

Abstract
Introduction and importance:
Gaucher disease is a rare autosomal recessive lysosomal storage disorder marked by a substantial reduction in beta-glucocerebrosidase activity. Historically, supportive treatments such as splenectomy and orthopedic interventions were employed, whereas recent advances have led to the approval of Enzyme Replacement Therapy (ERT) and Substrate Reduction Therapy (SRT) as therapeutic options.
Case presentation:
The authors present the case of a 61-year-old female with chronic abdominal pain, abdominal fullness, pancytopenia, and hepatosplenomegaly, all indicative of Gaucher’s disease, later confirmed by histopathological examination. The patient was informed about newer treatment options like ERT and SRT, as well as the traditional approach of splenectomy. However, due to financial constraints, she opted for splenectomy in conjunction with conservative management.
Discussion:
Gaucher disease is defined by a deficiency of glucocerebrosidase, leading to the accumulation of Gaucher cells (pathognomonic of the disease), particularly in the spleen, liver, bone marrow, and lungs. Type 1 Gaucher disease (GD1) can manifest at any age, from childhood to late adulthood. Definitive diagnosis is confirmed by reduced beta-glucocerebrosidase activity. Traditionally, treatment options for GD1 have been supportive, including splenectomy, blood transfusions, and orthopedic procedures. However, SRT and ERT, though effective, remain prohibitively expensive and often inaccessible in low-resource settings.
Conclusion:
Early diagnosis of Gaucher disease is challenging due to its rarity and should be considered in patients presenting with hepatosplenomegaly, pancytopenia, and low glucocerebrosidase activity.
Keywords: case report, Gaucher disease, lysosomal storage disorder, Nepal, splenectomy
Introduction
Highlights
Gaucher disease is a rare lysosomal disorder of autosomal recessive inheritance.
Substrate infiltration in the macrophage-monocyte system presents a characteristic ‘crumpled tissue paper’ appearance.
Splenectomy remains an alternative approach to substrate reduction therapy and enzyme replacement therapy.
Gaucher disease (GD, OMIM #230800, ORPHA355) is a rare autosomal recessive lysosomal storage disorder with a birth incidence of ~0.39–5.38 per 100 000 general population, but as high as 1 per 850 among the Ashkenazi Jewish population1–3. It is the most frequent multisystemic sphingolipidosis involving the liver, spleen, bone marrow, as well as lymph nodes. Gaucher disease is characterized by a significant reduction in the activity of the lysosomal enzyme, glucocerebrosidase, which hydrolyzes glucosylceramide (GlcCer) into ceramide and glucose due to mutations in the GBA1 gene located on chromosome 1(1q21). Consequently, substrate infiltration occurs in the cytoplasm of macrophage-monocyte system cells, such as those in the liver, spleen, bone marrow, and lymph nodes, presenting a characteristic ‘crumpled tissue paper’ appearance1,2.
GD exhibits variable phenotypes and is clinically classified into three subtypes based on the presence or absence of neurological involvement; (i) Type 1- non-neuropathic form (ii) Type 2- acute neuropathic form; infantile-onset (iii) Type 3- a neuronopathic form of juvenile-onset4. Previously, splenectomy and orthopedic intervention were the supportive treatments for GD1. However, recent advances have led to the approval of Enzyme Replacement Therapy (ERT) and Substrate Reduction Therapy (SRT) for therapeutic management in both children and adults, significantly altering the natural history of the disease5.
We present the case of an elderly female with chronic abdominal pain, abdominal fullness, pancytopenia, and hepatosplenomegaly, along with decreased beta-glucocerebrosidase activity, diagnosed as Gaucher’s disease, and later confirmed through histopathological examination. This case report has been reported in line with the SCARE Criteria.
Case presentation
A 61-year-old woman from the hilly region of Nepal, a farmer by occupation with type 2 diabetes mellitus under medication, presented to our center with complaints of unbearable pain in her left abdomen, along with a history of chronic right upper quadrant abdominal pain. The pain started 6 months prior and gradually worsened, with no identifiable aggravating or relieving factor, scoring 5/10 on the universal pain scale. The patient reported decreased appetite and a progressively enlarging mass in the left upper quadrant. She visited a tertiary medical care center, where blood tests revealed pancytopenia. She was, then, referred to the surgery department for a suspected surgical abdomen, imaging revealed a massive spleen. Consultation with the interventional radiology team considered the possibility of embolizing the splenic artery to prevent further enlargement due to abnormal scavenging of blood cells by the spleen. The patient had a history of hysterectomy 1 year prior. There is no history of recurrent infection, recurrent history of blood transfusion, bone pain, petechial rashes, or recent travel history to the endemic region. There were no similar or other significant illnesses in the past or in the family. She did not smoke or drink alcohol. Her menstrual history was normal.
