

Abstract
Microbial symbionts associate with multicellular organisms on a continuum from facultative associations to mutual codependency. In the oldest intracellular symbioses there is exclusive vertical symbiont transmission, and co-diversification of symbiotic partners over millions of years. Such symbionts often undergo genome reduction due to low effective population sizes, frequent population bottlenecks, and reduced purifying selection. Here, we describe multiple independent acquisition events of closely related defensive symbionts followed by genome erosion in a group of Lagriinae beetles. Previous work in Lagria villosa revealed the dominant genome-eroded symbiont of the genus Burkholderia produces the antifungal compound lagriamide, protecting the beetle’s eggs and larvae from antagonistic fungi. Here, we use metagenomics to assemble 11 additional genomes of lagriamide-producing symbionts from 7 different host species within Lagriinae from 5 countries, to unravel the evolutionary history of this symbiotic relationship. In each host, we detected one dominant genome-eroded Burkholderia symbiont encoding the lagriamide biosynthetic gene cluster. However, we did not find evidence for host–symbiont co-diversification or for monophyly of the lagriamide-producing symbionts. Instead, our analyses support a single ancestral acquisition of the gene cluster followed by at least four independent symbiont acquisitions and subsequent genome erosion in each lineage. By contrast, a clade of plant-associated relatives retained large genomes but secondarily lost the lagriamide gene cluster. Our results, therefore, reveal a dynamic evolutionary history with multiple independent symbiont acquisitions characterized by a high degree of specificity and highlight the importance of the specialized metabolite lagriamide for the establishment and maintenance of this defensive symbiosis.
Keywords: lagriamide, Burkholderia, symbiosis, symbiont replacement, biosynthetic gene cluster, metagenomics, Lagriinae, chemical defense, secondary metabolism
Introduction
Eukaryotes have been associated with prokaryotic microbes at least since the initial endosymbiotic events that led to the acquisition of mitochondria and chloroplasts [1]. These organelles represent the presumed endpoint of ancient symbioses with α-proteobacteria and cyanobacteria, respectively, that over time led to a progressive shrinkage of the symbionts’ genomes and eventual transfer of genes from symbionts to host [1]. Although organelle acquisition appears to be a rare event [2], other more recent symbioses appear to be on a similar evolutionary trajectory of profound genome reduction and absolute dependence on host cells. For example, the acquisition of the intracellular symbiont Buchnera aphidicola in the common ancestor of aphids allowed them to diversify as sap-feeding insects as the symbiont synthesizes essential amino acids not found in plant sap, and this is evidenced by a rapid basal radiation of aphid species [3] and strict co-evolution of aphids and Buchnera [4]. Buchnera aphidicola has been vertically transmitted for at least 200 million years [4] and has a profoundly reduced chromosome, ~11% of the size of Escherichia coli [5].
Through comparison of various symbionts, a model of genome reduction has emerged whereby host restriction initially weakens purifying selection on formerly essential genes, through both host-provided metabolites and symbiont population structure, with low effective population sizes and isolation within individual hosts [6]. When symbionts are vertically transmitted, population bottlenecks occur during every transmission event, causing the fixation of deleterious mutations within the population [6]. These factors combine to first cause an increase in pseudogenes in the genome [6] and then deletion of those pseudogenes due to a known deletion-bias within bacteria [7]. The most reduced genomes lose even central functions such as DNA repair pathways [6], which leads to an increased rate of evolution and further gene loss, as well as increased AT-bias in many cases [8, 9]. In the cases of symbionts living inside host cells, it is likely that this process is exacerbated due to a lack of opportunity or ability to horizontally acquire functional genes. However, genome reduction is also known to occur without genetic isolation. For instance, free-living bacteria living in nutrient-poor environments, such as Prochlorococcus spp., are thought to have reduced genomes as a consequence of selection pressure to streamline their metabolism [10], potentially explained through the Black Queen hypothesis [11], which posits that selection drives pathways to be lost when the respective metabolites are produced by another species in the ecosystem as “public goods.” There are also genome-reduced symbionts which seemingly are not genetically isolated. Burkholderia symbionts that reside extracellularly in leaf nodules in plants are mainly transmitted vertically because the symbiosis is mutually co-dependent [12], although horizontal transfer may have occurred occasionally between plants, the soil microbiota, and insects [13]. This suggests a lack of genetic isolation, and indeed there is evidence of repeated horizontal transfers of biosynthetic genes for defensive molecules among leaf nodule symbionts of Rubiacaeae plants [13]. Such systems may provide an opportunity to study the evolutionary pressures that lead to the process of genome reduction and the mechanisms of symbiosis that underlie it.
The dichotomy of vertical versus horizontal transfer of symbionts may be one determinant of genome reduction. A relatively clear-cut example is the two symbionts of the tunicate Lissoclinum patella, i.e. the extracellular cyanobacterium Prochloron didemni [14] and the intracellular “Candidatus Endolissoclinum faulkneri” [15]. The former is capable of horizontal transmission, which is reflected in its almost clonal genome among very divergent hosts and a lack of genome reduction [14], whereas the latter is vertically transmitted, as evidenced by its co-divergence with its hosts across cryptic speciations and profound genome reduction [15, 16]. However, the mode of transmission also exists on a continuum from strict vertical to strict horizontal, with mixtures of vertical and horizontal transmission in between [17]. For instance, the tsetse fly symbiont Sodalis glossinidius shows some signs of genome-reduction such as rampant pseudogenes, but remains culturable in the laboratory, meaning that horizontal transmission cannot be excluded [18]. Likewise, symbionts long thought to be exclusively vertically transmitted, such as the bryozoan symbiont “Ca. Endobugula sertula,” which is packaged with the hosts’ larvae, show no signs of genome reduction [19], indicating that there is no compelling reason why it should not be able to transmit horizontally between hosts. Indeed, “Ca. E. sertula” has been found in genetically divergent but proximal bryozoan individuals, suggesting horizontal transmission [20].
The Lagria and Ecnolagria beetles belong to the subfamily Lagriinae within the family Tenebrionidae (order Coleoptera). Lagria villosa, a known soybean pest [21], is a source of lagriamide, an antifungal polyketide, produced by a Burkholderia symbiont (Burkholderia sp. LvStB) [21]. The compound is made via a trans-AT polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) hybrid biosynthetic gene cluster (BGC), termed lga, which due to a nucleotide signature (k-mer frequency) distinct from the chromosome is predicted to have been horizontally acquired [21]. The symbiont is present in glandular structures associated with the ovipositor of female beetles and secreted on its eggs as they are laid [21], and this symbiont has been shown to have a defensive role against fungi in the egg [21] and larval stages [22]. Previously, we showed that the genome of Burkholderia sp. LvStB is reduced and has lost several essential genes including some genes involved in the DNA repair pathways and primary metabolism [23]. The genome has a low coding density, and a high number of pseudogenes and transposases, indicative of genome erosion [23]. These characteristics are consistent with host restriction and vertical transmission of LvStB. However, there is evidence that Burkholderia symbionts from L. villosa can be transferred to plant tissues and survive for several days and that bacteria can be acquired by the beetle from the plant and soil environment [24].
As several Lagriinae beetles harbor symbionts in special structures that likely evolved between 55 and 82 million years ago based on fossil evidence, positioned to deposit symbionts on the eggs [25, 26], we hypothesized that lagriamide-producing Burkholderia symbionts might have co-evolved with their hosts in a manner similar to other vertically transmitted insect symbionts. However, the possibility for transmission of the symbionts to and from plants, and the accessibility of the symbionts’ habitat on the surface of eggs and within adult females suggested that horizontal symbiont acquisition may be possible. As the beetles harbor complex microbiomes with multiple related Burkholderia strains as well as other bacteria [22, 24], both genome-reduced and not, an alternative hypothesis is that the lagriamide BGC has been repeatedly horizontally transferred among environmental strains and symbionts. Moreover, partnerships in defensive symbionts are usually more dynamic as compared with intracellular nutritional symbionts [27]. It is also possible that the lagriamide-producing strain is restricted to L. villosa and that different Lagriinae species have symbionts with different BGCs, as this would allow the association to react much more flexibly to changes in antagonist communities. To clarify this evolutionary picture, we analyzed the metagenomes of 12 beetle samples, spanning 7 species belonging to the genera Lagria and Ecnolagria across 5 different countries (four continents) (Table 1). We recovered the metagenome-assembled genomes (MAGs) of several different Burkholderia bacteria and confirmed the presence of the lagriamide BGC in each beetle specimen. We also report a complete genome of the genome-reduced, lagriamide-producing Burkholderia sp. LvStB symbiont, obtained through long-read Nanopore sequencing. We compared the phylogeny of the recovered Burkholderia MAGs, the lagriamide BGCs, and the host beetles to determine whether co-cladogenesis occurred in this system and to further explore the evolutionary relationships in the symbiosis. The results indicate that the lagriamide BGC was likely only acquired once in the common ancestor of beetle-associated Burkholderia symbionts and subsequently lost in the majority of the descendent free-living strains. As all the lagriamide-bearing symbionts are genome-reduced but do not form a monophyletic clade, do not correspond to host phylogeny, and the pattern of gene conservation is different in the component clades, they likely represent multiple symbiont acquisition events, followed by independent genome reduction processes. The common factor of lagriamide production might be one of the reasons for selection by and dependency on hosts. This would suggest that a single group of natural products caused several independent symbioses to be established over evolutionary time.
Table 1.
