

Abstract
Maternal mRNAs and proteins are produced during oogenesis by more than 60% of zebrafish genes. They are indispensable for fertilization and early embryogenesis. Generation and analysis of the maternal mutant is the most direct way to characterize the maternal function of the specific gene. However, due to the lethality of zygotic mutants, the maternal function of most genes in zebrafish remains elusive. Several methods have been developed to circumvent this obstacle, including mRNA rescue, germ-line replacement, oocyte microinjection in situ, mosaic mutation, and bacterial artificial chromosome (BAC)-mediated conditional rescue. Here, we provide an alternative approach to generate zebrafish maternal mutants rapidly and efficiently by introducing four tandem sgRNA expression cassettes into Tg(zpc:zcas9) embryos. This method is more technically feasible and cost- and time-effective than other established methods.
Key features
• This protocol can circumvent the lethality or infertility of the zygotic mutants to obtain maternal mutants of the target gene.
• This protocol is time-saving (one fish generation).
• Using this protocol, double-gene maternal mutants can be obtained in a single generation.
• Stable lines can be established to continuously produce maternal mutant embryos for the gene of interest.
Keywords: Zebrafish, Maternal mutant, Conditional knockout, Oocyte, Cas9, sgRNA
Graphical overview

Background
Maternal factors are mRNAs and proteins deposited in the oocyte. They play vital roles in oocyte maturation, fertilization, and blastocyst development. Approximately 66% of zebrafish genes are expressed maternally [1]. As zygotic genome activation (ZGA) starts at around the 1k-cell stage, maternal factors are dominant to function in the developmental events before this time point. In addition, even after zygotic genome activation (ZGA), maternal factors still function in later embryogenesis, including axis formation, germ layers differentiation, morphogenesis, etc. [2–4].
Generation of the corresponding maternal mutant is the most straightforward way to address the maternal function of a gene in zebrafish. The genotype of primary oocyte is the same as that of somatic cells. Hence, viable and fertile female zygotic mutants are indispensable for giving birth to maternal mutants or maternal and zygotic mutants. However, zygotic mutants usually cannot survive to adulthood or spawn because of the crucial and uncompensable functions of the mutated genes [5–7]. This obstacle represents the major bottleneck to studying maternal factors. Several methods have been developed to bypass this technical hurdle. The first is mRNA rescue. Through microinjection of in vitro–synthesized wild-type mRNA, the zygotic mutant can be rescued to live through a critical period, in which the zygotic products contribute to normal development [4]. Considering that the ectopic expression may disrupt normal embryogenesis and transient expression of mRNA cannot support long-term gene function, this method is not applicable to most genes. In 2002, Ciruna et al. developed the germ-line replacement technology. They injected dead end1 morpholino into wild-type embryos to block primordial germ cell development and then transplanted germ cells from zygotic mutants into these primordial germ cell (PGC)-free hosts. This ensured that the germ lines of the host were entirely replaced by donor PGCs. Upon maturation, all the embryos laid by the female chimera were maternal mutants [8]. Transplantation of PGCs is technically demanding and the chimeric embryos tend to develop into males because of the limited number of transplanted PGCs. Wu et al. established a surgery-based method [9]. Following the opening of the female fish abdomen, the fluorescent lineage tracer and the morpholino targeting the specific gene were co-injected into the stage- oocyte. After recovery, the laid fluorescent embryos were the desired maternal morphants [9]. However, the intricate operative skills required for the application of this method are significantly challenging. In 2018, Xing et al. generated mosaic mutants containing homozygous mutant cells through secondary genome editing in heterozygotes [2]. This approach was designed to circumvent the lethality associated with double zygotic mutants of dvl2 and dvl3a. Some of these homozygous mutant cells were incorporated into the germline, eventually resulting in maternal mutants after fertilization. However, this method is inefficient and time-consuming, requiring at least three generations. In addition to the methods mentioned above, the bacterial artificial chromosome-rescue-based knockout (BACK) is another method to bypass the zygotic lethality and obtain maternal mutants [10]. To obtain such mutants, first, the authors generated mutants of the target gene through nuclease-mediated genome editing. Second, they rescued the zygotic mutants by introducing a bacterial artificial chromosome (BAC) containing the target gene expression cassette flanked by loxP sites via Tol2-mediated transgenesis. Third, they crossed these fish with a germline-specific Cre line. This approach ultimately allowed them to obtain maternal mutants of the target gene. However, this strategy is time-consuming and labor-intensive, as it involves gene knock-out, transgenesis, and crossing with the Cre line.
Recently, we have developed a new strategy to generate single or double-gene maternal mutants through transgenic expression of Cas9 and sgRNA in oocytes [11,12]. Through I-Sce I-mediated transgenesis, we introduced an eGFP reporter and multiple tandem sgRNA expression cassettes into Tg(zpc:zcas9) transgenic fish, where Cas9 is specifically expressed in oocytes. This approach enables genome editing during the early stages of oogenesis. As a result, some of the GFP-positive embryos will have their maternal products completely ablated, thereby becoming maternal mutants. Consequently, we can analyze their phenotypes after identifying them through genotyping. Compared to other methods, it has several advantages. First, it is technically feasible. The main techniques are plasmid construction and transgenesis, which are conventional in most zebrafish labs. Second, it is time saving, taking one generation (2–3 months) for either single or double-genes maternal knockout. Third, the efficiency is generally stable despite individual variation. Through generating maternal mutants of nanog, ctnnb2, rbm24a, dvl2, and dvl3a, we find that the average ratio of the maternal mutant for a single gene is approximately 25% and the maximum efficiency can reach 63.3%. For every single founder fish, the ratio of maternal mutants remains stable in each spawning, so that we can obtain maternal mutants repeatedly once we get a founder. Finally, this approach can be utilized to generate double-gene maternal mutants, which takes nearly the same time.