On physical examination, all vitals were within normal limits, and the general examination revealed pallor; however, there was no koilonychia, flapping tremors, or jaundice. Abdominal examination revealed a palpable spleen just below the left subcostal margin and mild hepatomegaly. Similarly, neurological examination and other systemic findings were unremarkable. Her laboratory investigations (Table 1) revealed leukopenia, anemia, thrombocytopenia, and elevated LDH levels. Likewise, serology for HIV, syphilis, and hepatitis B and C were negative. Also, the test for sputum acid-fast smear was negative with no growth of organisms on the sputum culture. Similarly, stool routine and microscopy showed normal gastrointestinal flora.
Table 1.
Laboratory investigation profile of the patient
| Parameters | Units | Reference normal range | Reported |
|---|---|---|---|
| Lactate dehydrogenase (LDH), serum | U/l | 120–246 | 312.00 |
| Total protein, serum | g/dl | 6.3–8.2 | 7.60 |
| Albumin, serum | g/dl | 3.5–5.0 | 4.40 |
| Alanine amino transferase (ALT) | U/l | <35 | 14 |
| Aspartate amino transferase (AST/SGOT) | U/l | 14–36 | 25 |
| Bilirubin (Total), serum | mg/dl | 0.2–1.3 | 1.30 |
| Bilirubin conjugated | mg/dl | 0.0–0.3 | 0.2 |
| Bilirubin unconjugated | mg/dl | 0–1.1 | 1.1 |
| Alkaline phosphatase | U/l | 38–126 | 89 |
| Serum urea | mg/dl | 15–45 | 46 |
| Serum creatinine | mg/dl | 0.52–1.04 | 0.6 |
| Serum sodium | mmol/l | 137–145 | 143 |
| Total leukocyte count | cells/cumm | 4000–11 000 | 3220 |
| Red blood cells | 106 cells/cumm | 4.5–5.5 | 4.51 |
| Packed cell volume (PCV) | % | 40–50 | 32.9 |
| Hemoglobin | gm% | 11.9–14.6 | 10.8 |
| Platelet count | 103 cells/cumm | 150–450 | 146 |
Computed tomography (CT) of the abdomen and pelvis revealed massive splenomegaly and mild hepatomegaly, along with hypertrophied splenic arteries. A contrast-enhanced CT of the abdomen showed a dilated main portal vein, splenic vein, and superior mesenteric vein; a few enlarged periportal and celiac lymph nodes, multiple sub-centimeter aortocaval and left para-aortic lymph nodes; and mildly calcified plaques in the abdominal aorta and bilateral common iliac arteries (Fig. 1).
Figure 1.
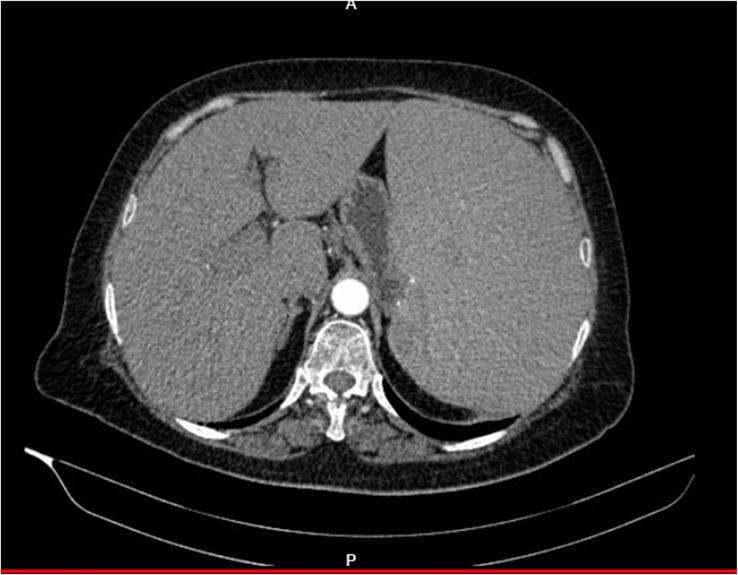
Contrast-enhanced computed tomography showing grossly enlarged liver and spleen.
