

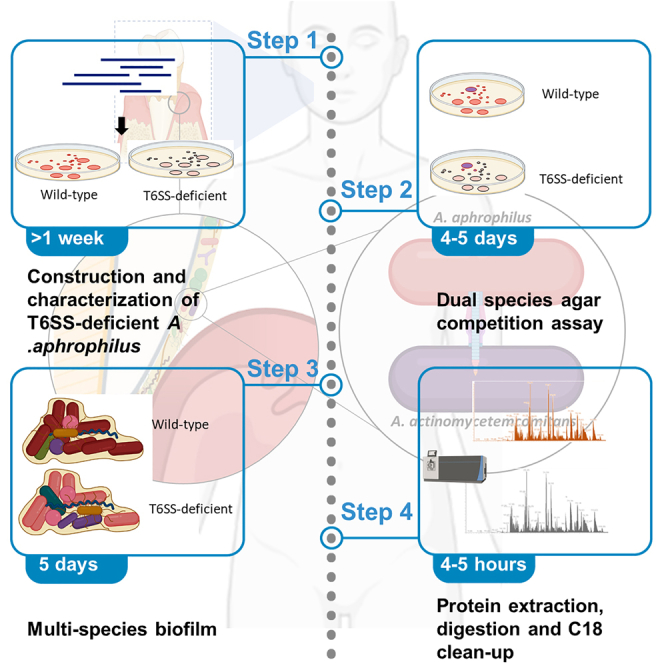
Summary
Here, we present a protocol for evaluating type VI secretion system (T6SS)-dependent fitness of the oral symbiont A. aphrophilus using biofilm competition assays and metaproteomics. We describe steps for designing T6SS-specific mutants. We then detail procedures for using them in competition assays with the pathobiont A. actinomycetemcomitans and in biofilm models, analyzing metaproteomes to assess the impact of the T6SS on multiple pathobionts. The biofilm model is designed to mimic the oral plaque ecosystem and includes seven species.
For complete details on the use and execution of this protocol, please refer to Oscarsson et al.1
Subject areas: genomics, microbiology, molecular biology
Graphical abstract
Highlights
-
•
Design and test T6SS-specific mutants in biofilm and competition assays
-
•
Perform dual-species competition assays with A. actinomycetemcomitans as prey
-
•
Use a seven-species biofilm model mimicking the dental plaque ecosystem
-
•
Analyze biofilm metaproteomes to assess T6SS impact on oral pathobionts
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a protocol for evaluating type VI secretion system (T6SS)-dependent fitness of the oral symbiont A. aphrophilus using biofilm competition assays and metaproteomics. We describe steps for designing T6SS-specific mutants. We then detail procedures for using them in competition assays with the pathobiont A. actinomycetemcomitans and in biofilm models, analyzing metaproteomes to assess the impact of the T6SS on multiple pathobionts. The biofilm model is designed to mimic the oral plaque ecosystem and includes seven species.
Before you begin
The following protocols outline the precise procedures for PCR-based generation of type VI-secretion-specific mutants of A. aphrophilus, using dual-species agar competition assays, the multi-species oral biofilm model, and subsequent metaproteome analyses to assess the role of this nanomachinery in the fitness of this organism against pathobionts. In these procedures, the bacterial species and multispecies biofilms are kept in incubators at 37°C with 5% CO2. For transformation assays, prepare in advance, as described below, Trypticase soy broth (TSB), and TSB agar plates containing heat-inactivated horse serum (sTSB), with and without 100 μg/mL kanamycin (final concentration). For dual-species agar competition it is necessary to isolate spontaneous streptomycin- and rifampicin-resistant derivatives of the A. aphrophilus and A. actinomycetemcomitans model strains, respectively as detailed below, which are needed in these assays. Moreover, for multispecies biofilms, we recommended inoculating all needed strains from agar plate to broth 3 days (on the Friday before the experiments) ahead of the experiment.
Note: If not stated otherwise, reagents can be substituted with similar alternatives from different vendors.
Institutional permissions
All procedures were conducted in accordance with the guidelines of the local ethics committee at the Medical Faculty of Umeå University, which are in compliance with the Declaration of Helsinki (64th WMA General Assembly, Fortaleza, October 2013). No live vertebrates or higher invertebrates are involved in this work.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal antiserum specific for V. cholerae Hcp (used at 1:5,000 final concentration) | Ishikawa et al.2 | RRID: AB_2313773 |
| Anti-rabbit horseradish peroxidase – conjugate (used at 1:10,000 final concentration) | Jackson ImmunoResearch, Newmarket, UK | RRID: AB_2313773 |
| Bacterial and virus strains | ||
| A. aphrophilus strains HK83 | Culture Collection University of Gothenburg (CCUG) | CCUG 49494 |
| A. aphrophilus strains CCUG 11575 | CCUG | CCUG 11575 |
| A. aphrophilus strains NJ8700 | CCUG | NJ8700 |
| A. aphrophilus strains Aap-4K | Isolated from a patient3,4 | Aap-4K |
| A. aphrophilus strains Aap-12K | Isolated from a patient3,4 | Aap-12K |
| A. aphrophilus strains Aap-13K | Isolated from a patient3,4 | Aap-13K |
| A. aphrophilus strains Aap-21K | Isolated from a patient3,4 | Aap-21K |
| A. aphrophilus strains Aap-29K | Isolated from a patient3,4 | Aap-29K |
| A. aphrophilus strains Aap-30K | Isolated from a patient3,4 | Aap-30K |
| A. aphrophilus strains Aap-32K | Isolated from a patient3,4 | Aap-32K |
| A. aphrophilus strains Aap-53K | Isolated from a patient3,4 | Aap-53K |