Materials and reagents
Biological materials
Zebrafish lines:
Wild-type AB zebrafish line was obtained from the China zebrafish resource center
Tg(zpc:zcas9) transgenic line was reported by Ming Shao’s lab and is available upon request [13]
Reagents
NaCl (Solarbio, catalog number: S8210)
KCl (Solarbio, catalog number: P9921)
CaCl2·2H2O (Solarbio, catalog number: C8370)
HEPES (Sigma-Aldrich, catalog number: H3375)
2× Taq Master Mix (Dye Plus) (Vazyme, catalog number: P112-01)
AxyPrep PCR Clean-Up Kit (Axygen, catalog number: AP-PCR-250)
T7 RNA polymerase, 5× transcription buffer (Thermo Scientific, catalog number: EP0111)
ATP/CTP/GTP/UTP, 10 mM each (Thermo Scientific, catalog number: R0481ribo); dilute the 100 mM solution with DNase/RNase-free H2O
RiboLock RNase inhibitor (Thermo Scientific, catalog number: EO0381)
TURBO DNase (Invitrogen, catalog number: AM2238)
Ammonium acetate, 5 M, RNase-free (Thermo Scientific, catalog number: AM9070G)
Phenol:chloroform:isoamyl alcohol 25:24:1, pH 5.2 (Thermo Scientific, catalog number: J62336.AE)
Cas9 protein: GenCrispr NLS-Cas9-NLS nuclease (GenScript, catalog number: Z03389-50)
AxyPrep Plasmid Miniprep Kit (Axygen, catalog number: AP-MN-P-250)
Acc65I (Asp718I) (Thermo Scientific, catalog number: ER0901)
Gibson assembly kit: Hieff Clone Plus One Step Cloning Kit (Yeasen, catalog number: 10911ES20)
CutSmart buffer (NEB, catalog number: B60004)
Agarose (BIOWEST, catalog number: 111860)
Glass capillaries (World Precision Instruments, catalog number: TWF-100F-4)
100× penicillin-streptomycin solution (Gibco, catalog number: 15140122)
cDNA synthesis kit (Transgene, catalog number: AT301)
pBackZero-T vector (Takara, catalog number: 3275)
-
Primers:
Universal primer:
5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGC
TATTTCTAGCTCTAAAAC-3′
Dest forward primer: 5′-TTCTTGTTTAAGCTTTTAATCTCAAAAAAC-3′
Dest reverse primer. 5′-GGCTGTTTACATCTGATAGTGG-3′
Ligation forward primer: 5′-gagtcggtgctttttttaaacctggTTCTTGTTTAAGCTTTTAATCTCAAAAAAC-3′
Ligation reverse primer: 5′-ATCCTGCACTGAATGCAC-3′
bmp2b sgRNA forward primer: 5′- taatacgactcactataGGGAGGCTGAGAGCAACCGGgttttagagctagaa-3′
M13 forward: 5′-GTAAAACGACGGCCAGT-3′
The plasmid system is available upon request to the authors Chong Zhang and Ming Shao
TRIzol (Thermo Scientific, catalog number: 15596026)
PBST (Solarbio, catalog number: P1031)
Chloroform (Sinopharm Chemical Reagent, catalog number: 10006862)
Isopropanol (Sinopharm Chemical Reagent, catalog number: 40064360)
Glycogen (Thermo Scientific, catalog number: R0561)
Solutions
Ringer's buffer (see Recipes)
Recipes
-
Ringer's buffer
Reagent Final concentration Quantity NaCl 116 mM 6.779 g KCl 2.9 mM 0.216 g CaCl2·2H2O 1.8 mM 0.265 g HEPES 5 mM 1.192 g H2O n/a 1,000 mL Total n/a 1,000 mL Dissolve all ingredients in 900 mL of deionized H2O. Adjust pH to 7.2 using NaOH. After being autoclaved, it can be long-term stored at room temperature.
Equipment
VeritiTM Dx 96-well thermal cycler (Thermo Fisher, catalog number: 4452300)
NanoDrop 2000 spectrophotometer (Thermo Scientific, model: ND-2000)
Block heater (Yiheng, model: TU-100C)
Electrophoresis system (Bio-Rad, catalog number: 1640302)
Centrifuge (Eppendorf, model: 5424R)
Pico-injector (Warner Instrument, model: PLI-100A)
Puller (NARISHIGE, model: PC-100)
Tweezer (WPI, catalog number: 500341)
Microforge (NARISHIGE, model: MF2)
Microinjector (Harvard Apparatus, model: PLI-100A)
Software and datasets
CRISPRScan, http://www.crisprscan.org/, for designing sgRNAs with high on-target activity and minimal off-target effects
Synthego, https://ice.synthego.com/#/, for analyzing sequencing results and assessing CRISPR editing efficiency
Procedure
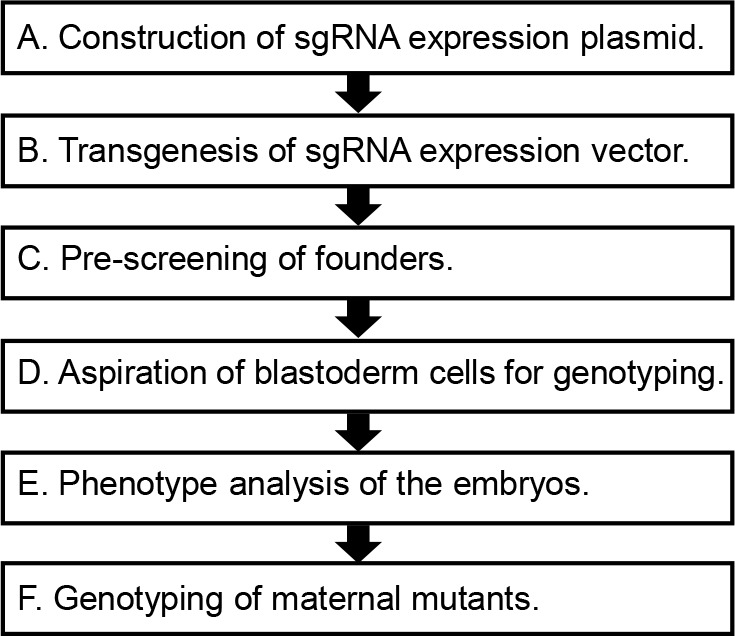
The summary of this protocol is illustrated in Figure 1.
Figure 1. Main steps of this protocol.
-
Construction of sgRNA expression plasmid
-
Prediction of sgRNA targeting sites.
For single-gene maternal knockout, we need 3–4 sgRNAs targeting different sites with high genome editing efficiency. To achieve this, we predict at least six different sgRNA targeting sites using the online CRISPRscan software (http://www.crisprscan.org/) [14]. After submitting the gene name, the website will predict several sgRNAs (Figure 2). The CRISPRscan model was designed and tested on zebrafish [15], and the authors recommend the following rules for selecting appropriate sgRNAs:
1) High CRISPRscan score: The CRISPRscan score represents the potential activity of the sgRNA. A score of at least 55 is required, with a score above 70 being recommended.
2) Low CFD (cutting frequency determination) score: The CFD score indicates the potential off-target cutting efficiency, so a lower CFD score is preferable.
In addition to these criteria, we also recommend the following rules for achieving higher knockout efficiency:
1) Avoid overlaps of selected sgRNA-targeting sites: Independent targeting sites increase the probability of successful gene disruption.