Metadata for different beetles collected for this study.
| Sample | Location | Statistics for Burkholderia symbiont MAG with lga BGC | |||||
|---|---|---|---|---|---|---|---|
| MAG ID | Genome size (Mbp) | N 50 (bp) | No. of contigs | Longest contig (bp) | Coverage | ||
| Lagria villosa 2019 (Lv19)a | São Paulo, Brazil | LvStB | 2.07 | 8138 | 294 | 99 474 | 1983.67 |
| Lagria villosa 2020 (Lv20) | São Paulo, Brazil | Lv20_9 | 1.88 | 11 699 | 200 | 99 421 | 281.97 |
| Lagria villosa 2023 (Lv23) | São Paulo, Brazil | LvStB_2023 | 2.50 | 1 892 292 | 3 | 1 892 292 | 2263.86 |
| Lagria rufipennis (Lruf1) | Osaka and Ibaraki, Japan | Lruf1_1 | 1.93 | 5267 | 368 | 29 589 | 636.05 |
| Lagria rufipennis (Lruf2) | Tokushima, Osaka, and Kogashima, Japan | Lruf2_2 | 2.66 | 7451 | 387 | 55 249 | 582.66 |
| Lagria okinawana (Loki) | Okinawa, Japan | Loki_2 | 2.14 | 6360 | 360 | 60 396 | 928.22 |
| Lagria hirta (LhSB) | Hessen, Germany | LhSB_1 | 1.17 | 5434 | 212 | 88 724 | 261.74 |
| Lagria hirta (LhHG) | Rhineland Palatinate, Germany | LhHG_2 | 1.14 | 6598 | 170 | 127 478 | 868.51 |
| Lagria hirta (LhG) | Galicia, Spain | LhG_1 | 2.70 | 8034 | 376 | 91 830 | 557.11 |
| Lagria grenieri (Lgren) | Huelva, Spain | Lgren_6 | 1.14 | 5417 | 205 | 91 929 | 83.02 |
| Lagria atripes (Latri) | Rhineland-Palatinate, Germany | Latri_1 | 2.20 | 7903 | 280 | 93 629 | 29.63 |
| Ecnolagria sp. (Ecno) | New South Wales, Australia | Ecno_3 | 3.16 | 8701 | 419 | 87 409 | 87.61 |
Materials and methods
Insect collection
Specimens were collected between 2009 and 2023 in Spain, Germany, Brazil, Japan, and Australia in the locations listed in Table S4. Female adults were dissected either directly after chilling for ca. 15 min at −20°C or preserved in 70% ethanol or acetone until dissected. The accessory glands were removed and preserved in 70% ethanol at −80°C until further processing. For species in which we suspected the presence of symbiont-harboring compartments within the ovipositor in addition to the glands, the ovipositor was also dissected and preserved along with the accessory glands.
DNA isolation and metagenomic sequencing
Given that the specimens used in this study were collected throughout multiple years and were available at different times during the project, we carried out DNA extractions in different batches. We used short-read sequencing (Illumina) for the majority of samples and long-read sequencing (Oxford Nanopore) to complement the metagenomic data for two of the species (Table S4).
Short-read sequencing
Genomic DNA from the preserved organs was extracted per individual after removing the fixative and homogenizing the tissue in liquid nitrogen. The MasterPure complete DNA and RNA isolation Kit (Epicentre Technologies) was used as indicated by the manufacturer, including an additional incubation step at 37°C for 30 min with 4 μl lysozyme (100 mg ml−1) before protein precipitation. The nucleic acids were re-suspended in low TE buffer (1:10 dilution of TE) and pooled by species. Metagenomic sequencing was carried out in two batches. The first batch included the samples corresponding to Lagria atripes, Lagria grenieri, Lagria hirta G, L. hirta SB, L. hirta HG, and L. villosa 2020. This first batch was sent for DNA library preparation using a Nextera XT DNA Library Prep Kit (Illumina) and metagenomic sequencing on a NovaSeq 6000 platform (Illumina), using a paired-end approach (2 × 150 bp) to a depth of 30 M reads (9 Gbp) by CeGaT GmbH (Tübingen, Germany). Samples from the second batch including Largus rufipennis 1 and 2, Lagria okinawana, and Ecnolagria sp., were sequenced using a NextSeq 2000 (Illumina, paired end 2 × 150 bp) to a depth of 28–44 million reads at the Max Planck Genome Centre (Cologne, Germany). The data from sample Lv19 corresponds to that described previously [21]. Taxonomic assignment of individual specimens of L. rufipennis was first done morphologically according to a previously described method [28] very similar to how the sympatrically occurring females of Lagria nigricollis, and the specimens used for sample Lruf2 were originally identified as L. nigricollis [26]. Due to the uncertainty associated with morphological identification, we, therefore, additionally barcoded the specimens after their metagenomes had been sequenced (Supplementary methods) and compared their cytochrome oxidase I sequences to those of male specimens of L. rufipennis and L. nigricollis that can be more easily distinguished based on their morphology. All 19 L. rufipennis and 10 L. nigricollis COI sequences that we obtained turned out to be very similar and formed a sister group to the L. rufipennis sequence available in NCBI (MW802588). However, the L. nigricollis sequences formed a distinct subclade, with the exception of the sample that had been used for metagenomics (Lruf2), which grouped within L. rufipennis. Hence, we reassigned Lruf2 to L. rufipennis, resulting in two replicate metagenomes for this species. Unfortunately, the L. nigricollis samples were males (which do not contain symbionts), preventing us from sequencing a metagenome of this species.
Long-read sequencing
We selected samples from L. villosa and L. hirta for long-read sequencing, aiming to improve the assembly of the lga-containing MAGs in these species obtained with short-read sequencing. For the L. hirta HG population, genomic DNA was extracted from a pool of six egg clutches (20–30 eggs per clutch) using the Genomic-tip 20/G Kit (Qiagen) following the instructions from the manufacturer. For L. villosa, the symbiotic organs of six female adults were dissected and gently homogenized to release bacterial symbionts. The residual host tissue was separated from the bacterial suspension, and both samples were frozen at −20°C. Later, both samples were thawed and centrifuged for 2 min at 3000 rpm + 2 min at 5000 rpm to pellet the tissue and bacteria, respectively. The supernatant was removed, and 20 μl sterile 1× PBS was added to both samples. Genomic DNA was extracted using the Nanobind CBB Big DNA kit (Circulomics, Baltimore, USA) followed by enrichment for HMW (high molecular weight) DNA using the Short Read Eliminator kit XS (Circulomics). Isolated HMW DNA purity and concentrations were measured using a Qubit (Thermo Fisher).
These samples, as well as an aliquot of the L. villosa 2020 genomic DNA sample, underwent end-DNA repair and library preparation using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, Ipswich, USA) and the Ligation Sequencing Kit V14 (SQK-LSK114; Oxford Nanopore Technologies, Oxford, UK) followed by a clean-up step with AMPure XP beads (Beckman Coulter). Sequencing was performed on a MinION platform (Oxford Nanopore Technologies) and MinION flow cells (vR10.4.1) with 100 ng of the library during a 72-h run.
Metagenomic sequence assembly and binning
Short-read sequences
Sample L. villosa 2019 represented data previously assembled and analyzed [21, 23]. Sequence data generated from L. atripes, L. grenieri, L. hirta G, L. hirta SB, L. rufipennis 1, L. rufipennis 2, L. okinawana, and Ecnolagria sp., consisted of only short-read Illumina sequence data. Sequences were trimmed using Trimmomatic v0.39 [29] using TruSeq3-PE as reference, and sequences shorter than 25 bp being discarded. The trimmed sequences were assembled using SPAdes v.3.14.1 [30] and binned using Autometa [31]. Sequence data generated from L. villosa 2020 and L. hirta HG consisted of both short-read Illumina sequence data and long-read Nanopore sequence data, as mentioned above. After trimming, reads were assembled with SPAdes v.3.14.1 as a hybrid assembly with the nanopore flag enabled. Assembled contigs were binned using Autometa [31]. The quality of all MAGs was assessed using CheckM2 v1.0.1 [32] and each MAG was classified using GTDB-Tk v.2.3.2 against database release 214. Coverage reported by SPAdes for each contig was used to calculate the MAG coverage, except for LvStB_2023 where coverage was calculated by read aligned using minimap2 [33].
Long-read sequences
After sequencing, super-high-accuracy base calling of the raw reads was performed with Guppy v6.3.8 (Oxford Nanopore Technologies) (dna_r10.4.1_400bps_sup.cfg model; split-read function enabled), resulting in a total of 9 Gb sequence data. The resulting reads were de novo assembled using Flye v2.9.1 [34, 35] with setting minimum overlap as 10 kb and with the “--meta” option, followed by four rounds of polishing with Racon v1.3.3 [34] starting from the Flye assembly with option (−m 8 -x − 6 -g − 8 -w 500). After each polishing round, reads were re-aligned to the resulting assembly with minimap2 v2.17 [33]. A final round of polishing was performed using Medaka v1.2.0 (https://github.com/nanoporetech/medaka) with the r941_min_high_g344 model using the MinION raw reads. After polishing, haplotype redundancies and overlaps in the assembly based on read depth were purged using Purge_Dups v1.2.6 [36]. The relative contig coverage, GC content, and contig taxonomic classification were scanned after each genome assembly using Blobtools and TaxonAnalysis to enable the identification of potential microbial symbiont contigs. We subsequently performed several rounds of Flye assemblies, using only subsets (e.g. 25%) of the complete MinION data and/or read length size-cutoffs (5 kb) to optimize symbiont genome assembly.