| A. aphrophilus strains AHI-3151 | Isolated from a patient5,6 | AHI-3151 |
| A. aphrophilus strains IH-90256 | Isolated from a patient5,6 | IH-90256 |
| A. aphrophilus strains IH-90274 | Isolated from a patient5,6 | IH-90274 |
| A. actinomycetemcomitans strain D7SS | Isolated from a patient7 | D7SS |
| A. actinomycetemcomitans strain JP2 | Bao et al.8 | OMZ 295 |
| Actinomyces oris | Bao et al.,8; Bostanci et al.9 | OMZ 745 |
| Fusobacterium nucleatum subsp. nucleatum KP-F2 | Bao et al.8 | OMZ 598 |
| Streptococcus oralis SK248 | Bostanci et al.9 | OMZ 607 |
| Streptococcus mutans UA159 | Thurnheer et al.10 | OMZ 918 |
| Veillonella dispar ATCC 17748T | Bostanci et al.,9 Bao et al.11 | OMZ 493 |
| Chemicals, peptides, and recombinant proteins | ||
| Tryptone | Difco | Cat#0123-01-1 |
| Yeast extract | Difco | Cat#0127-01-7 |
| Horse serum | Håtunalab AB | Product numbers 150, or 153 depending on the amount that is ordered (100 or 500 mL) |
| Blood agar plates | Laboratoriemedicin, Region Västerbotten, Technical Biochemical section | Product number TBK 14507 |
| KNO3 | Sigma-Aldrich | Cat#P6083 |
| NaCl | Sigma-Aldrich | Cat#S7653 |
| Glucose | Sigma-Aldrich | Cat#G7528 |
| Cysteine. HCl | Sigma-Aldrich | Cat#30078 |
| KH2PO4 | Sigma-Aldrich | Cat#S7795 |
| Na2HPO4.2 H2O | Sigma-Aldrich | Cat#P5655 |
| Iodoacetamide | Sigma-Aldrich | Cat#71507 |
| Urea | Sigma-Aldrich | Cat#I1149 |
| Clarity western ECL substrate | Sigma-Aldrich | Cat#U0631 |
| Tris/HCl | Sigma-Aldrich | Cat#T6666 |
| triethylammonium bicarbonate | Thermo Fisher Scientific | Cat#90114 |
| Trifluoroacetic acid | Sigma-Aldrich | Cat#1081780050 |
| Trypsin | Promega | Cat#V511C |
| Methanol | Fisher Scientific | Cat#10031094 |
| Formic acid | VWR | Cat#84865.180 |
| Acetonitrile | VWR | Cat#14261 |
| Critical commercial assays | ||
| Ready-To-Go PCR beads | Cytiva | Cat#407513-STR |
| Microcon YM-30 centrifugal filter unit | Sigma-Aldrich | Cat#UFC503008 |
| QIAquick PCR purification kit | QIAGEN | Cat# 28104 |
| C18 disk core | Fisher Scientific | Cat#13110016 |
| Deposited data | ||
| Proteomic raw files | ProteomeXchange | PXD042723 |
| Oligonucleotides | ||
| The A. actinomycetemcomitans-specific primers 5′- CTAGGTATTGCGAAACAATTTG -3 (forward) and 5′- CCTGAAATTAAGCTGGTAATC -3′ (reverse) | Kirakodu et al.12 | N/A |
| The A. aphrophilus-specific primers 5′-CCTACACCAGCGTTTATTTC-3′ (forward) and 5′-CTGAGGTTTACGCCAGTC -3′ (reverse) |
Lindholm et al.4 | N/A |
| Cyanine 3 -labeled A. actinomycetemcomitans 16S rRNA oligonucleotide probe Act639 5′-CTCCAGACCCCCAGTATG-3′ | Thurnheer et al.13 | Act639 |
| The hcp gene replacement in A. aphrophilus upstream fragment forward primer hcp_F2 (5′-CGAGCGCAGGATTATAGCAGCT-3′) | This work | N/A |
| The hcp gene replacement of hcp in A. aphrophilus upstream fragment reverse primer hcp_R2 (5′- AAACGCTGGTGGATCCATAGAATTCTC-3′) | This work | N/A |
| The hcp gene replacement of in A. aphrophilus downstream fragment hcp_F3 (5′-GATGACTGGCGGATCCCTCAGGTT-3′) | This work | N/A |
| The gene replacement of hcp in A. aphrophilus downstream fragment hcp_R3 (5′-CACCGCTTGTGTATTGGCAGTGGC-3′) | This work | N/A |
| The kanamycin resistance cassette primer H7R (5′-GGACGGCGGCTTTGTTGAATAAATCG-3′), | This work | N/A |
| FAM-labeled A. aphrophilus 16S rRNA oligonucleotide 5′-CTCTAGACCCCCAGTCTG-3′ | This work | Aaph639 |
| pUC4K | Vieira et al.14 | GenBank X06404; https://www.ncbi.nlm.nih.gov/genbank/ |
| Software and algorithms | ||
| Quantable packages (version 0.3.8) | N/A | https://github.com/protViz/quantable |
| Progenesis QI for proteomics (version 4.1 Nonlinear Dynamics) | Nonlinear Dynamics | https://www.nonlinear.com/progenesis/qi-for-proteomics/ |
| Other | ||
| Candida albicans | OMZ | OMZ 110 |
Materials and equipment
We recommend preparing Solutions A and B the week before the experiment, while SNB (including SNB1 and SNB2) can be prepared a few weeks in advance and stored at 4°C. The final FUM/Sö./0.3%G medium should be prepared the afternoon before the experiment.
FUM + 0.3% Glucose in Sörensen’s buffer (pH 7.2)
| Reagent | Final concentration | Amount |
|---|---|---|
| Solution A | N/A | 200 mL |
| Solution B | N/A | 12.5 mL |
| SNB Solution | N/A | 37.5 mL |
| Total | N/A | 250 mL |
Prepared the afternoon before the experiment. Store at 4°C until the end of the experiment.