2) Select sites close to the start codon ATG: Targeting the front two-thirds of the coding sequence is recommended, as frameshift mutations closer to ATG will more thoroughly disrupt gene function. The Oligos containing the T7 promoter, the predicted sgRNA sequence, and a 16 nt sgRNA scaffold sequence are synthesized for amplification of the sgRNA IVT DNA template.
-
Amplification and purification of each sgRNA DNA template.
Prepare the Fill-in PCR mixture on ice to ensure enzyme stability and activity, as shown in Table 1.
Put the mixture into the thermocycler and run the program as shown in Table 2.
Take 2 μL of PCR products and subject them to electrophoresis; the fragment size should be 117 bp.
Purify the rest of the PCR products with the AxyPrep PCR Clean-Up Kit. Follow the manual book and elute the DNA with 20 μL of deionized H2O.
Measure the concentration of the purified DNA template using a NanoDrop 2000 spectrophotometer. Ensure that the DNA concentration is within the optimal range for subsequent steps and store the DNA at -20 °C until use.
Note: To meet the requirement of the following transcription, scale up the PCR mixture to make the final concentration of purified DNA higher than 40 ng/μL.
-
In vitro transcription of sgRNA.
Thaw frozen reagents, mix thoroughly, and centrifuge briefly. Place the reaction buffer at room temperature and other components on ice.
Set up the in vitro transcription reaction mixture on ice to maintain enzyme activity and prevent degradation, as shown in Table 3.
Incubate at 37 °C for 2 h.
To remove the DNA template, add 1 μL of TURBO DNase (2 U/μL). Mix, briefly centrifuge, and incubate at 37 °C for 20 min.
Add 10 μL of ammonium acetate (5 M, RNase-free) and vortex to stop the reaction.
-
sgRNA purification and precipitation.
Add 70 μL of RNase-free H2O and mix thoroughly. Then, add an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1, pH 5.2). Vortex the mixture briefly and centrifuge at 12,000× g for 30 s to ensure proper mixing and pellet any debris. Transfer the aqueous phase to a new tube.
Add an equal volume of chloroform (100 μL). Vortex for 5 s and centrifuge at 12,000× g for 30 s. Transfer the aqueous phase to a new tube.
Add an equal volume of isopropanol (100 μL) and mix well.
Chill the mixture for at least 30 min at -20 °C or 5 min at -80 °C. Maintaining a lower temperature and extending the incubation time can improve sgRNA precipitation efficiency. Centrifuge at 12,000× g for 15 min at 4 °C to pellet sgRNA. Remove the supernatant liquid carefully (we can preserve some liquid to avoid taking sgRNA incidentally).
Rinse the sgRNA pellet with 1 mL of pre-chilled 70% ethanol. Centrifuge at 12,000× g for 15 min at 4 °C and discard the liquid supernatant.
Dissolve sgRNA with 10 μL of RNase-free H2O. Measure the concentration with NanoDrop 2000 spectrophotometer and dilute sgRNA to 100 ng/μL by adding RNase-free H2O. Aliquot the sgRNA to 1 μL per tube and store at -80 °C.
Note: Scale up the transcription mixture to yield more sgRNA.
-
Test sgRNA efficiency.
Dilute Cas9 protein to 200 ng/μL (add 1 μg/μL stock solution to 4 μL of RNase-free H2O and mix well). It can be stored at -20 °C for at least 30 days.
Mix 1 μL of sgRNA with 1 μL of Cas9 protein by pipetting gently on ice. The final concentrations should be 50 ng/μL for sgRNA and 100 ng/μL for Cas9 protein. Ensure thorough mixing to form sgRNA-Cas9 complexes.
Inject 2 nL mixture into each 1-cell stage embryo.
At 24 h post-fertilization (hpf), collect 10 embryos into a single microcentrifuge tube and carefully remove the medium. Use a pipette to ensure embryos are not damaged during collection. Then, add 100 μL of NaOH (50 mM, 10 μL each embryo) and heat at 95 °C in a metal bath or thermocycler for 15 min. Then, neutralize the lysate with 1/10 volume of Tris pH 8.0 (10 μL, 1 M).
-
Directly use the embryo lysate as the DNA template for PCR amplification of the fragment containing the sgRNA targeting site. Ensure the lysate is thoroughly mixed before use to avoid uneven template distribution.
Note: The flanking sequence of the sgRNA targeting site should be over 100 bp. After electrophoresis to ensure the proper amplification, we subject PCR products to Sanger sequencing, performing gel extraction when needed.
Analyze the sequencing results using the ICE (Inference of CRISPR Edits) web tool at https://ice.synthego.com/#/. Using this tool, we could quantify the sgRNA efficiency [16] to select the highly efficient ones. Upload the sequencing data and follow the tool's instructions for accurate analysis of editing efficiency The sgRNA efficiency will be normalized with uninjected group sequencing data. Choose the sgRNA with an indel percentage ≥ 30% for the following steps (Figure 3).
-
Construction of separate sgRNA expression plasmids (Figure 4).
-
According to the sgRNA sequence, synthesize sgRNA annealing primer through a commercial company.
Forward primer: TTCGN(19)
Reverse primer: AAAC[N(19)]*
GN(19) is the target sequence of sgRNA, starting as GG. Specifically, sgRNA primer given by CRISPRScan:
5′-TAATACGACTCACTATA[GN(19)]GTTTTAGAGCTAGAA-3′
[N(19)]* = Reverse complement of N(19).
For example, TAATACGACTCACTATAGGCGGGAGTGCGTGCAACACGTTTTAGAGCTAGAAATAGC represents the long primer sequence provided in the table, with the target sequence (protospacer) highlighted in bold. Therefore, the forward primer is ttcGGCGGGAGTGCGTGCAACAC, and the reverse primer is aaac GTGTTGCACGCACTCCCGC . The sequence italicized and in bold is the reverse complement of the forward primer sequence underlined.
-
Annealing of primers.
i. Set up the reaction as shown in Table 4.
ii. Put the mixture into a thermocycler and run the program as shown in Table 5.
-
Ligation to U6 vectors.
i. The number of sgRNAs in the final transgenic plasmid determines the choice of U6 vectors [17] and following pGGDestISceIEG-XsgRNA. We list each combination in Table 6.
To achieve high efficiency of gene disruption, four different sgRNAs are recommended. We ligate these four different sgRNAs to pU6a:sgRNA#1, #2, #3, #4, which contain different U6 promoters driving sgRNA expression and sgRNA scaffold. Even if we do not have four different sgRNAs, we can ligate single sgRNA repeatedly into different U6 vectors. Then, we assemble these four U6 vectors containing four different sgRNA oligos into pGGDestISceIEG-4sgRNA.
ii. Ligate sgRNA oligos to corresponding U6 vectors (Figure 4). Set up the ligation reaction as shown in Table 7.
iii. Place the mixture into the thermocycler and run the program as shown in Table 8.
iv. Transformation.
