weighting parameters are introduced to refine the potential structural alignment scores. Considering that sequences with high similarity tend to align identical nucleotides, the contact matrices corresponding to identical bases are integrated into the score map with a specified weight as the following equation
| 1 |
where is the sum of all contact matrices with identical bases. Based on the score map , the structural alignment for RNAs can be constructed by maximizing the overall score through dynamic programming. Specifically, we use the Needleman-Wunsch algorithm [34], which includes a gap weight to penalize gap insertion in the optimization. The algorithm ensures that the optimal alignment is derived by considering all possible alignments and selecting the one with the highest score. The resulting optimal structural alignment is then applied to the final aligned RNA sequences.
REDalign efficiently implements RNA structural alignment by utilizing the encoder-decoder structure with residual skip connections and dynamic programming techniques. The computational complexity of REDalign is , where L is the sequence length and N is the number of parameters in the network. Additionally, REDalign can take advantage of parallel computing to accelerate the calculations, thereby increasing the overall throughput. In comparison to traditional Sankoff-style algorithms that require a time complexity of , REDalign stands out as a highly efficient method for RNA structural alignment.
Datasets for evaluation
To evaluate the performance of the proposed structural alignment method, REDalign, the BRAliBase 2.1 K2 dataset [35] was used as the test benchmark for performance evaluation and comparison. BRAliBase 2.1 provides comprehensive benchmarking RNA sequences and their structural alignments based on various sequence identity levels. It has been widely utilized to fairly assess the performance of RNA structural alignment algorithms. Sequences containing unknown or uncertain bases were excluded from the assessment. The benchmark comprises 36 RNA structural families, accounting for a total of 8,587 RNA sequence pairs with an average sequence identity of 0.667. For the training of the REDalign learning network, we utilized the corresponding 39 RNA families in the Rfam 14.3 database [36] to construct the training set. Sequence pairs were randomly selected according to family size, ensuring no identical samples matched with those in the test benchmark. Specifically, we took 12,000 sequence pairs from 5 S rRNA family and sourced a total of 95,345 samples from the database. The composition of the samples with respect to the specific family groups of ncRNA in the training dataset is listed in Table S1 (Additionl file 1). During the training phase, we employed the Adam optimizer with a learning rate of 0.001 and used the binary cross-entropy with logits loss function. The training process ran for a maximum of 400 epochs, taking approximately 146 h on a system equipped with Intel x86-64 8-core CPUs clocked at 3.5 GHz and an NVIDIA RTX 3070 GPU.
Results and discussion
The RNA structural alignment performance was mainly assessed in terms of the sensitivity (SEN)= and positive predictive value (PPV)=. Here, True Positives (TP) indicate correctly aligned homologous bases, False Positives (FP) represent misaligned bases, and False Negatives (FN) account for homologous bases that were not aligned. In addition to the base metrics, the harmonic metric F-score = is also utilized for performance evaluation.
Parameters for structural alignment using REDalign
REDalign provides the ability to refine structural alignment by adjusting the weight parameters. Given sequences with a high degree of sequence identity (SI), increasing the weight for identical bases ( in Eq. 1) can enhance the scores for aligning the same bases. This correlation is depicted in Fig. 4(a), showing the effect of adjusting weight on the structural alignment performance. When dealing with sequences having high sequence identity (SI), increasing the weight can slightly improve alignment performance. In cases involving sequences with low SI, adjustments to the weight could negatively impact the performance, potentially causing misalignments of identical bases. While a negative might prevent incorrect alignments of identical bases, it also leads to missing more correct alignments and significantly degrades TP and sensitivity. Based on our experimental results, the weight ranging from 0 to 0.4 is considered an appropriate range for REDalign.
Fig. 4.