Phylogeny of beetles
Mitochondrial genomes (mitogenomes) were recovered from the Eukaryote kingdom bins from each respective sample. Mitogenomes used previously [37] to produce a beetle phylogenetic tree were selected for references and outgroups. Mitogenomes from all metagenomic datasets and reference mitogenomes were annotated using the MITOS2 webserver [38] against the RefSe89 Metazoan database, using genetic code 5 (invertebrate mitochondrial). Amino acid sequences of the 13 protein coding genes (PCGs) from each mitogenome were collected and aligned using muscle v5.1 [39]. Nucleic acid sequences of the corresponding PCGs were aligned using pal2nal v14 [40] with the -codontable 5 flag. Nucleic acid alignments were concatenated and a partition file was generated using the pxcat command from the phyx package [41]. Phylogenetic analysis was performed by partitioning each codon position for each gene. An AICC model predicted by ModelTest-NG v0.1.7 [42, 43] was used to construct the phylogenetic tree using RAxML-NG v1.2.1 [44] with the parameters —all and —tree pars{25},rand{25}. The alignment file in FASTA format was converted to nexus format using Geneious Prime 2023.2.1 (www.geneious.com). Bayesian analysis was performed by partitioning each codon position for each gene using MrBayes v3.2 [45] with seed and swapseed equal to 42 and using the following parameters lset applyto = (all) nst = 6 rates = invgamma; and unlink statefreq = (all) revmat = (all) shape = (all) pinvar = (all); using 10 million generations, and sample frequency of 500. The final average standard deviation of split frequencies (ASDSF) was 0.0042.
Burkholderia symbiont phylogeny
Prokka [46] was used to annotate the open reading frames (ORFs) of the genomes/MAGs. Pseudogenes were removed from the MAGs and orthofinder v2.5.5 [47] was run on amino acid sequences of the genomes/MAGs. A custom script was used to extract the genes with single-copy heirarchical orthogroups (HOGs) that are present in more than 95% (23 HOGs), 90% (126 HOGs), 80% (336 HOGs), 70% (656 HOGs), and 60% (888 HOGs) of the genomes/MAGs. Muscle v5.1 [39] was used to align the amino acid sequences of the selected HOGs, followed by pal2nal v14 [40] to align the corresponding nucleic acid sequences using -codontable 11. Subsequent steps were similar to those performed for constructing beetle phylogeny. Bayesian analysis was performed using MrBayes v3.2 [45] following the steps and parameters described in beetle mitogenome tree construction. The final ASDSF was 0.0002.
Amino acid sequences of MAGs were blasted (diamond blastP) [48, 49] (with parameters -k 1 --max-hsps 1 --outfmt 6 qseqid stitle pident evalue qlen slen) against a local copy of the NCBI nr database, where previously identified Burkholderia sequences [21, 23] were removed. Genes where the top blastP hits had percent identity <50% or those without “Burkholderia” in the subject sequence title of the top hit were classified as putative horizontally transferred genes. These genes along with any pseudogenes were removed from MAGs and orthofinder was used to detect HOGs present in more than 95% (16 HOGs), 90% (98 HOGs), 80% (304 HOGs), 70% (632 HOGs), and 60% (884 HOGs) of the genomes/MAGs. Subsequent steps were similar to those mentioned in the above paragraph.
Lagriamide BGC phylogeny
Lagriamide BGC genes from lgaA to lgaI were extracted. Protein sequences were aligned with muscle v5.1 [39] followed by alignment of DNA sequences using pal2nal v14 [40] using -codontable 11. The pxcat command in the phyx package [41] was used to concatenate the DNA alignments and generate a partition file. A maximum likelihood tree was made using RAxML v8.2.12 (raxmlHPC-PTHREADS-SSE3) [50], with the parameters -f a -# 1000 -p 1989 -x 1989. For the GTRGAMMAI model each gene was partitioned for each codon position, whereas using the GTRCAT -V model partitioning was only performed per gene as it resulted in higher bootstrap values than partitioning for each codon position in each gene. Bayesian analysis was performed as described in the beetle phylogeny with the final ASDSF being 0.0001.
Results and discussion
Beetle phylogeny
We sequenced and assembled the metagenomes of the collected Lagria and Ecnolagria beetle populations (Table 1), and beetle mitogenomes (see Table S1 for mitogenome statistics) were extracted and annotated to infer host beetle phylogeny (Fig. 1). In line with previous studies, mitogenomes belonging to the tenebrionid subfamilies Lagriinae, Blaptinae, Pimeliinae, Stenochiinae, and Alleculinae were found to be monophyletic, whereas Diaperinae and Tenebrioninae were found to be para- or polyphyletic [37, 50–52]. Maximum likelihood analysis using RAxML [53] (Fig. S1) and Bayesian analysis using MrBayes [45] (Fig. 1) gave similar results.
Figure 1.

Beetle mitogenome phylogenetic tree using 13 mitochondrial protein coding genes constructed using MrBayes [45]. Branch values represent posterior probabilities. Mitogenomes recovered in this study are highlighted with bold lettering. Pictures depicting a representative species of each subfamily are included (Pimeliinae: Pimelia obsoleta; Lagriinae: Lagria hirta; Diaperinae: Trachyscelis aphodioides; Tenebrioninae: Tenebrio molitor; Blaptinae: Blaps lethifera; Stenochiinae: Strongylium cultellatum; Alleculinae: Cteniopus sulphureus). Photography credits: Udo Schmidt [54] (CC BY-SA 2.0).
All collected Lagria beetle mitogenomes clustered into four distinct subclades: All L. hirta beetle mitogenomes were clustered in a single clade, alongside a closely related clade of Lagria species (L. rufipennis and L. okinawana) from Japan. The L. atripes and L. grenieri beetles formed another clade more distantly related to the L. hirta and Japanese Lagria species. Finally, the L. villosa and Ecnolagria sp. beetles formed a fourth clade along with Chrysolagria sp. (JX412760), distinct from the other Lagria beetles. A small distinction was noted here, wherein the Bayesian phylogeny (Fig. 1) suggested that the Cerogria beetles belonged to the clade with L. villosa and Ecnolagria species, whereas the maximum-likelihood phylogeny showed the Cerogria to be in a clade with all other Lagria beetles (Fig. S1). However, in both cases the branch support values are too low to make any definite conclusions. Publicly available sequences of L. hirta (OX375806) clustered with collected L. hirta samples from Rhineland-Palatinate, Germany (LhHG), and L. rufipennis (MW802588) clustered with the two L. rufipennis (Lruf1 and Lruf2) samples.
Recovery of lagriamide BGCs
A complete, or mostly complete, lga BGC was found, using antiSMASH v7 [55, 56], in 11 of the 12 samples, with the exception of L. rufipennis 1 where only small fragments of the lga BGC could be recovered. The BGC recovered from L. rufipennis 2 was found over two contigs and could not be manually joined following inspection of the assembly graph. The missing data for this region spans from approximately halfway through the lgaB gene to approximately halfway through the lgaC gene (Fig. 2A).
Figure 2.

Analysis of representative lga BGCs extracted from eleven Lagriinae beetle metagenomes. (A) Comparison of representative lga BGC gene organization. Individual genes in the lga BGCs are represented by arrows oriented in the predicted direction of transcription and colored according to identity. Pairwise amino acid similarity between BGCs is indicated in the shaded areas between genes, although we have omitted these numbers for the smallest genes. A scale bar is provided for gene size. Dashed lines indicate fragments missing from the respective assemblies. (B) Comparison of predicted enzyme domain organization in the representative lga BGCs, where genes are ordered according to biosynthetic order. Boxes around the domains indicate differences between the BGCs.
Analysis of representative BGCs revealed two differences in gene organization of the lga BGC across the different Lagriinae beetle species (Fig. 2A). The first difference observed regarded the lgaC gene. The lgaC gene from the BGCs recovered from the L. hirta G and L. hirta SB samples appeared to be split into two, denoted lgaC1 and lgaC2 for clarity. Alignment of the lgaC gene from the three L. hirta samples revealed that there was a perfect alignment of the nucleotide sequences save for a 37 bp deletion in the BGCs from L. hirta G and L. hirta SB, which introduced a frameshift (Fig. S2A). This frameshift consequently introduced a premature stop codon, which split the lgaC gene into two ORFs in L. hirta G and L. hirta SB (Fig. S2B).
The second difference we observed was in the lga BGC from the LvStB genome extracted from the 2023 L. villosa metagenome (Lv23). One split in lgaB and two splits in lgaC were seen. However, the assembly of the L. villosa 2023 metagenome was based solely on long-read data, which is error prone [57], and the splits may not be a true reflection of the BGC in this sample. Normally Sanger sequencing would be the solution to validate these questionable regions, but unfortunately, there was no remaining DNA after the long-read sequencing runs for this particular sample. For this reason, we left the BGC with the splits but were cautious not to over-interpret the apparent breaks in the genes in this BGC.
We then considered the domain organization within the lga BGC genes (Fig. 2B). The domain organization is largely congruent across the lga BGCs recovered from the metagenomes. We did note, however, an additional annotated “DHt” domain in lgaB, which is defined as “Dehydratase domain variant more commonly found in trans-AT PKS clusters,” in the lga BGCs from all L. hirta samples and the L. rufipennis 2 sample. Similarly, we detected an additional carrier protein domain (phosphopantetheine acyl carrier protein group) near the N-terminus of the lgaC protein in the BGCs from the L. grenieri and L. okinawana samples. In all cases, close inspection of the primary sequence of these additional dehydratase and carrier domains revealed mutations in the sequences that would likely render the encoded domain nonfunctional (Supplementary Methods).
