

Abstract
Background
Family history of Parkinson's disease (PD) is a common finding in PD patients. However, a few studies have systematically examined this aspect.
Objectives
We investigated the family history of PD patients, comparing demographic and clinical features between familial PD (fPD) and sporadic PD (sPD).
Methods
A cross‐sectional study enrolling 2035 PD patients was conducted in 28 Italian centers. Clinical data and family history up to the third degree of kinship were collected.
Results
Family history of PD was determined in 21.9% of patients. fPD patients had earlier age at onset than sporadic patients. No relevant differences in the prevalence of motor and nonmotor symptoms were detected. Family history of mood disorders resulted more prevalently in the fPD group.
Conclusions
fPD was found to recur more frequently than previously reported. Family history collection beyond the core family is essential to discover disease clusters and identify novel risk factors for PD.
Keywords: familial and sporadic Parkinson's disease, family history, hyposmia, cognitive impairment, depression, bipolar disorder
Parkinson's disease (PD) is a neurodegenerative disorder clinically characterized by the presence of bradykinesia, variably associated with resting tremor, muscle rigidity, and postural instability. Concerning its controversial etiopathology, both genetic and environmental factors are likely involved. 1 , 2 , 3
Positive family history for PD is an established risk factor for the development of the disease, especially in first‐degree relatives, whose risk for the disease is estimated to increase by 2‐ to 3‐fold. 4 , 5 Previous studies reported familial recurrence of PD in 5% to 15% of patients. 6 , 7
This study aimed to analyze the family history of PD patients up to the third degree of kinship, assessing the frequency of familial forms of PD and comparing demographic and clinical features between familial (fPD) and sporadic (sPD) patients. The prevalence of other neurological and psychiatric disorders across family members was also assessed.
Patients and Methods
This is a cross‐sectional study performed in 28 Parkinson's disease and movement disorders centers, located in 14 Italian regions.
Patients were consecutively recruited from outpatient clinics during a 30‐month period, from April 1, 2020, to November 30, 2021.
The following inclusion criteria were applied to establish patient eligibility:
Confirmed diagnosis of PD made by a movement disorder specialist, according to the Movement Disorder Society clinical diagnostic criteria 4
Signed informed consent to participate in the study
Age over 18 years at the time of assessment
Exclusion criteria were defined as follows:
Secondary or atypical parkinsonism
Lack of sufficiently comprehensive biographical, clinical and anamnestic information
All data were entered into an electronic database. For each patient, family history for PD, essential tremor (ET), cognitive impairment, and major depressive and bipolar disorders was assessed up to the third degree of kinship using a structured family history interview.
The occurrence of one of the aforementioned conditions in PD family members was classified as follows:
Certain, if the disease was diagnosed by a neurologist or a psychiatrist for what concerns movement disorders and psychiatric conditions/mood disorders, respectively
Possible, if the disease was reported by patients or caregivers without formal assessment by a physician
Negative, if the disease never occurred in the family
Unknown, if data on family history were incomplete/missing (eg, in the case of adoption, early death, abandoned)
Clinical and genetic data were obtained through neurological examination and local databases. Descriptive statistic was employed to characterize the population demographics. Categorical variables have been reported as count (percentage) and continuous variables as mean (standard deviation). Statistical comparisons between groups were performed using Fisher's exact test for categorical variables and Wilcoxon 2‐sample test for continuous variables. Statistical significance was set at the α = 0.05 level, 2 sided. All statistical analyses were performed using SAS software, version 9.4 (SAS Institute, Cary, NC, USA).
The entire study population was considered for demographic and epidemiological analyses. Only patients with certain (fPD) or negative (sPD) PD family history were included in comparison analyses.
Results
Family History for PD and Other Neuropsychiatric Disorders in PD Patients
According to inclusion/exclusion criteria, 2035 PD patients were included in the study.
A male predominance was observed, with a male‐to‐female ratio of 1.5:1.0. The mean age at evaluation was 68.9 ± 10.6 years, the mean age at onset of motor symptoms was 60.2 ± 11.5 years, and the average disease duration was 8.7 ± 5.9 years.
A family history of any degree for PD was reported by 34.5% of PD patients (n = 703, certain plus possible). However, a formal diagnosis of PD could be demonstrated in the relatives of 21.9% of cases (n = 446, certain) (Fig. 1).