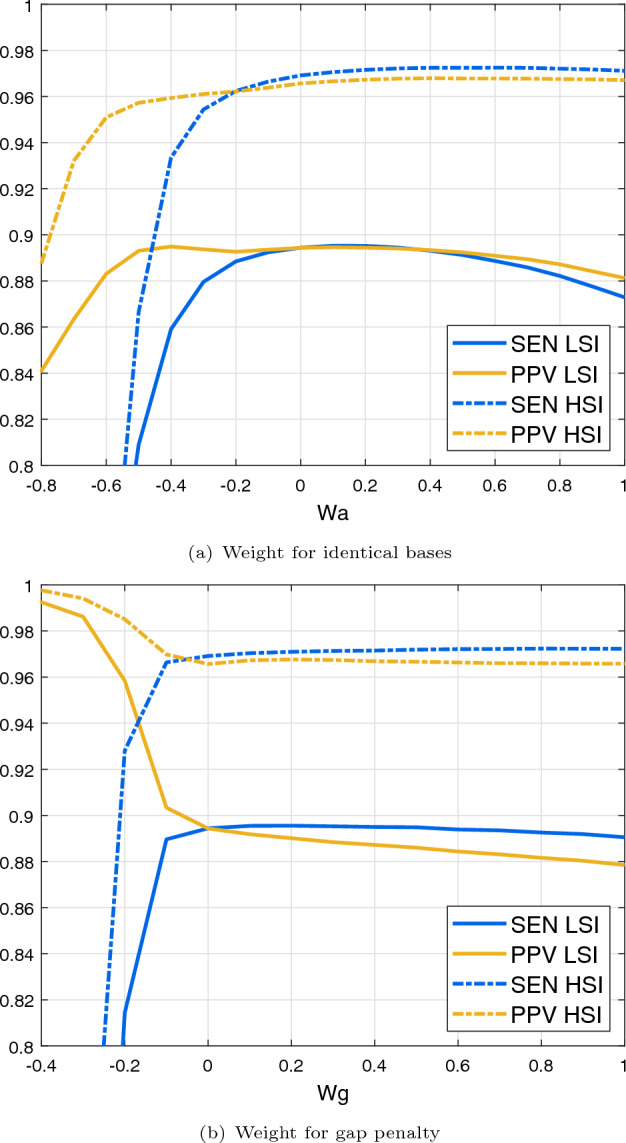
The effect of the weight parameters for the structural alignment. The blue line indicates sensitivity (SEN), and the yellow line denotes PPV. Sequences with a high sequence identity (HSI: SI>0.75) are represented by the dashed line, whereas the solid line depicts sequences with a low sequence identity (LSI). a Effect of the weight for identical bases . b Effect of the weight for gap penalty
Another adjustable weight parameter pertains to the gap penalty in the Needleman-Wunsch algorithm, employed to penalize the gap insertion during alignment. The effect of varying weight on the structural alignment performance is illustrated in Fig. 4b. For sequences with high SI, increasing the penalty weight can avoid gap insertion and encourage matching bases, resulting in a minor enhancement. Conversely, for sequences with low SI, modifying the penalty weight could potentially degrade the performance. Specifically, the negative values of can dramatically increase FN and decrease FP, leading to high PPV but potentially failing in the alignment, which can cause a sharp decrease in SEN. When considering both low and high sequence identity cases, a reasonable parameter setting for the weight ranges from 0 to 0.2. Given that these weight adjustments offer only marginal improvements, the default weight parameters of REDalign are set to zero. However, users can still appropriately adjust the weight parameters as needed for their specific applications.
Benchmarking results using BRAliBase
To draw a comprehensive comparison, we assessed the performance of several prominent Sankoff-style structural alignment algorithms using the same dataset. We also included the benchmarking results of REDalign against state-of-the-art neural network-based algorithms. The structural alignment algorithms assessed in this evaluation are summarized in Table 1. All experiments were conducted on a 64-bit server running the Linux 5.8.0 kernel, equipped with 8-core CPUs operating at 3.5 GHz and 32 GB RAM. Table 2 presents the overall prediction performance and total runtime based on the BRAliBase benchmark. While traditional Sankoff-style algorithms have their respective advantages, deep learning-based structural alignments stand out in terms of accuracy. As demonstrated in Table 2, REDalign yields highly precise structural alignment results, surpassing previous methods in both accuracy and computational efficiency.
Table 1.
Table of the RNA structural alignment algorithms under consideration for performance assessment in this study
| Program | Package/Version | References |
|---|---|---|
| PARTS | RNAstructure 6.3 | [17] |
| Dynalign2 | RNAstructure 6.3 | [15] |
| Foldalign | 2.5.3 | [37] |
| LocARNA | LocARNA 1.9.2 | [21] |
| SPARSE | LocARNA 1.9.2 | [22] |
| RNABERT | 1.0.0 | [28] |
| RNAmountAlign | 1.0.0 | [19] |
| LinearTurboFold | 1.0.0 | [23] |
†All algorithms were evaluated with default configurations, and the network model of RNABERT has been pretrained using the full Rfam 14.3 dataset [28]
Table 2.
