

Abstract
Natural killer (NK) cells, initially identified for their rapid virus-infected and leukemia cell killing and tumor destruction, are pivotal in immunity. They exhibit multifaceted roles in cancer, viral infections, autoimmunity, pregnancy, wound healing, and more. Derived from a common lymphoid progenitor, they lack CD3, B-cell, or T-cell receptors but wield high cytotoxicity via perforin and granzymes. NK cells orchestrate immune responses, secreting inflammatory IFNγ or immunosuppressive TGFβ and IL-10. CD56dim and CD56bright NK cells execute cytotoxicity, while CD56bright cells also regulate immunity. However, beyond the CD56 dichotomy, detailed phenotypic diversity reveals many functional subsets that may not be optimal for cancer immunotherapy. In this review, we provide comprehensive and detailed snapshots of NK cells’ functions and states of activation and inhibitions in cancer, autoimmunity, angiogenesis, wound healing, pregnancy and fertility, aging, and senescence mediated by complex signaling and ligand-receptor interactions, including the impact of the environment. As the use of engineered NK cells for cancer immunotherapy accelerates, often in the footsteps of T-cell-derived engineering, we examine the interactions of NK cells with other immune effectors and relevant signaling and the limitations in the tumor microenvironment, intending to understand how to enhance their cytolytic activities specifically for cancer immunotherapy.
Subject terms: Innate immunity, Cancer microenvironment, Immunotherapy
Introduction
Natural killer (NK) cells were first described as killer lymphocytes that induce rapid leukemia cell death without requiring soluble factors1 and separately, in the same year as lymphocytes distinct from T-cells but capable of killing tumors caused by viruses.2 The knowledge accumulated since then has revealed the complexity of NK cell biology and interactions with cancer cells and virus-infected cells. It also revealed their role in autoimmunity,3–5 angiogenesis,6 wound healing,7–9 pregnancy and fertility,10 aging,11 disease, and senescence.12,13 NK cells are large granular lymphocytes sharing a common lymphoid progenitor with two pillars of adaptive immunity: lymphocytes B and T-cells. However, NK cells do not express CD3, B-cell receptor, or T-cell receptor. A defining feature of NK cells is their high cytotoxicity, rapid recognition, and elimination of threats, suggesting a strong evolutionary pressure in organisms without adaptive immunity to have fast-acting and efficient NK cells with an adequate array of activating receptors to survive insults such as viral infections and intrusion by non-self. NK cells are unique among innate immune cells since they use tools similar to adaptive immunity to resolve these insults. Eliminating these cells by NK cells is achieved, as in the case of T-Cells, by using pore-forming perforin14 designed to create pores with an inner diameter of ~16 nm15 in the target cell membrane and delivery of proteolytic granzymes16 that activate Caspase-3 and 10 to trigger apoptosis and Granulysin (GNLY). This saponin-like toxin lyses bacteria such as Mycobacterium tuberculosis,17 preventing intracellular bacteria’s escape.18 During pregnancy, decidual NK (dNK) cells can deliver GNLY via nanotubes to surgically kill bacteria inside the infected trophoblast without harming it.19 Similar delivery of GLNY is also performed by peripheral blood (PB) NK cells in infected macrophages and dendritic cells (DCs). NK cell’s cellular granularity is due to cytoplasmic vesicles filled with perforin and several granzymes. These granules and the Golgi apparatus all become polarized toward the point of contact with the targeted cell, called synapse, where the cargo is concentrated and delivered.20,21 However, despite this arsenal, NK cells may not eliminate large tumors or systemic viral infections. Their role appears to have been defined by evolution as first responders to deal with emerging threats in collaboration with other components of innate immunity, such as macrophages until adaptive immunity is fully deployed. NK cells are at the center of innate immunity with a presence in strategic organs that constitute barriers, such as the skin, gut, lungs, liver, uterus, breasts, and blood, where NK cells represent 5–15% of the lymphocyte population. In these organs, NK cells could play either an inflammatory role or, counterintuitively, an immunosuppressive one. In the first scenario, they increase inflammation after activation by tumor and virus-infected cells by secreting inflammatory cytokines such as INFγ,22,23 which activates macrophages,24 T-cells,25 and B-cells.26 However, cancer cells treated with IFNγ become resistant to NK cells, suggesting that NK secretion of IFNγ is also designed to involve other immune cells.27 NK cells are also the only lymphocytes that constitutively secrete TGFβ28 to reduce inflammation and inhibit T-cells cytotoxicity and proliferation,29 allowing tissue repair.30 Additionally, there is an increased frequency of autocrine TGFβ signaling by TGFβ-producing NK cells in patients with breast cancer.31 NK cells can also secrete immunosuppressive IL-10 in an early response to systemic, but not local infection.32,33 This secreted IL-10 indirectly limits T-cell activation by blocking APCs secretion of IL-12 and producing factors involved in antigen presentation34 and T-cell anti-viral response,35 thus promoting T-cell exhaustion36 and reducing immune-mediated damage to the host. IL-10, however, improves the effector functions and metabolism of NK cells via the mTOR pathway.37 Therefore, NK cells also have an immunomodulatory role and can influence innate and adaptive immunity through these anti- and pro-inflammatory roles.
The expression level of NK marker CD56 commonly defines the oversimplistic distinction between NK cells mediating these two functions. CD56dim NK cells are efficient killers and produce more perforin and granzymes, while CD56bright NK cells, which produce INFγ, TNFβ, IL-10, IL-13, and GM-CSF, also have immunomodulatory and suppressive functions.38–40 A new refinement of this classification has recently delineated three major populations of NK cells in PB.41,42 However, mass cytometry analysis considering twenty-eight NK cell receptors revealed an astounding 6000 to 30,000 phenotypic populations within an individual, where inhibitory receptors are determined by genetics and activating receptors are by the environment.43 Most circulating NK cells, ~90%, are CD56dim, suggesting that circulating NK cells primary function is to eliminate rapidly targeted cells. Most CD56dim cell subset also expresses CD16 (FcγRIII, Fc gamma receptor III),40 which is necessary for ADCC, again bridging innate and adaptive immunity.
A brief history of five decades of progress in natural killer cell research
In 1971, even before NK cells formal identification, radioresistant lymphoid cells in lethally irradiated mice were reported to reject allogenic bone marrow,44 and the cytolytic activity attributed to PB lymphocytes was reported in 1973.45 In 1975, the term NK “Natural Killer” was coined1 (Fig. 1), and the discovery of IL-2 the same year, later revolutionized NK cell studies.46 In 1986, the “Missing-self” hypothesis was advanced to explain how NK cells pull the trigger.47 Also, in 1986, impaired activity of NK cells in HIV patients was reported.48 In 1988, NK cells were found to express CD16 and to mediate ADCC.49 In 1989, two CD56 subsets (dim and bright) were identified,50 and “interferon-inducing” IL-1251 and IL-18, crucial for NK activity, were discovered. Also, in 1989, the CD3ζ chain was discovered52 and shown to transduce CD16 signaling.53 In 1990, surface antigens with a role in cell activation and regulation of cytolytic function (later called KIRs) in NK cells were reported.54 In 1992, the first NK cell-activating receptor, 2B4, was discovered.55 The “Missing-self” hypothesis implied the existence of inhibitory receptors such as Ly49,56 first found in 1992 in mice, then in 1995 in humans, the KIRs57–59 that bind to MHC I were cloned/identified. A year earlier (1994), Klingemann published the NK cell line NK-92,60 established in 1992 and later used as a model in many NK studies. Cytokine IL-15, necessary for NK cell development, was also discovered in 1994.61,62 In 1996 NK cell activator DNAM-1 was discovered, first in T-cells.63 Natural cytotoxicity receptors (NCRs) will be discovered in succession: NKp4664 in 1997, NKp4465 in 1998, and in 1999, NKp3066 and adapter DAP12.67 In 1998 the inhibitory NKG2A and activating NKG2C receptors interactions with HLA-E were identified.68 In 1999, NKG2D receptor and adapter DAP10 activation by MICA69 and later in 2000 with ULBP70 and Retinoic acid early inducible gene (Rae1)71 were reported. Also, in 2000, IL-21 was discovered and found to expand NK cells.72 In 1999, the role of NK cells emerged in lowering the rates of leukemia relapse in MHC class I and KIR mismatch between the donor and recipient of hematopoietic stem cell transplants in a transplant setting.73 In 2002, the interactions between NK and DC cells were discovered.74–76 In 2003, TGFβ1 was found to impact the interaction between DCs and NK cells by suppressing NKp30 and NKG2D.77 In the same year, PVR and Nectin 2 were identified as ligands for DNAM-1.78 In 2005, Miller et al. pioneered the first use of NK cells in a non-transplant setting and showed the benefit of lymphocyte depletion preconditioning on NK cell expansion and persistence in vivo.79 In 2006, a component of the TME, Tryptophan metabolite, L-Kynurenine was reported to inhibit surface expression of NKp46 and NKG2D.80 In 2008, NK-92’s first phase I clinical trial was published.81 In 2009, NK cell secretion of IL-10 was reported to regulate CD8+T cells to prevent damage35 and another mucosal NK cell subset was found to produce IL-22.82 In 2010, NK cell interaction with macrophages was identified,83 and later in 2012, NK cells were reported to kill Neutrophils.84 In 2012, memory-like human NK cells that expand after transplantation are described.85 In 2015, evidence of adaptive or memory NK cells emerged after epigenetic changes (hypermethylation of Syk gene promoter) were found in NK cells in response to CMV infection.86,87 Also, in 2015, the first clinical trial using feeder-expanded NK cells showed safety and efficacy.88 In 2016, cytokine-induced memory-like NK cells were used in a phase-I clinical trial to show safety and efficacy.89 In 2020, CAR-NK (CD19) cells were used in a landmark clinical study to show safety and efficacy.90 In 2020, severely impaired NK cells were found in severe COVID-19 patients, and these NK cells were unable to kill overactive and inflammatory macrophages.91 Also in 2020, NK cells were discovered to specifically deliver Ganulysin, via nanotubes, to bacteria-infected trophoblasts, DCs and macrophages, without harm.19 In 2022, long-lasting NK cell clonal expansion from HCMV+ patients was reported.92 In early 2024, three major populations of NK cells are identified in PB.41,42 In mid-2024, CAR-NK cells are offered as an experimental option for cancer treatment at MD Anderson, and two NK cell lineage progenitors are identified in two seminal papers.93,94 Also, by mid-2024, NK cells were reported to kill, via NKp30, activated T-cells and CAR-CD19 T-cells expressing B7H6.95
Fig. 1.

Historical narrative of important milestones in NK cell research. Interrogation of the historical record of natural killer cell research from PubMed using the keywords “Natural Killer cells”, “Natural Killer cells and Immunotherapy”, “Natural Killer cells and CAR-NK”, and “Natural Killer cells and Covid”. We provide in the main text of the review a year-by-year narrative of the progress/discovery culminating in the offering of CAR-NK as an “experimental therapy” against cancer at MD Anderson cancer center. In 2021, the number of publications related to “NK cells” is 3.57-fold less than “T-Cells” and the research record of “CAR-NK” is even more minuscule. Both fields show a subsequent slump in research publications in the period 2020–2023 which may be due to the Covid-19 pandemic
NK interaction with the major histocompatibility complex class I molecules
A critical regulator of NK cell reactivity is the major histocompatibility complex (MHC I). NK and T-cells interact and interrogate MHC I complexes from different angles with different outcomes. In several examples, the outcome of these interactions is that target cells that are sensitive to killing by NK cells are resistant to killing by T cells, and the opposite is true, leading to the seminal observation of the “missing self-hypothesis” by Karre.47,96 T-cells, via their TCRs, recognize foreign peptides presented by MHC I complexes and get an activation signal (signal-1) from antigen-presenting cells (APCs) and cancer cells or virus-infected cells. T-cells ignore MHC I-presenting self-peptide or cells with low MHC I expression, such as some virus-infected cells, and delegate this function to NK cells. Additionally, T-cells do not tolerate polymorphism in the HLA groups that compose MHC I complexes and, as a consequence, mediate tissue rejection and destruction. In contrast, NK cell interaction with MHC I induces a tolerogenic signal via inhibitory signals from interacting killer cell immunoglobulin-like receptors (KIRs), and NKG2A/CD94.97,98 Additionally, NK cells tolerate allogeneic variability and polymorphism in HLA99,100 to a certain degree. However, they always interpret MHC I absence in scrutinized cells as non-self that must be destroyed.47,101 Indeed, NK cells were shown to kill preferentially cells lacking MHC I.102,103 NK cells will also destroy cells presenting certain empty MHC I complexes lacking a self-peptide.104 Others reported protection from lysis by empty MHC I.105 However, empty MHC I is unlikely to be expressed at the cell surface as loading the peptide onto MHC I complex is a requirement for quality control before export to the cell membrane,106 and this expression is only seen at temperatures near 26 °C in the absence of TAP (transporter associated with antigen processing).107 NK cells may also kill cells due to mutations in the peptides presented by certain HLA molecules, which may affect the interaction between KIRs and target cells, influencing NK cell activity.108–110
The inhibitory arm involves primarily KIRs with long cytoplasmic domains KIR-2DL, KIR-3DL, or C-type lectin receptors CD94/NKG2A/B interacting with MHC I complex. Inhibitory receptors CD94/NKG2A/B in normal cells recognize HLA-E molecules presenting the leader sequence peptides of the HLA-A, HLA-B, and HLA-C groups. Furthermore, HLA-E becomes only expressed at the cell surface when occupied by these peptides. This recognition of normalcy in cells inhibits NK cells only when they express normal levels of classical HLA class I molecules, effectively preventing NK cell-mediated cytotoxicity against normal cells.
Therefore, MHC I recognition is the primary and default inhibitory mechanism through which NK cells decide to engage scrutinized cells. Thus, the lack of MHC I recognition by KIRs, which exposes the missing self,96 is one of the main and default regulators of NK cell killing (Fig. 2). NK cells achieve optimal functionality through KIRS interactions with the four MHC I classes during their development when NK cells are educated or licensed.111 Tumorigenesis is characterized by reduced MHC I expression.112,113 MHC I deficient cancer cells can escape T-cells, but not NK cells, as these are MHC I unrestricted cells. However, MHC I deficient cancer cells may still escape NK cell surveillance due to other dysfunctions. This escape is mainly mediated through the anergy of NK cells due to weak activation or exhaustion, which can be reversed by cytokines such as IL-18 and IL-12.114
Fig. 2.