Note: Solution B and RTF solution are produced separately to avoid precipitation.
Solution A
| Reagent | Final concentration | Amount |
|---|---|---|
| Tryptone (Difco, 0123-01-1) | N/A | 2.5 g |
| Yeast Extract (Difco, 0127-01-7) | N/A | 1.25 g |
| KNO3 | N/A | 250 mg |
| NaCl | N/A | 500 mg |
| Sörensen’s buffer | N/A | 200 mL |
| Total | N/A | 200 mL |
Autoclave 20 min at 121°C.
Prepare a few weeks in advance and stored at 4°C up to 1 month.
Solution B
| Reagent | Final concentration | Amount |
|---|---|---|
| Glucose | N/A | 750 mg |
| Cysteine. HCl | N/A | 125 mg |
| Na2CO3 | N/A | 125 mg |
| Sörensen’s buffer | N/A | 12.5 mL |
| Total | N/A | 12.5 mL |
Filter sterilization.
Prepare a few weeks in advance and stored at 4°C up to 1 month.
Sörensen’s buffer, pH 7.2
| Reagent | Final concentration | Amount |
|---|---|---|
| KH2PO4 | 9.078 g/L | 330 mL |
| Na2HPO4.2 H2O | 11.876 g/L | 670 mL |
| Total | N/A | 1 L |
Store at 4°C for up to 1 year or until precipitates appear.
Salt Nutrient Buffer (SNB)Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| SNB 1 stock solution | N/A | 18.75 mL |
| SNB 2 stock solution | N/A | 18.75 mL |
| Total | N/A | 37.5 mL |
Filter sterilization. Prepare just before use.
SNB 1 stock solution
| Reagent | Final concentration | Amount |
|---|---|---|
| K2HPO4 | 6 g/L | 6 g |
| distilled H2O | N/A | 1000 mL |
| Total | N/A | 1 L |
Store at 4°C for up to 1 year or until precipitates appear.
SNB 2 stock solution
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 12 g/L | 12 g |
| (NH4)2SO4 | 12 g/L | 12 g |
| KH2PO4 | 6 g/L | 6 g |
| MgSO4 7H2O | 2.5 g/L | 2.5 g |
| distilled H2O | N/A | 1000 mL |
| Total | N/A | 1 L |
Store at 4°C for up to 1 year or until precipitates appear.
-
•
lysis buffer (4% [w/v] SDS, 100 mM Tris/HCl pH 8.2, 0.1 M dithiothreitol [DTT]).
Prepare just before use. Avoid light.
-
•
UA buffer (8 M urea in 100 mM Tris/HCl pH 8.2).
Prepare just before use.
-
•
The IAA solution (0.05 M IAA in UA buffer).
Prepare just before use. Avoid light.
-
•
0.05 M triethyl-ammoniumbicarbonate (TEAB).
Prepare a few weeks in advance and store at 4°C for up to 1 year or until precipitates appear.
Step-by-step method details
Primer design and PCR conditions to generate DNA fragments for gene replacement of hcp in A. aphrophilus
Timing: 4–5 h
This section describes the steps to generate DNA fragments for gene replacement of hcp in A. aphrophilus.
-
1.Prepare the DNA template.
-
a.Boil a loopful of A. aphrophilus colonies (we picked generally 10 colonies) from fresh agar plates in 100 μL Milli-Q ultrapure water for 10 min.
-
b.Centrifuge the sample for 1 min at 14,000 rpm using a benchtop Eppendorf centrifuge.
-
a.
-
2.For each PCR reaction (final volume 25 μL).
-
a.Add 22 μL Milli-Q ultrapure water to Ready-To-Go PCR beads.Note: The reaction used Illustra PuReTaq Ready-To-Go PCR beads and Milli-Q ultrapure water, and was optimized for A. aphrophilus strains HK83 and CCUG 11575.
-
b.Add 1 μL of each primer (from stock concentrations of 10 μM).Note: Primers were designed using OligoCalc (http://biotools.nubic.northwestern.edu/OligoCalc.html) to ensure compatible melting temperatures and prevent hairpin formation, with BamHI restriction sites added to minimize alterations to the target sequences.
-
c.Add 1 μL of the prepared DNA template.
-
a.
-
3.Run the PCR with the following settings.
-
a.10 min at 95°C.
-
b.35 cycles of:
-
i.95°C for 30 s.
-
ii.56°C for 30 s.
-
iii.72°C for 1 min.
-
i.
-
c.Followed by 7 min at 72°C.
-
a.
-
4.
Purify the obtained PCR products, directly after the PCR, using reagents that remove the reaction components. We have used the Qiaquick PCR Purification Kit according to the manufacturer’s instructions.
Preparation of sTSB agar plates
Here we outline the preparation of sTSB agar plates, including sterilization and the addition of heat inactivated horse serum, providing a nutrient-rich medium for bacterial growth.
-
5.
For one liter of sTSB agar medium (enough for ≈ 30 plates) suspend 45 g Trypticase soy broth (TSB; Bacto Tryptic Soy Broth, Becton Dickinson, Heidelberg, Germany), 1 g yeast extract, and 15 g agar in 1000 mL of distilled water.
-
6.
Sterilize by autoclaving at 121°C for 15 min.
-
7.
Cool to agar medium to 45°C–50°C.
-
8.
Add aseptically 50 mL of heat inactivated horse serum.
-
9.
Mix and then pour the plates.
DNA ligation and transformation procedures for hcp gene replacement in A. aphrophilus
This section describes the steps for DNA digestion, purification, ligation, and bacterial transformation. DNA fragments and plasmid are digested with BamHI, and the kanamycin resistance cassette is purified via gel electrophoresis. The PCR fragments and kanamycin cassette are then ligated, i.e., generating a construct hcp upstream – kanamycin – hcp downstream, followed by bacterial transformation using A. aphrophilus. Finally, transformants are selected for kanamycin resistance and confirmed via PCR.