Evaluation results of RNA structural alignment algorithms using the BRAliBase 2.1 K2 benchmark. Performance is assessed using SEN, PPV, and F-Score metrics, with the computation time recorded for aligning all sequences in the benchmark (in seconds)
| Program | SEN | PPV | F-Score | (Time) |
|---|---|---|---|---|
| REDalign | 0.929 | 0.927 | 0.928 | 2.724 |
| PARTS | 0.860 | 0.850 | 0.855 | 5.951 |
| Foldalign | 0.860 | 0.847 | 0.854 | 5.572 |
| Dynalign2 | 0.707 | 0.714 | 0.711 | 5.212 |
| LocaRNA | 0.820 | 0.865 | 0.842 | 3.901 |
| SPARSE | 0.848 | 0.848 | 0.848 | 3.306 |
| RNABERT | 0.870 | 0.860 | 0.865 | 4.389 |
| RNAmountAlign | 0.854 | 0.840 | 0.847 | 2.800 |
| LinearTurboFold | 0.881 | 0.872 | 0.876 | 3.335 |
To investigate the effect of RNA sequence similarity on the alignment accuracy across different algorithms, we categorized sequence pairs based on their sequence identity. As shown in Fig. 5, REDalign consistently outperforms other structural alignment algorithms across all SI levels, with the exception of instances around 90% SI, where a minor gap is observed. The results indicate that the performance gaps between different algorithms are negligible for sequence pairs with high sequence identity. Additionally, most algorithms successfully predict the correct structural alignments for RNA sequence pairs with high SI, achieving high SEN and PPV. Hence, achieving higher performance for the mid and low sequence identity would be more appropriate from a practical standpoint. REDalign clearly outperforms other competing algorithms for the sequence pairs with a 50% SI. Moreover, for the challenging cases with SI lower than 30%, REDalign achieves SEN and PPV values that are at least 0.1 higher than those of the best runner-up algorithms and 0.3 higher than the next groups. In low sequence identity benchmarks, the remarkable performance gap provides clear evidence for the effective learning capability of the proposed deep learning framework for identifying secondary structural similarities among different RNA sequences. Though the deep learning-based RNABERT is able to align sequences based on the base embedded information, the performance of structural alignment is similar to the traditional Sankoff-style algorithm such as PARTS. In contrast, REDalign is capable of analyzing consensus structures, ensuring accurate alignment even for sequences with low similarity. Table 3 illustrates the alignment performance for tRNAs (D31785, J04815) with an SI of 0.246 and 5 S rRNAs (X54477, X02731) with an SI of 0.376 from the BRAliBase benchmark. As REDalign adopts the residual encoder-decoder network, similar to the REDfold structure prediction, it can effectively analyze RNA secondary structure and perform RNA structural alignment. Figure 6 shows the predicted structures using REDfold, which successfully reconstructs the RNA structures for tRNAs and 5 S rRNAs. Since the residual encoder-decoder network has the ability to analyze the corresponding RNA structures, REDalign is able to recognize the corresponding base pairs and loops among RNAs. Even for RNA sequences with low sequence identity, REDalign can achieve high alignment accuracy, whereas conventional approaches struggle to perform such alignments. Furthermore, for highly similar sequence alignment, performance can be refined by adjusting weight parameters to enhance the matching of identical bases.
Fig. 5.
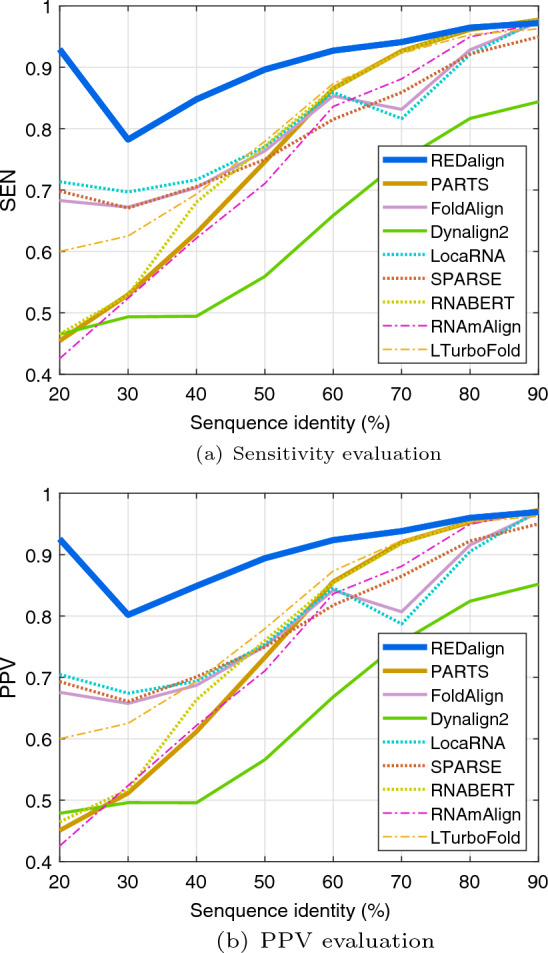
Performance evaluation results based on the BRAliBase 2.1 K2 dataset. a Sensitivity (SEN) of different algorithms are shown as a function of SI. b Positive predictive value (PPV) of different algorithms are shown as a function of SI
Table 3.