Considering the rare pathology, we decided to measure beta-glucocerebrosidase activity, which was found to be low at 0.82 nmol/h/mg protein (normal: 8.7 nmol/h/mg protein). Thus, in the background of the absence of neurological components, clinical spectrum, and laboratory findings, Gaucher type I was diagnosed. The patient was informed about the newer treatment modalities such as ERT and SRT, along with the traditional treatment option of splenectomy, for managing her condition and its potential complications if left untreated. Due to the patient’s financial constraints, she opted for splenectomy along with conservative treatment modalities.
Preoperatively vaccinations for Pneumococci, Meningococcal, and H influenza were provided. It was then decided to undergo a splenectomy with biopsy by a multidisciplinary team. The gross specimen of the spleen measured 23×22×11 cm. The spleen was markedly enlarged, and the capsule was intact. The cross-section showed areas of congestion. Likewise, the microscopy report of the specimen revealed autolytic and ischemic changes along with the marked expansion of the red pulp with a large number of histiocytic cells with finely fibrillary cytoplasm, particularly in the splenic cords. The white pulp was intact and preserved (Fig. 2). Then differential diagnosis was discussed again with the multidisciplinary team, and the team of pathologists decided it was consistent with Gaucher’s disease, type 1, with ischemic changes. Postoperatively, recovery was uneventful, and there were no complications during follow-up at 2 and 4 weeks.
Figure 2.

Histopathological images of the spleen showing a large number of histiocytic cells with finely fibrillary cytoplasm suggestive of Gaucher’s disease.
Discussion
Gaucher disease is a rare autosomal recessive lysosomal storage disorder, characterized by a deficiency of glucocerebrosidase (a lysosomal enzyme), which breaks down glucosylceramide into glucose and ceramide6,7. Deficiency of glucocerebrosidase enzyme results in the accumulation of glycolipids in macrophages (Gaucher cells), especially in the spleen, liver, bone marrow, and lungs6. The Gaucher cell is the pathognomonic feature of Gaucher disease, which was first recognized in 1882 by CPE Gaucher in a woman with an enlarged spleen6,8,9.
Based on early-onset neurological involvement, Gaucher disease can be classified into neuronopathic Type 1 and non-neuronopathic Types 2 and 38,10. Type 2 is an acute form manifesting in early childhood while type 3 is a subacute form that manifests in adolescence11. Type 1 Gaucher disease is the most common form among the three types, accounting for ~94% of all cases followed by type 3 (5%) and type 2 (1%)8. Here, we report a case of Type 1 Gaucher disease.
In the Gaucher registry, the mean age of diagnosis was 17.4 years, the majority (49%) receiving a diagnosis before 10 years of age, and Jews were the most commonly affected ethnic group12. In contrast to the data from the Gaucher registry, our case involved a 61-year-old Asian woman, indicating that Gaucher disease can occur at any age regardless of ethnicity as supported by a case of Gaucher disease reported in a 75-year-old Korean woman7. Type 1 GD can present at any age from childhood to old age and can have near-normal to normally expected survival13.
It can be asymptomatic in mild cases. Patients can present with massive organomegaly (splenomegaly more common than hepatomegaly), cytopenia, and bone lesions6,8,10. Symptomatic splenomegaly with associated pain, early satiety, and cytopenia can be the presenting features of Gaucher disease13, as observed in our case. Splenomegaly and hepatomegaly, in our case, could be attributed to the infiltration of Gaucher cells in the spleen and liver, while pancytopenia could be due to splenic sequestration or bone marrow infiltration by Gaucher cells, leading to bone marrow failure10. Early satiety and abdominal pain, in our case, could be attributed to splenomegaly13. Skeletal involvement in Gaucher disease can manifest as an acute painful bone crisis (due to infarcts) and chronic pain due to avascular necrosis10,13. However, the bone changes noticed in our case, lumbar spondylosis could also be age-related.
The definitive diagnosis of Gaucher disease is confirmed by reduced beta-glucocerebrosidase activity14. The enzyme level was reduced in our case too. Radiological imaging in Gaucher disease assesses liver and spleen morphology, and effects on bone10. Abnormal liver function tests may indicate cholestasis; however, they are typically normal in Gaucher disease, as observed in our case. The most common hematological abnormality in Gaucher disease is thrombocytopenia, followed by anemia and leukopenia10. Our case also exhibited pancytopenia. Gaucher cells can be identified in bone marrow aspiration, however, it is not done routinely for the diagnosis of GD10.