-
10.Digestion of DNA Fragments and Plasmid with BamHI:
- a.
-
b.Use 500 ng of each DNA sample in a 30 μL reaction mixture.
-
c.Incubate the reactions at 37°C for up to 2 h.
-
11.Purification of Kanamycin (Kmr) Gene Cassette from Plasmid:
-
a.Perform agarose gel electrophoresis to separate DNA fragments.
-
b.Excise and purify the band containing the 1364 base pair kanamycin resistance cassette using the QIAquick Gel Extraction Kit (Cat. No. 28704, QIAGEN), following the manufacturer’s instructions.
-
a.
-
12.Ligation of PCR Fragments and Kanamycin Cassette:
-
a.Prepare ligation reactions by mixing equal amounts of PCR fragments (obtained by using the primers hcp_F2 plus hcp_R2, and hcp_F3 plus hcp_R3, respectively) flanking hcp and the purified kanamycin cassette.
-
b.For each ligation reaction, combine the DNA fragments with 1 μL of T4 DNA ligase (Promega) in a total volume of 11 μL.
-
c.Achieve a final DNA concentration of approximately 100 μg/mL.
-
d.Incubate the ligation reactions at RT overnight.
-
a.
-
13.Preparation of Bacterial Cells and Transformation:
-
a.Harvest fresh A. aphrophilus from cultures grown on sTSB agar plates to a concentration of approximately 5 × 109 CFU/mL in TSB broth.
-
b.Spread 20 μL aliquots of bacterial solution on pre-warmed (37°C) blood agar plates in small areas (approximately 10 mm in diameter).
-
c.Incubate the plates at 37°C for 2 h.
-
d.Mix the bacterial cells on the agar plates with the ligation mixture(s) prepared in step 3.
-
e.Incubate the plates at 37°C with 5% CO2 for 6 h.
-
a.
-
14.Selection and Confirmation of Transformants:
-
a.Scrape the bacterial cells from the sTSB agar plates using a loop.
-
b.Resuspend the cells in 100 μL of TSB.
-
c.Plate the cell suspension onto sTSB agar plates containing 100 μg/mL kanamycin.
-
d.Incubate the plates at 37°C for 48 h.
-
e.Test the obtained colonies for kanamycin resistance.
-
f.Confirm transformants by PCR using the following primers:
-
i.The hcp upstream (hcp_F2).
-
ii.The downstream (hcp_R3).
-
iii.A primer specific for the kanamycin resistance cassette.
-
i.
-
g.Run the PCR reaction following the PCR cycling conditions as described in step 2 of the PCR protocol.
-
h.Confirm the presence of the kanamycin cassette and successful transformation by comparing the sizes of the PCR products to the expected values.
-
a.
Note: Depending on the orientation of the kanamycin cassette, a PCR product will be obtained with primer H7R either in combination with hcp_F2 (≈1,100 base pairs [bp]), or with hcp_R3 (≈1,270 bp).
Dual species agar competition assay
The protocol below describes the steps for assessing T6SS-dependent killing of A. actinomycetemcomitans by A. aphrophilus, on agar. Bacterial strains to be used in this protocol need to be cultivated on agar prior to experiments.
-
15.Preparation of Co-culture and Controls (See the schematic in our graphic abstract):
-
a.Harvest A. aphrophilus from blood agar plates to achieve OD600nm = 1.7 in Tryptic Soy Broth (TSB).Note: Due to the similar colony morphology of the bacterial strains used in these experiments, spontaneous streptomycin- and rifampicin-resistant derivatives ensure that both species, despite being mixed in the experiments, can be independently enumerated on the blood agar plates containing the respective antibiotic selection. For obtaining spontaneous streptomycin-resistant derivatives of A. aphrophilus (strains HK83, and HK83Δhcp, and CCUG 11575 and CCUG 11575 Δhcp), and rifampicin-resistant derivatives of A. actinomycetemcomitans strain D7SS, respectively. Spread aliquots equivalent to approximately (OD600nm ≈ 1.7–2.0) resuspended in TSB onto blood agar plates containing 100 μg/mL of streptomycin or rifampicin. Isolated obtained resistant colonies to use in the following steps. We have confirmed the species identity by PCR, using the PCR beads, and specific oligonucleotide primers targeting A. actinomycetemcomitans12 and A. aphrophilus,4 respectively using the PCR cycling conditions described in the original sources for these primers as indicated in the key resources table:
-
b.Harvest A. actinomycetemcomitans from blood agar plates to achieve OD600nm = 1.3 in TSB.
-
c.Maintain monocultures of both strains in TSB medium as controls. We re-streaked them to fresh blood agar plates, which were then placed in the 37°C incubator with 5% CO2 until use.
-
d.Mix A. aphrophilus and A. actinomycetemcomitans at a ratio of 3:1 (vol/vol).
-
e.Spot 40 μL aliquots of the mixed culture and each monoculture on blood agar plates.
-
f.Incubate the plates overnight at 37°C with 5% CO2.
-
a.
-
16.Harvest and resuspension of bacteria from plates:
-
a.Scrape bacteria from the blood agar plates using a loop or sterile swab.
-
b.Transfer the scraped material into TSB to resuspend the bacterial cells.
-
a.
-
17.Enumeration of colony-forming units (CFU) and Calculation of Killing Index:
-
a.Perform serial dilutions of the bacterial suspension.
-
b.Spread appropriate dilutions on blood agar plates containing:
-
i.Streptomycin (for A. aphrophilus selection).
-
ii.Rifampicin (for A. actinomycetemcomitans selection).
-
i.