MHC I and the balance of stimulations and inhibitions dictate rules of engagement with cancer and stressed cells. Cancer and stressed cells expressing MHC I usually have multiple triggering ligands and can only escape NK cells if the balance of inhibitory signals is higher than activation. Cancer cells deficient in MHC I are killed through the “missing self” rule and are unlikely to escape NK cells, especially if the signaling balance favors activation. Unlicensed NK cells cannot kill through the “missing self” rule because they lack KIRS /CD94/NKG2A/B but are most likely to kill cancer and stressed cells that induce reasonable stimulation of NK cells due to the missing MHC I inhibition. Exhausted NK cells, usually having a dominance of inhibitory receptors, are less likely to kill cancer and stressed cells
Overall, the interactions of NK cells and T-cells with MHC I are quite similar but yield different outcomes. NK cell interrogation of MHC I creates a tolerance signal that accepts HLA polymorphisms unless HLA is completely missing, very polymorphic, or from another species. This tolerance signal is relevant to fetus implantation, transplantation, and rejection and is evident in the urochordate Botryllus. Schlosseri, the closest invertebrate to vertebrates, which has only NK cells with no T or B-cells.115–117 Each B. Schlosseri individual transplants daily with others to form chimeras, and each need only one common allele of Botryllus histocompatibility factor118–120 to transplant with another individual successfully. The B. Schlosseri histocompatibility complex allowing this transplantation has extensive polymorphism119 and the mechanism that controls the tolerance signal and success of transplantation is mediated by BsCD94-1gene, a CD94-related transmembrane receptor of vertebrate NK cells, expressed on the surface of a subpopulation of Botryllus blood cells and upregulated during the allorecognition process.121 CD94 is expressed in modern NK and CD8 T-cells to interact with non-classical MHC I HLA-E, presenting the leader sequence peptides of HLA-A, HLA-B, and HLA-C groups. CD94 associates as a heterodimer with NKG2C and DAP12 to activate NK and T-cells or with NKG2A to inhibit them. This suggests first that NK cells are more ancient than T-cells and second that original NK cells via MHC I may have been designed initially to identify the self but also to regulate asexual reproduction and tolerance between two close individuals.
NK strategies to identify the self
NK cells utilize two strategies to identify the self through MHC I. In one strategy, they recognize polymorphic MHC I proteins using polymorphic KIRs. In another strategy, they utilize the CD94-NKG2 receptor to query HLA-E, presenting conserved peptides derived from all HLA-A, B, and C classes. Both signals synergize to further prevent NK cells from killing normal cells. KIRs interactions with the four MHC I classes have been solved by crystallography.122–124 Structural analysis shows the two immunoglobulin-like extracellular domains of KIRs, D1 and D2 (in KIR2D receptors), to be arranged, depending on KIR members, like two hands (V-shaped) with angles between 66° and 81° and with each hand slightly twisting (along the axis of D1 or D2) at the wrist (hinge). This opening of the angle was found to affect the affinity of KIRs to HLA-C ligands.125 Near the KIR’s wrist is placed the presented peptide in a groove between the α1 and α2 helices of HLA. At this KIR-peptide-HLA interface, on the KIR side, D2 interacts with a well-conserved docking region of the HLA α2 helix spanning from amino acids 145 to 151. On the other hand, the regions of interaction between D1 of KIR and α1 helix are variable and seem to determine the specificity for each KIR. KIRs exhibit a high degree of polymorphism in humans, with a number of 2238 alleles reported in 2024 (https://www.ebi.ac.uk/ipd/kir/). This genetic diversity is “the single most important factor that shapes functional NK cell repertoires”.126 As an example of KIRs diversity in a defined population, a recent study reported using 1173 individuals of Japanese descent, 118 KIR alleles in 13 genes.127 The high diversity of the 16 different KIR genes on chromosome 19q13.4 is promoted by their head-to-tail orientation, which facilitates the deletion or duplication of KIR genes. KIRs are categorized into two haplotypes: A, which mainly encodes inhibitory KIRs, and B, which encodes stimulatory KIRs. The number of KIR genes per individual varies on different haplotypes and ranges from six to sixteen genes. As a rule, a particular KIR gene in an individual will be expressed stochastically in some NK cells, leading to subsets of NK cells within a person expressing different combinations of KIR receptors, with a majority not exceeding two. This stochastic expression increases the diversity of NK cells, with some NK cell subsets having only inhibitory and other subsets only stimulatory KIRs.128 KIR2DL4 is present in all haplotypes and is exceptionally expressed in all individuals. HLA-G, a non-classical HLA class I molecule, specifically expressed in extravillous trophoblasts is the only known ligand of KIR2DL4, and as we will see later, plays a major regulatory role in maternal-fetal immune tolerance and is also highly expressed in tumors.
An important difference between activating and inhibitory KIRs is that despite the high homology of their extracellular domains their binding to MHC I is weaker compared to inhibitory KIRs. KIRs that transmit inhibitory signaling have longer intracellular domains containing an immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which associate with phosphatases like SHP-1. In contrast, KIRs that transmit activating signaling have a short intracellular domain containing an immunoreceptor tyrosine-based activating motif (ITAM) that associates with activating adapter DAP12 to signal through Syk/ZAP-70 tyrosine kinases. An exception to this rule is KIR2DL4, which is a long-tailed but activating KIR that associates with FcεRI-γ instead of DAP12.129 Although KIR2DL4 is defined as an activating KIR, its association with ligand HLA-G does not lead to more NK cells cytotoxicity but rather to cytokine secretion130 as do dNK cells. KIR2DL4 expression at the cell surface is restricted to cytokine-producing CD56bright and is not detected on CD56dim NK cells surface but interestingly did so after cell culture in vitro.131 However, KIR2DL4 is also located intracellularly in the endosomes of CD56dim primary NK cells, where it can be activated by soluble HLA-G.132 This endosomal signaling by KIR2DL4 activates NF-κB and AKT, leading to IFNγ secretion.133 HLA-G can also be transferred to NK cells via endocytosis134 and trogocytosis, leading to a state of tolerance without compromising the antiviral response.135 This induced state of tolerance could also drive tumor resistance to therapies and affect the tumor microenvironment.136 KIR2DL4 fulfills its inhibitory receptor role when bound by HLA-G (soluble, membrane-bound bound, or trogocytosed). This triggers the phosphorylation of KIR2DL4, ITIM domain, leading to the recruitment of SHP-2 and the dephosphorylation of downstream signaling activating molecules and decreasing NK cell cytotoxicity. However, due to a positively charged Arginine on its transmembrane domain, KIR2DL4 can associate with FcεRI-γ.129 This association leads to the phosphorylation of the ITAM on FcεRI-γ, thus allowing NK cells to produce cytokines, including IFNγ, even though the cytotoxic response is generally suppressed due to the ITIM in KIR2DL4. The role of inhibitory KIRs is to interpret a “do not kill me” signal from HLA presenting a self-peptide, while activating KIRs are to interpret a “kill me” signal from HLA presenting specific viral peptides137 or open HLA with no peptides.138,139 The protective role of activating KIRs against certain viral infections has been reported for KIR3DS1+ NK cells against HIV-1140 and H1N1 influenza.141 However, activating KIRs could also prolong inflammation and injury, as in chronic hepatitis,142 and as we will see later, KIR composition could also affect autoimmunity.
Maturation and education of NK cells (the pre-2024 view)
The earliest NK progenitor was described in the bone marrow of mice143; consequently, bone marrow ablation results in NK cell deficiency. NK cells mature and receive an “education” or “license” early in the bone marrow (Fig. 3). This process is designed to increase their reactivity threshold by experiencing inhibitory signals from self-MHC I. Indeed, the capacity of a future mature NK cell to respond to stimulation is quantitatively determined by the strength of inhibitory signals received from MHC I molecules during NK cell education.144 Uneducated NK cells respond to inhibitory signals with strong production of phosphatase SHP-1, leading to their rapid inactivation, while educated, licensed NK cells have reduced SHP-1 production when encountering these inhibitory ligands, allowing them to remain activated.145 Therefore, educated NK cells are more cytolytic, and their maturation starts from a CD34+ human hematopoietic stem cell or mouse Sca+, CD117+ to the common lymphoid progenitor, which expresses IL2Rβ, responds to IL-15,146 and maintains this expression throughout the maturation stages, branching into an intermediary natural killer precursor (NKP) committed to developing into NK lineage which develops first into an immature iNK cell and then a mature NK cell with a CD56bright phenotype that upon further maturation becomes CD56dim 147 in humans. In the mouse, the NKP precursor develops into an immature iNK-a then an iNK-b stage, which is closer to human CD56bright stage with further maturation by acquisition of Ly49. In humans, this phylogeny is supported by the longer telomeres found in CD56bright compared to CD56dim.148,149 Although they are functionally similar in their interaction with MHC I. There are significant differences between mouse and human NK cells at the level of markers, residency, and longevity. For example, human NK cells can be expanded in vitro for extended periods of time, while mouse NK cells always die after a few weeks in culture. Similarly, opposite to humans, mouse NK cells are seldom found in the lymph nodes and mouse NK do not express CD56. Gradually, during their development, human NK cells acquire their receptors, starting with inhibitory CD161, then adhesion molecule CD56, inhibitory CD94/NKG2A, and activation receptors NKp46 and NKG2D. Acquisition of inhibitory and activating KIRs and later, CD16 complete their maturation.150–152 Transcription factor EOMES plays a role in early NK cell maturation and enhances CD16 expression, while T-BET controls maturation markers and induction of KIR expression.153 However, it is essential to note that maturation and education could be carried out in lymph nodes, thymus, uterus, liver, and mucosal lymphoid tissues, probably for cells that drop early of the bone marrow education before the maturation of CD56bright to CD56dim and the acquisition of CD16 and KIRs. Interestingly, the proportion of CD56bright CD16neg is higher in fetal tissues,154 and this population also decreases with age while CD56dim CD16pos increases.155 This suggests that maturation and education of “dropout” NK cells at the CD56bright stage and earlier is high at a young age in the bone marrow and is reduced in the elderly. This might be due to the age-related decline of the secondary lymphoid sites, such as the thymus156 and lymph nodes.157,158 Possibly, at a younger age, these secondary sites might be more able to recruit less mature NK cells and induce them to exit the bone marrow early.
Fig. 3.

Development and maturation of NK cells. A pre-2024 view. Starting from a CD34+ hematopoietic stem cell in the bone marrow to a fully functional and mature CD56pos CD16pos NK cell. NK cell development and maturation (left to right axis) is marked by the acquisition of cytokine receptors responding to IL-15 transcription factors EOMES, T-bet and AhR and the acquisition of inhibitory receptors KIRs and antibody-mediated cytotoxicity receptor CD16. Due to increased adhesion proteins, NK cells could be released earlier than expected and migrate to secondary lymphoid organs to continue their varied maturations and education. NK cells released earlier CD56bright KIRNeg and CD56dim KIRNeg are unlicensed and their proportions in humans and mice are substantial, suggesting an evolutionary advantage to unlicensed NK cell release from the bone marrow, which is frequent at younger age and subsides to favor fully mature NK cells in older adults
Like the stromal cells of the bone marrow, which provide necessary cytokines such as IL-15 and IL-7 for NK maturation,159 the stromal cells found in secondary lymphoid sites such as the spleen can also provide these cytokines.160 However, secondary lymphoid sites contain other monocyte populations like DCs, which might provide additional cytokines such as IL-2 and IL-15.161 Since mouse models have shown that bone marrow ablation results in NK cell deficiency, it can be assumed that any NK cell maturing in a secondary lymphoid organ is originally from the bone marrow regardless of its maturation stage. Indeed, upon exiting the bone marrow at the earliest NKP stage, these cells can be found transiting in PB among the CD34+ hematopoietic stem cell population. Not surprisingly, CD34+ NKP cells in lymph node highly express surface proteins, CD62L, lymphocyte function-associated antigen 1 (LFA-1), and α4β7 integrins, allowing cell migration, high binding, and rolling adhesion.162 It is unclear if NK cells that exit the bone marrow at early stages can be licensed elsewhere or if they remain unlicensed without acquiring KIRs. Both humans and mice present a large population of unlicensed NK cells without KIRs or Ly49, respectively. In humans, 62% of CD56 bright NK cells lack KIRs, while 26% of CD56 dim NK cells don’t express them, suggesting a large population of circulating NK cells is unlicensed163 and that more CD56bright exit the bone marrow earlier. Similarly, 50% of NK cells in mice are Ly49 negative and unlicensed.111
Interleukins IL-12, IL-15, and IL-18 play a significant role in NK cell maturation and can reeducate unlicensed NK cells to enhance their functionality and exert stronger responses.164 KIRs acquisition by unlicensed human KIRNeg that are CD56bright and CD56 dim NK cells can be obtained after stimulation with IL-15 in the presence of stromal cells.165 Similarly, de novo expression of KIRs and NKG2A in unlicensed NK cells can be obtained using IL-2, IL-15, or IL-12 only.164,166 These observations have an important impact on immunotherapies using primary NK cells. Moreover, NK cells infiltrating solid cancers have been reported to be predominately CD56bright.167 Therefore, it is essential to understand how these unlicensed NK populations operate compared to licensed ones and if licensing is required for NK cells to carry out their functions.
In a tumor environment characterized by reduced MHC I expression,112,113 the fate of cancer cells facing licensed NK cells is almost certainly death and will be influenced by the balance between activators and inhibitors on their surface (Fig. 2). If NK cell activation by MHC I deficient cancer cells is weak or the balance of inhibitory signals is high, leading to anergy and exhaustion of NK cells, then activation by cytokines such as IL-18 and IL-12 may restore their activation.114 However, licensed NK cells in an MHC I sufficient environment will be inhibited, especially without activation or with increased inhibition from cancer cells. This exact experiment was reported using MHC I deficient cell line RMA-S and MHC I sufficient RMA cell lines grown subcutaneously in the same mouse. It showed better control of MHC I deficient RMA-S tumors.47 This suggests that the MHC I expression could offer an escape mechanism from licensed NK cells in the absence of a convincing activation that could override MHC I inhibition. However, this escape is unlikely with unlicensed NK cells that don’t express KIRs. Indeed, KIR-deficient unlicensed NK cells are more efficient than licensed NK cells at killing MHC I sufficient RMA cells.168 Similarly, the blockade of KIRs enhanced ex-vivo patient-derived NK cell cytotoxicity against multiple myeloma.169 Therefore, unlicensed NK cells offer an evolutionary advantage against the narrow NK specialization and broaden the spectrum of action for NK cells instead of relying on one rule regarding MHC I status. This is even more obvious in the case of viral infection against which NK cells are essential, where particularly unlicensed NK cells offer an edge. Viruses can alter MHC I antigen presentation in an attempt to escape T-cells.170 MHC I alteration leading to its downregulation does not escape licensed NK cells. However, few viruses, such as MCMV, express mimics of MHC I that bind to Ly49, the equivalent of KIRs in mice, and mediate repression of NK cell function.171 Immunological synapses initiated by NK cells when in contact with cancer cells are inhibited by KIRs.172 Since unlicensed NK cells do not express inhibitory KIRs but express activating KIRs, the binding by the viral MHC I mimics to activating KIRs leads to the activation of NK cells, making them instrumental in resisting MCMV infection. There is an evolutionary advantage to having polyfunctional populations of licensed and unlicensed NK cells that can be CD56dim or CD56bright with numerous phenotypes estimated in the thousands, maturing and receiving different “educations” in the bone marrow or second lymphoid organs. This advantage is apparent when facing threats that use evolution as a mechanism to adapt.