FIG. 1.
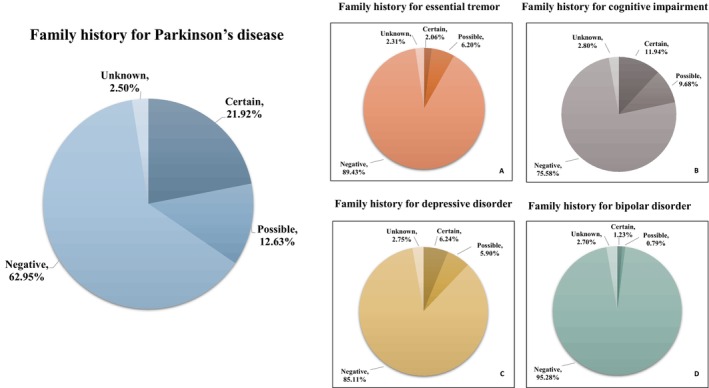
Pie charts showing the diseases (PD, ET, Cognitive Impairment, Depressive Disorder, Bipolar Disorder) for which the family history was investigated and the percentages of certain, possible, negative, and unknown family history.
Considering PD patients with certain or possible family history for PD, 67.9% (477/703) reported at least 1 first‐degree affected relative, whereas 32.1% (226/703) described only second‐ or third‐degree affected family members.
As concerns other neurological and psychiatric disorders, 11.9% (n = 243) of PD patients reported certain family history for cognitive impairment, 2% (n = 42) for ET, and 6.2% (n = 127) and 1.2% (n = 25) for depression and bipolar disorders, respectively (Fig. 1A–D). The inclusion of possible cases in the estimation of prevalence significantly increased numbers as follows: cognitive impairment = 21.6% (n = 440), ET = 8.2% (n = 168), depression = 12.1% (n = 247), and bipolar disorder = 2% (n = 41).
Comparison between fPD and sPD
No differences in mean age and sex distribution between fPD (n = 446) and sPD (n = 1281) groups were observed. The mean age at onset was significantly lower in fPD patients (58.5 ± 11.5 years) than in sPD (60.8 ± 11.5 years) (P = 0.0001), and mean disease duration was longer in the fPD group (9.6 years) than in the sPD group (8.4 years) (P = 0.0002). No relevant difference between the 2 groups in the modified Hoehn and Yahr Staging Scale scores corrected for disease duration was observed.
The distribution of motor and nonmotor symptoms between fPD and sPD was similar, except for hyposmia that resulted more frequently in the fPD group (Table 1).
TABLE 1.
Comparison of clinical characteristics between patients with fPD and sPD
| fPD (n = 446) | sPD (n = 1281) | Statistical significance | |
|---|---|---|---|
| Motor symptoms | |||
| Rest tremor, % (n) | 68.6 (306) | 67.2 (861) | P = 0.5977 |
| Rigidity, % (n) | 86.1 (384) | 85.1 (1090) | P = 0.6413 |
| Postural instability, % (n) | 24.0 (107) | 25.3 (324) | P = 0.6116 |
| Freezing, % (n) | 20.4 (91) | 17.4 (223) | P = 0.1755 |
| Dystonia, % (n) | 12.6 (56) | 11.0 (141) | P = 0.3874 |
| Pisa syndrome, % (n) | 7.6 (34) | 8.1 (104) | P = 0.8393 |
| Camptocormia, % (n) | 21.8 (97) | 22.2 (284) | P = 0.8946 |
| Nonmotor symptoms | |||
| Hyposmia, % (n) | 42.6 (190) | 35.4 (453) | P = 0.0075 |
| Constipation, % (n) | 53.4 (238) | 49.6 (635) | P = 0.1698 |
| Orthostatic hypotension, % (n) | 16.1 (72) | 17.1 (219) | P = 0.6603 |
| Urinary symptoms, % (n) | 41.3 (184) | 39.0 (499) | P = 0.3994 |
| Sialorrhoea, % (n) | 15.9 (71) | 13.5 (173) | P = 0.2075 |
| Rem behavior disorder, % (n) | 46.0 (205) | 43.3 (555) | P = 0.3467 |
| Depression, % (n) | 31.6 (141) | 31.2 (399) | P = 0.8589 |
| Bipolar disorder, % (n) | 0.9 (4) | 0.6 (8) | P = 0.5195 |
| Cognitive decline, % (n) | 13.5 (60) | 16.6 (212) | P = 0.1314 |
| Psychosis, % (n) | 8.5 (38) | 10.2 (131) | P = 0.3105 |
| Anxiety, % (n) | 33.0 (147) | 29.4 (377) | P = 0.1692 |
| Pain, % (n) | 23.8 (106) | 19.8 (253) | P = 0.0781 |
| Family history of neuropsychiatric disorders | |||
| Essential tremor, % (n) | 3.7 (15) | 2.0 (24) | P = 0.0647 |
| Cognitive impairment, % (n) | 15.6 (61) | 13.8 (160) | P = 0.4027 |
| Depression, % (n) | 9.8 (40) | 5.7 (68) | P = 0.0058 |
| Bipolar disorder, % (n) | 1.4 (6) | 1.3 (16) | P = 0.8082 |
Statistically significant differences are indicated in blue, whereas trends of clinical interest are highlighted in gray.