Finally, as with the originally described lga BGC recovered from the L. villosa 2019 sample [21], we found mutations in the catalytic or conserved motifs of lgaG DH2, lgaG KS6, lgaB KR3, and lgaC KS5 domains, that we believe may render these domains inactive (Supplementary Methods). As a result, the domain architecture of all representative lga BGCs from all samples appear functionally identical.
Together, the conservation of the lga BGC in at least seven different species of Lagriinae beetles, across four geographically distant countries, implies that the production of lagriamide is an important factor for the host beetle and that the lga BGC is under strong selective pressure. The presence of additional domains in the lga BGC in several samples, even though they are likely inactive, is intriguing as it suggests that these domains may have previously been present in all lga BGCs but may have decayed over time and were lost. The reason as to why these domains were selected against would be speculative at best and all lagriamide-like compounds produced in the different beetle populations would need to be characterized to truly infer differences that the domain architecture may have on the resulting chemistry. Conserved production of other bioactive compounds has been observed, such as pederin, across Staphylinidae beetle species (Paederus and Paederidus genera) [58], which are host to a Pseudomonas symbiont that produces pederin [59].
The two systems have several parallels: both pederin and lagriamide are produced by a trans-AT PKS NRPS hybrid BGC, the former in a Pseudomonas bacterium [58], and the latter in a Burkholderia sp., where the compound is concentrated in the host’s (female) oviposition organs, coated onto the eggs and serves to protect juveniles [60]. Further, both pederin and lagriamide are the sole insect-associated compounds in suites of compounds otherwise associated with marine invertebrates. Groups of pederin analogs, such as the onnamides, mycalamides, psymberins, and theopederins, have been isolated from a variety of marine sponges [61–65] and ascidians [66], whereas bistramide, the most structurally similar compound to lagriamide, was isolated from an ascidian [67]. The question remains, however, as to the evolutionary origins of this BGC and how it came to be present in such diverse ecological niches.
Complete genome of the lga-carrying LvStB symbiont
Long-read sequencing of the L. villosa 2023 (Lv23) metagenome allowed us to assemble a complete genome of an lga-carrying Burkholderia strain (referred to as LvStB_2023 from hereon). LvStB_2023 was found to have a 2.5-Mbp long genome with a GC percentage of 58.63%. It has two circular chromosomes—chromosome 1 is 1.89 Mbp, chromosome 2 is 0.55 Mpb in size, and there is a plasmid 59.77 kbp long. The genome is estimated to be 97.1% complete (98.8% with “specific” mode) and 0.02% contaminated as per CheckM2 [32] and thus is a high-quality MAG according to the MIMAG standards [68]. Assembly graph analysis of LvStB_2023 verified that we have the complete sequence of two circular chromosomes and a plasmid. However, the CheckM2 estimate did not reflect a fully complete genome, at 98.8%, and we believe that this small discrepancy in predicted completeness may be a result of ongoing genome reduction [69].
LvStB_2023 has a coding density of 78% and 59.1% with and without pseudogenes, respectively. A large percentage (43.87%) of the ORFs in LvStB_2023 were identified as pseudogenes (1613 out of 3676), the highest of any lga-carrying Burkholderia symbiont. However, this estimate may be artificially high as pseudogenes were identified purely based on their length relative to their closest BLASTP match and these counts are derived from an assembly generated from only long-read data, which can be prone to errors [70–72], particularly homopolymeric runs. However, coding density and frequency of pseudogenes is not very different from LvStB MAGs assembled from short-read data (see Table S2 for complete genome characteristics of recovered MAGs). Having multiple chromosomes is a common phenomenon in Burkholderia [73, 74]. Generally in multi-chromosome bacteria, the majority of the genes for essential functions are located on one larger or primary chromosome, whereas the smaller or secondary chromosome has much fewer essential genes and it mostly carries genes for niche-specific functions [75]. In the case of LvStB_2023, chromosome 1 appears to be the primary chromosome as it is much larger in size, and has 77 out of 84 core genes (including multiple copies) (Fig. 3A). Functional analysis revealed chromosome 1 to have the highest number of genes for all essential COG categories (Fig. 3B), including categories L (replication, recombination and repair), J (translation, ribosomal structure, and biogenesis), M (cell wall/membrane/envelope biogenesis), and H (coenzyme transport and metabolism).
Figure 3.
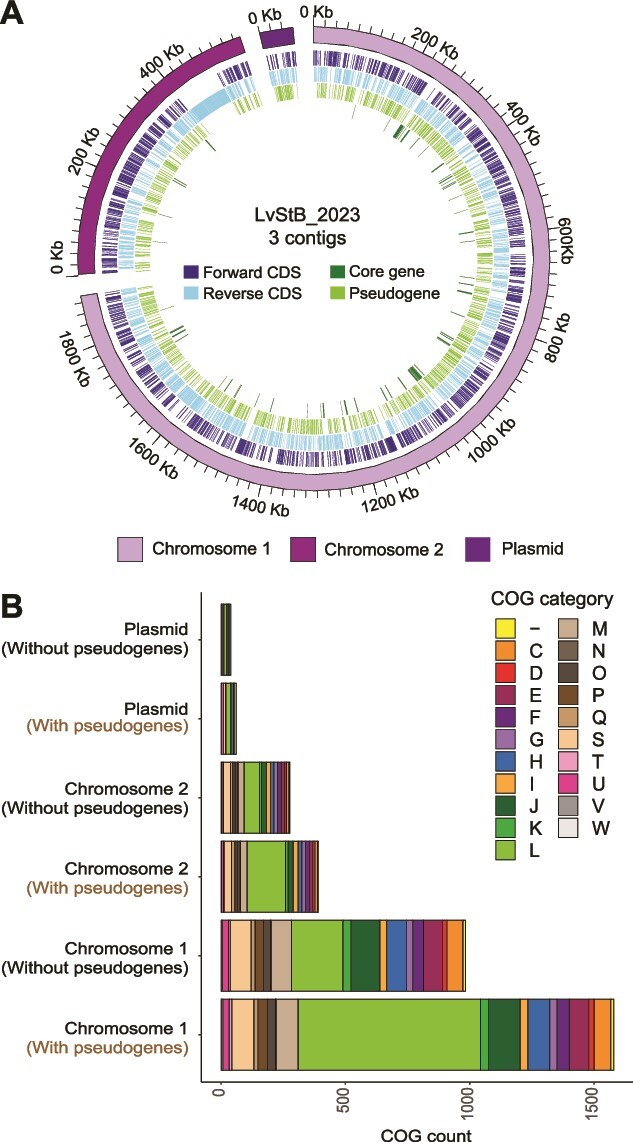
(A) Circular representation of LvStB_2023 genome from the L. villosa 2023 sample. Individual chromosomes are indicated by separate continuous arcs in the outer ring (ring 5). Coding sequences (CDS) which are core genes or pseudogenes are shown in the innermost ring (ring 1) and next innermost ring (ring 2), respectively, whereas the rest are indicated the next two rings (rings 3 and 4). (B) Raw count of COG categories present on different contigs of the LvStB_2023 genome (with and without pseudogenes) from the L. villosa 2023 sample.
The lga BGC is on chromosome 2 (0.55 Mbp long) and can be distinguished by the continuous block of coding sequences on the reverse strand (Fig. 3A,Fig. S3). Chromosome 1, chromosome 2, and the plasmid have 44.75%, 37.59%, and 42.34% of their coding capacity taken up by pseudogenes, respectively. The similar abundance of pseudogenes in each of the contigs indicates that the whole genome is undergoing reduction simultaneously. The chromosome with lga (chromosome 2) has the smallest percentage of pseudogenes, which may be a reflection of the required conservation of the lga BGC in combination with the presence of large genes in lga.
Diversity of beetle-associated Burkholderia symbionts
Recovery and analysis of MAGs
Following assembly, the 12 beetle metagenomes were binned, and the resultant bins were manually refined. A total of 77 MAGs were recovered from all samples, of which 24 MAGs were of high quality, 30 of medium quality, and 23 of low quality (Table S2) in accordance with published MIMAG standards [68]. Only medium and high-quality MAGs were used for downstream analysis, with the exception of one low-quality bin carrying the lga BGC (LhHG_2). Genome erosion, such as that already observed for the lga-carrying symbiont Burkholderia sp. LvStB [23], can skew the completeness metric. To determine if a lower quality MAG was incomplete or genome reduced, we also considered several other metrics, including core gene presence, number of pseudogenes [23], and coding density (Table S2), and concluded that this particular MAG (LhHG_2) was likely both reduced and incomplete.
For each beetle population, a single MAG belonging to the genus Burkholderia with a single copy of the lga BGC was identified. Previous studies on the lagriamide-carrying symbiont strain Burkholderia gladioli LvStB [21, 23, 76], showed that this strain was significantly more abundant than all other bacteria associated with L. villosa, and had a reduced genome. Consistent with this, all newly recovered MAGs that included lga BGCs were the most abundant MAGs in each sample, had reduced genomes with an abundance of pseudogenes and transposases, and had lower coding densities relative to other B. gladioli genomes (Table S2). In standing with previous studies of Lagria beetles, where both reduced and non-reduced B. gladioli genomes were recovered, additional B. gladioli MAGs (Latri_2, LhHG_3, and LhSB_5) were recovered that did not carry the lagriamide BGC and showed no evidence of genome erosion. We also recovered three small B. gladioli MAGs (Lgren_7, Lv19_6_18, Lv20_2) and one small Burkholderia MAG (Lv19_6_14), as well as MAGs classified as Burkholderia lata (Lv19_4_0) and Burkholderia arboris (Lv20_1).