Maturation and education and the new view on the origin of NK lineages
The Common lymphoid progenitor (CLP) can generate, in addition to committed NK cells, Innate lymphoid cells173 (ILCs) (Fig. 4). These are mostly tissue-resident innate immune cells without cytolytic activity and are subdivided into three groups. The ILC1s group when stimulated by IL-12, IL-15, and proinflammatory IL-1b will produce IFNγ, without cytolytic function, termed type 1 immunity, and participate in viral and bacterial infection. ILC2 group function is type 2 immunity and responds to parasites such as helminths and allergens when stimulated by IL-25, IL-33, and TSLP. The ILC3 group mediates type 3 immunity in response to microbes, such as bacteria, by producing, among others, antimicrobial peptides when stimulated by IL-1b and IL-23. In mice both ILC1s and NK cells produce IFNγ, are both NK1.1+, NKp46+, CD3ε− and express transcription factor T-bet.174 Commitment to an ILC progenitor (ILCP) lineage but not NK lineage requires the expression of transcription factor PLZF.175 However, ILCP co-expressing PLZF and Inhibitor of DNA binding 2 (ID2) retain the potential to produce an NK cell lineage suggesting a common ancestor of ILC1s and NK cells.176 Both ILC1s and NK cells express T-Bet. However, ILC1s do not express EOMES, while it is essential for NK cell development in the bone marrow.177,178 Using single-cell sequencing, two very recent studies aimed to understand how NK cells that appear after birth, originate and differentiate from ILC1s group, which are present in fetal life and beyond. In one study, Liang et al. show the expression of both PLZF and EOMES to confer both an NK and ILC1s potential and that NK-committed precursor cells express Eomeshigh 93 but not PLZF and that the expression of Eomes transcription factor precludes the development of ILC2 and ILC3 groups. In the other study, Ding et al.94 identified two NK-committed lineages. One from an early NK progenitor (ENKP), developing into Ly49H+ NK cells and an ILCP-derived NK lineage with low expression of Ly49H. Both studies identify NK-committed lineages in the bone marrow, which may represent different stages of NK progenitor development. Eomes expression is, therefore, intrinsic to the NK phenotype, and the higher Eomes expression is, the closer to the mature phenotype NK cells are.
Fig. 4.

Development and maturation of NK cells. A 2024 view. NK cells originate from two lineages. An early natural killer progenitor (ENKP), which produces the CD56dim population, and another progenitor deriving from an innate lymphoid progenitor (ILCP), which produces both CD56bright and also ILCs. Both ENKP and ILCP would originate from a common lymphoid progenitor (CLP). The ENKP derived CD56dim population matures, after an intermediary stage NKint, into an NK1 subset composed of three subsets: NK1A, NK1B, and NK1C with increased maturation but differing phenotypes related to response to surface receptors (NK1B), cytokine response and increased cytolytic activity (NK1C). A later more mature stage NK3 is characterized by increased CD57 expression, suggesting an adaptive phenotype with high NKG2C and antiviral potential that may lead to clonal expansion of adaptive/memory cells or may lead to senescence if no viral event occurs. The CD56bright less mature population (NK2) is characterized by enhanced chemotaxis and is unlicensed with no KIRs and no CD16. NK2 subset is probably the source of dNK cells in pregnancy after migrating to the uterus. NK1B subset’s high response to TGFβ, IL-10, and IL-12 suggests it may contribute to building dNK populations with the potential to reduce the NK1C subset
In human PB, Vivier et al.41 delineated three major subsets of NK cells discernible through single-cell transcriptomic analysis. One subset, called NK2, is CD56bright and CD16neg, along with ID2 expression, and lacks KIRs, suggesting an immature phenotype. This subset showed markers of tissue residency. The most abundant subset in the blood, called NK1, is CD56dim CD16pos, which expresses KIRs, GZMA, GZMB, and PRF1, a phenotype that suggests maturity. A third subset, termed NK3, is NKG2Chigh, CD16dim, CD57pos, suggesting further maturation and an adaptive phenotype. Of note, adaptive NKG2ChighCD57+ cells expand in humans infected with HCMV.179,180 The study concludes that the two populations, NK1 and NK3, are originating from ENKPs, and that NK2 cells originate from ILCPs.
It is unclear whether these three populations exhibit plasticity and can convert into one another. However, there are reports of conversion from a CD56dim to CD56bright phenotype under IL-12.181 Interestingly, TGFβ can convert PB CD16pos into a CD16neg decidual like NK cells,182 and NK cells exposed to TGFβ or its relative Activin, acquire a gene signature and phenotype similar to the less cytotoxic ILCs, becoming unable to control tumor growth in mice.183–185 This suggests IL-12 and TGFβ1 may be possible mechanisms for converting NK1 to an NK2-like state or NK2 to an ILC state. Of the three subsets composing NK1 (NK1A, NK1B, and NK1C), NK1B appears the most likely to convert to an NK2-like or decidual phenotype as it has a strong response signature to IL-12, TGFβ, and IL-10.
The NK1 subset with further maturation leads to the NK3 phenotype, with increased KIRs and high CD57 expression. CD57 is associated with more experienced and terminally differentiated NK cells, possibly on the verge of senescence186 with higher frequency in older age.187 CD57 is also a marker of senescent T-cells that have short telomeres and low replication potential.188,189 NK3 population might specialize in highly effective and adaptive properties with memory-like features if they encounter an event such as viral infection. In the absence of such an event, they could become terminally senescent. This antiviral phenotype is suggested by the gradual increase from NK1 to NK3 of Granzyme H, which destroys critical adenoviral viral proteins that inhibit granzyme B, which is also present in NK3.190 Granzyme H also destroys the La-mediated HCV-IRES translational activity.191 Similarly, the exclusive expression of CCL5 in NK3 suggests antiviral defenses against Influenza A virus.192 Moreover, IL-32, which is elevated in NK3, plays a crucial role in responding to infections caused by viruses like HIV-1 and influenza. Additionally, it provides protection against cell death induced by the vesicular stomatitis virus. Notably, IL-32 exhibits antagonistic effects against the DNA virus HSV-2 in both epithelial Vero cells and human umbilical cord endothelial cells, thereby influencing the production of HSV-2,193 Finally, NK3 increased NKG2C expression reinforces the antiviral defense194 and NKG2C as well as ADCC mediated responses are enhanced by co-stimulatory molecule CD2195 which is also induced in NK3 subset.
Vivier et al. examined whether any subset is preferentially found in patients’ tumors and found the proportion of NK2 cells was increased in most tumors tested. NK2 population was characterized by higher CXCR3 expression, in agreement with better homing into tumors of CXCR3+ NK cells in a CXCL10-dependent fashion, leading to improved survival.196 However, CXCR2 and CXCR4 (distinguishing NKint and NK1A, respectively) were reported to enhance the migration of human primary NK cells to tumors expressing their ligands.197 NK1B cells high potential to respond to activation through increased surface receptors, suggest their potential in immunotherapeutic strategies. However, the NK1C subset’s pronounced cytoskeletal activity and cell-killing signature suggest it is the most cytotoxic. Overall, these studies suggest that an NK phenotype that is optimal for cancer immunotherapies may be within reach but still awaits further confirmation. Therefore, the lineage ENKP to NK1 might be the phase with the highest antitumor activity, while the further mature state NK3 excels in antiviral defenses. The lineage ILCP to NK2 appears to be mainly tasked with cytokine production and immunoregulatory functions like dNK. We can also infer that NK2 subset which is CD56bright CD16neg and KIRneg is probably the seed of dNK cells that migrate to decidua in pregnancy, to mature and gain KIRs without gaining CD16.
NK cell activation mechanisms that trigger killing
NK cells exhibit rapid activation and launch cytotoxic attacks on stressed, senescent, virus-infected, and cancer cells, bypassing the need for prior antigen presentation by MHC I. Unlike T and B-cells, which express specific activating receptors, NK cells express all activating and inhibitory receptors, creating an intricate and complicated equilibrium between multiple activating (Fig. 5) and inhibitory signals (Fig. 6) arising from their interaction with ligands on target cells with, however, a dominance of inhibitory receptors.198 It is important to note that except CD16, no other single activating receptors, including NKp46, NKG2D, 2B4, DNAM-1 (CD226), or CD2, are sufficient to activate NK cells on their own.199,200 Additionally, unlike most inhibitory receptors, many activating receptors, including KIRs, have no proper cytoplasmic signaling domain and rely on associations with adapter molecules that have ITAMs, allowing the creation and transmission of activating signals.
Fig. 5.

Dynamics of activation signaling in NK cells in contact with cancer and stressed cells. Activation signaling from Slam family 2B4, NTB-A, and CRACC. Upon ITSM phosphorylation, following ligand binding, an activation signal can be generated depending on the recruitment of EAT-2 and SLAM-associated protein (SAP), thereby blocking the binding site of lipid phosphatases SHP-1 and SHP-2. SAP recruits the Src-family kinase Fyn, leading to downstream PLCγ1, PLCγ2, and PI3K signaling. 2B4 can also recruit after phosphorylation, another adapter protein 3BP2, which activates VAV1 and ERK pathway upon phosphorylation. DNAM-1 engaged with ligand PVR or nectin-2 is tyrosine phosphorylated by Src kinases. This phosphorylation enables the binding of adapter Grb2 to DNAM-1, leading to VAV1, PI3K, SLP-76, and PLCγ1 activation, thereby increasing calcium fluxes and activating ERK and AKT pathways leading to FOXO1 degradation. DNAM-1 activating signal has a synergetic effect with LFA-1, to which it can be associated physically to induce tyrosine kinase Fyn to phosphorylate DNAM-1. NKG2D associates with adapter DAP10 after binding to ligand UL16-binding proteins (ULBP)1–6 and to ligands MICA and MICB, whose expression is regulated by the heat shock stress pathway or by DNA damage induced by chemotherapy and radiotherapy. NKG2D activation is triggered upon ligand engagement, leading to assembly with adapter DAP10 and phosphorylation of its motif followed by recruitment of PI3K, growth factor receptor-bound protein 2 (Grb2), VAV1, SLP-76, GTPase Rac1-dependent actin cytoskeleton rearrangement, thereby leading to MAPK signaling pathway activation and Pak1–Mek–Erk cascade signaling pathway. This culminates in granule polarization, calcium influx, cytokine production, synapse formation, and clustering of receptors. A parallel activation pathway triggered by PI3K is the AKT/mTOR pathway activation. NCR activation and killing depend on Src and Syk kinase activities. Engagement of NCRs with their cognate ligands induces associations with adapter CD3ζ, FCRγ or DAP12 whose ITAMs are phosphorylated by members of the Src kinase family: Lck, Fyn, Lyn, Fgr, Src, and Yes. The phosphorylated ITAMs will then attract and activate the tyrosine kinases Syk and ZAP70. These kinases will then phosphorylate other adapters, such as LAT, to recruit more downstream adapters and signaling complexes, such as PLCγ and PI3K, VAV1/2/3. Under PLCγ, Ca2+ influx increases, and PI3K will recruit p85, leading to phosphorylation of AKT and VAV1, which promotes GTPase Rac1-dependent actin cytoskeleton rearrangement, thereby activating the MAPK signaling pathway, leading to the Pac1–Mek–Erk cascade signaling pathway. The PI3K/AKT/mTOR pathway triggers a parallel activation pathway. CD16 is the only receptor in NK cells that can trigger alone and with the homodimer of adapters CD3ζ or FCRγ, an effective activation signal mediating antibody-dependent cellular cytotoxicity (ADCC). Only CD16 activation can lead to phosphorylation of both tyrosines (Y128) and tyrosine (Y113) on SLP-76. This double phosphorylation allows the binding of two VAV1 and more robust downstream signaling. Complexed Crk is required for CD16 signaling and the movement of microclusters of CD16 ligands on the lipid bilayer
Fig. 6.