1). Thaw DH5α competent cells on ice. Add the ligation mixture to 50–100 μL of DH5α cells and gently mix by flicking the tube. Avoid vortexing to maintain cell viability.
2). Incubate on ice for 30 min.
3). Heat shock at 42 °C for 60 s.
4). Chill on ice for 2 min.
5). Spread the mixture on a solid medium plate containing 100 μg/mL spectinomycin.
6). Culture at 37 °C for more than 16 h.
v. Colony PCR.
1). Pick a single bacterial colony using a sterile pipette tip and rinse it into 10 μL of deionized H2O. Ensure the colony is fully resuspended for subsequent PCR analysis.
2). Set up the PCR reaction as shown in Table 9.
3). Place the mixture into the thermocycler and run the program as shown in Table 10.
4). Subject PCR products to electrophoresis. The PCR product size of the positive colony is 482 bp.
vi. Bacteria growth and extraction of plasmids.
1). Add the rest of the colony liquid into 3 mL of LB medium containing 100 μg/mL spectinomycin.
2). Shake at 200 rpm and 37 °C for 14 h.
3). Extract plasmids using the AxyPrep Plasmid Miniprep Kit and measure the concentration.
4). Subject plasmids to Sanger sequencing with M13 forward and align with the U6 vector map.
-
-
Construction of pGGDestISceIEG-XsgRNA vector.
According to the number of constructed sgRNA vectors, choose the appropriate pGGDestISceIEG-XsgRNA. Different pGGDestISceIEG-XsgRNA can receive different sets of sgRNA expression cassettes. Figure 5A shows an example of four sgRNA U6 vectors ligation to pGGDestISceIEG-4sgRNA through Golden Gate cloning.
Set up the Golden Gate reaction mixture as shown in Table 11.
Place the mixture into the thermocycler and run the following program (Table 12):
Take out 5 μL of mixture to do transformation. The solid LB medium plate is ampicillin-positive.
Perform colony PCR with Dest forward primer and Dest reverse primer.
Bacteria growth and extraction of plasmids: Subject plasmids to Sanger sequencing using Dest forward primer and Dest reverse primer. Then, align the sequence results with that of the predicted vector.
-
Assembly sgRNA expression cassettes for two genes (Figure 5B).
We have reported that the oocyte-specific conditional knockout strategy can be utilized to obtain double-gene maternal mutants. We just need to insert the sg RNA expression cassette of one gene into another through Gibson assembly. For convenience, we termed the pGGDestISceIEG-XsgRNA of two different genes as pISceI-XsgRNA-gene a and pISceI-XsgRNA-gene b.
Check the sequence of two plasmids to find whether there is an Asp718(Acc65) digesting site. If not, select anyone as the backbone. Here, we assume pISceI-XsgRNA-gene a as the backbone.
-
Amplify the sgRNA expression sequence of pISceI-XsgRNA-gene b.
i. Set up the PCR reaction as shown in Table 13.
ii. Run the following PCR program (Table 14):
iii. After gel visualization and purification with the AxyPrep PCR Clean-Up Kit, measure the concentration and store it at -20 °C.
Linearize pISceI-4sgRNA-1 with Asp718 following the instruction. Perform electrophoresis and gel visualization to confirm sufficient digestion. Then, purify the linearized pISceI-4sgRNA-1 and measure the concentration.
Perform Gibson assembly of purified PCR products and linearized backbone vector using Hieff Clone Plus One Step Cloning Kit.
Perform transformation, colony PCR, bacteria amplification, and plasmids extraction. Next, subject plasmids to Sanger sequencing to ensure the correct ligation. Store plasmids at -20 °C.
-
-
Transgenesis of sgRNA expression vector
Assemble the injection solution on ice as shown in Table 15.
-
Collect embryos from wild-type female mating with Tg(zpczcas9) homozygous male.
Inject the solution immediately after collecting embryos. We puncture into the blastodisc instead of the yolk and inject 2 nL of solution. To ensure efficient transgenesis, only 1-cell stage embryos are used.
After injection, incubate the rest of the injection solution at 37 °C for 30 min to test the I-Sce I activity. Then, subject all solutions to electrophoresis to check whether I-Sce works efficiently (Figure 6). If I-Sce I works well, it should completely cut the plasmids into two fragments at this condition. If we can still find the original band, it means the I-Sce I is not effective. This result can be considered as a quality control for I-Sce I-mediated transgenesis.
At 24 hpf, pick up the embryos with strong and ubiquitous GFP signals. In general, 200 alive embryos will contain 10–20 efficiently transgenic ones. Raise the selected embryos to adulthood.
-
Pre-screening of founders
Due to the individual variation in the transgenic expression of Cas9, we need to perform pre-screening of the founders. To facilitate a rapid assessment, we inject bmp2b sgRNA into the embryos of the founder. This is because bmp2b is crucial for early embryo dorsal-ventral patterning, and its mutant phenotype becomes visible as early as 10 hpf. At 12 hpf, the dorsalized embryos should display a long elliptical shape, while the normal embryos are spherical (Figure 7).
Synthesize bmp2b sgRNA as described above.
Mate Tg(zpc:zcas9;U6X:sgRNAX) female individuals with wild-type males.
Collect embryos and inject 2 nL bmp2b sgRNA (50 ng/μL) per embryo at 1-cell stage.
Observe the embryos at 12 hpf. Quantify the dorsalized embryos (long elliptical shape). If the ratio exceeds 50%, the fish is considered a putative founder.
-
Aspiration of blastoderm cells for genotyping
Zebrafish show regulative development. Hence, we can isolate some cells for examination of maternal mRNA and the rest of the embryo can still develop, so that we can achieve genotyping and phenotype analysis simultaneously. Maternal products originate from multiple duplicated alleles in the oocyte, including the one transmitted. Hence, mutated transmitted alleles could not represent the elimination of wild-type maternal products of the target gene. To identify the maternal mutants, we will examine the maternal mRNAs through RT-PCR.
Add 1.5% agarose in water and boil it to melt. Pour it into the 90 mm Petri dish and put the mold onto the molten agarose. Bend the solidified agarose and remove the mold gently, then the plate is ready to use.