Evaluation results of structural alignment for RNA sequences tRNAs (D31785, J04815) and 5 S rRNAs (X54477, X02731)
| tRNAs | 5S rRNAs | |||||
|---|---|---|---|---|---|---|
| SEN | PPV | F-Score | SEN | PPV | F-Score | |
| REDalign | 0.985 | 0.985 | 0.985 | 0.982 | 0.982 | 0.982 |
| PARTS | 0.642 | 0.623 | 0.632 | 0.526 | 0.513 | 0.519 |
| Foldalign | 0.433 | 0.426 | 0.430 | 0.816 | 0.802 | 0.809 |
| Dynalign2 | 0.418 | 0.431 | 0.424 | 0.289 | 0.282 | 0.286 |
| LocaRNA | 0.433 | 0.439 | 0.436 | 0.623 | 0.612 | 0.617 |
| SPARSE | 0.433 | 0.426 | 0.430 | 0.211 | 0.240 | 0.224 |
| RNABERT | 0.567 | 0.551 | 0.559 | 0.789 | 0.783 | 0.786 |
| RNAmountAlign | 0.478 | 0.464 | 0.471 | 0.526 | 0.513 | 0.519 |
| LinearTurboFold | 0.642 | 0.623 | 0.632 | 0.553 | 0.563 | 0.558 |
Fig. 6.

The illustration of the predicted RNA secondary structures for tRNAs and 5 S rRNAs. The structures of tRNAs (D31785, J04815) and 5 S rRNAs (X54477, X02731) were created using REDfold[29] and drawn using VARAN[38]. The RNA sequences are arranged from 5’ to 3’, and the base pairs are linked with blue lines
Secondary structure alignments for RNA sequences with pseudo-knot
To further assess the performance of structural alignment for RNAs with pseudoknots, sequence pairs of RNA families listed in Table 4 were randomly selected from Rfam 14.3 for evaluation. The benchmark comprises 9 RNA structural families with pseudoknots, encompassing a total of 8,926 RNA sequence pairs that exhibit an average sequence identity of 0.614. Test sample counts were set proportional to the size of each RNA family, while the training sample size was approximately six times larger than the test samples. Table 5 summarizes the alignment results for these RNA families with pseudoknots. Given that predicting RNA secondary structures with pseudoknots is generally more challenging, most Sankoff-style algorithms tend to show degraded performance in structural alignment. Nonetheless, as depicted in Table 5, REDalign consistently outperforms other alignment algorithms, emphasizing its speed and precision even with pseudoknot structures.
Table 4.
List of the RNA family in the Rfam 14.3 database containing pseudoknots that were used for performance evaluation in this study
| RNA family | Test sample size | Training sample size |
|---|---|---|
| SAM | 565 | 3392 |
| Downstream-peptide | 777 | 4665 |
| SAM-I-IV-variant | 542 | 3251 |
| DUF805b | 1521 | 9128 |
| skipping-rope | 1764 | 10586 |
| drum RNA | 574 | 3445 |
| raiA RNA | 601 | 3608 |
| twister-P1 | 2000 | 12000 |
| c-di-GMP-II-GAG | 582 | 3489 |
Table 5.