Previously, the treatment options for GD1 were supportive, including splenectomy, blood transfusions, and orthopedic procedures15,16. However, the approach has now shifted to enzyme replacement therapy (ERT) and substrate reduction therapy (SRT)13,15. ERT functions by hydrolyzing glycolipids accumulated in macrophages, while SRT (miglustat, eliglustat) reduces glycolipid synthesis by inhibiting the glucosylceramide synthase enzyme13,15. Our case was managed with splenectomy since advanced treatments such as ERT and SRT are expensive and unavailable in our setting.
Conclusion
Gaucher’s disease can be overlooked clinically due to its rarity; therefore, it should be considered if a patient presents with hepatosplenomegaly, pancytopenia, and low glucocerebrosidase enzyme activity. Splenectomy remains a viable alternative to substrate reduction therapy and enzyme replacement therapy.
Ethical approval
Not applicable.
Consent
Written informed consent was obtained from the patient for the publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Source of funding
Not applicable.
Author contribution
S.B.1, S.B.2, M.K.: conceptualization, resources, writing – original draft, and writing – review and editing; D.G. and G.A.: writing – review and editing and data curation; N.P. and S.K.: resources, writing – review, and editing; S.S.D,: writing – review and editing and supervision.
Conflicts of interest disclosure
The authors declare no conflicts of interest.
Guarantor
Sujan Bohara.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Published online 11 September 2024
Contributor Information
Sujan Bohara, Email: mjsujan777@gmail.com.
Sanjeet Bhattarai, Email: sanjeetbhattarai@gmail.com.
Manoj Khadka, Email: khadkamanoj432@gmail.com.
Deepak Ghimire, Email: deepakghimire101@gmail.com.
Samikshya Karki, Email: smilyg938@gmail.com.
Nahakul Poudel, Email: naacool999@gmail.com.
Gopi Aryal, Email: gopiaryal1@gmail.com.
Sunil S. Dhakal, Email: samknp63@gmail.com.
References
- 1. Baris HN, Cohen IJ, Mistry PK. Gaucher Disease: The Metabolic Defect, Pathophysiology, Phenotypes, And Natural History. [PMC free article] [PubMed]
- 2. Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation, and treatments. Int J Mol Sci 2017;18:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weinreb NJ, Goker-Alpan O, Kishnani PS, et al. The diagnosis and management of Gaucher disease in pediatric patients: where do we go from here? Mol Genet Metab 2022;136:4–21. [DOI] [PubMed] [Google Scholar]
- 4. Rigante D, Cipolla C, Basile U, et al. Overview of immune abnormalities in lysosomal storage disorders. Immunol Lett 2017;188:79–85. [DOI] [PubMed] [Google Scholar]
- 5. Gary SE, Ryan E, Steward AM, et al. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev Endocrinol Metab 2018;13:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cox TM. Gaucher disease: understanding the molecular pathogenesis of sphingolipidoses. J Inherit Metab Dis 2001;24:107–123. [DOI] [PubMed] [Google Scholar]
- 7. Han M, Byun JM, Koh Y, et al. Gaucher disease type 1: unexpected diagnosis in a 75-year old patient presenting with splenomegaly. Curr Probl Cancer 2021;45:100708. [DOI] [PubMed] [Google Scholar]
- 8. Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Crit Rev Oncog 2013;18:163–175. [DOI] [PubMed] [Google Scholar]
- 9. Burns GF, Cawley JC, Flemans RJ, et al. Surface marker and other characteristics of Gaucher’s cells. J Clin Pathol 1977;30:981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation, and treatments. Int J Mol Sci 2017;18:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dandana A, Ben Khelifa S, Chahed H, et al. Gaucher disease: clinical, biological and therapeutic aspects. Pathobiology 2016;83:13–23. [DOI] [PubMed] [Google Scholar]
- 12. Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 2000;160:2835–2843. [DOI] [PubMed] [Google Scholar]
- 13. Thomas AS, Mehta A, Hughes DA. Gaucher disease: hematological presentations and complications. Br J Haematol 2014;165:427–440. [DOI] [PubMed] [Google Scholar]
- 14. Charrow J, Esplin JA, Gribble TJ, et al. Gaucher disease: recommendations on diagnosis, evaluation, and monitoring. Arch Intern Med 1998;158:1754–1760. [DOI] [PubMed] [Google Scholar]
- 15. Gary SE, Ryan E, Steward AM, et al. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev Endocrinol Metabol 2018;13:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: an expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis 2018;68:203–208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.