Abbreviations: fPD, familial Parkinson's disease; Rem, rapid eye movements; sPD, sporadic Parkinson's disease.
Family history of depressive disorder (9.8% vs. 5.7%, P = 0.0058) was more common in fPD. A similar but not significant trend was observed for ET (3.7% vs. 2.0%, P = 0.0647), whereas no differences were observed in the recurrence of cognitive and bipolar disorders.
Genetics
Genetic testing was performed in 21.8% (443/2035) of the patients. In all cases a minimal gene set (ie, SNCA, LRRK2, GBA1, PRKN, and PINK1, including relevant dosage assays) was analyzed.
Considering the 372 subjects tested with certain positive or negative family history, a positive genetic result was more frequent in fPD (55/151, 36.4%) than in sPD (57/221, 25.8%) (P = 0.0295). The prevalence of pathogenic variants in the most common PD genes (GBA1, LRRK2, and PRKN) did not differ between sPD and fPD patients (Table S1). Most of the enrolled patients were not genetically tested, thus precluding further analyses and possibly enriching the rate of positive results by having selected more likely genetic cases (eg, due to family history, clinical characteristics or geographical origin).
Discussion
The frequency of family history of PD is known to be higher among PD cases than the general population. 5 , 6 Previous studies reported that about 10% to 15% of PD patients have at least 1 affected relative. 7 , 8 In this study, more than one‐third of PD patients (34.5%) presented a positive family history for PD (21.9% when considering only certain cases, thus eliminating the limitation due to the anamnestic report of possible cases). The higher rate of positive family history encountered may suggest either a major role played by genetic factors in the Italian PD population or a possible underestimation in previous studies. A plausible explanation may be the limitation of data collection to first‐degree relatives. Interestingly, a remarkable 32.1% of PD patients reported only second‐ or third‐degree affected family members, which should prompt clinicians to investigate the family history more thoroughly.
As a matter of fact, given the incidence of PD in the general population, the presence of more than 1 family member affected may not be related to genetic factors.
In line with previous studies, 6 , 9 , 10 fPD exhibited younger age at onset. This observation could be due to a higher prevalence of genetic forms in fPD or to an earlier recognition of PD symptoms by patients with affected relatives. 11
No major differences in motor phenotype between fPD and sPD were observed, confirming previous observations, 9 , 12 with a few exceptions. 10
Among nonmotor features, hyposmia was more represented in the fPD group. A clear explanation of this finding is still elusive. The higher prevalence may be due to PD genetic risk factors predisposing also to PD‐related hyposmia in familial cases. 13 However, confounding factors (eg, cigarette smoking, allergies, drugs) that were not collected in this study may also play a role.
We then investigated the occurrence of neurological and psychiatric features in relatives of PD patients. To this aim, we collected data on whether PD relatives had a diagnosis of disorders previously associated with PD (ie, ET, bipolar disorder, and depression) or presented clinical features (ie, tremor, cognitive impairment, and mood disorders), which may occur in prodromal PD.