Evaluation results of structural alignment for RNA families with pseudoknot. Performance is assessed using SEN, PPV, and F-Score metrics, with the computation time recorded for aligning all sequences in the benchmark
| Program | SEN | PPV | F-Score | (Time) |
|---|---|---|---|---|
| REDalign | 0.966 | 0.958 | 0.962 | 2.498 |
| PARTS | 0.806 | 0.787 | 0.796 | 5.689 |
| Foldalign | 0.779 | 0.762 | 0.770 | 4.821 |
| Dynalign2 | 0.546 | 0.546 | 0.546 | 4.980 |
| LocaRNA | 0.787 | 0.766 | 0.776 | 3.535 |
| SPARSE | 0.761 | 0.753 | 0.757 | 3.000 |
| RNABERT | 0.838 | 0.819 | 0.828 | 4.827 |
| RNAmountAlign | 0.772 | 0.751 | 0.762 | 2.711 |
| LinearTurboFold | 0.828 | 0.810 | 0.819 | 3.331 |
Conclusions
RNA structural alignment is a pivotal process in identifying conserved structural motifs among RNAs, playing a key role in shedding light on new RNAs through comparative genomic analysis. A range of computational algorithms and tools for RNA structural alignment have been developed, each with its own merits and limitations. The Sankoff-style algorithms, which concurrently predict optimal foldings and alignments, have gained popularity due to their accuracy. However, these algorithms often come with a high computational cost, requiring significant time and computational resources. In response to these challenges, this paper introduces REDalign, a novel and efficient approach for RNA structural alignment based on a residual encoder-decoder learning network. This deep learning approach incorporates ResNet with the encoder-decoder network, enhancing the efficiency and effectiveness of the model for RNA structural alignment. Extensive performance comparisons, including tests on the BRAliBase 2.1 K2 dataset, reveal that REDalign offers competitive efficiency with a significant advantage in accuracy compared to Sankoff-style algorithms and base embedded learning method. Unlike traditional methods, which could degrade when dealing with RNA structures containing pseudoknots, REDalign effectively aligns RNA sequences with complex pseudoknots. While the deep learning approach employed by REDalign requires substantial training data, its predictive accuracy exceeds that of traditional methods. As RNA databases continue to grow due to ongoing research and discoveries, it is expected that larger, more comprehensive datasets will become available for training deep learning models like REDalign, further enhancing their utility and effectiveness in the rapidly evolving field of RNA research.
Supplementary Information
Additional file 1. Table S1. List of RNA family groups and the number of sequences in the ncRNA training dataset. The families are grouped according the Rfam ID in the Rfam database 14.3.
Acknowledgements
The authors thank the anonymous reviewers for their valuable suggestions.
Author Contributions
CC, YC, and HJ conceived the method. CC developed the algorithm and performed the simulations. CC, YC, and HJ analyzed the results and wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by Incheon National University (International Cooperative) research grant in 2021. This work was also supported by MOST of Taiwan under project 110-2222-E-415-001-MY2.
Availability of data and materials
The REDalign web server is freely available at https://REDalign.ee.ncyu.edu.tw. The dataset used for analyzing ncRNAs with pseudoknots in REDalign is accessible at https://github.com/aky3100/REDalign.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
No Conflict of interest is declared.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chun-Chi Chen, Email: aky3100@mail.ncyu.edu.tw.
Hyundoo Jeong, Email: hdj@inu.ac.kr.
References
- 1.Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew Chem Int Ed. 1999;38:2326–43. [DOI] [PubMed] [Google Scholar]
- 2.Tinoco I Jr, Bustamante C. How RNA folds. J Mol Biol. 1999;293:271–81. [DOI] [PubMed] [Google Scholar]
- 3.Flamm C, Fontana W, Hofacker IL, Schuster P. RNA folding at elementary step resolution. RNA. 2000;6:325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathews DH. Predicting RNA secondary structure by free energy minimization. Theoret Chem Acc. 2006;116:160–8. [Google Scholar]
- 5.Giedroc DP, Theimer CA, Nixon PL. Structure, stability and function of RNA pseudoknots involved in stimulating ribosomal frameshifting. J Mol Biol. 2000;298:167–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peselis A, Serganov A. Structure and function of pseudoknots involved in gene expression control. RNA. 2014;5:803–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosig A, Zhu L, Stadler PF. Customized strategies for discovering distant ncRNA homologs. Brief Funct Genomic Proteomic. 2009;8:451–60. [DOI] [PubMed] [Google Scholar]
- 9.Washietl, S. Sequence and structure analysis of noncoding RNAs. Data Mining Techniques for the Life Sciences 285–306 (2010). [DOI] [PubMed]