Dynamics of inhibitory signaling to block NK cell activation. Inhibitory receptors, including MHC class I-specific inhibitory receptors, target VAV1 for dephosphorylation by Src homology 2 domain-containing protein tyrosine phosphatase 1 SHP1. Another potent inhibitory relay is Crk dissociation mediated by c-Abl phosphorylation of Crk, which in its active form (non-phosphorylated) is associated with the complexes c-Cbl/Crk/C3G and p130CAS/Crk/C3G. C-Abl phosphorylation of Crk causes its dissociation from these complexes. Inhibitory signaling by CD94-NKG2A binding to HLA-E uses the E3 ubiquitin ligase c-Cbl to enhance the degradation of phosphorylated VAV1 and its downstream signaling PLCg2. Receptor tyrosine kinases TAM receptors (Tyro3, Axl, and Mertk) are expressed by multiple immune cells, including NK cells. TAM receptors phosphorylate ubiquitin ligase Cbl-b and dampen NK-cell activation signaling by promoting the degradation of LAT1, thus blocking VAV1-dependent signaling and, blocking, among others, glutamine transport and the fueling of the tricarboxylic cycle. DNAM-1 inhibition occurs when PD-1 recruits SHP2 to inhibit DNAM-1 phosphorylation via its intracellular domain signaling. TIGIT induces inhibitory signaling, while on the cancer cell side, PVR interaction with ligands TIGIT or DNAM-1 leads to tyrosine phosphorylation of the PVR’s ITIM domain by Src kinases and recruitment of SHP-2 followed by dephosphorylation of focal adhesion kinase and paxillin thereby reducing adhesion, increasing motility, survival, and proliferation of cancer cells. PD-1, CTLA-4, and TIGIT all recruit SHP-1 and SHP-2 leading to VAV1, PIP3 and SLP76 dephosphorylation. TIM-3 inhibition leads to Bat-3 release, which inhibits Lck and Zap70 activation and promotes with P300 the transcription of antiproliferative genes. LAG3 inhibition blocks STAT5 activation and reduces mitochondrial mass. Lair-1 inhibition by tumor collagen leads to SHP-1 and SHP-2 docking, VAV1 dephosphorylation and inactivation of NK cells
Natural cytotoxicity receptors
Among the most potent activating receptors in NK cells, CD16 is the only receptor in NK cells that can trigger alone, in association with the homodimer of adapters CD3ζ or FCRγ, an effective activation signal mediating antibody-dependent cellular cytotoxicity (ADCC). A process where NK cells destroy target cells coated with antibodies.201 Other potent activating receptors for NK cells lacking an activating cytoplasmic tail include the natural cytotoxicity triggering receptors (NCRs) (NKp46, NKp30, and NKp44)202 (Fig. 5). However, some NKp44 isoforms contain a cytoplasmic ITIM-like motif.203 NCR ligands are not expressed in normal cells but are induced in pathological conditions.204 NKp3066 is critical for NK interactions with DCs and binds to ligand B7H6 expressed exclusively on tumor cells,205 but is also transiently expressed by activated T-cells.95 NKp46 receptor206 was recently found to recognize externalized calreticulin (ecto-CRT) expressed during ER stress, virus infection, and senescence.207 NKp46 prevents metastasis208,209 and mediates cytotoxicity against cells that are otherwise resistant to NK cells through the secretory pathway and TRAIL.210
Both NKp30 and NKp46 use activating adapters CD3ζ or FCRγ. NKp44211 interacts with ligand NKp44L,212 and uses homodimers of activating adapter DAP12.203 NKp44 exists in three isoforms (NKp44-1, 2, and 3), with the cytoplasmic domain of NKp44-1 containing an ITIM-like domain (EILYHTVA). The expression of ITIM-bearing NKp44-1 inhibitory isoform has been reported to be detrimental to the survival of acute myeloid leukemia patients.213 However, its expression during pregnancy in dNK cells214 allows decidua vascularization, maternal-fetal tolerance, and antiviral resistance. In this context, trophoblasts expression of NKp44L proliferating cell nuclear antigen (PCNA)215 and ligation to NKp44 through HLA or exosomes inhibits dNK cells through the ITIM-like domain, inhibits IFNγ secretion, and reduces their toxicity. Similarly, three forms were described for NKp30 (A-C) with different cytoplasmic sequences due to alternative splicing. Forms A and B induce IFNγ, TNFα, and IL-12B, while form C induces IL-10.216 Additionally, soluble B7H6 (sB7H6)217 and BAG-6 (sBAG-6)218 downregulate or inhibit NKp30 signaling. sBAG-6 is detectable in high levels in Chronic lymphocytic leukemia patients at advanced disease stages. Surprisingly, NK cells were activated when BAG-6 was presented on the surface of exosomes.219 This suggests an imbalance between soluble and exosomal BAG-6 could promote CCL evasion. Moreover, NKp30 and NKp44 engagement with cancer cells can induce NK cell death via the upregulation of Fas Ligand in certain tumors.220 Surprisingly, overexpression of NKp44 in NK-92 was shown to inhibit activation after binding of NKp44 to PCNA, which is widely overexpressed in tumor cells.221,222
NCR activation and the ensuing killing largely depend on Src and Syk kinase activities.223,224 The engagement of NCRs with their cognate ligands will induce associations with adapter CD3ζ, FCRγ or DAP12 whose ITAMs are phosphorylated by many redundant members of Src kinase family: Lck, Fyn, Lyn, Fgr, Src and Yes. The Phosphorylated ITAMs will then attract and activate the tyrosine kinases Syk and ZAP70 (Fig. 5). These kinases will then phosphorylate other adapters, such as LAT (linker for activation of T cells or P36). LAT is tyrosine phosphorylated upon stimulation of NK cells through FcγRIII receptors following contact with target cells to recruit more downstream adapters and signaling complexes, such as phospholipase C (PLCγ), phosphatidyl- inositol-3-OH kinase (PI3K), and guanine nucleotide exchange factor VAV1/2/3. Under PLCγ, Ca2+ influx increases, and PI3K will recruit p85, leading to phosphorylation of AKT, and VAV1, which promotes GTPase Rac1-dependent actin cytoskeleton rearrangement, thereby activating the MAPK signaling pathway, leading to the Pac1–Mek–Erk cascade signaling pathway. Since AKT is a major downstream target of PI3K,225 a parallel activation pathway is triggered by the PI3K/AKT/mTOR pathway. All these events culminate in granule polarization, calcium influx, cytokine production, synapse formation, and clustering of receptors. CD59 is another activating receptor physically associated with NKp46 and NKp30. Its activation leads to tyrosine phosphorylation of CD3ζ chains associated with these NCRs.226
NKG2D receptor
Another pivotal receptor involved in NK cell tumor and senescence surveillance, a member of the NKG2 family of receptors, is NKG2D. In humans, due to the lack of an activation domain in its cytoplasmic tail, NKG2D associates with adapter DAP10227 after binding to ligand UL16-binding proteins (ULBP)1–6228 and to ligands MICA and MICB,69 whose expression is regulated by the heat shock stress pathway229 or by DNA damage induced by chemotherapy and radiotherapy.230 NKG2D ligands are absent in normal tissues but widely expressed in many cancers, including colorectal and ovarian cancers.231,232 Experimental evidence shows that the inducible expression of surface NKG2D ligands in tumors effectively controlled their initiation or growth233 and that mice deficient in NKG2D could not control tumors.234 However, just as it is common for other receptors such as NCRs, NKG2D ligands are also shed in soluble forms: sMICA and sULBP2, which have inhibitory properties.235 This inhibition is exerted even in the presence of membrane NKG2D ligands. Soluble NKG2D ligands shedding by tumors is metalloproteinases-dependent236 and could lead to high levels of NKG2D ligands in the sera and the tumor microenvironment to the point that NKG2D ligands inhibition with antibodies could enhance CTLA-4 and PD-1 immune checkpoint blockades.237,238 Soluble sMICA and sULBP2 levels in the serum of patients with oral squamous cell carcinoma, melanoma, and NSCL correlated with disease progression.239–241
NKG2D activation is triggered upon ligand engagement leading to assembly with adapter DAP10 and phosphorylation of its motif Tyr-ILe-Asn-Met at Tyrosine followed by recruitment of PI3K, growth factor receptor-bound protein 2 (Grb2), VAV1, SLP-76, GTPase Rac1-dependent actin cytoskeleton rearrangement, thereby leading like in the case of NCRs to MAPK signaling pathway activation and Pak1–Mek–Erk cascade signaling pathway. This culminates in granule polarization, calcium influx, cytokine production, synapse formation, and clustering of receptors. Similarly to NCR activation, a parallel activation pathway triggered by PI3K is the AKT/mTOR pathway activation.
The SLAM family of receptors
Other critical receptors initiating NK cell responses upon binding to specific ligands on target cells are receptors of the signaling lymphocytic activation molecule family (SLAM) that possess one or more immunoreceptor tyrosine-based switch motif (ITSM) in their cytoplasmic tails. These are 2B4 (CD244), which is activated by ligand CD48,242,243 self-ligand NK-T-B-Antigen (NTB-A),244 and self-ligand CRACC.245 Upon ITSM phosphorylation, following ligand binding, either an activating or an inhibitory signal can be generated depending on the recruitment of EAT-2244 and SLAM-associated protein (SAP), thereby blocking the binding site of lipid phosphatases SHP-1246 and SHP-2,247 which generally inhibit NK effector functions and cytokine release. SAP is also able to recruit the Src-family kinase Fyn.248 CRACC, however, can associate only with EAT-2 but not SAP, leading to an effective downstream PLCγ1, PLCγ2, and PI3K signaling.249 2B4 can also recruit after phosphorylation, another adapter protein 3BP2, which upon phosphorylation, activates VAV1 and the ERK pathway.250
DNAM-1 receptor
DNAM-1(CD226)63 is a crucial co-stimulatory receptor for NK cells with a prominent role in anti-tumor and anti-viral surveillance. DNAM-1 cytoplasmic tail contains an ITT-like motif (YVNY), which upon DNAM-1 engagement with ligand PVR or nectin-2 is tyrosine phosphorylated by Src kinases. This phosphorylation enables the binding of adapter Grb2 to DNAM-1, leading to the activation of VAV-1, PI3K, SLP-76, and PLCγ1, thereby increasing calcium fluxes and activating ERK and AKT pathways.251 DNAM-1 activating signal has a synergetic effect with LFA-1, to which it can be associated physically, to induce tyrosine kinase Fyn to phosphorylate CD226.252 Association with LFA-1 is important for DNAM-1 clustering in the immune synapse,253 after LFA-1 interaction with PTA-1, which, in turn, associates with actin-binding protein 4.1G, to associates with membrane-associated guanylate kinase homolog protein leading to clustering and transport of DNAM-1 to lipid rafts.254 DNAM-1 does not have an exclusive ligand and must compete for PVR (CD155) and nectin-2 (CD112)78 against other inhibitory receptors, including TIGIT, TACTILE (CD96), and PVRIG (CD112R). The dynamics of this fierce competition will be discussed later in some detail. However, by virtue of DNAM-1 having a higher affinity to PVR than to nectin-2, it is safe to assume that NK cytotoxicity will largely depend on PVR expression level, and indeed PVR is widely expressed in human cancers.255–257 Other important activating receptors include NKp80, which binds to activation-induced C-type lectin (AICL), CD28 which binds to CD80 and CD86, CD2 which binds to CD48 (also a partner of 2B4) and CD58; the KIRs with short cytoplasmic domains, KIR-2DS and KIR-3DS, and C-type lectin receptors CD94/NKG2C, and NKG2E/H/2F.
The synergy between activating signals
2B4 activation can synergize with NKG2D or DNAM-1 at the level of PLC-γ and ERK phosphorylation (Fig. 5). This synergy was shown to be required to overcome the inhibitory signaling by CD94-NKG2A binding to HLA-E that controls VAV1 phosphorylation and its downstream signaling, PLCγ2.258 It was later discovered that SLP-76 needed to be phosphorylated once by NKG2D or DNAM-1 in one tyrosine (Y128) and a separate phosphorylation by 2B4 at tyrosine (Y113). Only CD16 activation can lead to phosphorylation of both tyrosines on SLP-76. This double phosphorylation allows the binding of two VAV1 molecules259 with more robust downstream signaling. An interesting aspect of NKG2D and DNAM-1 signaling is that the activation of NKG2D can block DNAM-1 activation through the induction of TIGIT expression and the inhibition of DNAM-1 signaling.260 This phenomenon was explained by the reduction in Pyk2 and Erk1/2 phosphorylation upon DNAM-1 engagement. However, AKT and VAV1 activation remained unaffected.260 This observation is substantiated by another group that reported a lack of synergistic effects when co-expressing both DNAM-1 and NKG2D in NK-92.261 However, the fact that VAV1 and AKT activations were not affected or, more accurately, not increased suggests that the early event of DNAM-1 activation did not proceed. Another recently described mechanism of DNAM-1 inhibition occurs when PD-1, via its intracellular domain signaling, recruits SHP-2 to inhibit DNAM-1 phosphorylation262 (Fig. 6). Since TIGIT is induced by NKG2D activation260 and since PD-1 and TIGIT were found to be co-expressed in CD8 T-cells of NSLCC patients,262 it is possible that both TIGIT and PD-1 induced by NKG2D activation, conspire together to inhibit DNAM-1 signaling in NK cells. The inability of DNAM-1 to synergize with NKG2D signaling and NK cell cytotoxicity suggests an overlap or a rheostat mechanism accepting an “either-or” pathway, which could be designed to avoid exhaustion when two pathways could hyperactivate NK cells. Indeed, co-activator 2B4, which synergizes with NKG2D, can also synergize with DNAM-1, but not simultaneously.258,259 These findings have profound implications for cancer immunotherapy aiming to exploit NKG2D and DNAM-1 and suggest that it is better to combine each one of them with other modalities, such as immune checkpoints, especially in the case of loss of expression of one of them.262–264
Many synergetic activating signaling in NK cells, such as NKG2D, DNAM-1, 2B4, NTB-A, and CRACC, converge on the phosphorylation of VAV1. And inhibitory receptors, including MHC I-specific inhibitory receptors, target VAV1 for dephosphorylation by SHP-1.265 Another potent inhibitory relay is Crk dissociation mediated by c-Abl phosphorylation of Crk, which in its active form (non-phosphorylated) is associated with the complexes c-Cbl/Crk/C3G and p130CAS/Crk/C3G. c-Abl phosphorylation of Crk causes its dissociation from these complexes (Fig. 6). Complexed Crk is required for CD16 signaling and the movement of microclusters of CD16 ligands on the lipid bilayer.266 Additionally, the inhibitory signaling by CD94-NKG2A binding to HLA-E uses the E3 ubiquitin ligase c-Cbl to enhance the degradation of phosphorylated VAV1 and its downstream signaling PLCγ2.258 Therefore, Cbl-b inhibition affecting Vav1 can only be overcome by synergistic signaling of multiple activating receptors.258 Receptor tyrosine kinases TAM receptors (Tyro3, Axl, and Mertk) are expressed by multiple immune cells, including NK cells. TAM receptors phosphorylate ubiquitin ligase Cbl-b and dampen NK-cell activation signaling by promoting the degradation of (Large Amino-acid Transporter 1) LAT1, thus blocking VAV1-dependent signaling267 and blocking, among others, glutamine transport and the fueling of the tricarboxylic cycle. It is accepted that VAV1 might be the point of convergence for various activating and inhibitory pathways, offering a rational and strategic switch to turn off NK activation and prevent the downstream activation cascade.268 Therefore, preventing VAV1 deactivation could provide a potent means to activate NK cells, with, however, the potential risk of higher toxicity to normal tissues.
The interplay of inhibitory and activating signals: The TIGIT/PVR/DNAM-1 axis
Most successful cancer immunotherapies are achieved using activating cytokines and activating receptors or their activation domains. This suggests that additional activation signals can be integrated into preexisting ones to strengthen them and reduce existing inhibitions. At the cell surface, activating and inhibitory receptors interact with their cognate ligands. Often, these ligands are unique to an activator or an inhibitory receptor. However, multiple instances exist where both the activating and inhibitory receptors compete for the same ligand, often to the benefit of the inhibitory receptor signaling. For example, the competition for HLA-E, the most ancient of the six functional HLA class I genes, by the inhibitory receptor CD94/NKG2A (Kd = 0.8 μM) and activating receptor CD94/NKG2C (Kd = 5.2 μM).269 Similarly, the competition for CD80 between immune checkpoint CTLA-4 (Kd = 0.46 μM) and CD28 (Kd = 4 μM)270 or for CD86 (CD86–CD28 ~ 20 μM and CD86–CTLA-4 ~ 2 μM). Another more complex and striking example is illustrated by immune checkpoint TIGIT and activating receptor DNAM-1, which compete for PVR (CD155) and nectin2 (CD112). In this race, DNAM-1 loses as TIGIT has a higher affinity for PVR (Kd = 1–3 nM) than DNAM-1 (Kd = 119 nM).271 TIGIT extends its inhibitory dominance by interacting with other inhibitory ligands, Nectin2, Nectin3,272 and Nectin4.273 In addition to TIGIT, CD112R(PVRIG) also competes with DNAM-1 for Nectin2,274 while CD96275 and KIR2DL5276 compete for PVR against DNAM-1.