A certain diagnosis of ET was present in family members of 2% of the patients, whereas in 6.2% the diagnosis of ET was reported as possible. The remarkable difference between the certain and possible ET diagnoses is likely reflecting the uncertainty in categorizing tremor in terms of diagnosis and phenomenology. Indeed, the coexistence of PD and ET has been reported in PD patients, and ET was consistently found with a higher prevalence in family members of PD patients compared to those of controls. 14
However, the relationship between ET and PD remains controversial, and most studies failed to find a significant connection. 15 These observations should prompt the collection of family history of ET in PD patients, encouraging further assessments and clinical studies to better understand these associations.
A diagnosis of cognitive impairment was reported from 12% (certain) to 21% (certain + possible) of PD families. This prevalence did not include relatives affected by PD dementia. A precise characterization of the cognitive disorders (Alzheimer's disease, frontotemporal dementia, dementia with Lewy bodies, vascular dementia, etc.) was often not retrievable due to missing anamnestic data. Such a prevalence of cognitive impairment in PD family members may be explained by shared molecular mechanisms between PD and other neurodegenerative disorders, as demonstrated by the presence of common genetic risk factors (ie, GBA1, C9ORF72, MAPT, PSEN1). 16 , 17 , 18 , 19 , 20
Depression and anxiety were referred in 32% and 31% of PD patients, respectively (Table 1). This finding is higher compared to the reported prevalence of such symptoms in the Italian population (3%–6% and 2%–5%, respectively). 21 , 22 Notably, bipolar disorder, which has been proposed as a risk factor for PD, was quite rare in this cohort, occurring in 0.7% of patients. 23 , 24 Finally, a higher prevalence of mood disorders was observed in family members of fPD compared to sPD relatives. This observation may suggest a role of genetic factors in the predisposition of mood disorders, likely representing prodromal symptoms of PD.
In conclusion, PD patients have a higher prevalence of family members affected than previously reported, reaching up to one‐third of cases after recording information on second‐ and third‐degree relatives. This observation should stimulate further studies evaluating the risk of PD in family members to improve the counseling in PD patients and their families. Moreover, the identification of familial clusters will help to dissect the genetic and nongenetic contributions to the pathogenesis of PD and other neurodegenerative disorders.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical analysis: A. Design, B. Execution, C. Review and critique; (3) Manuscript preparation: A. Writing of the first draft, B. Review and critique.
F.A.: 1A, 1B, 1C, 2A, 2B, 3A
G.C.: 2A, 2B, 2C
G.F., G.L., E.M., A.D.M.: 1C, 2A, 3B, 3B
A.D.F.: 1A, 2C, 3B
C.C., L.L., M.Z., A.R., F.S., P.D.M., N.T., A.A., A.A., M.C., L.A., R.M., F.D.B., T.B.M., M.C., A.R., R.C., G.P., A.F., A.C., N.T., E.M., G.C., L.M., C.C., E.C., R.C., R.E., P.B., M.P., C.S., V.F., C.L., M.C.M., M.P., C.L., O.D., R.D.G., A.P., A.P., A.L., A.I., M.C.S., G.C., A.B., C.S., G.D.L., A.B., P.N., F.T., E.C., P.V.M., S.T., A.T., R.D.M., S.A., M.T., I.D., S.O., P.T., F.D.B.; A.D'A.; F.V., F.C., G.T., V.F., M.A.V., L.C., S.G., G.D.N., D.V., M.Z., E.M.V., F.B.: 1C, 3B
All authors contributed equally to the manuscript and read and approved the final version of the manuscript.
Disclosures
Ethical Compliance Statement: The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Milan (Comitato Etico Milano Area 2, parere 1106_2019). Written informed consent was obtained for each patient participating in this work. All authors have read and complied with the journal's ethical publication guidelines. We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: Andrea Pilotto received speaker's honoraria from AbbVie, Angelini, Bial, Lundbeck, Roche, and Zambon Pharmaceuticals. Alessandro Padovani received speaker's honoraria and personal compensation as a consultant/scientific advisory board member for Biogen, Eisai Eli Lilly, General Healthcare (GE), Lundbeck, Nestlè, and Roche. Alberto Albanese received speaker's honoraria from Merz and Ipsen; served as advisor to Boston Scientific, and is specialty chief editor for Frontiers in Neurology and president of the International Association for Parkinsonism and Related Disorders. Carlo Colosimo has provided consultancy services for Ipsen and Bial. Leonardo Lopiano received honoraria for lecturing and travel grants from Medtronic, UCB Pharma, and AbbVie. M. Zibetti received honoraria from Medtronic. A. Rini received consultancy fees from Sanofi, Biogen, Neuraxpharma, and Novartis. Francesca Spagnolo has provided consultancy services for Zambon and has served on the scientific advisory board of Sanofi. P. De Marco received consultancy fees from Biogen. Nicola Tambasco received speaker's honoraria from AbbVie and travel grant from Sanofi, AbbVie, and Ipsen. Roberto Cilia has received speaker's honoraria from Zambon Italia, Zambon SAU, and Bial Italia Srl; advisory board fees from Bial; research support from the Italian Ministry of Health; honorarium from MDPI as editor in chief for Brain Sciences section neuromuscular and movement disorders.