-
c.Incubate the plates for two days at 37°C with 5% CO2.
-
d.Counting CFUs.
-
e.After incubation, count the number of CFU for:
-
i.A. aphrophilus on streptomycin-containing plates.
-
ii.A. actinomycetemcomitans on rifampicin-containing plates.
-
i.
-
f.Calculate the killing index as described,1 i.e., the ratio of A. aphrophilus CFU numbers in the co-culture (mixed culture) divided by the CFU numbers when A. aphrophilus is in monoculture (alone).
-
a.
Note: The killing index provides a measure of bacterial competition or antagonism, where a ratio less than 1 indicates potential killing or inhibition executed by the donor strain (A. aphrophilus) towards the recipient strain (A. actinomycetemcomitans).
Multi-species biofilm
The protocol below describes the steps for developing multi-species biofilm models to mimic the killing activity of A. aphrophilus against A. actinomycetemcomitans in their natural habitat with other oral species.
-
18.Day 1:
-
a.Prepare two 15 mL tubes for each cell strain with labeled group A and group B.Note: The experiment lasts for 5 days. Thus, we recommended starting the experiment on a Monday while preparing the strains on Friday the week before, as stated in the “before you begin” section.
-
b.Aliquot 9 mL FUM with 0.3% Glucose in Sörensen’s buffer (FUM/Sö./0.3%G; please see the materials table below) medium in tubes of group A and 5 mL of the same medium in tubes of group B.Note: The medium used in the experiment is FUM + 0.3% glucose in Sörensen's buffer (FUM/Sö./0.3%G), as described in the “materials and equipment” section above, along with added saliva and human serum. The FUM/Sö./0.3%G medium consists of three parts: Solution A, Solution B, and the Salt Nutrient Buffer (SNB) solution. These components should be prepared separately to avoid precipitation. We recommend preparing Solutions A and B the week before the experiment, while SNB (including SNB1 and SNB2) can be prepared a few weeks in advance and stored at 4°C. The final FUM/Sö./0.3%G medium should be prepared the afternoon before the experiment, with the saliva and human serum added on the same day of the experiment.
-
c.Inoculate strains by adding 0.5 mL cell suspensions with OD550nm values around 1 AU (from Friday).
-
d.Incubate overnight.Note: Unless stated otherwise, the incubators were aerobic, operating at 37°C with 5% CO₂.
-
e.Label 24 well plates for the later experiment.
-
a.
-
19.Day 2:
-
a.Aliquot 0.5 mL of solutions from group A to ground B and incubate in incubators at 37°C.
-
b.Check the turbidity of each bacterial culture, and store those with OD550nm values around 1 AU in a 4°C fridge.
-
c.Place 24 hydroxyapatite discs (Clarkson Chromatography Products Inc) into the 24-well plate.
-
d.Prepare 20 mL of initial medium (10 mL saliva, 7.5 mL dH2O and 2.5 mL 0.9% NaCl).
-
e.Apply 0.8 mL initial medium to each well (with disc).
-
f.Leave a 24-well plate on the shaker at 95 rpm for 4 h at room temperature (roughly 20°C–22°C).
-
g.Meanwhile, check the turbidity of the strains. Place strains in the 4°C fridge when their OD arrives at around 1.
-
h.Prepare 44 mL of growth medium (28 mL saliva, 4 mL human serum and 12 mL FUM/Sö./0.3%G mixture (783.3 μL in 50 mL FUM/Sö./0.3%G medium) and apply 1.6 mL in each well.
-
i.Equilibrated 24-well plate in an incubator for at least 45 min before use.
-
j.Adjust the OD550nm value to 1 AU (acceptable error range of 0.05 AU).
-
k.Check the purity of each medium by microscope.Note: This is a preliminary quality check process. We expect to see only one type of species observed under the microscope (no cross-contamination) and that each species displays its typical morphology. We recommend maintaining a notebook with figures of the typical morphology for all species as a standardized reference for comparison. If any signs of contamination or abnormal morphology are observed, the experiment should be terminated at this stage.
-
l.Add 0.5 mL of each different strain to make 5 different mixtures.Mixture A: Non-A. aphrophilus control.Mixture B: All the other bacterial species with A. aphrophilus strain HK83.Mixture C: All the other bacterial species with A. aphrophilus strains CCUG 11575.Mixture D: All the other bacterial species with A. aphrophilus strain HK83 Δhcp.Mixture E: All the other bacterial species with A. aphrophilus strains CCUG 11575 Δhcp.
-
m.Transfer discs (after 4 h shaking) to a pre-equilibrated 24-well plate.
-
n.Add 200 μL mixture medium into the corresponding well.
-
o.Incubate 24-well plates overnight.
-
a.
-
20.Day 3 and Day 4:
-
a.Make 44 mL of growth medium (28 mL saliva, 4 mL human serum and 12 mL FUM/Sö./0.3%G mixture (783.3 μL in 50 mL FUM/Sö./0.3%G medium) and apply 1.6 mL in each well. Equilibrated in an incubator for at least 45 min before use.
-
b.Fill the first row of the wash plate with 1 mL of 0.9% NaCl.
-
c.Move hydroxyapatite discs to the first row of the wash plate.
-
d.Gently shake plates for 1 min (count after moving the first disc into the wash plate).
-
e.Dip each disc 3 times in 3 wells that are filled with 2 mL 0.9% NaCl.
-
f.Move dipped discs to a pre-equilibrated medium.
-
g.Keep the plate in the incubator for 4 h.
-
h.Repeat steps b to d.
-
i.Move the dipped discs back into the plate and incubate for another 4 h.
-
j.Repeat steps b and d.
-
k.Move the dipped discs back into the plate and incubate overnight.
-
a.
-
21.Day 5.
-
a.Wash discs 3 times in 0.9% NaCl.