Pull glass capillaries to make tip-closed needles using the puller. To prepare the needles suitable for cell transplantation, we use two light weights, set at Lv1: 60 and Lv2: 90, and select Step2 procedure. Using tweezers, we cut the capillary tips to achieve an opening diameter of 30–40 μm. It is crucial to ensure a clean and precise cut to facilitate effective cell aspiration. Then, make a spike at the tip of the pipette using a microforge. The spike facilitates easier penetration and reduces damage to embryos. The pipette for cell isolation is now ready to use.
Mate putative founder with wild-type fish and collect embryos. Pick up the GFP-positive embryos at 1-cell stage. At 3 hpf, put embryos onto the agarose plate (Figure 8). Reorient the embryos to make the blastomere point to the tip of the capillary. Use a microinjector to aspirate 20–40 cells from the embryo. Carefully control the pressure to avoid damaging the cells [18]. Then, release these cells into 2 μL of deionized H2O at the opening edge of different tubes. Clean the capillary through suction and ejection of deionized H2O three times before each aspiration to prevent cross-contamination and ensure accurate cell collection. Add 200 μL of TRIzol to wash down the cells. The tube can be put on ice temporarily. Put the embryos into 24-well plates separately. To prevent infection, change the medium to 1/3× Ringer’s solution containing 1× penicillin-streptomycin solution to allow the development of the embryos to proper stages for phenotyping. We normally examine 24 embryos at once, which will usually take approximately 1 h. Aspiration of dozens of cells will not be harmful to embryo development. Importantly, zygotic transcription does not commence extensively during this interval, allowing us to specifically examine maternal transcripts in these cells. The cells in TRIzol can be stored at -20 °C until the aberrant phenotype of the corresponding embryos is observed. Subsequently, the genotypes of cells from embryos with or without developmental defects are examined by RT-PCR. However, if no developmental defects are present, cells from all embryos should be genotyped to confirm the presence of maternal mutant embryos.
-
Phenotype analysis of the embryos (Figure 8)
When the embryos develop to the desired stage, maternal mutants may be directly recognized by live imaging. Otherwise, they can be separately fixed with 4% PFA at 4 overnight for in-depth analysis. Wash the samples with 1 mL of PBST three times (10 min for each time), and then subject them to whole-mount in situ hybridization, immunofluorescent staining, or other chemical staining assays. Follow established protocols for each assay to ensure accurate and reproducible results. Optionally, we can dehydrate the embryos by incubating three times in 1 mL of 100% methanol for 5 min each. The dehydrated embryos can be stored at -20 for an extended period. Before the next staining assay, we need to rehydrate the embryos with 1 mL of 75%, 50%, and 25% methanol in PBST for 5 min each, and rinse them with PBST three times to remove the methanol completely.
-
Genotyping of maternal mutants
For genotyping, first extract the total RNA of isolated cells of corresponding embryos separately as follows: Add 60 μL of chloroform and vortex for 5–10 s. Centrifuge at 12,000× g for 15 min at 4 and transfer supernatant to a new tube. Add an equivalent volume of isopropanol and 1 μL of glycogen (20 mg/mL, facilitating the precipitation of RNA), then mix it and incubate at -80 for 30 min to precipitate RNA. Centrifuge at 12,000× g for 15 min at 4 . Discard the supernatant and rinse the pellet with pre-chilled 70% ethanol. Centrifuge at 12,000× g for 5 min at 4 . Remove the supernatant and resolve the pellet with 8 μL of deionized H2O (the volume depends on the cDNA synthesis reaction so that the total RNA can be applied for cDNA synthesis). Then, reverse-transcribe the cDNA following the cDNA synthesis kit manual.
Use the cDNA as the template to amplify the coding sequence of the target gene. The PCR mixture is as shown in Table 16.
The PCR procedure is as shown in Table 17.
Take 2 μL of PCR products to perform electrophoresis and purify the rest using a clean-up kit.
Ligate the purified PCR products to the pBackZero-T vector following the kit manual.
Mix the ligation mixture with 100 μL of DH5α competent cell. Incubate on ice for 30 min and heat shock for 60 s at 42 . Immediately after heat shock, chill the cells on ice for 2 min. Then, spread it on the ampicillin-resistant solid LB medium and incubate for 14 h at 37 .
Pick up 40–60 colonies randomly, stab them with autoclaved tips separately, and pipette into LB medium in each test tube. After growth at 37 for 14 h, extract plasmids using commercial kits and subject them to Sanger sequencing. Align the sequencing results with the wild-type target gene sequence. If there is no wild-type sequence, we consider the corresponding embryo as a maternal mutant.
Figure 2. Example output from CRISPRScan.

Zebrafish-Danio rerio, Cas9-NGG, Gene, in vitro T7 promoter, and 4 mismatches should be chosen, and the gene symbol is then submitted to “Get sgRNAs”. This protocol uses Cas9 for genome editing, so we select Cas9-NGG to specify the prediction of sgRNA for Cas9. Allowing for “4 mismatches” enables the sgRNA site to tolerate up to four mismatches in off-target sequences. This criterion helps to identify a broader range of potential target sites for consideration.
Table 1. PCR reaction for amplification of sgRNA DNA template.
| Reagent | Volume |
|---|---|
| 2× Taq Master Mix (Dye Plus) | 25 μL |
| sgRNA primer (10 μm) | 2 μL |
| Universal primer (10 μm) | 2 μL |
| Deionized H2O | 21 μL |
| Total volume | 50 μL |
Table 2. PCR program for amplification of sgRNA DNA template.
| Temperature | Time | Cycle number |
|---|---|---|
| 94 °C | 3 min | 1 cycle |
| 94 °C | 15 s | 45 cycles |
| 50 °C | 15 s | |
| 72 °C | 30 s | |
| 72 °C | 5 min | 1 cycle |
Table 3. In vitro transcription reaction for synthesis of sgRNA.
| Reagent | Volume |
|---|---|
| 5× Transcription buffer | 4 μL |
| ATP/GTP/CTP/UTP, 10 mM each | 4 μL |
| DNA template | 400 ng |
| RNase inhibitor | 0.5 μL |
| T7 RNA polymerase | 1 μL |
| DNase/RNase-free H2O | to 20 μL |
Figure 3. Example output of ICE analysis.

We consider the value of “indel %” as the efficiency of the sgRNA.
Figure 4. Schematics illustrating the construction of U6 sgRNA expression vector.

All U6 vectors have a U6 promoter and a sgRNA scaffold. Digestion of BsmBI makes U6 vectors expose the overhangs recognizing the annealed sgRNA primer. Digestion of PstI and SalI can disrupt the vector undigested by BsmBI to improve positive cloning.