No financial disclosures were documented for the following authors: F. Arienti, G. Casazza, G. Lazzeri, G. Franco, E. Monfrini, A. Di Maio, A. Antonini, M. Carecchio, L. Avanzino, R. Marchese, F. Di Blasio, T. Benzi Markushi, M. Canesi, A. Ranghetti, R. Ceravolo, G. Palermo, A. Francesconi, A. Ciammola, N. Ticozzi, E. Manfroi, G. Cossu, L. Magistrelli, C. Comi, E. Contaldi, R. Cantello, R. Eleopra, R. Erro, P. Barone, M. Picillo, C. Sorrentino, V. Fetoni, C. Ledda., M.C. Malaguti, M. Pellegrini, C. Longo, O. Donatella, R. Di Giacopo, A. Pilotto, A. Padovani, A. Luppini, A. Imarisio, M.C. Sensi, G. Carroli, A. Braccia, C. Sorbera, G. Di Lorenzo, A. Brigandì, P. Nigro, F. Tamma, E. Caputo, P.V. Mancino, S. Tagliente, A. Tessitore, R. De Micco, S. Aramini, M. Tinazzi, I. Divico, S. Ottaviani, P. Tocco, F. Di Blasio, A. D'Andreagiovanni, F. Valzania, F. Cavallieri, G. Toschi, V. Fioravanti, M.A. Volontè, L. Cacciaguerra, S. Galantucci, G. Di Napoli, D. Volpe, M. Zappia, E.M. Valente.
Supporting information
Table S1. Distribution of GBA1, LRRK2 and PARK2 mutations in fPD and sPD with positive results in genetics analysis.
Acknowledgments
We thank the Associazione Centro Dino Ferrari and the Fresco Institute for their support and the Italian Study Group on Family History in PD for their collaboration and commitment in data collection. Open access funding provided by BIBLIOSAN.
Italian Study Group on Family History in PD:
M. Picillo (Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana,” Neuroscience Section, University of Salerno, Baronissi [SA], Italy)
C. Sorrentino (Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana,” Neuroscience Section, University of Salerno, Baronissi [SA], Italy)
P.V. Mancino (Department of Neurology, “F. Miulli” General Hospital, Acquaviva delle Fonti, Italy)
S. Tagliente (Department of Neurology, “F. Miulli” General Hospital, Acquaviva delle Fonti, Italy)
S. Galantucci (Department of Neurology, IRCCS San Raffaele Scientific Institute, Milan, Italy)
G. Di Napoli (Department of Neurology, IRCCS San Raffaele Scientific Institute, Milan, Italy)
A. Luppini (Neurology Unit, Department of Clinical and Experimental Sciences, University of Brescia, and Department of Continuity of Care and Frailty, ASST Spedali Civili Brescia Hospital, Italy Brescia, Italy)
A. Imarisio (Neurology Unit, Department of Clinical and Experimental Sciences, University of Brescia, and Department of Continuity of Care and Frailty, ASST Spedali Civili Brescia Hospital, Italy Brescia, Italy)
E. Contaldi (Department of Translational Medicine, Section of Neurology, University of Piemonte Orientale, Novara, Italy)
R. Cantello (Department of Translational Medicine, Section of Neurology, University of Piemonte Orientale, Novara, Italy)
G. Toschi (Neurology Unit, Neuromotor and Rehabilitation Department, Azienda USL‐IRCCS di Reggio Emilia, Reggio Emilia 42124, Italy)
V. Fioravanti (Neurology Unit, Neuromotor and Rehabilitation Department, Azienda USL‐IRCCS di Reggio Emilia, Reggio Emilia 42124, Italy)
F. Di Blasio (IRCCS Ospedale Policlinico San Martino—UOC Genetica Medica, Largo R. Benzi 10, 16132 Genova, Italy)
T. Benzi Markushi (Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal and Child Health, University of Genoa, Largo P. Daneo 3, 16132 Genova, Italy)
A. Braccia (Department of Neuroscience and Rehabilitation, Azienda Ospedaliera‐Universitaria S. Anna, Ferrara)
A. Rini (Neurological Department, A. Perrino's Hospital, Brindisi, Italy)
P. De Marco (Neurological Department, A. Perrino's Hospital, Brindisi, Italy)
S. Aramini (Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”)