-
b.Transfer discs into a 50 mL tube (with 1 mL 0.9% NaCl).
-
c.Vortex discs 2 min.
-
d.Sonicate bacteria for 5 s.
-
e.Mix 15 μL bacterial suspensions with 0.3 μL live/dead dye (Invitrogen LIVE/DEAD BacLight Bacterial Viability Kits or other fluorescent staining kits commonly used for assessing the viability of bacterial cells) on cover slides.
-
f.Incubate slides in a dark environment for 15 min at room temperature.
-
g.Check slides under the fluorescence microscope.
-
h.Dilute bacterial suspension into different concentrations.
-
i.Plate dilution for CFU counting.
-
a.
Protein extraction, digestion, and C18 clean-up
The protocol below describes the steps for extracting proteins from the biofilms before being subjected to the mass spectrometer (MS). In our previously published paper,1 we employed reversed-phase HPLC coupled with electrospray ionization (ESI)–MS for our analyses. The HPLC system used comprised a Thermo Scientific EASY-nLC 1200, coupled with a 15 cm-long, 75 μm-diameter silica emitter and ReproSil-Pur C18-AQ 120 Å, 1.9 μm resin (Dr. Maisch HPLC GmbH). We employed a three-step gradient of acetonitrile/water (with 0.1% formic acid) at a flow rate of 300 nL/min: initially, the gradient increased from 2% to 30% acetonitrile over 60 min; followed by an increase from 30% to 97% over 10 min; and finally, maintained at 97% for an additional 10 min. The mass spectrometer, an Orbitrap Fusion (Thermo Fisher Scientific), was operated in data-dependent mode with automatic switching between MS and MS/MS using Xcalibur software (Thermo Fisher Scientific). The Orbitrap analyzer was configured to scan within a mass range of 300–1500 m/z. However, tryptic peptides can also be used in other shotgun proteomics protocols with varying analysis modes, columns, mobile phases, flow rates, and gradients, depending on the specific requirements of the user’s application.
-
22.Protein extraction from biofilm.
-
a.Vortex bacterial suspension, spin down 13,000 rpm for 15 min.
-
b.Resuspend pellets with 30 μL lysis buffer.Note: The lysis buffer (4% [w/v] SDS, 100 mM Tris/HCl pH 8.2, 0.1 M dithiothreitol [DTT]) used in the experiment should be prepared on the day of the experiment. Since DTT is sensitive to light, we recommend wrapping them in aluminum foil after preparation.
-
c.Incubate at 95°C and 900 rpm for 5 min (at a thermomixer [Eppendorf], or in a water bath without shaking).Note: Use Eppendorf Safe-Lock Tubes to avoid lids popping off or holes on lids (which may cause water to come to the tube during sonification).
-
d.Cool the tubes on ice for 40 s.
-
e.Resuspend well by pipetting or vox on a bench-top vortex mixer, and spin down the medium to the bottom.
-
f.Sonification using 65%–75% amplitude, 0.5 cycles, with floating ice bath for 3 min.
-
g.Cool the tubes at room temperature for 3 min.
-
h.Repeat the sonication step twice.
-
i.Vortex, centrifuge at 14,000 rpm for 20 min.
-
a.
-
23.
Measure the protein concentrations using Qubit.
Note: A typical biofilm on an 8 mm hydroxyapatite disc will yield 10–30 μg of protein (0.3–1 μg/μL in lysate).
-
24.Protein digestion.
-
a.Aliquot sample with 20 μg protein with 200 μL UA buffer (8 M urea in 100 mM Tris/HCl pH 8.2) and load to the filter unit.Note: The UA buffer (8 M urea in 100 mM Tris/HCl pH 8.2) used in the experiment should be prepared on the day of the experiment.Note: The recommended digestion amount for FASP is 20 μg. However, lower protein weight (down to 10 μg) could be possible, with a risk of reducing proteome coverage or creating outliers for label-free quantification. Analyzing samples with lower protein abundance is not recommended. We suggest first verifying that the biofilm growth is normal. If it is, consider combining multiple biological replicates into a single protein extraction sample.
-
b.Sample volumes should be smaller than 30 μl, otherwise divided into different times according to the proportions.
-
c.Centrifuge at 14,000 rpm for 20 min at RT or 35°C.
-
d.Discard flow-through.
-
e.Add 200 μL of UA to the filter unit.
-
f.Centrifuge at 14,000 rpm for 20 min at RT or 35°C.
-
g.Discard flow-through.Note: No need to change the medium since the collection tube can hold up to 500 μL.
-
h.Add 100 μL iodoacetamide (IAA) solution (0.05 M IAA in UA) to a Microcon YM-30 centrifugal filter unit (Millipore).Note: The IAA solution (0.05 M IAA in UA buffer) used in the experiment should be prepared on the day of the experiment. Since IAA is sensitive to light, we recommend wrapping in aluminum foil after preparation. Mix at 600 rpm in the thermo-mixer for 1 min.
-
i.Incubate without mixing for 5 min.
-
j.Centrifuge at 14,000 rpm for 20 min at RT or 35°C.
-
k.Add 100 μL of UA to the filter unit.
-
l.Centrifuge at 14,000 rpm for 20 min at RT or 35°C.
-
m.Repeat the step two more times.Note: Do not shorten this step – the total volume of wash must be 300 μL.
-
n.Add 100 μL of 0.5 M NaCl to the filter unit.
-
o.Centrifuge at 14,000 rpm for 17 min at RT or 35°C.
-
p.Repeat step one more time – the total volume of wash is 200 μl.
-
q.Transfer the filter units to new collection tubes.
-
r.Resuspend 0.4 μg trypsin in 120 μL 0.05 M TEAB (a 1:50 ratio of trypsin to the substrate) and load this 120 μL mixture to filter unite.Note: The 0.05 M TEAB solution for trypsin digestion and solutions used for the C18 cleanup method can be prepared a few weeks in advance and stored under cool conditions.