Table 4. Reaction for annealing of sgRNA oligos.
| Reagent | Volume |
|---|---|
| Forward primer (100 μM) | 1 μL |
| Reverse primer (100 μM) | 1 μL |
| 10× NEB buffer 2.1 | 2 μL |
| Deionized H2O | 16 μL |
Table 5. Program for annealing of sgRNA oligos.
| Temperature | Time |
|---|---|
| 95 °C | 15 min |
| 50 °C | 10 min (ramp rate: 0.1 °C/s) |
| 4 °C | 10 min |
Table 6. Combination of U6 vectors and pGGDestISceIEG-XsgRNA for different number of sgRNAs.
| sgRNA number | U6 vectors | pGGDestISceIEG-XsgRNA |
|---|---|---|
| 1 | pU6a:sgRNA#1 | pGGDestISceIEG-1sgRNA |
| 2 | pU6a:sgRNA#1, pU6a:sgRNA#2 | pGGDestISceIEG-2sgRNA |
| 3 | pU6a:sgRNA#1, pU6a:sgRNA#2, pU6b:sgRNA#3, | pGGDestISceIEG-3sgRNA |
| 4 | pU6a:sgRNA#1, pU6a:sgRNA#2, pU6b:sgRNA#3, pU6c:sgRNA#4 | pGGDestISceIEG-4sgRNA |
Table 7. Reaction for ligating sgRNA oligos to U6 vectors.
| Reagent | Volume |
|---|---|
| 10× Cutsmart buffer | 1 μL |
| 10× T4 ligase buffer | 1 μL |
| T4 ligase | 0.3 μL |
| BsmB | 0.3 μL |
| Pst | 0.2 μL |
| Sal | 0.2 μL |
| H2O | 1 μL |
| Annealed sgRNA primer | 1 μL |
| pU6x:sgRNA#x (20 ng/μL) | 5 μL |
| Total volume | 10 μL |
Table 8. Program for ligating sgRNA oligos to U6 vectors.
| Temperature | Time | Cycle number |
|---|---|---|
| 37 °C | 20 min | 6 cycles |
| 16 °C | 15 min | |
| 37 °C | 10 min | 1 cycle |
| 55 °C | 15 min | 1 cycle |
| 80 °C | 15 min | 1 cycle |
Table 9. PCR reaction for identification of positive colony.
| Reagent | Volume |
|---|---|
| 2× Taq Master Mix (Dye Plus) | 5 μL |
| Colony liquid | 2 μL |
| M13 forward (10 μm) | 0.4 μL |
| Reverse primer (10 μm) | 0.4 μL |
| Deionized H2O | 2.2 μL |
| Total volume | 10 μL |
Table 10. PCR program for identification of positive colony.
| Temperature | Time | Cycle number |
|---|---|---|
| 94 °C | 3 min | 1 cycle |
| 94 °C | 15 s | 23 cycles |
| 55 °C | 15 s | |
| 72 °C | 30 s | |
| 72 °C | 5 min | 1 cycle |
Figure 5. Golden Gate cloning of tandem sgRNA expression vector and Gibson assembly of double-genes sgRNA expression cassettes.

A. BsaI digestion leaves specific overhangs, which are designed for tandem ligation into pGGDestISceIEG-4sgRNA. B. The amplified sgRNA expression cassette targeting gene b can be cloned into the vector of gene a through Gibson assembly.
Table 11. Reaction for Golden Gate assembly.
| Reagent | Volume |
|---|---|
| 10× Cutsmart buffer | 2 μL |
| 10× T4 ligase buffer | 2 μL |
| pU6X:sgRNA#X | 100 ng each type |
| pGGDestISceIEG-XsgRNA | 50 ng |
| T4 DNA ligase | 1 μL |
| Bsa | 1 μL |
| H2O | To 20 μL |
Table 12. Program for Golden Gate assembly.
| Temperature | Time | Cycle number |
|---|---|---|
| 37 °C | 20 min | 3 cycles |
| 16 °C | 15 min | |
| 80 °C | 15 min | 1 cycle |
Table 13. PCR reaction for amplification of sgRNA expression sequence.
| Reagent | Volume |
|---|---|
| 5× Phusion HF buffer | 10 μL |
| dNTPs (10 mM) | 1 μL |
| Ligation forward primer | 2 μL |
| Ligation reverse primer | 2 μL |
| pISceI-XsgRNA-2 (0.1 ng/μL) | 1 μL |
| Phusion high-fidelity DNA polymerase | 0.5 μL |
| H2O | To 50 μL |
Table 14. PCR program for amplification of sgRNA expression sequence.
| Temperature | Time | Cycle number |
|---|---|---|
| 98 °C | 30 s | 1 cycle |
| 98 °C | 10 s | 35 cycles |
| 58 °C | 15 s | |
| 72 °C | 1 min | |
| 72 °C | 5 min | 1 cycle |
Table 15. Injection mixture for I-SceI-mediated transgenesis.
| Reagent | Volume |
|---|---|
| pGGDestISceIEG-XsgRNA (10 ng/μL) | 7.5 μL |
| 10× CutSmart buffer | 0.5 μL |
| I-Sce | 0.5 μL |
| Total volume | 10 μL |
Figure 6. Representative electrophoresis result of I-Sce efficiency test.
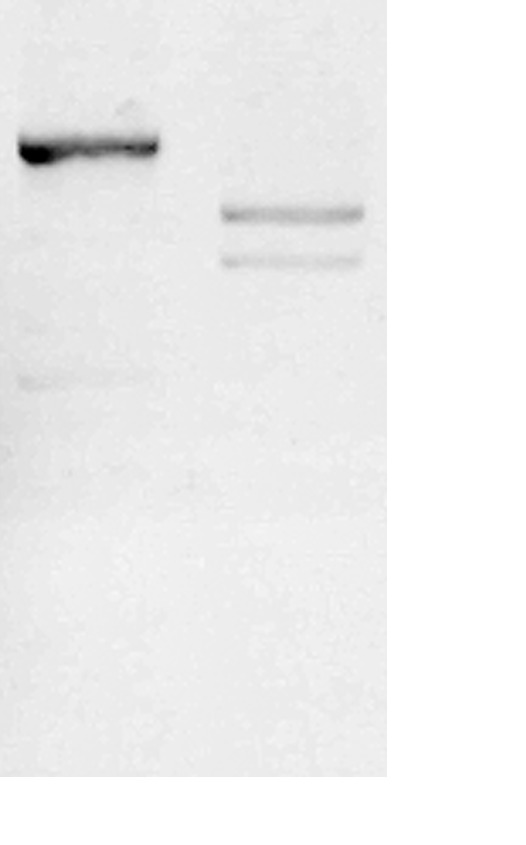
I-Sce I digestion can split the original plasmid into two fragments. No original plasmids should be found when the I-Sce1 works well.