A. Francesconi (Center for Neurodegenerative Diseases—Parkinson's Disease and Movement Disorders, Unit of Neurology, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy)
M. Pellegrini (Neurology Unit, Trento Hospital, Azienda Provinciale per i Servizi Sanitari [APSS] di Trento, Trento 38122, Italy)
C. Longo (Neurology Unit, Trento Hospital, Azienda Provinciale per i Servizi Sanitari [APSS] di Trento, Trento 38122, Italy)
M. Zibetti (Department of Neuroscience “Rita Levi Montalcini,” University of Turin, Via Cherasco 15, 10126 Turin, Italy; Neurology 2 Unit, A.O.U. Città della Salute e della Scienza di Torino, Corso Bramante 88, 10126 Turin, Italy)
C. Ledda (Department of Neuroscience “Rita Levi Montalcini,” University of Turin, Via Cherasco 15, 10126 Turin, Italy; Neurology 2 Unit, A.O.U. Città della Salute e della Scienza di Torino, Corso Bramante 88, 10126 Turin, Italy)
F. Di Blasio (Neurology and Stroke Unit, Pescara Hospital, Pescara, Italy)
A. D'Andreagiovanni (Neurology and Stroke Unit, Pescara Hospital, Pescara, Italy)
G. Di Lorenzo (Neurorehabilitation Unit IRCCS Centro Neurolesi “Bonino Pulejo,” Messina, Italy)
A. Brigandì (Neurorehabilitation Unit IRCCS Centro Neurolesi “Bonino Pulejo,” Messina, Italy)
I. Divico (Department of Neurosciences, Biomedicine and Movement Sciences, University of Verona, Verona, Italy)
S. Ottaviani (Department of Neurosciences, Biomedicine and Movement Sciences, University of Verona, Verona, Italy)
N. Ticozzi (Department of Neurology and Laboratory of Neuroscience, Istituto Auxologico Italiano IRCCS, Milan, Italy)
R. Di Giacopo (Neurology Unit, Rovereto Hospital, Azienda Provinciale per i Servizi Sanitari [APSS] di Trento, Trento, Italy)
A. Ranghetti (Department of Parkinson's Disease, Movement Disorders and Brain Injury Rehabilitation, Moriggia Pelascini Hospital, Gravedona, Italy)
E. Manfroi (Department of Neurology, Santa Maria University Hospital, Terni, Italy)
P. Nigro (Movement Disorders Center, Neurology Department, Perugia General Hospital and University of Perugia, Perugia, Italy)
References
- 1. Kieburtz K, Wunderle KB. Parkinson's disease: evidence for environmental risk factors. Mov Disord 2013;28(1):8–13. 10.1002/mds.25150. [DOI] [PubMed] [Google Scholar]
- 2. Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: Progress and therapeutic implications. Mov Disord 2013;28(1):14–23. 10.1002/mds.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cm T, Sm G. Epidemiology of Parkinson's disease. Neurol Clin 1996;14:2–335. 10.1016/S0733-8619(05)70259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30(12):1591–1601. 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 5. Marder K, Tang MX, Mejia H, et al. Risk of Parkinson's disease among first‐degree relatives: a community‐based study. Neurology 1996;47(1):155–160. 10.1212/wnl.47.1.155. [DOI] [PubMed] [Google Scholar]
- 6. Bonifati V, Fabrizio E, Vanacore N, De Mari M, Meco G. Familial Parkinson's disease: a clinical genetic analysis. Can J Neurol Sci 1995;22(4):272–279. 10.1017/s0317167100039469. [DOI] [PubMed] [Google Scholar]
- 7. de Lau LML, Breteler MMB. Epidemiology of Parkinson's disease. Lancet Neurol 2006;5(6):525–535. 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 8. Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol 2020;27(1):27–42. 10.1111/ene.14108. [DOI] [PubMed] [Google Scholar]
- 9. Papapetropoulos S, Adi N, Ellul J, Argyriou AA, Chroni E. A prospective study of familial versus sporadic Parkinson's disease. Neurodegener Dis 2007;4(6):424–427. 10.1159/000107702. [DOI] [PubMed] [Google Scholar]