-
s.Mix at 600 rpm in the thermo-mixer for 1 min.
-
t.Incubate the units overnight on the bench in a wet cell.
-
u.Centrifuge the filter units at 14,000 rpm for 17 min to collect the digested peptide.
-
a.
-
25.C18 clean up.
-
a.Acidify with 5% TFA Solution to the final concentration of 0.5% trifluoroacetic acid (TFA) in new Eppendorf tubes.
-
b.Make sure the final solution is acidic (pH 3 or lower) with a universal indicator (MQuant).
-
c.Active StageTips (200 μL tip with a C18 disk core (Thermo Scientific)) by load 200 μL mL 100% methanol.
-
d.Equilibrate columns by load 200 μL mL 60% acetonitrile (ACN), 0.1% TFA.
-
e.Equilibrate columns by load 200 μL 3% ACN, 0.1% TFA.
-
f.Load samples on the columns, collect flow through and load it on columns again.
-
g.Wash columns by load 1.2 mL 3% ACN, 0.1% TFA.
-
h.Elute samples from columns with 200 μL 60% ACN, 0.1% TFA.
-
i.Store at −20°C until subject to the mass spectrum.
-
a.
Note: The peptide samples are now ready for LC-MS/MS analysis. We recommend resuspending the dried peptides with 30 μL of loading buffer (e.g., 3% ACN, 0.1% formic acid) and injecting 2–4 μL of the resuspended sample into the LC-MS/MS. In our paper,1 label-free quantitative proteomics identified and quantified 3,286 proteins, with a protein false discovery rate of 0.091%. You can also get more details from our previously published protocol15 Quality control (QC) is essential for tracking variations between biofilm samples. However, since this protocol primarily focuses on sample preparation rather than the LC-MS/MS process, QC is not the main emphasis of our paper. We recommend following best practices for sample preparation as outlined earlier,16 in accordance with the standards of your lab or local proteomics center. Additionally, we strongly suggest spiking in a known peptide standard (e.g., digested bovine serum albumin) every 4–5 samples to ensure consistent quantification. It is also crucial to ensure that ≥80% of proteins have no missed cleavages (in our study, this was over 90%) to confirm proper tryptic digestion. Furthermore, the coefficient of variation within each biofilm condition should be less than 50% (in our case, it ranged from 10%–30% across five different biofilm conditions).
Expected outcomes
The protocol evaluates the role of the T6SS in the fitness of the oral symbiont A. aphrophilus and its antagonistic effects on the pathobiont A. actinomycetemcomitans using a multi-species biofilm model that mimics the oral dental plaque ecosystem. Expected outcomes include the successful design, construction, and validation of T6SS-deficient strains. Additionally, co-culturing A. aphrophilus and A. actinomycetemcomitans on agar (dual-species agar competition assays) and within the multispecies biofilm (containing these two and five additional organisms on a hydroxyapatite disc) confirms the antagonistic effect of T6SS on A. actinomycetemcomitans. This analysis identifies and quantifies proteomic changes in response to T6SS activity, characterizing its influence on microbial interactions. The study provides evidence of T6SS’s active role in shaping the oral microbial community, enhancing the understanding of anti-bacterial strategies employed by oral symbionts. This protocol can also be extended to study other antagonistic effects between oral species in the oral cavity.
For example, variations of biofilms, such as a 10-species 'subgingival' biofilm model (including Prevotella intermedia, Campylobacter rectus, V. dispar, F. nucleatum, S. oralis, Treponema denticola, Actinomyces oris, Streptococcus anginosus, Tannerella forsythia, and Porphyromonas gingivalis) or its derivative that includes A. actinomycetemcomitans, were also published and could potentially be used for analysis.
Limitations
The present assays have certain limitations that must be considered. Firstly, although the genomics of the species used in the model is fully annotated, the current database for oral species lacks comprehensive coverage and detail, which can affect the accuracy and completeness of the metaproteomic analysis. This limitation may impact the annotation of quantified bacterial proteins, potentially skewing the characterization of T6SS activity and its effect on microbial interactions within the biofilm. Secondly, while previous studies have shown that it is possible to introduce selected species not part of the normal oral microbiota into an oral biofilm, doing so can dramatically alter the bacterial proportions within the biofilm.17 Such changes can affect the dynamics and interactions among the species, potentially leading to misleading results. Therefore, it is crucial to carefully evaluate whether introducing other species is necessary and consider the potential impacts on the composition and behavior of the biofilm before proceeding with such modifications.
Troubleshooting
Problem 1
Low reproducibility between multispecies biofilms.
Potential solution
-
•
Ensure the purity of each strain at the beginning of the experiment (Day 2 step k).
-
•
Ensure consistency in the washing steps for biofilms (Day 3 and Day 4 steps b–e). Prepare extra discs in case of dropping or other accidents during the experiment.
-
•
Remove the “apparent outliers” with very low live bacteria counts (Day 5 step g).
-
•
Vortex the disc thoroughly to ensure proper swirling.
Problem 2
Protein digestions can be complex, leading to excessive mis-cleavage and generating unusual chromatogram results.
Potential solution
-
•
Increase the incubation time during protein extraction from the biofilm (step 1) and the sonication duration (steps f–h).
-
•
Always use freshly prepared iodoacetamide.
-
•
Ensure that each centrifugation step completely removes the medium without over-drying the filter during protein digestion.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jan Oscarsson (jan.oscarsson@umu.se).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contact, Kai Bao (kai.bao@ki.se).