Figure 7. Representative images of normal and dorsalized embryos at 12 hpf.
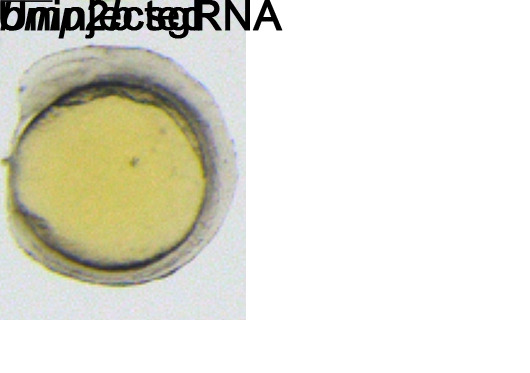
Scale bar: 200 μm.
Figure 8. Illustration of procedures of phenotyping and genotyping individual embryos [11,12,18].

Table 16. PCR reaction for amplification of the coding sequence of the target gene.
| Reagent | Volume |
|---|---|
| 2× Phanta Max Mixture | 25 μL |
| cDNA | 1 μL |
| Forward primer (10 μm) | 2 μL |
| Reverse primer (10 μm) | 2 μL |
| deionized H2O | 20 μL |
| Total volume | 50 μL |
Table 17. PCR program for amplification of the coding sequence of the target gene.
| Temperature | Time | Cycle number |
|---|---|---|
| 95 °C | 30 s | 1 cycle |
| 95 °C | 15 s | 35 cycles |
| 58 °C | 15 s | |
| 72 °C | 30 s/kb | |
| 72 °C | 5 min | 1 cycle |
Data analysis
Using this protocol, we have successfully obtained ctnnb2, nanog, and rbm24a maternal mutants [12]. Further, we also constructed dvl2 and dvl3a double-gene maternal mutants [11]. Here, we will show the data from these two articles as examples.
We designed three sgRNAs with high efficacy to target the coding sequence of ctnnb2. The maternal expression of ctnnb2 is vital for initiating maternal Wnt signaling and is indispensable for the development of the dorsal organizer [19,20]. Interference with maternal ctnnb2 expression leads to zebrafish embryos exhibiting a ventralized phenotype. Adhering to this protocol, we screened 10 female zebrafish and identified two that produced embryos positive for GFP. Analysis of these embryos at 1-day post fertilization (dpf) showed that, on average, 25.6% of GFP-positive embryos displayed various degrees of ventralization (Figure 9A and B). These phenotypes were classified into four categories (V1 to V4) according to established criteria [19]. Additionally, the ventralized phenotype was successfully rescued by injecting wild-type ctnnb2-myc mRNA at the one-cell stage (Figure 9B). We then randomly selected four embryos representing the V1–V4 phenotypes and collected cell samples from each to construct a plasmid library. Sequencing of 116 colonies confirmed that they all contained mutations in the ctnnb2 coding sequence (Figure 9C).
Figure 9. Generation of ctnnb2 maternal mutants using this protocol.

These data were reproduced from our previous publication [12]. A. Phenotypes of the GFP-positive embryos expressing three sgRNAs targeting ctnnb2. B. Injection of wild-type ctnnb2-myc mRNA at the 1-cell stage could efficiently rescue the ventralized phenotypes. C. Schematics shows the mutation types by examining colonies.
In zebrafish, dvl2 and dvl3a play redundant roles in regulating Wnt/PCP and zygotic Wnt/beta-catenin signaling pathways [2,21]. Mutations in either dvl2 or dvl3a alone do not result in noticeable phenotypes. However, the simultaneous loss of maternal and zygotic expression of both dvl2 and dvl3a leads to anteriorization and impaired convergence and extension cell movements [2]. We selected four highly efficient sgRNAs each for targeting the coding sequences of dvl2 and dvl3a. After constructing the sgRNA expression vectors for dvl2 and dvl3a and performing transgenesis via I-Sce I, we obtained two F0 founder females capable of producing a high percentage of GFP-positive progeny. Among the GFP-positive embryos from these founders, 5.4% exhibited a shortened body axis and expanded dorsal structures at 12 h post-fertilization (hpf). By 30 hpf, these embryos showed pronounced yolk extension defects. Crossbreeding these two female founders with dvl2 +/-;dvl3a +/- mutant males resulted in embryos with broad notochords and severely shortened body axes at 12 hpf. At 30 hpf, these embryos presented posterior truncation, closely resembling the phenotypes described for MZdvl2;MZdvl3a mutants (Figure 10A). To genotype the dvl2 and dvl3a maternal mutants, we amplified the coding sequences of both genes. In comparison to wild-type and other GFP-positive control embryos, one double-gene maternal mutant (#1) lacked the wild-type coding sequences for both dvl2 and dvl3a (Figure 10B). Sanger sequencing revealed substantial deletions in the coding regions of both dvl2 and dvl3a (Figure 10C).
Figure 10. Generation of dvl2 and dvl3a maternal mutants using this protocol.

These data were reproduced from our previous publication [11]. A. Phenotypes of wild-type, Mdvl2;Mdvl3a and MZdvl2;MZdvl3a embryos at 12 hpf and 30 hpf. B. Gel analysis after amplifying the coding sequence of dvl2 and dvl3a. C. Large deletions happened both in dvl2 and dvl3a coding sequences.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Zhang et al. [11]. A Time-Saving Strategy to Generate Double Maternal Mutants by an Oocyte-Specific Conditional Knockout System in Zebrafish. Biology (Basel) (Figures 2–3).
Zhang et al. [12]. Rapid generation of maternal mutants via oocyte transgenic expression of CRISPR-Cas9 and sgRNAs in zebrafish. Science Advances (Figures 2–4).
General notes and troubleshooting
| Problem | Solution |
|---|---|
| No commercial Cas9 protein for sgRNA test. | In vitro transcription and purification of Cas9 mRNA and use it to test sgRNA efficiency. |
| Low maternal mutant frequency. | Prescreen Tg(zpc:zcas9) by injecting bmp2b sgRNA into the embryos and select the fish with injected embryos displaying a high ratio of dorsalized phenotypes. |
| Failure in amplification of the coding sequence. | Use nested PCR. |
Acknowledgments
We are grateful to Bo Zhang and Wenbiao Chen for providing zcas9 and pU6X:sgRNA#X plasmids. This work was supported by Guangdong Basic and Applied Basic Research Foundation [2023A1515110960], Central People’s Hospital of Zhanjiang Startup Project of Doctor Scientific Research [2020A12], and Zhanjiang Science and Technology Project [2022A01076 and 2022A01079]. This protocol was adapted from our previous work [11,12]. We thank all the authors who conducted the original work.