- 10. Vibha D, Sureshbabu S, Shukla G, Goyal V, Srivastava AK, Singh S, Behari M. Differences between familial and sporadic Parkinson's disease. Parkinsonism Relat Disord 2010;16(7):486–487. 10.1016/j.parkreldis.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 11. Pagano G, Ferrara N, Brooks DJ, Pavese N. Age at onset and Parkinson disease phenotype. Neurology 2016;86(15):1400–1407. 10.1212/WNL.0000000000002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baba Y, Markopoulou K, Putzke JD, Whaley NR, Farrer MJ, Wszolek ZK, Uitti RJ. Phenotypic commonalities in familial and sporadic Parkinson disease. Arch Neurol 2006;63(4):579–583. 10.1001/archneur.63.4.579. [DOI] [PubMed] [Google Scholar]
- 13. Doty RL. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol 2012;8(6):329–339. 10.1038/nrneurol.2012.80. [DOI] [PubMed] [Google Scholar]
- 14. Geraghty JJ, Jankovic J, Zetusky WJ. Association between essential tremor and Parkinson's disease. Ann Neurol 1985;17(4):329–333. 10.1002/ana.410170404. [DOI] [PubMed] [Google Scholar]
- 15. Jiménez‐Jiménez FJ, Alonso‐Navarro H, García‐Martín E, Agúndez JAG. The relationship between Parkinson's disease and essential tremor: review of clinical, epidemiologic, genetic, neuroimaging and neuropathological data, and data on the presence of cardinal signs of parkinsonism in essential tremor. Tremor Hyperkinetic Mov 2012;2:tre‐02‐75‐409‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shiner T, Mirelman A, Rosenblum Y, et al. The effect of GBA mutations and APOE polymorphisms on dementia with Lewy bodies in Ashkenazi Jews. J Alzheimers Dis 2021;80(3):1221–1229. 10.3233/JAD-201295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsuang D, Leverenz JB, Lopez OL, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 2012;79(19):1944–1950. 10.1212/WNL.0b013e3182735e9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bourinaris T, Houlden H. C9orf72 and its relevance in parkinsonism and movement disorders: a comprehensive review of the literature. Mov Disord Clin Pract 2018;5(6):575–585. 10.1002/mdc3.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tobin JE, Latourelle JC, Lew MF, et al. Haplotypes and gene expression implicate the MAPT region for Parkinson disease. Neurology 2008;71(1):28–34. 10.1212/01.wnl.0000304051.01650.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang Y, Bagyinszky E, An SSA. Presenilin‐1 (PSEN1) mutations: clinical phenotypes beyond Alzheimer's disease. Int J Mol Sci 2023;24(9):8417. 10.3390/ijms24098417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Girolamo G, Polidori G, Morosini P, et al. Prevalence of common mental disorders in Italy: results from the European study of the epidemiology of mental disorders (ESEMeD). Soc Psychiatry Psychiatr Epidemiol 2006;41(11):853–861. 10.1007/s00127-006-0097-4. [DOI] [PubMed] [Google Scholar]
- 22. Report_Salute_mentale.Pdf. Accessed December 12, 2021. https://www.istat.it/it/files/2018/07/Report_Salute_mentale.pdf.
- 23. Pontone GM, Koch G. An association between bipolar disorder and Parkinson disease: when mood makes you move. Neurology 2019;92(24):1125–1126. 10.1212/WNL.0000000000007641. [DOI] [PubMed] [Google Scholar]
- 24. Risk of Developing Parkinson Disease in Bipolar Disorder: A Systematic Review and Meta‐analysis | Bipolar and Related Disorders | JAMA Neurology | JAMA Network. Accessed January 10, 2023. https://jamanetwork.com/journals/jamaneurology/article-abstract/2752486. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Distribution of GBA1, LRRK2 and PARK2 mutations in fPD and sPD with positive results in genetics analysis.