DNAM-1 does not have an exclusive ligand for its activation, thus giving competing inhibitory receptors a clear advantage. This example illustrates the roadblocks for efficient NK cell activation at the level of competing extracellular domains for ligands. However, an additional layer of complexity is added by the fact that TIGIT will disrupt DNAM-1 homodimer assembly at the cell membrane, preventing its activation.277 This thug of war continues at the level of intracellular domains signaling with PVR/TGIT signaling blocking AKT phosphorylation, thus stabilizing transcription factor FOXO1, which inhibits NK and T-cell activation and enhances immunosuppressive functions of T-regulatory cells.278 The exact opposite is produced by PVR/DNAM-1 signaling, which phosphorylates AKT and destabilizes FOXO1 by phosphorylation, promoting its nuclear exclusion and degradation, thus enhancing NK and T cell activation.279
It is safe to assume that if these signals are present in the same cell, the inhibitory PVR/TIGIT axis will probably dominate the PVR/DNAM-1 axis. Another recently described mechanism of DNAM-1 inhibition occurs when PD-1, via its intracellular domain signaling, recruits SHP-2 to inhibit DNAM-1 phosphorylation.262 This finding is critical since PD-1 and TIGIT were found to be co-expressed in CD8 T-cells of NSLCC patients, suggesting the need for dual inhibition of PD-1 and TIGIT immune checkpoints.262 In addition, several tumors develop strategies to downregulate activators, including DNAM-1 expression in NK cells.280–282 Overall, inhibition and activation signals are regulated first through fierce competition for ligands with different intrinsic affinities at the cell surface. However, the axis PVR/TIGIT signaling between NK cells and cancer cells is bidirectional. On the NK cell side, TIGIT induces inhibitory signaling. In contrast, on the cancer cell side, PVR interaction with ligands TIGIT or DNAM leads to tyrosine phosphorylation of the PVR’s ITIM domain by Src kinases and recruitment of SHP-2 followed by dephosphorylation of focal adhesion kinase and paxillin thereby reducing adhesion, increasing motility, survival, and proliferation of cancer cells.283–285 Therefore, it is conceivable that if exhausted NK cells cannot kill cancer cells, they could make them stronger through stimulation of PVR or other immune checkpoints, especially with the ability of some NK cell subsets to support angiogenesis.286
Kinetics of killing
The rapid killing of cancer and virus-infected cells suggests that all effectors are available in NK cells and ready for immediate delivery. This killing largely depends on Src and Syk kinase activities.223,224 However, whether NK cells can kill multiple cancer cells at once or over time will depend on the presence of activating signals and sustaining cytokines. In a six-h assay, NK-92MI cell line, which produces a membrane-bound IL-2, can kill ten cancer cells serially.287 The authors noted that the first kill was slower than subsequent ones and that if cells are denser, the following killings are executed more rapidly, suggesting possible simultaneous killings. Short distances between target cells might encourage disengagement with the killed cell and engagement with a new target. We reported in NK-92 expressing IL-2 tethered to its receptor IL2Rβ a replenishment of granzyme and perforin stores after 3 h of exposure to PC-3 cells, suggesting serial killing.288 Cytotoxic T-cells have been reported to polarize lytic granules toward different cells and interact with multiple targets simultaneously.289,290 Another study found that human primary NK can kill four cancer cells serially but cannot engage simultaneously with two or more cancer cells.291 This suggests that primary NK cells activated by IL-2 cannot multitask and must disengage from a killed cell to kill a second one. This might be due to a missing component that allows multiple polarizations. The same study also reported increased killing by ADCC using Rituximab. However, this may be due to the efficient synapse formation initiated by antibody Fc binding. Without a novel cell target nearby, NK cells can remain attached to the dead cancer cells, which could deepen its activation via prolonged contact of activating receptors with their ligand in a manner already observed in the case of T-cells.290
Migration patterns
Release of activated NK cells from the bone marrow following inflammation or infection allows NK cells to migrate to affected tissues to kill abnormal cells and create inflammatory conditions in preparation for an adaptive immune response.292 The first step in the extravasation of NK cells into tissues requires tethering to endothelial cells, and this is accomplished by LFA-1, expressed on CD56bright and CD56dim subsets, and L-selectin (CD62L), which is only expressed in CD56bright subset.293 Therefore, L-selectin is a significant determinant in CD56bright delocalization from PB towards tissues. CD56bright cells migration in tissues is decelerated by downregulation of L-selectin by IL-2, IL-15, or TGFβ1 and accelerated by increased L-selectin expression under IL-12, IL-10, or IFNα.293 Chemokine ligands play a role in this relay by exerting attraction functions by binding to G protein‐coupled chemokine receptors. They play a significant role in immune cell recruitment into tissues, including tumors, by attracting cells expressing their cognate chemokine receptor. Depending on their resting or activated states, NK cells express heterogeneously the four groups of chemokine receptors for ligands CXC, CC, CX3C, and C. NK cells express receptors CXCR1, CXCR2, and CX3CR1.294–298 In the bone marrow, specific chemokines, such as CCL3, which binds to receptors CCR1, CCR4, and CCR5, regulate NK cell localization and induce migration to PB. In contrast, CXCL12 induces the accumulation of NK cells expressing high CXCR4.299 Breast cancer cells and tumor-associated stromal cells express high levels of CXCL12 to stimulate their proliferation and invasiveness in autocrine and paracrine modes.300 Tumors also secrete chemokines ligands to attract pro-tumorigenic cells such as myeloid-derived suppressor cells (MDSCs),301 T-regulatory cells,302 Tumor-associated macrophages,303 and tumor-associated neutrophils.304 Monocyte chemoattractant CCL2 (MCP-1), which interacts with CCR2, plays a prominent role in tumor angiogenesis, tumor cell survival, and the recruitment of immunosuppressive cells that will challenge immune cells, including NK cells in the tumor microenvironment.305 These pro-tumorigenic cells will be recruited through the CCR2, CXCR1 and CXCR2 axes. These cells create a tumor microenvironment that suppresses immune cell invasion of the tumor cells’ chemokine ligand secretion, which will directly enhance the growth and survival of cancer cells in the tumor microenvironment and promote metastasis.306 However, chemokines play a dual role and could promote anti-tumorigenic effects by attracting NK and T-cells expressing chemokine receptors CXCR3 and CXCR4. For example, overexpression of CXCR4 in NK cells improved tumor eradication of U87-MG glioblastoma secreting CXCL12.307 Migration of human primary NK cells to CXCR1, CXCR2, and CXCR4 ligands was reported.197 However, CXCR4 is also overexpressed in more than 23 human cancers and contributes to tumor growth, angiogenesis, and metastasis. This overexpression would naturally capture CXCL12 at the surface of cancer cells, an effect that would distort the gradient that attracts typically immune cells to tumors.308
Studies showed that CXCR3+ NK cells infiltrate tumors in a CXCL10-dependent fashion, leading to improved survival,196 while NK cells from CXCR3−/− mice show impaired tumor infiltration.309 Similarly, inhibiting pro-tumorigenic chemokine signaling enhances the potential of anti-tumorigenic chemokines, as exemplified by the knockdown of transcription factor Snail, reducing the expression of CXCR2 ligands (CXCL1 and CXCL2), and MDSCs attraction to the tumor via CXCR2, leading to increased T-cell and NK cell numbers in tumors.310
CD56bright and CD56dim primary NK cells express CXCR1, CXCR3, and CXCR4.311 However, it is clear that PB NK cells probably have different subsets with different chemokine phenotypes and migration abilities and that there are differences between individuals in these populations.311 For example, the CD56bright CD16+ NK cells were the predominant population responding to IL-8 (CXCR1,2) and fractalkine (CX3CR1),197 while others reported CXCR1 and CXCR2 to be highly expressed by cytotoxic CD56dim NK cells.296,312
In addition to chemokine receptors, NK cells express other chemotactic receptors, such as ChemR23313 and CCRL2,314 which, by attraction to chemerin, recruit NK cells to colocalize with DCs in inflammatory sites. ChemR23 is also expressed on macrophages, adipocytes, and endothelial cells,315–317 suggesting they all colocalize with NK cells.
Human NK cells activated by IL-2 express SIPR1,4 and 5, a G-coupled receptor proteins chemoattracted to bioactive lipid Sphingosine 1-phosphate (S1P).318,319 Receptor SIPR5 is expressed by NK and DCs, suggesting their colocalization.320 In inflamed tissues, S1P levels increase to promote the retention of immune cells.321 NK cells were also shown to directly recruit conventional type-1 dendritic cells (cDC1), which are critical for antitumor immunity through the secretion of CCL5 and XCL1.322 Senescent cells in aging tissues secrete senescence-associated secretory phenotype (SASP) proteins, which are inflammatory cytokines with chemokines GM-CSF, CCL2, 3, 4, and 5, CXCL1, 9, 10, and 11, which attract immune cells including NK cells, macrophages, neutrophils, and DCs. These immune cells will remove senescent cells but may also kill neighboring cancer cells in the same inflammatory environment. Chemokines binding to chemokine receptors is followed by internalization and degradation, which reduces homing. This could be alleviated by upregulating chemokine receptors.323–325
NK cells role in autoimmunity
Two major subsets of NK cells can be distinguished. CD56bright CD16negative, which secrete cytokines, and CD56Dim CD16positive, which are highly cytotoxic. However, NK cells that secrete IL-10 and possess immunosuppressive functions could form a third group with immunoregulatory functions. Autoimmune diseases arise from autoreactive T-cells and autoantibody-producing B-cells (plasma cells) against self-antigens. Autoreactive T-cells that escape thymic deletion326,327 are present in most healthy humans, and 55–75% of the repertoire generated by random immunoglobulin G gene rearrangement during early B cell development in the bone marrow is autoreactive and removed by two checkpoints.328 In the case of T-cells, central to autoimmune diseases is the role played by DCs,329 which migrate to lymphoid organs to present pathogen-derived antigens to antigen-specific T-cells. NK cells, particularly CD56bright NK cells, can, by production of GM-CSF and CD154, induce CD14+ monocyte differentiation into DCs in RA and psoriatic arthritis but not osteoarthritis OA patients.330 Therefore, RA NK cells provide a local milieu for monocytes to differentiate into DCs and sustain the disease. This could also be exacerbated by IFNγ secretion, which promotes Th1 polarization of CD4+ T. Similarly, the interaction of NK cells with DCs induces IFNγ, especially from the CD56bright subset, which expresses surface molecules CD62L, CCR7, and CXCR3.331 This suggests this subset may colocalize with DCs in secondary organs and other inflamed tissues. However, several studies showed that although NK cells can increase in RA, they are less cytotoxic and have decreased IFNγ production.332,333 Since these NK cells also produce pro-inflammatory cytokine GM-CSF, it has been proposed that NK contribution to inflammation in RA might be due to the attraction of neutrophils, thereby upregulating pro-inflammatory CXCL2, CCL3, and LTb4, that sustain immune cell recruitment into inflamed joints.334 The involvement of NK cells in other autoimmune diseases remains contentious. For example, in multiple sclerosis, it is thought that NK cells fail to remove myelin-reactive T-cells and fail to suppress autologous CD4 + T cells compared to healthy controls.335,336 In Systemic Lupus Erythematosus (SLE), notable reductions in peripheral NK cell number and cytotoxicity were observed.337,338 However, the role of NK in developing SLE has been established through a bidirectional interaction between NK and peripheral DCs. NK cells augment IFNα production by activated DCs,339 in turn, IFNα increases NK cell production of IFNγ,340 thereby establishing highly inflammatory conditions. Incidentally, SLE patients have higher levels of IL-15, which is also conducive to increased inflammation.341 In Type 1 diabetes mellitus (T1DM), which is due to the destruction of pancreatic β cells by CD8 T-cells, a systematic reduction in the number and cytotoxicity of peripheral NK cells was observed.342,343 It is noteworthy that NK cells are also impaired in type 2 diabetes, suggesting their reduced activity in both diabetes types is mainly related to glucose levels and that the prevalence of infectious diseases and malignancy in type-2 diabetes patients may be associated with NK cell impairment.344 However, NK cells that infiltrate inflamed islet cells345 might have a sinister role in the development of T1DM by killing virus-infected pancreatic β cells,346 which reduce their HLA-1 expression to escape T-cells but become targets of NK cells. The subsequent β cell killing by NK cells could lead to the exposure of autoantigens recognized by CD8 T cells.
We have mentioned earlier that KIRs are categorized into two haplotypes: A, which mainly encodes inhibitory KIRs, and B, which encodes stimulatory KIRs. A study examining the role of the KIR haplotype on NK cells reported that KIR A1 haplotypes were positively associated with T1D in the subset of patients without the high T1D risk HLA genotype.347 In these patients, inhibitory KIR A2 haplotypes were over-transmitted, and the stimulatory KIR B haplotypes were under-transmitted, suggesting haplotypes A are predisposing and stimulatory haplotypes B confer protection. From our perspective, we interpret this result as due to the restricted ability of NK cells with inhibitory KIR A2 haplotype to kill or suppress overactive CD8 T-cells thus promoting T1D.