Average nucleotide identity (ANI) analysis of B. gladioli MAGs carrying the lga BGC showed that MAGs from different beetle species and/or different locations were likely different bacterial species due to shared ANI values <95% [77]. However, previous studies have suggested that ANI alone is not a sufficient metric for species delineation and that the aligned fraction (AF) must also be taken into account [77–80]. Following recent cutoffs adopted for species delineation [78], we opted to use AF ≥ 60% along with ANI ≥ 95% as a cutoff for species assignment. Subsequently, we found that the Burkholderia MAGs carrying the lagriamide BGC appeared to be split into at least five novel species (Table S3).
Phylogenetic analysis of recovered MAGs
In order to elucidate the evolutionary history of the association between Lagriinae beetles and Burkholderia symbionts, we reconstructed phylogenies of the Burkholderia symbionts and free-living relatives based on shared single-copy genes. A priori, we hypothesized that the lga-encoding, genome-eroded symbionts would form a monophyletic clade showing co-diversification with the hosts, given that such patterns have been previously described across many ancient and co-evolved symbioses.
The phylogeny of the beetle-associated Burkholderia symbionts, relative to other Burkholderia species, was inferred using 126 single-copy hierarchical orthogroups (HOGs) (non-pseudogenes) present in more than 90% of the genomes using both RAxML and a Bayesian approach (Fig. 4, Figs S4 and S5). Burkholderia symbionts without the lga BGC were broadly present across the phylogeny containing B. gladioli, B. lata, and B. arboris strains. By contrast, and consistent with our expectation, symbionts of different host species carrying the lga BGC were closely related. However, these genome-eroded, lga-encoding symbionts did not form a monophyletic clade. Because the tree indicates that the common ancestor of the genome-reduced lga-encoding symbionts also gave rise to a lineage of non-genome-reduced descendants, this result indicates a non-reduced free-living common ancestor and subsequent multiple independent acquisition events by Lagriinae beetles. To test for the robustness of our phylogenetic analysis, we repeated the analysis using single-copy HOGs present in 95%, 80%, 70%, and 60% (Fig. S6) of the genomes, as well as after removing any putative horizontally transferred genes (Figs S7 and S8). Other than minor discrepancies in the terminal nodes, we obtained highly similar phylogenetic trees, supporting the lack of monophyly of the lga BGC carrying Burkholderia symbionts. Thus, all our analyses support a phylogeny that contains a clade of mostly free-living Burkholderia (plus some beetle-associated symbionts with non-eroded genomes) that groups within the lga BGC-containing Lagriinae symbionts (Fig. 4 and Figs S4–S8). Concerning the evolutionary history of the symbiosis, this leaves us with two alternative scenarios: (i) an ancestral association of the whole clade of bacteria with beetles and a certain degree of genome erosion on the deep branches, and a subsequent reversal to a free-living stage of the presently extant clade containing many plant-associated B. gladioli strains; or (ii) at least four independent transitions from a free-living (or plant-associated) to a symbiotic lifestyle, each of which was followed by genome erosion.
Figure 4.

RAxML phylogenetic tree (left) and shared hierarchical orthogroups (HOGs) (non-pseudogenes) between different Burkholderia genomes (matrix on the right). Each shaded block in the matrix indicates a shared HOG. HOGs have been hierarchically clustered on the x-axis. Bootstrap values are indicated on nodes. Genome size and coverage is represented in brackets next to MAG ID. Outgroups include—Paraburkholderia acidiphila (GCF_009789655.1), Cupriavidus necator (GCF_000219215.1), Herbaspirillum seropedicae (GCF_001040945.1). The branches of other Burkholderia and outgroups have been collapsed for the sake of clarity.
To unravel which of these scenarios is more likely, we analyzed shared HOGs between different Burkholderia spp., after removing any pseudogenes from MAGs. We observed higher conservation of orthogroups between the potentially free-living Burkholderia spp. than among the lga-containing symbionts, with the free-living strains sharing a large core genome (Fig. 4). If the shared ancestor of all lga-encoding symbionts and the free-living strains would have been tightly associated with beetles and experienced some degree of genome erosion (scenario i), this observation would postulate a substantial increase in the genome size of the bacteria after the reversal to the free-living/plant-associated lifestyle and before the clade split into the different taxa. Even though theoretically possible, this scenario seems highly unlikely, because acquisition of a large number of genes would have to have happened quickly and early in order for extant strains in this clade to have such a degree of gene overlap. Instead, it appears much more plausible that the common ancestor of the entire clade had a full-sized genome similar to the presently free-living and plant-associated members and that genome erosion occurred later. Because the sequenced genome-reduced symbionts are significantly diverged in terms of sequence, and the extent (and therefore perhaps the age) of genome reduction appears to vary, we posit that there were at least four independent transitions to a symbiotic lifestyle with beetles, each of which was followed by genome erosion (Fig. 5). This is consistent with the observation that the genomes of the eroded strains retain distinct sets of genes, many of which represent subsets of the free-living strains’ core genomes (Fig. S9), as gene loss from independent host-restriction events would be expected to be largely stochastic. This distinct set of genes can, however, also be due to symbiont replacement events followed by genome reduction. Furthermore, the lack of synteny observed in the genes flanking the lga BGC (Fig. S10) is indicative of genomic rearrangement that is often observed in the early stages of genome erosion. Both of these further support the independent acquisitions of symbionts followed by genome erosion. This conclusion, however, is based on the current data and may change as we obtain more samples and long-read metagenomes that allow for synteny analyses across the entire genome.
Figure 5.
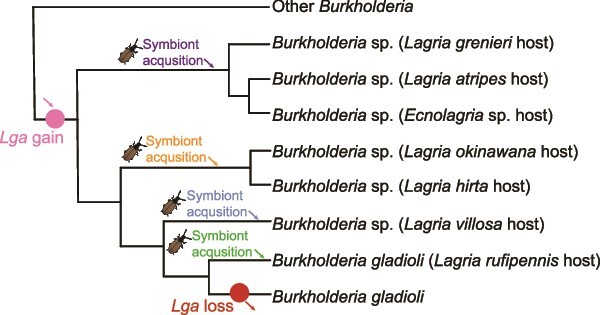
Schematic representation of proposed evolutionary scenario. The lga BGC was acquired by the common ancestor but lost in the free-living relatives. Burkholderia carrying the lga BGC were independently acquired multiple times by the beetle hosts.
Consistent with the scenario of multiple independent transitions to a symbiotic lifestyle, the phylogeny of the lga BGC-carrying Burkholderia symbionts was found to be incongruent with the beetle phylogeny (Fig. 6A), except for the symbionts grouping together for individuals of the same host species, i.e. L. hirta and L. rufipennis, respectively. The incongruence between host and symbiont phylogenies suggests both multiple symbiont acquisition and possibly host switching events that lead to symbiont replacements. Symbiont replacement has often been reported in nutritional symbionts as a way for the hosts to replace a genetically degraded symbiont with a more complete and effective one and to acquire new adaptations for expanding into different niches [81]. Burkholderia symbionts related to B. gladioli in Lagria beetles have been reported to evolve from plant-associated bacteria [26] capable of transfer from beetles to plants with subsequent survival [24]. It is possible that the horizontal acquisition might occur in the egg and larval stages, where the symbionts are localized on the surface (eggs) or in cuticular invaginations (larvae and pupae) that remain connected to the external surface via a small duct [82]. As the closely related Burkholderia strain LvStA can be acquired horizontally from the environment [24], and there is evidence of free-living bacteria carrying lagriamide-like BGCs [83], we propose that there are lga-carrying Burkholderia strains persevering in the environment (e.g. in plants or soil) [24] that can be horizontally acquired by the beetle host.
Figure 6.

Congruence between phylogenies of beetle host, Burkholdria symbionts and lga BGCs in all samples. (A) Tanglegram between lga-carrying symbionts and beetle host phylogeny. (B) Tanglegram between lga-carrying symbionts (center) and the lga BGC, as inferred via two models GTRCAT (left) and GTRGAMMAI (right). In all panels, the four conserved clades are highlighted in purple, green, blue, and orange. Gray dots on nodes indicate congruence between the compared phylogenies, whereas red dots indicate incongruence.
As we previously observed that lga has distinct nucleotide composition to the Lv19 genome [21], suggestive of a recent horizontal transfer, we sought to determine if it has been independently transferred to the corresponding symbiont in different beetle hosts. Phylogenetic analysis of the representative lga BGCs from all samples resulted in two possible topologies using GTRCAT-V and GTRGAMMAI models (Fig. 6B). Both topologies included conserved clades. However, the relative positions of the three clades are poorly supported (Fig. 6B), resulting in the two alternative topologies. A Bayesian tree was also constructed (Fig. S11), which is congruent with the GTRGAMMAI tree topology. The inconsistent topology likely stems from limited resolution of the phylogeny affecting deep nodes in the trees. The GTRCAT-V topology is perfectly congruent with the symbiont phylogeny based on genome-wide marker genes, whereas the GTRGAMMAI topology shows one discrepancy at one of the deep nodes. Thus, these analyses do not provide evidence for additional horizontal transfer events of the lga cluster, so it is likely that there was a single acquisition of lga in the common ancestor of the symbiont and B. gladioli clade, with subsequent loss in the free-living group (Fig. 5). It appears that lagriamide production was highly selected for in symbiotic settings and hence retained, whereas it was lost in the larger genomes (assumed to be free-living) where it was not selected for. However, there is likely to be at least some strains in the environment or associated with plants that harbor lga, as relatives in different lineages have been discovered in free-living strains [83], that served as sources for these independent symbiont acquisitions. Our findings indicate that the lga BGC is important in the symbiosis, either for symbiont establishment (e.g. competition with other symbionts) and/or because lagriamide is an effective host-defensive molecule. Furthermore, the fact that different Burkholderia species with lga were identified across different Lagriinae beetles indicates that symbiont acquisition is highly selective.