- 10.Borozan I, Watt S, Ferretti V. Integrating alignment-based and alignment-free sequence similarity measures for biological sequence classification. Bioinformatics. 2015;31:1396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raué H, Klootwijk J, Musters W. Evolutionary conservation of structure and function of high molecular weight ribosomal RNA. Prog Biophys Mol Biol. 1988;51:77–129. [DOI] [PubMed] [Google Scholar]
- 12.Johnsson P, Lipovich L, Grandér D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochimica et Biophysica Acta (BBA)-General Subjects. 2014;1840:1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J Appl Math. 1985;45:810–25. [Google Scholar]
- 14.Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J Mol Biol. 2002;317:191–203. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y, Sharma G, Mathews DH. Dynalign II: common secondary structure prediction for RNA homologs with domain insertions. Nucleic Acids Res. 2014;42:13939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Havgaard JH, Lyngsø RB, Stormo GD, Gorodkin J. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics. 2005;21:1815–24. [DOI] [PubMed] [Google Scholar]
- 17.Harmanci AO, Sharma G, Mathews DH. PARTS: Probabilistic Alignment for RNA joinT Secondary structure prediction. Nucleic Acids Res. 2008;36:2406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabei Y, Tsuda K, Kin T, Asai K. Scarna: fast and accurate structural alignment of rna sequences by matching fixed-length stem fragments. Bioinformatics. 2006;22:1723–9. [DOI] [PubMed] [Google Scholar]
- 19.Bayegan AH, Clote P. RNAmountAlign: efficient software for local, global, semiglobal pairwise and multiple RNA sequence/structure alignment. PLoS ONE. 2020;15: e0227177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofacker IL, Bernhart SH, Stadler PF. Alignment of RNA base pairing probability matrices. Bioinformatics. 2004;20:2222–7. [DOI] [PubMed] [Google Scholar]
- 21.Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol. 2007;3: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Will S, Otto C, Miladi M, Möhl M, Backofen R. SPARSE: Quadratic time simultaneous alignment and folding of RNAs without sequence-based heuristics. Bioinformatics. 2015;31:2489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, et al. LinearTurboFold: linear-time global prediction of conserved structures for RNA homologs with applications to SARS-CoV-2. Proc Natl Acad Sci. 2021;118: e2116269118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C-C, Jeong H, Qian X, Yoon B-J. TOPAS: network-based structural alignment of RNA sequences. Bioinformatics. 2019;35:2941–8. [DOI] [PubMed] [Google Scholar]
- 25.Mamoshina P, Vieira A, Putin E, Zhavoronkov A. Applications of deep learning in biomedicine. Mol Pharm. 2016;13:1445–54. [DOI] [PubMed] [Google Scholar]
- 26.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 27.Eraslan G, Avsec Ž, Gagneur J, Theis FJ. Deep learning: new computational modelling techniques for genomics. Nat Rev Genet. 2019;20:389–403. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama M, Sakakibara Y. Informative RNA base embedding for RNA structural alignment and clustering by deep representation learning. NAR genomics and bioinformatics. 2022;4:lqac012. [DOI] [PMC free article] [PubMed]
- 29.Chen C-C, Chan Y-M. REDfold: accurate RNA secondary structure prediction using residual encoder-decoder network. BMC Bioinformatics. 2023;24:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang G, Liu Z, Van Der Maaten L, Weinberger K Q. Densely connected convolutional networks 2017;4700–4708.
- 31.Li G, Zhang M, Li J, Lv F, Tong G. Efficient densely connected convolutional neural networks. Pattern Recogn. 2021;109: 107610. [Google Scholar]
- 32.He K, Zhang X, Ren S, Sun J. Deep residual learning for image recognition. 2016;770–8.
- 33.Jégou S, Drozdzal M, Vazquez D, Romero A, Bengio Y. The one hundred layers tiramisu: Fully convolutional densenets for semantic segmentation 2017;11–19.
- 34.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–53. [DOI] [PubMed] [Google Scholar]
- 35.Wilm A, Mainz I, Steger G. An enhanced RNA alignment benchmark for sequence alignment programs. Algorithms for molecular biology. 2006;1:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalvari I, et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021;49:D192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.multithreaded implementation for pairwise structural RNA alignment. Sundfeld D, d. M. A., Havgaard JH & J., G. Foldalign 2.5. Bioinformatics. 2016;32:1238–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darty K, Denise A, Ponty Y. Varna: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table S1. List of RNA family groups and the number of sequences in the ncRNA training dataset. The families are grouped according the Rfam ID in the Rfam database 14.3.
Data Availability Statement
The REDalign web server is freely available at https://REDalign.ee.ncyu.edu.tw. The dataset used for analyzing ncRNAs with pseudoknots in REDalign is accessible at https://github.com/aky3100/REDalign.