NK cells pro-angiogenic role in tumors and pregnancy
Angiogenesis is supported by transcription factor HIF-1α, which is induced under hypoxia to promote the expression of pro-angiogenic factors to stimulate blood vessel growth through the HIF-1α/VEGF axis.348 Hypoxia dramatically affects NK cells, as demonstrated in vitro and in cancer patients. One week after being exposed to hypoxia (1%O2), peripheral NK cells were enriched in CD56brightCD16Neg phenotype and became capable of secreting VEGFA in the media that could increase HUVEC cell’s angiogenic capacity.286 A clear demonstration of the NK cell’s conversion to a pro-angiogenic phenotype was shown in renal cell cancer patients who had peripheral NK cells with a CD56pos CD16pos phenotype, but NK cells infiltrating renal cancer with a CD56pos CD16Neg phenotype, like dNK, with enrichment in genes of the hypoxia-inducible factor HIF-1α pathway.349 To understand the role of these NK cells in the tumors, it is important to learn from another conversion of NK cells to a pro-angiogenic phenotype observed in another normal physiological phenomenon, pregnancy. NK cells CD56brightCD16Neg, also called dNK, secrete an array of pro-angiogenic factors that regulate trophoblasts invasion and actively produce IL-8 and interferon-inducible protein-10 chemokines, CXCL10. dNK cells are anergic non-cytotoxic despite expressing NK activating receptors, including NKp44, NKp46, NKp30, and NKG2D.350–352 However, dNK cells express high levels of GNLY and are capable of killing virus infected stromal cells of the mother after activation,353 but do not kill the bacteria infected trophoblasts. Instead, they deliver GNLY to specifically kill the bacteria without harming the trophoblast19 or damaging the maternal-fetus interface.
dNK emerge from immature uterine NK cells originally from PB and which upon stimulation with IL-15, acquire KIRs and mature.354 The implantation of the embryo is an inflammatory process of the uterus primed by ovarian hormones to secrete IL-8, IL-15, IL-6, CXCL10, and CXCL11.355 These cytokines and chemokines attract decidual immune cells of which 70% are uterine NK cells. Survival of the embryo with its semiallogenic genetic stock in the uterus will depend on the tolerance of maternal immune cells. dNK cells at the maternal–fetal interface express inhibitory receptors such as KIR2DL1, KIR2DL2, L3, and Leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1), which recognizes HLA-G to inhibit NK-cell cytotoxicity132 and inhibitory receptors CD94/NKG2A which interact with and HLA-E.356 Indeed, NKG2A genetic ablation in female mice caused suboptimal maternal vascular remodeling in pregnancy, reduced fetal weight, and abnormal brain development resembling the human syndrome pre-eclampsia.357 At the onset of pregnancy, the high expression of KIR2D in dNK and the upregulation of HLA-C in the stromal cells of the endometrium, which transform into decidua, are crucial. At the maternal-fetal interface, NK cells represent the majority of immune resident cells as they expand in uterus spiral arteries. Therefore, dNK cells have a productive role in pregnancy by regulating key developmental processes, including angiogenesis at the human fetal-maternal interface.358 dNK cells also appear to control oxygen levels by regulating uterine spiral artery development. Indeed, the absence of NK at the fetal-maternal interface increases hypoxia.359 Therefore, NK cells maintain an oxygen and nutrient-rich environment, influence trophoblasts, and promote the development of the invasive trophoblast lineage necessary for optimal blood supply between mother and fetus through the mother KIRs and fetal HLA interactions.360,361 Going back to tumor physiology, strikingly, the deletion of HIF-1α in NK cells reduced their recruitment into tumors, while it did not affect that of CD4 or CD8 T-cells. The lack of NK cell recruitment led to a reduction in tumor size through non-productive angiogenesis. This later is characterized by increased hypoxia and a high density of immature hemorrhagic blood vessels,362 suggesting that NK cells are required to mature blood vessels during the remodeling of tumor vasculature as in pregnancy. Krzywinska et al. showed that HIF-1α KO-NK cells prefer to reside in well-oxygenated areas, thus ignoring hypoxic regions that need their presence. Most importantly, HIF-1α was found to be required for the cytotoxicity of NK cells.362 The authors concluded that NK cells will balance excessive angiogenic tumor efforts by providing the angiostatic soluble VEGFR1 (sVEGFR1) to control VEGF bioavailability in an HIF-1α-dependent manner. While the role of HIF-1α in tumor angiogenesis is established in the above study and is in line with the events during pregnancy, the conclusions regarding NK cytolytic functions might depend on the tumor model used in the study. Another study showed that HIF-1α deletion unleashed NK cells cytolytic activity, but only against MHC I deficient tumors, and that this required IL-18.363 Single-cell analysis showed that HIF-1α inhibits IL-18 signaling, thus reducing NFkb signaling and IFNγ, reducing NK cell infiltration in tumors. Indeed, deletion of HIF-1α allows IL-18 secreted by myeloid cells to activate NK cells against MHC I deficient tumors. Of note, hypoxia also induces IL-18 to promote angiogenesis364 and it might be needed for the initial phase of gestation, but its upregulation in the decidua of patients was associated with recurrent miscarriages.365 IL-18 role in tumor hypoxia and pregnancy is complicated by its pleiotropic effect and its ability to induce more than 1000 genes in NK cells, as well as, the partial overlap with IL-2, IL-12, and IL-15 functions.366 Additionally, we have seen earlier that IL-18 and IL-12 can reverse the anergy of NK cells in MHC I deficient tumors,114 suggesting this cytokine is critical for NK-mediated immunotherapy. Another intriguing effect of IL-18 is its ability to convert CD56dim to a helper CD56bright CD16Neg phenotype,367 which is potentially more pro-angiogenic. In summary, the presence of dNK cells at the maternal-fetal interface is driven by CD56bright migration in response to cytokines and chemokines and probably hypoxia, sensed through HIF-1α. Interaction with trophoblasts triggers a pro-angiogenic dNK phenotype that helps in building spiral arteries, creates better and balanced oxygenation and brings more nutrients to the interface. Trophoblasts through HLA-E and HLA-G, represses dNK cytolytic activity and further promotes their pro-angiogenic role. Reduced dNK at the interface has been reported in pre-eclampsia,368 suggesting that the dNK to trophoblasts ratio is crucial for balanced angiogenesis. In this regard an intriguing question regarding the role of HIF-1α in initiating or maintaining this dynamic must be studied through the conditional knockout of HIF-1α, before and after pregnancy is established. Knockout of HIF-1α would prevent dNK cells migration to the interface. These investigations could confirm if HIF-1α KO dNK’s inability to correctly sense hypoxia is an important factor in pre-eclampsia or even parturition. The same animal model could also evaluate the impact of hypoxia sensing by NK cells in tumor initiation, metastasis, and angiogenesis. HLA-G found in trophoblasts of the placenta, plays a crucial role in maternal-fetal tolerance, acting as an immune checkpoint.369 Expression of HLA-F and HLA-G on migrating trophoblast support their invasion and interactions with uterine natural killer cells.370 HLA-G is also highly expressed in a variety of tumors and is involved in their immune escape, which is mediated by the interaction with immune cells, including NK cells.371 In tumors, HLA-G interacts with LILRB1/2 and KIR2DL4 to suppress cytotoxic T-cells and NK cells and promotes the expansion of immunosuppressive cells, Treg cells and MDSCs, creating an immunosuppressive microenvironment that aids tumor cells in evading the immune system. Moreover, KIR2DL4 expression is enhanced by IFNγ,372 suggesting a role in immune response regulation. Therefore, for a common purpose, KIR2DL4, by interacting with HLA-G, participates in pacifying the maternal-fetus interface and allows tumors to escape immunity.
NK cells in senescence and developing cancers
Most established cancers have already escaped surveillance by immune cells, including NK cells. There is an emergent consensus around the decidualization of NK cells in the tumor microenvironment as in the maternal/fetal interface and even of some circulating NK cells in cancer patients, leading to anergy and even subservient status in tumors. The CD56bright, CD16dim/neg NK cells could become pro-angiogenic, possibly hijacked and reprogrammed to benefit the tumor progression.373,374 Peripheral NK cells, which are mostly CD56Dim CD16positive, are likely to intercept transiting metastatic cells. However, the less active CD56BrightCD16dim/neg NK cells that localize in tissues are intrinsically less likely to achieve that. In a human of 73 kg, the total number of NK cells in the bone marrow where they are continuously produced is 4 × 109. The blood and skin each harbor 2 × 109, while a large majority (30%) of 5 × 109 NK cells are found in the liver, and only 1 × 109 can be found in the lymphatic system or the lungs.375 The most likely initial mechanism a developing cancer cell uses in the very initial stage would be the most potent inhibitory tool against NK cells, the MHC I complex. Interestingly, senescence, which shares many precursor states with tumorigenesis, such as accumulation of DNA damage or defective signaling and which is now proposed as an enabling hallmark of cancers,376 also leads to overexpression of MHC I.377,378 This could further inhibit the already subdued CD56Bright, CD16dim/neg NK cells. Therefore, it stands to reason that because of the large population of senescent cells accumulating in aging tissues,379,380 there will be more inhibitory forces against NK cell populations. This is compounded in the elderly by the cross-the-board decline of immune cell functions that normally support NK cells by providing cytokines. Notably, macrophages’ reduction in numbers and bactericidal capacity,381 the decreased antigen presentation function in DC cells,382 the dwindling numbers of B-cells and their capacity to properly produce a diverse immunoglobulin repertoire,383 as well as the reduced stemness of hematopoietic stem cells, producing less lymphocytes such as T-cells.384,385 All these events may lead to reduced clearance of senescent cells and their accumulation in aging tissues and age-associated diseases.386 Senescent cells overexpress MHC I and their HLA-E expression consistently increases in aging human skin and melanocytic nevi compared to young skin. Blocking HLA-E interaction with ligand NKG2A on NK and CD8 T-cells allowed the killing of senescent cells by NK cells.387 A clear link between senescent and cancer cells was demonstrated by the reduction of spontaneous tumorigenesis and cancer-related death after the depletion of senescent cells in aging mice.388 An immediate question arises regarding why senescent cells accumulate in the elderly but not in the young. This could originate from the increased number of cells entering senescence in the elderly compared to the young. However, a study in mice showed that the expression of MHC I ligands and KIRs on NK cells also increases in the elderly,389 suggesting that NK cells also become less responsive to senescent cells. Another study in elderly humans showed a reduction of NKp30 and NKp46 expression in NK cells, suggesting reduced interactions with DCs and functions,390 with increased KIR expression in the CD56bright population.155 However, the same study found evidence of some NK cells subset compensating for these deficiencies. For example, CD56dim population increased and CD94 expression declined in the elderly in both NK subsets. Nonetheless, more evidence of reduced NK activity in the elderly is suggested by their reduced response to IL-2 and impaired cytokine signaling.391 It is plausible that senescence’s increased rate at older age is only due to the lack of immune cell reactivity, including from NK cells, leading to reduced clearance of senescent cells. This could lead, in turn to a critical mass of proinflammatory senescent cells with a SASP, which produce inflammatory cytokines like IL-1α/β, IL-6, IL-8, TNFα, chemokines, DNA, microRNAs, proteases such as matrix metalloproteinases, wound healing factors PDGF-AA, endothelial vascular factor VEGF and senescence promoting factor IGFBP4/7,392–395 extracellular vesicles and exosomes containing cytokines such IL-15396 or Heat shock proteins.397 Additionally, SASP from senescent cells can induce the senescence of neighboring cells,398 leading to a vicious cycle of senescent cell accumulation. However, this conversion could also transform neighboring cancer cells into senescent non-replicating cells.399 This effect is thought to be protective, reducing cancer and providing an evolutionary explanation of the benefit of senescent cells. However, as mentioned earlier, depleting senescent cells in animal models reduced cancer frequency.388 Senescent cells overexpress decoy receptor 2, allowing them to escape the FasL death pathway. The mechanism through which NK cells remove senescent cells involves granular exocytosis, mostly through overexpressed ligands MICA/B, ULBP1-3, PVR, and nectin-2 binding to activating receptors NKG2D and DNAM-1.400–402 Additionally, senescent cells in aging tissues secrete SASP with chemokines GM-CSF, CCL2, 3, 4, and 5, CXCL1, 9, 10, and 11, which attract immune cells, including NK cells.
Crosstalk with other immune cells
NK cells increase inflammation after activation by tumor and virus-infected cells by secreting inflammatory cytokines such as IFNγ,22,23 which activates macrophages and neutrophils,24 T-cells,25 and B-cells.26 However, cancer cells treated with IFNγ become resistant to NK cells, suggesting that NK cells secretion of IFNγ may be designed to involve other immune cells27 to remedy their deficiency, suggesting a redundancy mechanism.
Macrophages
Macrophages derive from circulating monocytes and reside in tissues where they adopt different phenotypes, such as unpolarized M0 and two polarized states (M1), which is anti-tumor and pro-inflammatory, and (M2) which is pro-tumor and anti-inflammatory. Due to their abundance in tissues, they are most likely the first to discover sites of infections by viruses, bacteria, and parasites. Overactivated or infected macrophages will be killed by bystander-activated NK cells.83 Initially, NK cells, in direct contact with these macrophages, increase their degranulation marker CD69 and IFNγ expression403 and collaborate closely with macrophages to control the infection and inflammation (Fig. 7a). NK cells and IFNγ are required and sufficient for the polarization of tumor-associated macrophages (TAMs) to M1, which protect against tumor growth even in the absence of adaptive immunity.404 Depending on the infectious agent, NK cells will express specific receptors to specific ligands, the involved macrophages express. Macrophages stimulated or infected by cytomegalovirus will stimulate co-activating receptors 2B4, NKp46, and DNAM-1 on NK cells.405,406 At the same time, stimulation by Streptococcus pneumonia induces clearance through activation of NKp46407 and stimulation by bacterial moieties such as lipopolysaccharides (LPS), an outer membrane component of gram-negative bacteria, as well as Mycobacterium tuberculosis, Sendai or Influenza A virus,408 induce the ligands, retinoic acid early inducible-1 (RAE-1),409 ULBP1-3,405 MICA, and MICB,410 which will activate the NKG2D receptor. The developing tumor microenvironment, with its increased inflammation, acidic metabolism, hypoxia, and chemokines, attracts monocytes to seed the tumor with what is to become TAMs. Monocyte chemoattractant protein CCL2 (MCP-1), which interacts with CCR2, plays a prominent role in this recruitment.305 Recruited monocytes are either polarized into M1 macrophages characterized by IL-12high IL-23high IL-10low and have phagocytic and antitumor activity, or M2, which are IL-12low IL-23low IL-10high and TGFβ1high with no phagocytic activity and secrete TGFβ1 to inhibit NK cells anti-tumor activity.411 However, these two states are interchangeable, depending on the balance between immunosuppression and immunostimulation.412 For example, stimulation by LPS can revert M0 and M2 macrophages to an M1 phenotype, leading to NK cell activation.83 This activation could help restore anergic NK activity by cytokines such as IL-18 and IL-12.114 Similarly, IL-15 trans presented by M1 macrophages after contact with bacterial moieties leads to strong NK cell activation.413 Virus-infected Macrophages are killed by NK cells. In patients with severe COVID-19, a surge in many proinflammatory cytokines leads to acute respiratory disease syndrome originating from macrophage-activation syndrome. In these patients, the number of NK cells was dramatically reduced, and their activation by K562 leukemia was impaired compared to healthy controls. Additionally, these patients had very low levels of IL-12, IL-15, and IL-21 needed to activate NK cells. These findings suggest that in severe COVID-19 patients, NK cells are highly exhausted and fail to kill virus-infected macrophages that produce proinflammatory cytokines.91 To evaluate the efficacy of engineered allogenic cord blood NK cells, clinical trial NCT04324996 (Table 1) is evaluating NKG2D-ACE2 CAR-NK targeting the S protein of SARS-CoV-2 and NKG2DL on the surface of infected cells with ACE2 and NKG2D, respectively.
Fig. 7.

Crosstalk with other immune cells. a NK cells secreted IFNγ can help polarize macrophages (M0, M2) to antitumor M1 phenotype. Macrophages reciprocate by IL-12 and IL-15 trans-presentation to increase IFNγ production by NK. Virus-infected Macrophages are killed by NK cells, and anergic NK cells may be reactivated by macrophages IL-12 and IL-18. b Neutrophils enhance tumor defense by triggering TRAIL-mediated apoptosis. They release IL-12 to boost IFNγ and perforin in NK cells but also downregulate NK cell receptors via PD-L1 upregulation induced by G-CSF. To support neutrophil function, NK cells reciprocate by secreting IFNγ, GM-CSF, and TNFα. However, neutrophils can inhibit NK cells through NET-mediated NKp46 cleavage, while tumor-associated neutrophils suppress immune responses via ARG1 release and ROS production. c MDSCs and Tregs suppress NK cell function through direct contact or secretion of TGFβ1 and IL-2 depletion, alongside IL-10 production. MDSCs also employ TIGIT to inhibit NK cells, reducing CD3ζ and impairing NK cell receptors. They can also hinder NK cells through direct interaction with NKp30. However, MDSCs can induce IFNγ release in NK cells via NKG2D activation by RAE-1 ligand. MDSCs promote the trans-differentiation of naive CD4+ T cells into Foxp3+ Tregs. Additionally, MDSCs and Tregs convert extracellular ATP and ADP to cAMP and adenosine by CD39 and CD73, inhibiting NK cell antitumor responses via A2AR binding. d T-cell production of IL-2 activates NK cells, which, by the production of IFNγ, activates DCs. DCs reciprocate by IL-12 to reinforce IFNγ production and stimulate CD8 T-cells. NK cells producing GM-CSF and CD154 can induce CD14+ monocyte differentiation into DCs. TAF production of PGE2 and IDO can exhaust NK cells, thereby blocking their mutual activation with DCs and subsequent CD8 T-cell activation
Table 1.