Lagriamide seems to be highly conserved, despite the dynamics of the system, where multiple species of bacteria associate with each beetle host, and several lga-producing Burkholderia have apparently been independently acquired. A dynamic association in defensive symbioses has been previously hypothesized, to allow for rapid adaptation to a changing community of antagonists, or to individual co-evolving pathogens [27]. We expected to see changes in the defensive chemistry used in a symbiotic context, akin to the rapid evolution of immune genes in animals [84–86]. However, despite the dynamic nature of many defensive symbioses, with symbiont replacements on ecological or evolutionary timescales, several examples of defensive symbioses highlight that the same bioactive compounds can be used over long evolutionary timescales. In case of beewolf wasps, Streptomyces symbionts have been found to produce piericidin and streptochlorin for an estimated 68 million years [87, 88]. Both compounds are found in different beewolf species and across different geographic locations. Similarly, as discussed above, pederin is produced across different species of Paederus and Paederidus beetles by Pseudomonas symbionts [58, 59]. Similarly, we now describe the production of lagriamide by a Burkholderia symbiont across several species of Lagria and Ecnolagria beetles. Thus, even though these defensive symbioses are dynamic in the acquisition and replacement of microbial partners, the chemistry seems to be conserved. This suggests a limited diversity of chemical compounds that can be used for defense against eukaryotic antagonists (predators or fungi) in a symbiotic context, which is supported by the convergence on similar compounds in terrestrial and aquatic symbioses. It is possible that this might be due to the harmful side effects of the bioactive molecule on the eukaryotic host, analogous to the cytotoxic side effects of antifungal pharmaceuticals on humans, resulting in only limited diversity of such compounds.
To gain insights on the possible origin of the lga BGC, we performed an analysis of pentanucleotide (5-mer) frequencies of the beetle-associated, lga-carrying symbionts and their associated BGCs, along with the genomes of recently identified soil-borne Paraburkholderia species that carry the lagriamide B (lgb) BGC, which is highly similar to the lga BGC [83]. Visualization of 5-mer frequencies of the BGCs and the genomes revealed three clusters of BGCs: The BGCs from the two soil-borne Paraburkholderia strains, the BGCs from the Brazilian L. villosa-derived LvStB strains, and then a third cluster of all other lga BGCs (Fig. S12). A similar pattern was observed for the nucleotide composition of the respective genomes wherein LvStB and Lv20_9 form an isolated cluster, the two soil-borne Paraburkholderia form a second, distant cluster, and all other lga-carrying Burkholderia strains and cultured Lagria-associated genomes (LvStA and LhStG) form a third cluster. None of the BGCs share similar 5-mer composition with their respective genomes, providing additional evidence for the horizontal acquisition of the lga BGC.
We noted during the analysis of the COG annotated genes in LvStB_2023 that there appeared to be a particularly high number of pseudogenized genes in the L category (replication, recombination, and repair) (Fig. 3B). We assessed the percentage change of COG annotated genes in all lga-carrying Burkholderia and found that this pseudogenization of genes involved in DNA replication, recombination, and repair was particularly high in all the Brazilian L. villosa-derived LvStB strains, as well as the MAGs LhSB_1, LhG_1, Loki_2, and Lgren_6 (Fig. S13). Two of the three LvStB strains also exhibited high pseudogenization of the genes associated with cell motility (Category N). Even though COG annotation of genes does not provide a robust picture, as not all genes are successfully annotated, the increased pseudogenization of genes involved in DNA replication and repair may explain the divergence of the LvStB strains observed in both the phylogenetic analysis and the related 5-mer analysis. In particular, LvStB MAGs possessed highly truncated and psuedogenized polA genes, coding for DNA polymerase I used in many DNA-repair pathways and chromosome replication [89], whereas other lga-containing MAGs, except LhHG_2, had intact polA genes (Fig. S14). The loss of polA in the L. villosa symbionts explains their accelerated sequence evolution in the genome as a whole and also in the lga BGC compared with other lga-possessing symbionts (Fig. S12). The absence of polA in LhHG_2 could be due to its poor quality, as it is only 46% complete and has only 47.6% of core genes.
Previous studies have highlighted how symbionts can be conserved across host-speciation events and millions of years, leading to genome reduction in the symbiont [6, 16]. A disadvantage of such an exclusive relationship is that the symbiont inevitably suffers from increasing genome erosion that can result in reduced efficiency in providing benefits to the host [90]. Consequently, many long-term obligate symbioses have experienced symbiont replacement events that can provide an escape route for the insect host after its symbiont enters the irreversible phase of degenerative genome reduction [91]. Such replacement is a common phenomenon in Hemipteran symbionts [81]. In the present study, however, we are suggesting that the repeated replacement of symbionts may have happened with very closely related strains that carry the same BGC and hence likely provide the same functional benefit to the host. One reason we are suggesting multiple acquisitions and displacements may have happened is that all the lga-containing symbionts appear to be at different stages of genome reduction, with different genome sizes and gene complements, perhaps indicating that they have been symbionts for different amounts of time. That in combination with the apparent importance of lga specifically, the incongruence of symbiont and host phylogeny, and the fact that none of the symbionts is profoundly genome reduced suggests that although Lagriinae likely hosted lga-containing symbionts since the evolution of special symbiont storage structures, the current symbionts are not direct descendants of those original symbionts. The replaced symbionts were likely genome reduced to an extent that they were outcompeted by incoming lga-bearing strains from the environment. The lga BGC-containing Burkholderia strains were consistently the most abundant symbionts in the metagenomes across seven different Lagriinae species, indicating that the lga BGC or an as yet unknown genomic feature shared among the symbiont strains provides a key selective advantage in the beetles’ symbiotic organs. Possibly, lagriamide is uniquely suited to defend the symbionts’ niche against competitors and/or protect its host from antagonists. However, as lagriamide shows lower antifungal activity than some secondary metabolites of related Burkholderia strains [21, 26, 92], another intriguing possibility is that it only provides a moderate degree of defense but at the same time exhibits less harmful side effects on the host than other antifungal compounds. Further elucidating the relevance of lagriamide in establishing the symbiotic association with beetles will not only provide valuable insights into the ecological and evolutionary dynamics of defensive symbioses but may also unravel the mechanisms ensuring specificity in symbiotic alliances.
Supplementary Material
Acknowledgements
We would like to thank Jean Keller for providing python scripts that helped in creating the symbiont phylogeny. We are grateful to Rebekka Janke, Takema Fukatsu, Kiyoshi Ando, Kimio Masumoto, Juan José López, and Rolf Beutel for support with insect collection and to Domenica Schnabelrauch for carrying out Polymerase Chain Reactions, cloning and Sanger sequencing.
Contributor Information
Siddharth Uppal, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, Madison, WI 53705, United States.
Samantha C Waterworth, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, Madison, WI 53705, United States.
Alina Nick, Department of Insect Symbiosis, Max Planck Institute for Chemical Ecology, 07745 Jena, Germany.
Heiko Vogel, Department of Insect Symbiosis, Max Planck Institute for Chemical Ecology, 07745 Jena, Germany.
Laura V Flórez, Department of Plant and Environmental Science, University of Copenhagen, DK-1871 Copenhagen, Denmark.
Martin Kaltenpoth, Department of Insect Symbiosis, Max Planck Institute for Chemical Ecology, 07745 Jena, Germany.
Jason C Kwan, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, Madison, WI 53705, United States.
Author contributions
Siddharth Uppal (Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing—original draft, Writing—review & editing), Samantha C. Waterworth (Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing—original draft, Writing—review & editing), Alina Nick (Data curation, Formal analysis, Investigation, Methodology), Heiko Vogel (Data curation, Formal analysis, Investigation, Methodology), Laura V. Flórez (Conceptualization, Investigation, Writing—review & editing, Supervision, Funding acquisition), Martin Kaltenpoth (Formal analysis, Methodology, Conceptualization, Investigation, Writing—review & editing, Supervision, Funding acquisition), and Jason C. Kwan (Conceptualization, Investigation, Writing—review & editing, Supervision, Funding acquisition)
Conflicts of interest
The Kwan lab offers their metagenomic binning pipeline Autometa on the paid bioinformatics and computational platform BatchX in addition to distributing it through open source channels.
Funding
This work was supported by the U.S. National Science Foundation [DBI-1845890 to J.C.K., S.U., and S.C.W.], the Max Planck Society (to A.N., H.V., and M.K.), and the European Research Council through an ERC Consolidator Grant (ERC CoG 819585 “SYMBeetle,” to M.K.).
Data availability
The data associated with this study was deposited under BioProject accession no. PRJNA1054523. Metagenomic reads have been deposited in the Sequence Read Archive with accessions SRR27332963–SRR27332975. Representative lga BGC sequences have been submitted to Genbank with accession numbers PP034267–PP034277 and PP034279. All lagriamide BGC-carrying MAGs were deposited with the following accession numbers: Ecno_1, JAYFRU000000000; Latri_1, JAYFRV000000000; Lgren_6, JAYFRW000000000; LhG_1, JAYFRX000000000; LhHG_2, JAYFRY000000000; LhSB_1, JAYFRZ000000000; Lruf2_2, JAYFSA000000000; Loki_2, JAYFSB000000000; Lruf1_1, JAYFSC000000000; Lv20_9, JAYFSD000000000; LvStB_2023, CP144361-CP144363.