Materials availability
All A. aphrophilus strains generated in this study are available upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE18 partner repository with the dataset identifier PXD042723. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This work was funded by grants from the Swedish Research Council (2017-01198 and 2021-03528, to N.B.), strategic funds from Karolinska Institutet (to G.N.B. and N.B.), and TUA grants from Region Västerbotten, Sweden (7002667, to J.O.), and by funds from Insamlingsstiftelsen, Medical Faculty, Umeå University (to J.O.). We would like to thank Dr. Marek Basler and Dr. Karina Persson for valuable discussions and Manuela Flury, Elisabeth Granström, and Carina Öhman for their excellent technical advice and assistance.
Author contributions
All authors have made substantial contributions to the conception and design of the study. J.O., K.B., G.N.B., and N.B. were responsible for the study concept and design. J.O., K.B., A.S., J.G., W.W., K.M.A., M.L., A.J., F.R.M., and N.B. have been involved in data collection and data analysis. All authors have been involved in data interpretation, drafting the manuscript, and revising it critically and have given final approval of the version to be published.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Jan Oscarsson, Email: jan.oscarsson@umu.se.
Nagihan Bostanci, Email: nagihan.bostanci@ki.se.
References
- 1.Oscarsson J., Bao K., Shiratsuchi A., Grossmann J., Wolski W., Aung K.M., Lindholm M., Johansson A., Mowsumi F.R., Wai S.N., et al. Bacterial symbionts in oral niche use type VI secretion nanomachinery for fitness increase against pathobionts. iScience. 2024;27 doi: 10.1016/j.isci.2024.109650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishikawa T., Rompikuntal P.K., Lindmark B., Milton D.L., Wai S.N. Quorum sensing regulation of the two hcp alleles in Vibrio cholerae O1 strains. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindholm M., Min Aung K., Nyunt Wai S., Oscarsson J. Role of OmpA1 and OmpA2 in Aggregatibacter actinomycetemcomitans and Aggregatibacter aphrophilus serum resistance. J. Oral Microbiol. 2019;11 doi: 10.1080/20002297.2018.1536192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindholm M., Claesson R., Kemoli A., Mulli T., Oscarsson J., Haubek D., Johansson A. Aggregatibacter actinomycetemcomitans and Aggregatibacter aphrophilus in a Kenyan Maasai adolescent population and inhibition of leukotoxic activity by herbal plants used as part of oral hygiene procedures. J. Clin. Med. 2021;10 doi: 10.3390/jcm10225402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dogan B., Asikainen S., Jousimies-Somer H. Evaluation of two commercial kits and arbitrarily primed PCR for identification and differentiation of Actinobacillus actinomycetemcomitans, Haemophilus aphrophilus, and Haemophilus paraphrophilus. J. Clin. Microbiol. 1999;37:742–747. doi: 10.1128/jcm.37.3.742-747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paju S., Carlson P., Jousimies-Somer H., Asikainen S. Actinobacillus actinomycetemcomitans and Haemophilus aphrophilus in systemic and nonoral infections in Finland. APMIS. 2003;111:653–657. doi: 10.1034/j.1600-0463.2003.1110608.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y., Goodman S.D., Redfield R.J., Chen C. Natural transformation and DNA uptake signal sequences in Actinobacillus actinomycetemcomitans. J. Bacteriol. 2002;184:3442–3449. doi: 10.1128/JB.184.13.3442-3449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bao K., Bostanci N., Selevsek N., Thurnheer T., Belibasakis G.N. Quantitative proteomics reveal distinct protein regulations caused by Aggregatibacter actinomycetemcomitans within subgingival biofilms. PLoS One. 2015;10 doi: 10.1371/journal.pone.0119222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bostanci N., Meier A., Guggenheim B., Belibasakis G.N. Regulation of NLRP3 and AIM2 inflammasome gene expression levels in gingival fibroblasts by oral biofilms. Cell. Immunol. 2011;270:88–93. doi: 10.1016/j.cellimm.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Thurnheer T., Bostanci N., Belibasakis G.N. Microbial dynamics during conversion from supragingival to subgingival biofilms in an in vitro model. Mol. Oral Microbiol. 2016;31:125–135. doi: 10.1111/omi.12108. [DOI] [PubMed] [Google Scholar]
- 11.Bao K., Bostanci N., Thurnheer T., Belibasakis G.N. Proteomic shifts in multi-species oral biofilms caused by Anaeroglobus geminatus. Sci. Rep. 2017;7:4409. doi: 10.1038/s41598-017-04594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirakodu S.S., Govindaswami M., Novak M.J., Ebersole J.L., Novak K.F. Optimizing qPCR for the Quantification of Periodontal Pathogens in a Complex Plaque Biofilm. Open Dent. J. 2008;2:49–55. doi: 10.2174/1874210600802010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thurnheer T., Belibasakis G.N. Integration of non-oral bacteria into in vitro oral biofilms. Virulence. 2015;6:258–264. doi: 10.4161/21505594.2014.967608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vieira J., Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 1982;19:259–268. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 15.Bostanci N., Oztürk V.Ö., Emingil G., Belibasakis G.N. Elevated oral and systemic levels of soluble triggering receptor expressed on myeloid cells-1 (sTREM-1) in periodontitis. J. Dent. Res. 2013;92:161–165. doi: 10.1177/0022034512470691. [DOI] [PubMed] [Google Scholar]
- 16.Bittremieux W., Tabb D.L., Impens F., Staes A., Timmerman E., Martens L., Laukens K. Quality control in mass spectrometry-based proteomics. Mass Spectrom. Rev. 2018;37:697–711. doi: 10.1002/mas.21544. [DOI] [PubMed] [Google Scholar]
- 17.Thurnheer T., Belibasakis G.N. Incorporation of staphylococci into titanium-grown biofilms: an in vitro "submucosal" biofilm model for peri-implantitis. Clin. Oral Implants Res. 2016;27:890–895. doi: 10.1111/clr.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE18 partner repository with the dataset identifier PXD042723. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.