Competing interests
The authors declare no competing interests.
Ethical considerations
Zebrafish were raised under standard conditions. All experiments were designed and performed following the principles issued by the Ethics Committee for Animal Research of Life Science of Shandong University (permit number SYDWLL-2018-05).
References
- 1. White R. J., Collins J. E., Sealy I. M., Wali N., Dooley C. M., Digby Z., Stemple D. L., Murphy D. N., Billis K., Hourlier T., et al.(2017). A high-resolution mRNA expression time course of embryonic development in zebrafish. eLife. 6: e30860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xing Y. Y., Cheng X. N., Li Y. L., Zhang C., Saquet A., Liu Y. Y., Shao M. and Shi D. L.(2018). Mutational analysis of dishevelled genes in zebrafish reveals distinct functions in embryonic patterning and gastrulation cell movements. PLos Genet. 14(8): e1007551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hino H., Nakanishi A., Seki R., Aoki T., Yamaha E., Kawahara A., Shimizu T. and Hibi M.(2018). Roles of maternal wnt8a transcripts in axis formation in zebrafish. Dev Biol. 434(1): 96-107. [DOI] [PubMed] [Google Scholar]
- 4. Gritsman K., Zhang J., Cheng S., Heckscher E., Talbot W. S. and Schier A. F.(1999). The EGF-CFC Protein One-Eyed Pinhead Is Essential for Nodal Signaling. Cell. 97(1): 121-132. [DOI] [PubMed] [Google Scholar]
- 5. Shao M., Lu T., Zhang C., Zhang Y. Z., Kong S. H. and Shi D. L.(2020). Rbm24 controls poly(A) tail length and translation efficiency of crystallin mRNAs in the lens via cytoplasmic polyadenylation. Proc Natl Acad Sci USA. 117(13): 7245-7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang C., Huang R., Ma X., Chen J., Han X., Li L., Luo L., Ruan H. and Huang H.(2021). The Ribosome Biogenesis Factor Ltv1 Is Essential for Digestive Organ Development and Definitive Hematopoiesis in Zebrafish. Front Cell Dev Biol. 9: e704730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu J., Zhang D., Liu X., Yu G., Cai X., Xu C., Rong F., Ouyang G., Wang J., Xiao W., et al.(2019). Zebrafish prmt5 arginine methyltransferase is essential for germ cell development. Development. 146(20): e179572. [DOI] [PubMed] [Google Scholar]
- 8. Ciruna B., Weidinger G., Knaut H., Thisse B., Thisse C., Raz E. and Schier A. F.(2002). Production of maternal-zygotic mutant zebrafish by germ-line replacement. Proc Natl Acad Sci USA. 99(23): 14919-14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu X., Shen W., Zhang B. and Meng A.(2018). The genetic program of oocytes can be modified in vivo in the zebrafish ovary. J Mol Cell Biol. 10(6): 479-493. [DOI] [PubMed] [Google Scholar]
- 10. Liu Y., Zhu Z., Ho I. H. T., Shi Y., Xie Y., Li J., Zhang Y., Chan M. T. V. and Cheng C. H. K.(2017). Germline-specific dgcr8 knockout in zebrafish using a BACK approach. Cell Mol Life Sci. 74(13): 2503-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang C., Li J., Tarique I., Zhang Y., Lu T., Wang J., Chen A., Wen F., Zhang Z., Zhang Y., et al.(2021). A Time-Saving Strategy to Generate Double Maternal Mutants by an Oocyte-Specific Conditional Knockout System in Zebrafish. Biology. 10(8): 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang C., Lu T., Zhang Y., Li J., Tarique I., Wen F., Chen A., Wang J., Zhang Z., Zhang Y., et al.(2021). Rapid generation of maternal mutants via oocyte transgenic expression of CRISPR-Cas9 and sgRNAs in zebrafish. Sci Adv. 7(32): eabg4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y., Zhang C., Zhang Y., Lin S., Shi D. L. and Shao M.(2018). Highly efficient genome editing using oocyte-specific zcas9 transgenic zebrafish. J Genet Genomics. 45(9): 509-512. [DOI] [PubMed] [Google Scholar]
- 14. Moreno-Mateos M. A., Vejnar C. E., Beaudoin J. D., Fernandez J. P., Mis E. K., Khokha M. K. and Giraldez A. J.(2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 12(10): 982-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vejnar C. E., Moreno-Mateos M. A., Cifuentes D., Bazzini A. A. and Giraldez A. J.(2016). Optimized CRISPR–Cas9 System for Genome Editing in Zebrafish. Cold Spring Harb Protoc. 2016(10): 1101. [DOI] [PubMed] [Google Scholar]
- 16. Etard C., Joshi S., Stegmaier J., Mikut R. and Strähle U.(2017). Tracking of Indels by DEcomposition is a Simple and Effective Method to Assess Efficiency of Guide RNAs in Zebrafish. Zebrafish. 14(6): 586-588. [DOI] [PubMed] [Google Scholar]
- 17. Yin L., Maddison L. A., Li M., Kara N., LaFave M. C., Varshney G. K., Burgess S. M., Patton J. G. and Chen W.(2015). Multiplex Conditional Mutagenesis Using Transgenic Expression of Cas9 and sgRNAs. Genetics. 200(2): 431-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao M., Cheng X. N., Liu Y. Y., Li J. T. and Shi D. L.(2018). Transplantation of Zebrafish Cells by Conventional Pneumatic Microinjector. Zebrafish. 15(1): 73-76. [DOI] [PubMed] [Google Scholar]
- 19. Kelly C., Chin A. J., Leatherman J. L., and D. J. K. and Weinberg E. S.(2000). Maternally controlled β-catenin-mediated signaling is required for organizer formation in the zebrafish. Development. 127(18): 3899-3911. [DOI] [PubMed] [Google Scholar]
- 20. Bellipanni G., Varga M., Maegawa S., Imai Y., Kelly C., Myers A. P., Chu F., Talbot W. S. and Weinberg E. S.(2006). Essential and opposing roles of zebrafish β-catenins in the formation of dorsal axial structures and neurectoderm. Development. 133(7): 1299-1309. [DOI] [PubMed] [Google Scholar]
- 21. Shi D. L.(2020). Decoding Dishevelled-Mediated Wnt Signaling in Vertebrate Early Development. Front Cell Dev Biol. 8: e588370. [DOI] [PMC free article] [PubMed] [Google Scholar]