References
- 1. Dyall SD, Brown MT, Johnson PJ. Ancient invasions: from endosymbionts to organelles. Science 2004;304:253–7. 10.1126/science.1094884 [DOI] [PubMed] [Google Scholar]
- 2. Stephens TG, Gabr A, Calatrava Vet al. Why is primary endosymbiosis so rare? New Phytol 2021;231:1693–9. 10.1111/nph.17478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Von Dohlen CD, Moran NA. Molecular data support a rapid radiation of aphids in the cretaceous and multiple origins of host alternation. Biol J Linn Soc Lond 2000;71:689–717. 10.1111/j.1095-8312.2000.tb01286.x [DOI] [Google Scholar]
- 4. Nováková E, Hypša V, Klein Jet al. Reconstructing the phylogeny of aphids (Hemiptera: Aphididae) using DNA of the obligate symbiont Buchnera aphidicola. Mol Phylogenet Evol 2013;68:42–54. 10.1016/j.ympev.2013.03.016 [DOI] [PubMed] [Google Scholar]
- 5. Chong RA, Park H, Moran NA. Genome evolution of the obligate endosymbiont Buchnera aphidicola. Mol Biol Evol 2019;36:1481–9. 10.1093/molbev/msz082 [DOI] [PubMed] [Google Scholar]
- 6. McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 2011;10:13–26. 10.1038/nrmicro2670 [DOI] [PubMed] [Google Scholar]
- 7. Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet 2001;17:589–96. 10.1016/S0168-9525(01)02447-7 [DOI] [PubMed] [Google Scholar]
- 8. Hershberg R, Petrov DA. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet 2010;6:e1001115. 10.1371/journal.pgen.1001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dietel A-K, Merker H, Kaltenpoth Met al. Selective advantages favour high genomic AT-contents in intracellular elements. PLoS Genet 2019;15:e1007778. 10.1371/journal.pgen.1007778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dufresne A, Garczarek L, Partensky F. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol 2005;6:R14. 10.1186/gb-2005-6-2-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morris JJ, Lenski RE, Zinser ER. The black queen hypothesis: evolution of dependencies through adaptive gene loss. MBio 2012;3:e00036–12. 10.1128/mBio.00036-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinto-Carbó M, Gademann K, Eberl Let al. Leaf nodule symbiosis: function and transmission of obligate bacterial endophytes. Curr Opin Plant Biol 2018;44:23–31. 10.1016/j.pbi.2018.01.001 [DOI] [PubMed] [Google Scholar]
- 13. Pinto-Carbó M, Sieber S, Dessein Set al. Evidence of horizontal gene transfer between obligate leaf nodule symbionts. ISME J 2016;10:2092–105. 10.1038/ismej.2016.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Donia MS, Fricke WF, Partensky Fet al. Complex microbiome underlying secondary and primary metabolism in the tunicate-Prochloron symbiosis. Proc Natl Acad Sci USA 2011;108:E1423–32. 10.1073/pnas.1111712108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwan JC, Donia MS, Han AWet al. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci USA 2012;109:20655–60. 10.1073/pnas.1213820109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kwan JC, Schmidt EW. Bacterial endosymbiosis in a chordate host: long-term co-evolution and conservation of secondary metabolism. PLoS One 2013;8:e80822. 10.1371/journal.pone.0080822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ebert D. The epidemiology and evolution of symbionts with mixed-mode transmission. Annu Rev Ecol Evol Syst 2013;44:623–43. 10.1146/annurev-ecolsys-032513-100555 [DOI] [Google Scholar]
- 18. Toh H, Weiss BL, Perkin SAHet al. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res 2006;16:149–56. 10.1101/gr.4106106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller IJ, Vanee N, Fong SSet al. Lack of overt genome reduction in the bryostatin-producing bryozoan symbiont, ‘Candidatus Endobugula sertula’. Appl Environ Microbiol 2016;82:6573–83. 10.1128/AEM.01800-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linneman J, Paulus D, Lim-Fong Get al. Latitudinal variation of a defensive symbiosis in the Bugula neritina (Bryozoa) sibling species complex. PLoS One 2014;9:e108783. 10.1371/journal.pone.0108783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Flórez LV, Scherlach K, Miller IJet al. An antifungal polyketide associated with horizontally acquired genes supports symbiont-mediated defense in Lagria villosa beetles. Nat Commun 2018;9:2478. 10.1038/s41467-018-04955-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janke RS, Kaftan F, Niehs SPet al. Bacterial ectosymbionts in cuticular organs chemically protect a beetle during molting stages. ISME J 2022;16:2691–701. 10.1038/s41396-022-01311-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waterworth SC, Flórez LV, Rees ERet al. Horizontal gene transfer to a defensive symbiont with a reduced genome in a multipartite beetle microbiome. MBio 2020;11:e02430–19. 10.1128/mBio.02430-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wierz JC, Gaube P, Klebsch Det al. Transmission of bacterial symbionts with and without genome erosion between a beetle host and the plant environment. Front Microbiol 2021;12:715601. 10.3389/fmicb.2021.715601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jürgen SH. Die symbiose der Lagriiden (Coleoptera). Z Morph u Okol Tiere 1929;15:1–34. 10.1007/BF00403097 [DOI] [Google Scholar]
- 26. Flórez LV, Scherlach K, Gaube Pet al. Antibiotic-producing symbionts dynamically transition between plant pathogenicity and insect-defensive mutualism. Nat Commun 2017;8:15172. 10.1038/ncomms15172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flórez LV, Biedermann PHW, Engl Tet al. Defensive symbioses of animals with prokaryotic and eukaryotic microorganisms. Nat Prod Rep 2015;32:904–36. 10.1039/C5NP00010F [DOI] [PubMed] [Google Scholar]
- 28. Jung BH, Kim JI. Taxonomy of the genus Lagria Fabricius (Coleoptera: Tenebrionidae: Lagriinae) in Korea. Korean J Appl Entomol 2009;48:275–80. 10.5656/KSAE.2009.48.3.275 [DOI] [Google Scholar]
- 29. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bankevich A, Nurk S, Antipov Det al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller IJ, Rees ER, Ross Jet al. Autometa: automated extraction of microbial genomes from individual shotgun metagenomes. Nucleic Acids Res 2019;47:e57. 10.1093/nar/gkz148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chklovski A, Parks DH, Woodcroft BJet al. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 2023;20:1203–12. 10.1038/s41592-023-01940-w [DOI] [PubMed] [Google Scholar]
- 33. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34:3094–100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaser R, Sović I, Nagarajan Net al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 2017;27:737–46. 10.1101/gr.214270.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kolmogorov M, Bickhart DM, Behsaz Bet al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods 2020;17:1103–10. 10.1038/s41592-020-00971-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guan D, McCarthy SA, Wood Jet al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020;36:2896–8. 10.1093/bioinformatics/btaa025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wei Z, Shi A. The complete mitochondrial genomes of gour Lagriine species (Coleoptera, Tenebrionidae) and phylogenetic relationships within Tenebrionidae. PeerJ 2023;11:e15483. 10.7717/peerj.15483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Donath A, Jühling F, Al-Arab Met al. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res 2019;47:10543–52. 10.1093/nar/gkz833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgar RC. Muscle5: high-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat Commun 2022;13:6968. 10.1038/s41467-022-34630-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 2006;34:W609–12. 10.1093/nar/gkl315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown JW, Walker JF, Smith SA. Phyx: phylogenetic tools for Unix. Bioinformatics 2017;33:1886–8. 10.1093/bioinformatics/btx063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Darriba D, Posada D, Kozlov AMet al. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol 2020;37:291–4. 10.1093/molbev/msz189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flouri T, Izquierdo-Carrasco F, Darriba Det al. The phylogenetic likelihood library. Syst Biol 2015;64:356–62. 10.1093/sysbio/syu084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kozlov AM, Darriba D, Flouri Tet al. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019;35:4453–5. 10.1093/bioinformatics/btz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ronquist F, Teslenko M, van der Mark Pet al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 2012;61:539–42. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prokka ST. Rapid prokaryotic genome annotation. Bioinformatics 2014;30:2068–9. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- 47. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 2019;20:238. 10.1186/s13059-019-1832-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12:59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- 49. Buchfink B, Reuter K, Drost H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 2021;18:366–8. 10.1038/s41592-021-01101-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kergoat GJ, Soldati L, Clamens A-Let al. Higher level molecular phylogeny of darkling beetles (Coleoptera: Tenebrionidae). Syst Entomol 2014;39:486–99. 10.1111/syen.12065 [DOI] [Google Scholar]
- 51. Wu C, Zhou Y, Tian Tet al. First report of complete mitochondrial genome in the subfamily Alleculinae and mitochondrial genome-based phylogenetics in Tenebrionidae (Coleoptera: Tenebrionoidea). Insect Sci 2022;29:1226–38. 10.1111/1744-7917.12983 [DOI] [PubMed] [Google Scholar]
- 52. Ragionieri L, Zúñiga-Reinoso Á, Bläser Met al. Phylogenomics of darkling beetles (Coleoptera: Tenebrionidae) from the Atacama desert. PeerJ 2023;11:e14848. 10.7717/peerj.14848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312–3. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schmidt U. Kaefer der Welt—beetles of the world. Coleoptera picture collection. https://www.kaefer-der-welt.de/ (31 October 2024, date last accessed).