List of clinical trials using CAR-NK, TCR-NK, and BICAR-NK
| Target | Trial identifier | NK source | Disease | Phase | Study Status | First Posted |
|---|---|---|---|---|---|---|
| Clinical trials using CAR-engineered NK cell therapy in malignant tumors | ||||||
| CD5 | NCT05110742 | Cord blood | R/R hematological malignancy | I/II | Recruiting | 2021 |
| CD7 | NCT02742727 | NK-92 | CD7 + R/R leukemia and lymphoma | I/II | Unknown | 2016 |
| 5T4 | NCT05194709 | Not disclosed | Advanced solid tumors | I | Recruiting | 2022 |
| BCMA | NCT05182073 | iPSC | MM | I | Recruiting | 2022 |
| NCT06242249 | Not disclosed | R/R MM | I/II | Not yet recruiting | 2024 | |
| NCT05008536 | Cord blood | R/R MM | I | Unknown | 2021 | |
| NCT05652530 | Healthy donor | R/R MM | I | Recruiting | 2022 | |
| NCT03940833 | NK-92 | R/R MM | I/II | Unknown | 2019 | |
| NCT06045091 | Allogenic NK | R/R MM/ Plasma cell leukemia | I | Recruiting | 2023 | |
| CD123 | NCT06006403 | Not disclosed | AML/blastic plasmacytoid dendritic cell neoplasm/relapse leukemia or leukemia | I/II | Recruiting | 2023 |
| NCT05574608 | Allogenic NK | R/R AML | I | Recruiting | 2023 | |
| NCT06201247 | Healthy donor | R/R AML | I | Recruiting | 2024 | |
| CD19 | NCT05020678 | PB-NK | B-cell malignancies | I | Recruiting | 2021 |
| NCT05654038 | Allogenic NK | B-Cell LL/Lymphoma | I/II | Recruiting | 2022 | |
| NCT06334991 | NK-92 | R/R NHL | I | Not yet recruiting | 2024 | |
| NCT04887012 | Haploidentical donor | R/R B-cell NHL | I | Unknown | 2021 | |
| NCT01974479 | Haploidentical donor | B-cell ALL | I | Suspended | 2013 | |
| NCT05336409 | iPSC | R/R CD19 + B-Cell malignancies | I | Recruiting | 2022 | |
| NCT05739227 | Allogenic NK | ALL/B-cell lymphoma/CLL | I | Recruiting | 2023 | |
| NCT04639739 | Not disclosed | R/R B-cell NHL | I | Unknown | 2020 | |
| NCT05673447 | Allogenic NK | Diffuse large B cell lymphoma | I | Recruiting | 2023 | |
| NCT05472558 | Cord blood | B-cell NHL | I | Recruiting | 2022 | |
| NCT04887012 | Haploidentical donor | B-cell NHL | I | Unknown | 2021 | |
| NCT05336409 | iPSC | R/R CD19 + B-Cell malignancies/NHL | I | Recruiting | 2022 | |
| NCT05563545 | Not disclosed | ALL | I | Completed | 2022 | |
| NCT05410041 | Not disclosed | ALL/CLL/NHL | I | Unknown | 2022 | |
| NCT05645601 | Allogenic NK | R/R B-cell hematologic malignancies | I | Recruiting | 2022 | |
| NCT06464861 | Cord blood | R/R B cell lymphoma | I | Not yet recruiting | 2024 | |
| NCT03056339 | Cord blood | B-lymphoid malignancies | I/II |
Completed: Phase I Interim results reported 202090 Phase 1/2 results reported 2024579 |
2017 | |
| NCT05618925 | NK-92 | R/R NHL | I | Recruiting | 2022 | |
| NCT04796675 | Cord blood | B-cell lymphoid malignancies | I | Unknown | 2021 | |
| NCT04796688 | Not disclosed | ALL/CLL and B-cell lymphoma | I | Recruiting | 2021 | |
| NCT05379647 | Not disclosed | R/R B-cell ALL | I | Recruiting | 2022 | |
| NCT03690310 | iPSC | R/R B-Cell lymphoma | I | Recruiting | 2018 | |
| NCT03824951 | iPSC | R/R B-Cell lymphoma | I | Recruiting | 2019 | |
| NCT01974479 | Haploidentical donor | B-cell ALL | I | Suspended | 2013 | |
| NCT00995137 | Haploidentical donor | R/R ALL | I | Completed | 2009 | |
| NCT06206902 | Not disclosed | NHL | I | Recruiting | 2024 | |
| NCT02892695 | NK-92 | CD19+ leukemia/ lymphoma | I/II | Unknown | 2016 | |
| NCT05020015 | Not disclosed | R/R B-cell NHL | II | Not yet recruiting | 2021 | |
| NCT04245722 | iPSC | R/R B-NHL/CLL | I |
Recruiting Interim trial results 2021583 |
2020 | |
| CD22 | NCT03692767 | iPSC | R/R B-Cell lymphoma | I | Unknown | 2018 |
| CD33 | NCT05008575 | Not disclosed | AML | I | Unknown | 2021 |
| NCT02944162 | NK-92 | AML | I/II | Unknown | 2016 | |
| NCT05665075 | Allogeneic NK | AML | I | Recruiting | 2022 | |
| CD70 | NCT05092451 | Cord blood | R/R Hematological malignances | I/II | Recruiting | 2021 |
| NCT05703854 | Cord blood | Renal cancer/ mesothelioma/osteosarcoma | I/II | Recruiting | 2023 | |
| Claudin18.2 | NCT06464965 | Cord blood | Gastric cancer/pancreas adenocarcinoma | I | Not yet recruiting | 2024 |
| Claudin6 | NCT05410717 | Autologous PB-NK | CLDN6+ advanced solid tumors | I | Recruiting | 2022 |
| CLL1 | NCT06307054 | Patient or healthy donor | R/R AML | I | Recruiting | 2024 |
| NCT06027853 | iPSC | AML | I | Recruiting | 2023 | |
| DLL3 | NCT05507593 | Not disclosed | Extensive stage SCLC | I | Recruiting | 2022 |
| HER-2 | NCT04319757 | Not disclosed | Advanced or metastatic HER2+ solid tumors | I | Recruiting | 2020 |
| NCT03383978 | NK-92 | Recurrent HER2+ glioblastoma | I | Recruiting | 2017 | |
| Mesothelin | ChiCTR2100048100 | Autologous | Refractory epithelial ovarian carcinoma | 0 | Recruiting | 2021 |
| NCT03692637 | iPSC | Epithelial ovarian cancer | I | Unknown | 2018 | |
| MUC1 | NCT02839954 | Not disclosed | R/R MUC1+ solid tumors | I/II | Unknown | 2016 |
| NKG2D ligands | NCT05528341 | NK-92 | R/R solid tumors | I | Recruiting | 2022 |
| NCT05247957 | Cord blood | R/R AML | NA | Terminated | 2022 | |
| NCT03415100 | PB-NK | Metastic solid tumors | I |
Unknown Interim results reported 2019584 |
2018 | |
| NCT06478459 | Not disclosed | Non-resectable pancreatic cancer | I | Not yet recruiting | 2024 | |
| NCT05776355 | Not disclosed | Platinum-resistant recurrent ovarian cancer | NA | Recruiting | 2023 | |
| NCT05213195 | Not disclosed | Refractory metastatic colorectal cancer | I | Recruiting | 2022 | |
| NCT06379451 | Not disclosed | MM | I | Not yet recruiting | 2024 | |
| NCT04623944 | Allogeneic NK | Refractory MDS and AML | I | Not yet recruiting | 2020 | |
| NCT05734898 | Not disclosed | R/R AML | NA | Recruiting | 2023 | |
| PSMA | NCT03692663 | iPSC | Castration-resistant prostate cancer | I | Not yet recruiting | 2018 |
| TAA (Not disclosed) | NCT05856643 | Not disclosed | Ovarian epithelial carcinoma | I | Not yet recruiting | 2023 |
| NCT05686720 | Not disclosed | Advanced triple negative breast cancer | I | Not yet recruiting | 2023 | |
| NCT05845502 | Not disclosed | Advanced hepatocellular carcinoma | NA | Not yet recruiting | 2023 | |
| TROP2 | NCT06454890 | Not disclosed | NSCLC | I/II | Not yet recruiting | 2024 |
| NCT06358430 | Cord blood | Colorectal cancer/MRD | I | Not yet recruiting | 2024 | |
| NCT05922930 | Cord blood | Pancreatic cancer/ovarian cancer/adenocarcinoma | I/II | Recruiting | 2023 | |
| NCT06066424 | Cord blood | Solid tumors | I | Recruiting | 2023 | |
| PD-L1 | NCT04847466 | NK-92 | Gastroesophageal Junction (GEJ) Cancers/advanced HNSCC | II | Recruiting | 2021 |
| NCT06239220 | NK-92 | Recurrent and metastatic HNSCC | II | Recruiting | 2024 | |
| NCT04390399 | NK-92 | Pancreatic cancer | II | Recruiting | 2020 | |
| ROBO1 | NCT03940820 | Not disclosed | Solid tumor | I/II | Unknown | 2019 |
| CD19/CD22 | NCT03824964 | iPSC | Refractory B-Cell Lymphoma | I | Unknown | 2019 |
| CD33/CLL1 | NCT05215015 | Not disclosed | AML | I | Unknown | 2022 |
| ChiCTR2100047084 | Not disclosed | R/R AML | I | Recruiting | 2021 | |
| NCT05987696 | iPSC | AML/MRD | I | Not yet recruiting | 2023 | |
| CD33/DLL3 | NCT06367673 | iPSC | AML | I | Recruiting | 2024 |
| CD19/70 | NCT05667155 | Cord blood | R/R B-cell NHL | I | Recruiting | 2022 |
| NCT05842707 | Cord blood | R/R B-cell NHL | I/II | Recruiting | 2023 | |
| CD33/TIM3 | ChiCTR2100043081 | Cord blood | AML | 0 | Recruiting | 2021 |
| MICA/B | NCT06342986 | iPSC | Gynecologic cancer/ovarian cancer/fallopian tube cancer/primary peritoneal cavity cancer | I | Not yet recruiting | 2024 |
| CLL1 | NCT06027853 | iPSC | AML | I | Not yet recruiting | 2023 |
| Clinical trials using TCR-engineered NK cell therapy in malignant tumors | ||||||
| PRAME | NCT06383572 | Cord blood | Myeloid malignancies | I/II | Recruiting | 2024 |
| NY-ESO-1 | NCT06083883 | Healthy donor | Synovialsarcoma/ myxoid/round cell Liposarcoma | I/II | Suspended | 2023 |
| Clinical trials using Bi-CAR-engineered NK cell therapy in malignant tumors | ||||||
| CD33/FLT3 | NCT06325748 | Healthy donor | AML/MDS/CD33+and or FLT3+ Hematological Malignancies | I | Recruiting | 2024 |
| CD30/CD16A | NCT04074746 | Cord blood | R/R CD30 + HL and NHL | I/II | Not yet recruiting | 2019 |
| ROBO1 | NCT03941457 | Not disclosed | Pancreatic cancer | I/II | Recruiting | 2019 |
| ROBO1 | NCT03931720 | Not disclosed | Malignant Tumor | I/II | Recruiting | 2019 |
| Clinical trials using CAR-engineered NK cell therapy in Autoimmune Diseases or COVID19 | ||||||
| CD19 | NCT06464679 | Not disclosed | Autoimmune diseases | I | Not yet recruiting | 2024 |
| NCT06318533 | Not disclosed | Autoimmune diseases | I | Recruiting | 2024 | |
| NCT06208280 | Not disclosed | Autoimmune diseases | I | Recruiting | 2024 | |
| NCT06468683 | Not disclosed | Lupus erythematosis | I | Not yet recruiting | 2024 | |
| NCT06377228 | Not disclosed | Refractory lupus nephritis | I | Not yet recruiting | 2024 | |
| NCT06421701 | Not disclosed | SLE | I | Not yet recruiting | 2024 | |
| NCT06255028 | Not disclosed | SLE | I | Not yet recruiting | 2024 | |
| NCT06010472 | Not disclosed | SLE | I | Recruiting | 2023 | |
| NCT06337474 | Not disclosed | Thrombocytopenia Alloimmune | I | Not yet recruiting | 2024 | |
| NCT06469190 | Not disclosed | R/R Immune Nephropathy | I | Not yet recruiting | 2024 | |
| NKG2D ligands | NCT04324996 | Cord blood | COVID-19 | I/II | Unknown | 2020 |
ALL acute lymphoblastic leukemia, AML acute myeloid leukemia, BCMA B cell maturation antigen, CAR chimeric antigen receptor, CLL chronic lymphocytic leukemia, CR complete remission, CRS cytokine-release syndrome, HLA human leukocyte antigen, hnCD16 high-affinity non-cleavable CD16, HNSCC head and neck squamous cell carcinoma, iPSC induced pluripotent stem cell, MDS myelodysplastic syndrome, MRD minimal residual disease, MICA/B MHC class I chain-related protein A and B, NHL non-Hodgkin lymphoma, NK natural killer, NSCLC non-small cell lung cancer, ORR objective response rate, PB peripheral blood, PSMA prostate specific membrane antigen, ROBO1 roundabout homolog 1, R/R relapsed or refractory, SCLC small cell lung cancer, SLE systemic lupus erythematosus, 5T4 oncofetal trophoblast glycoprotein, TCR T-cell receptor, COVID-19 Coronavirus disease 2019, TIM3 T-cell immunoglobulin and mucin domain 3
Neutrophils
Neutrophils are required for NK cell development in mice and humans,414 and patients with chronic neutropenia have increased frequencies of CD56bright NK cells and lack mature CD56dim NK cells.415 Neutrophils have an anti-tumor effect mediated by TNF-related apoptosis-inducing ligand (TRAIL), which can induce apoptosis in leukemic cells416 (Fig. 7b). Additionally, neutrophils release IL-12, crucial for NK cells’ enhanced IFNγ and perforin production.417 However, in tumor-bearing animals, neutrophils downregulated chemokine receptor CCR1, NKp46, and NKG2D expression in NK cells through direct contact with NK cells, weakening their tumor infiltration and responsiveness.418 This immunosuppression was mediated by neutrophils’ increased PD-L1 expression, induced by G-CSF, the regulator of neutrophils’ generation and differentiation, and the STAT3 signaling. Since NK cells also produce pro-inflammatory cytokine GM-CSF, they might attract neutrophils, thereby upregulating pro-inflammatory CXCL2, CCL3, and LTb4, which sustain immune cell recruitment into inflamed tissues.334 It is unclear whether neutrophils have a beneficial role in NK cell’s antitumor activity. Still, the fact that their numbers are increased in cancer patients304 and that neutrophils are a critical component of the inflammatory process, which is now accepted as part of tumorigenesis,419 suggests that neutrophils may be mostly immunosuppressive forces in tumors, promoting angiogenesis, extracellular matrix remodeling, metastasis, and immunosuppression.420 By secretion of IFNγ, GM-CSF, and TNFα, NK cells can enhance neutrophil survival, activation,421,422 and the formation of Neutrophils Extracellular Traps (NET).423 However, neutrophils can inhibit NK cells through NKp46 cleavage by NETs enriched in cathepsin G.424 Tumor Neutrophils are the primary source of Arginase I (ARGI),425 which they store in granules. ARG1 depletion of L-Arginine by hydrolysis to L-ornithine and urea profoundly suppresses T-cell immune responses.426 Finally, neutrophils produce reactive oxygen species (ROS) such as H2O2, O2–, OH·, and HOCl, which reduce NK survival and cytotoxicity.427 Therefore, NK cells and neutrophils can modulate each other.