- 55. Blin K, Shaw S, Steinke Ket al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 2019;47:W81–7. 10.1093/nar/gkz310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blin K, Shaw S, Augustijn HEet al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res 2023;51:W46–50. 10.1093/nar/gkad344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hu Y, Fang L, Nicholson Cet al. Implications of error-prone long-read whole-genome shotgun sequencing on characterizing reference microbiomes. iScience 2020;23:101223. 10.1016/j.isci.2020.101223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Piel J, Höfer I, Hui D. Evidence for a symbiosis island involved in horizontal acquisition of pederin biosynthetic capabilities by the bacterial symbiont of Paederus fuscipes beetles. J Bacteriol 2004;186:1280–6. 10.1128/JB.186.5.1280-1286.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kador M, Horn MA, Dettner K. Novel oligonucleotide probes for in situ detection of pederin-producing endosymbionts of Paederus riparius rove beetles (Coleoptera: Staphylinidae). FEMS Microbiol Lett 2011;319:73–81. 10.1111/j.1574-6968.2011.02270.x [DOI] [PubMed] [Google Scholar]
- 60. Kellner RLL, Dettner K. Differential efficacy of toxic pederin in deterring potential arthropod predators of Paederus (Coleoptera: Staphylinidae) offspring. Oecologia 1996;107:293–300. 10.1007/BF00328445 [DOI] [PubMed] [Google Scholar]
- 61. Witczak ZJ, Bommareddy A, AL VanWertet al. Pederin, psymberin and the structurally related mycalamides: synthetic aspects and biological activities. In: Kim S-K (ed.), Handbook of Anticancer Drugs from Marine Origin. Cham: Springer, 2015, 683–700. 10.1007/978-3-319-07145-9_32 [DOI] [Google Scholar]
- 62. Sakemi S, Ichiba T, Kohmoto Set al. Isolation and structure elucidation of onnamide A, a new bioactive metabolite of a marine sponge. Theonella sp J Am Chem Soc 1988;110:4851–3. 10.1021/ja00222a068 [DOI] [Google Scholar]
- 63. Perry NB, Blunt JW, Munro MHGet al. Antiviral and antitumor agents from a New Zealand sponge, Mycale sp. 2: structures and solution conformations of mycalamides A and B. J Org Chem 1990;55:223–7. 10.1021/jo00288a037 [DOI] [Google Scholar]
- 64. West LM, Northcote PT, Hood KAet al. Mycalamide D, a new cytotoxic amide from the New Zealand marine sponge Mycale species. J Nat Prod 2000;63:707–9. 10.1021/np9904511 [DOI] [PubMed] [Google Scholar]
- 65. Simpson JS, Garson MJ, Blunt JWet al. Mycalamides C and D, cytotoxic compounds from the marine sponge Stylinos n. species. J Nat Prod 2000;63:704–6. 10.1021/np990431z [DOI] [PubMed] [Google Scholar]
- 66. Dyshlovoy SA, Fedorov SN, Kalinovsky AIet al. Mycalamide A shows cytotoxic properties and prevents EGF-induced neoplastic transformation through inhibition of nuclear factors. Mar Drugs 2012;10:1212–24. 10.3390/md10061212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gouiffes D, Juge M, Grimaud Net al. Bistramide A, a new toxin from the Urochordata Lissoclinum bistratum Sluiter: isolation and preliminary characterization. Toxicon 1988;26:1129–36. 10.1016/0041-0101(88)90297-8 [DOI] [PubMed] [Google Scholar]
- 68. Bowers RM, Kyrpides NC, Stepanauskas Ret al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 2017;35:725–31. 10.1038/nbt.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Manzano-Marín A, Kvist S, Oceguera-Figueroa A. Evolution of an alternative genetic code in the Providencia symbiont of the hematophagous leech Haementeria acuecueyetzin. Genome Biol Evol 2023;15:evad164. 10.1093/gbe/evad164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tvedte ES, Gasser M, Sparklin BCet al. Comparison of long-read sequencing technologies in interrogating bacteria and fly genomes. G3 (Bethesda) 2021;11:jkab083. 10.1093/g3journal/jkab083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marx V. Method of the year: long-read sequencing. Nat Methods 2023;20:6–11. 10.1038/s41592-022-01730-w [DOI] [PubMed] [Google Scholar]
- 72. Dong X, Du MRM, Gouil Qet al. Benchmarking long-read RNA-sequencing analysis tools using in silico mixtures. Nat Methods 2023;20:1810–21. 10.1038/s41592-023-02026-3 [DOI] [PubMed] [Google Scholar]
- 73. Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol 2005;3:144–56. 10.1038/nrmicro1085 [DOI] [PubMed] [Google Scholar]
- 74. Lessie TG, Hendrickson W, Manning BDet al. Genomic complexity and plasticity of Burkholderia cepacia. FEMS Microbiol Lett 1996;144:117–28. 10.1111/j.1574-6968.1996.tb08517.x [DOI] [PubMed] [Google Scholar]
- 75. Egan ES, Fogel MA, Waldor MK. Divided genomes: negotiating the cell cycle in prokaryotes with multiple chromosomes. Mol Microbiol 2005;56:1129–38. 10.1111/j.1365-2958.2005.04622.x [DOI] [PubMed] [Google Scholar]
- 76. Flórez LV, Kaltenpoth M. Symbiont dynamics and strain diversity in the defensive mutualism between Lagria beetles and Burkholderia. Environ Microbiol 2017;19:3674–88. 10.1111/1462-2920.13868 [DOI] [PubMed] [Google Scholar]
- 77. Goris J, Konstantinidis KT, Klappenbach JAet al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 2007;57:81–91. 10.1099/ijs.0.64483-0 [DOI] [PubMed] [Google Scholar]
- 78. Varghese NJ, Mukherjee S, Ivanova Net al. Microbial species delineation using whole genome sequences. Nucleic Acids Res 2015;43:6761–71. 10.1093/nar/gkv657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 2005;102:2567–72. 10.1073/pnas.0409727102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Parks DH, Chuvochina M, Rinke Cet al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res 2022;50:D785–94. 10.1093/nar/gkab776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sudakaran S, Kost C, Kaltenpoth M. Symbiont acquisition and replacement as a source of ecological innovation. Trends Microbiol 2017;25:375–90. 10.1016/j.tim.2017.02.014 [DOI] [PubMed] [Google Scholar]
- 82. Janke RS, Moog S, Weiss Bet al. Morphological adaptation for ectosymbiont maintenance and transmission during metamorphosis in Lagria beetles. Front Physiol 2022;13:979200. 10.3389/fphys.2022.979200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fergusson CH, Saulog J, Paulo BSet al. Discovery of a lagriamide polyketide by integrated genome mining, isotopic labeling, and untargeted metabolomics. Chem Sci 2024;15:8089–96. 10.1039/D4SC00825A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McTaggart SJ, Obbard DJ, Conlon Cet al. Immune genes undergo more adaptive evolution than non-immune system genes in Daphnia pulex. BMC Evol Biol 2012;12:63. 10.1186/1471-2148-12-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Erler S, Lhomme P, Rasmont Pet al. Rapid evolution of antimicrobial peptide genes in an insect host-social parasite system. Infect Genet Evol 2014;23:129–37. 10.1016/j.meegid.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 86. Behrman EL, Howick VM, Kapun Met al. Rapid seasonal evolution in innate immunity of wild Drosophila melanogaster. Proc Biol Sci 2018;285:20172599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Engl T, Kroiss J, Kai Met al. Evolutionary stability of antibiotic protection in a defensive symbiosis. Proc Natl Acad Sci USA 2018;115:E2020–9. 10.1073/pnas.1719797115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kroiss J, Kaltenpoth M, Schneider Bet al. Symbiotic Streptomycetes provide antibiotic combination prophylaxis for wasp offspring. Nat Chem Biol 2010;6:261–3. 10.1038/nchembio.331 [DOI] [PubMed] [Google Scholar]
- 89. Patel PH, Suzuki M, Adman Eet al. Prokaryotic DNA polymerase I: evolution, structure, and ‘base flipping’ mechanism for nucleotide selection. J Mol Biol 2001;308:823–37. 10.1006/jmbi.2001.4619 [DOI] [PubMed] [Google Scholar]
- 90. Bennett GM, Moran NA. Heritable symbiosis: the advantages and perils of an evolutionary rabbit hole. Proc Natl Acad Sci USA 2015;112:10169–76. 10.1073/pnas.1421388112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Michalik A, Bauer E, Szklarzewicz Tet al. Nutrient supplementation by genome-eroded Burkholderia symbionts of scale insects. ISME J 2023;17:2221–31. 10.1038/s41396-023-01528-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dose B, Niehs SP, Scherlach Ket al. Biosynthesis of sinapigladioside, an antifungal isothiocyanate from Burkholderia symbionts. Chembiochem 2021;22:1920–4. 10.1002/cbic.202100089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data associated with this study was deposited under BioProject accession no. PRJNA1054523. Metagenomic reads have been deposited in the Sequence Read Archive with accessions SRR27332963–SRR27332975. Representative lga BGC sequences have been submitted to Genbank with accession numbers PP034267–PP034277 and PP034279. All lagriamide BGC-carrying MAGs were deposited with the following accession numbers: Ecno_1, JAYFRU000000000; Latri_1, JAYFRV000000000; Lgren_6, JAYFRW000000000; LhG_1, JAYFRX000000000; LhHG_2, JAYFRY000000000; LhSB_1, JAYFRZ000000000; Lruf2_2, JAYFSA000000000; Loki_2, JAYFSB000000000; Lruf1_1, JAYFSC000000000; Lv20_9, JAYFSD000000000; LvStB_2023, CP144361-CP144363.