Myeloid-derived stem cells (MDSCs) and Tregs
These cells mediate NK cell function suppression by direct contact or secretion of TGFβ1428–430 and IL-2 depletion,431 respectively. However, both produce immunosuppressive IL-10. MDSCs could also suppress NK cells via the inhibitory receptor TIGIT (Fig. 7c), an effect abrogated by TIGIT blockade.432 Additionally, NK exposed to MDSCs have reduced CD3ζ, with impaired natural cytotoxicity receptors NKp46, NKp30, and CD16.433 MDSCs can also inhibit NK cells by direct interaction with NKp30.434 However, MDSCs were reported to stimulate NK cells to release IFN-γ by activating NKG2D by MDSCs ligand RAE-1.435 Retinoids and TGFβ produced by MDSC promote the trans-differentiation of naive CD4+ T cells into Foxp3+ Tregs.436 Interestingly, MDSCs and Tregs can convert extracellular ATP and ADP in the TME to cAMP by CD39 and, subsequently, CD73 dephosphorylates AMP to adenosine, which by binding to adenosine receptor (A2AR) on NK cells inhibits their antitumor response.437
Collaboration, suppression, and murder of T-cells
IL-2 released by activated T cells plays a role in NK cell activation and IFNγ production.438,439 Conversely, T helper cell type 1 (Th1) polarization requires IFNγ provided by activated NK cells.440 The same IFN-γ secreted by NK cells will also stimulate IL-12 production by DCs, which activates CD8 + T anti-tumor activity441 (Fig. 7d). Similarly, activation of DCs by Cetuximab-activated NK cells enhanced antigen-specific T-cell immune responses in patients with head and neck cancer.442 In another mutual collaboration, NK cells expressing the OX40 ligand and B7 will induce the proliferation of T-cells.443 Therefore, the presence of both NK and T-cells in tumors will be synergistic and beneficial, as shown in colorectal cancer patients where NK cells and CD8 + T cell infiltration is associated with prolonged patient survival.444 NK cells ability to constitutively secrete TGFβ128 may reduce inflammation and inhibit T-cell cytotoxicity and proliferation,29 allowing tissue repair.30 Additionally, NK cells secretion of immunosuppressive IL-10 in early response to systemic but not to local infection,32,33 indirectly limits T-cell activation by blocking DCs secretion of IL-12 and production of factors involved in antigen presentation34 and T-cell anti-viral response,35 thus promoting T-cell exhaustion36 and reducing immune-mediated damage to tissues. The involvement of NK cells in directly dampening T-cell activity by cytokines (IL-10, TGFβ1) and indirectly by blocking IL-12 cytokine secretion by DCs through NKp30 has now been extended to the direct kill of activated T-cells that express B7-H6.95 This finding also applies to CAR-CD19-T cells, which, upon knockout of their B7-H6, escape being killed and expand more. Concurrently to B7-H6-induced expression on activated T-cells, Kilian et al, observed downregulation of HLA-E and C-type lectin domain family 2 member D, perhaps further enhancing T-cells killing by NK cells. It is interesting to note that NK cells also kill immature DCs (see below) through NKp30 recognition and that this kill is prevented in mature DCs by enhanced expression of HLA-E.
Dendritic cells
NK cells enhance DCs maturation, IL-12 production, and priming of CD4(+) T-cell proliferation and IFNγ secretion445 (Fig. 7d). Immature DCs are killed by a subset of NK cells lacking KIRs446 and through signals mediated by NKp30,76 whereas mature DCs are protected from NK lysis by upregulation of MHC I molecules,447 HLA-E in particular.446 This DCs selection is important for the downstream development of adaptive immunity. CD56bright NK cells producing GM-CSF and CD154 can induce CD14+ monocyte differentiation into DCs, in RA and psoriatic arthritis patients.330 Therefore, NK cells promote monocyte differentiation into DC to sustain the disease. NK cells expression of ChemR23313 and CCRL2,314 which by attraction to chemerin recruit NK to colocalize with Chem23-expressing DCs in inflammatory sites. ChemR23 is also expressed on macrophages, adipocytes, and endothelial cells,315–317 suggesting they all colocalize with NK cells. As mentioned earlier, human NK cells activated by IL-2 express SIPR1,4 and 5, a G-coupled receptor protein chemoattracted to bioactive lipid S1P.318,319 Receptor SIPR5 expression by NK and DCs suggests their colocalization.320 Similarly, the induction of IFNγ from CD56bright subset interaction with DCs induces surface molecules CD62L, CCR7, and CXCR3331 in NK cells, thus increasing their potential to colocalize with DCs.
Tumor-associated fibroblasts (TAFs)
TAFs are heterogeneous populations derived from various cell types, including normal fibroblasts, smooth muscle cells, pericytes, and tumor epithelial cells transformed by the epithelial-mesenchymal transition. This heterogeneity creates a complex matrix in the tumor environment mainly focused on tissue remodeling by producing MMPs, VEGFA, and FAP. It also produces tumor-promoting factors, including FGF2, IGF, and HGF, and immunosuppressive factors TGFβ, PGE2, and IDO, as well as factors promoting inflammation, like chemokines CCL2, CXC, CXCL12, CXCL8, and IL-6. The concept that tumor-promoting inflammation by cancer cells and by the associated tumor microenvironment can support cancer progression is a well-established hallmark of cancer.376 TAFs production of inflammatory mediators Prostaglandin E2 (PGE2) and Indoleamine 2,3-dioxygenase (IDO) can suppress NK cells.448 PGE2, a significant product of cyclooxygenases, suppresses NK cell function by signaling through PGE2 receptors Ep(1-4),449–451 with Ep4 being the most potent at inhibiting IFNγ production.452 Additionally, tumor-derived PGE2 signaling through EP2 and EP4 receptors increases T-reg cell activity in lung cancer,453 further antagonizing NK cells. Similarly, PGE2 impairs the NK cell and DCs interactions, reducing IL-12 secretion by DCs and CD4 T-cell polarization.454 Tumor PGE2 was also reported to inhibit chemokine receptors on cDC cells, preventing their attraction by CCL5 and XCL1 secreted by NK cells.322 IDO metabolizes Tryptophan to L-kynurenine, which inhibits the upregulation of NKp46 and NKG2D under IL-2 stimulation. Therefore, IDO depletes tryptophan and starves, particularly T-cells,455 thus disrupting the cooperation between NK and T-cells, inhibiting CD4 and CD8 T-cells,456 and NK cell cytotoxicity.457 Knockdown of IDO in cancer cells enhanced their sensitivity to NK cells in vitro and promoted their accumulation in the tumors.458
Platelets
NK cells are essential for controlling metastasis. However, this task might be impeded by platelets, which are small non-nucleated fragments of megakaryocytes that aggregate with fibrin deposits on cancer cells’ surface in a process miming coagulation.459 Additionally, aggregated platelets could transfer MHC I to MHC I-deficient cancer cells, thereby interfering with the missing self-recognition by NK cells.460 In addition to the physical shielding of cancer cells, platelets are the richest source of TGFβ1, downregulating NKG2D in NK cells.461
Natural killer cells mediated cancer immunotherapies
Currently, NK cells used therapeutically are derived from PB,462 umbilical Cord blood (CB), and in vitro differentiated CD34+ progenitor cells,463 induced pluripotent stem cells (iPSCs),464 and immortalized NK cell lines, most notably NK-92 cell line which lacks most KIRs and is more likely to resist inhibition.465 NK cells isolated from PB are, by definition, mature with a complete armamentarium. However, they are stubbornly challenging to engineer, especially if repeated manipulations are needed to build on previous improvements. Like other continuously produced innate immune cells, primary NK cells lifespan is short (~2 weeks). This poses serious challenges for their use in immunotherapies. By definition, NK cells derived from CB are allogenic and were shown to induce monocyte-to-dendritic cell conversion in patient-derived cultures of primary and metastatic colorectal cancer.466 CB NK cells proliferate better than adult PB NK cells467 and can be obtained without the screening and leukapheresis required for adult PB NK. PB and CB NK cells can be expanded to large numbers using antigen-presenting feeder cells.468,469 Ex-vivo-activated autologous NK cells display less anti-tumor efficacy470 than allogeneic NK cells471 because self-class I HLA signaling suppresses NK cytotoxicity and cytokine release.472 Additionally, unlike allogeneic T-cells, allogeneic NK cells mediate anticancer effects without causing graft versus host disease.473–475 However, the effectiveness of allogeneic donor NK stimulated ex-vivo is reduced by competition for cytokines79,476 and approaches relying only on CAR technology, as CAR-NK cells suffer from tumor cell escape by HLA aberrant expression and epitope loss.477 Moreover, cytokine administration would be required to sustain NK cells after in vivo transfer,478 exposing patients to potential side effects. Therefore, developing novel NK cell activation strategies to reduce cytokine toxicity, increase resistance to immunosuppression, and enhance NK cell persistence is critical.
Arming NK cells with activating cytokine signaling
The activating signaling from cytokines serves a different purpose than the signaling from activating ligand/receptor interactions. Ligand/receptor activation signals mobilize the machinery for cell killing but can also trigger proliferation. On the other hand, cytokines signaling initially directs the NK cell’s maturation and later serves to enhance their survival and proliferation. This is achieved mainly through transcription factors STATs, which produce a battery of genes that will maintain the NK cellular homeostasis. Ligand-receptor activation signals mostly originate from aberrant cells, while cytokines are secreted and used mainly between immune cells. NK cells do not manufacture interleukins and depend on other immune cells for survival. NK cells will respond to IL-2 and IFNγ from T cells, IL-12, IL-15, IL-18, and IL-21 from DCs and macrophages. IL-2 stimulates both NK and T cells, including Tregs. For this reason, and to reduce IL-2 toxicity in immunotherapies, efforts were devoted to creating potent IL-2 forms that discriminate between NK cells and Tregs.479–481 We contributed to this effort by tethering IL-2 to its receptor IL2Rβ.288 Our strategy abrogated IL-2 toxicity and allowed enhanced NK cell activation and cytotoxicity in vitro and in vivo. IL-15 is the only cytokine capable of inducing NK cell proliferation in vivo. Embattled T-cells receive survival factors and cytokines in the tumor as trans-presented IL-15 by DCs.482 Surprisingly, the mature NK population could collapse in vivo when DCs are depleted, suggesting that most NK stimulation in vivo occurs through IL-15 trans presentation by DCs.483 IL-15 is among a few cytokines that can extend telomeres by enhancing telomerase activity in NK, NKT, and CD8 T-cells.484 However, telomeres erode at a rate of 50 bp/year in human T-cells,485 with old individuals having shorter telomeres than young subjects.486 This implies that differentiated primary NK cells used in immunotherapies will probably have similar shortcomings. In vitro, the viability of CB-NK expressing soluble IL-15 and CAR-CD19 declined precipitously from day four post-plating,487 suggesting telomeres loss due to insufficient activation from CARs. IL-15 substantially improved CB-NK use in NK cells, especially when combined with the knockout of the CIS gene.488 However, reports suggest that secreted IL-15 expands primary and CB-NK cells but causes severe487 to lethal toxicity and cytokine release syndrome in animal models.489 Others reported that NK cell chronic stimulation by IL-15 leads to exhaustion by a metabolic defect.490 IL-12 produced by DCs and macrophages stimulates NK cells and leads to IFNγ production, which enhances DCs activation and induces T-cell polarization. Additionally, we have seen earlier that IL-12 with IL-18 can reverse the anergy of NK cells in MHC I deficient tumors,114 suggesting IL-12 will have a critical role in NK-mediated immunotherapy. However, the use of IL-12 in the clinic is hampered by its induced neutropenia and thrombocytopenia.491
The generation of mouse cytomegalovirus-specific long-lived memory NK cells with higher responses compared to naïve NK cells was shown to be dependent on IL-12-STAT4 signaling.492 Short-term pre-activation with a combination of IL-12/15/18 can induce memory characteristics in human NK cells.462 These memory-like NK cells have prolonged expression of CD25, capable of responding to IL-2 at picomolar concentrations.493 Therefore, strategies to develop memory NK cells ex vivo for clinical therapy are worthy of investigation.
IL-18, when combined with IL-12, potentiates the production of IFNγ and TNF in NK cells.494 Alone, IL-18 induces NK cells with a helper phenotype expressing chemokine CCR7 that migrate to secondary lymphoid organs,367 where they could potentially synergize with adaptive immunity. However, IL-18 pleiotropic effect, role in tumor hypoxia and pregnancy, and its ability to induce more than 1000 genes in NK cells, as well as its overlap with IL-2, IL-12, and IL-15 functions366 render its use in immunotherapy problematic. Another intriguing effect of IL-18 is its ability to convert CD56dim to helper CD56bright CD16 Neg phenotype,367 which is potentially more pro-angiogenic. IL-18 is normally inactivated by binding to serum IL-18 binding protein. However, a remarkable recent advance was able to circumvent this hurdle.495
IL-21 induces the transcription of many genes,496 including suppressors of cytokine signaling, Socs1, and Socs3, which downregulate the JAK–STAT pathway and inhibit IL-2 signaling.497,498 IL-21 activates Stat3,499,500 and Stat1.501 This latter leads to IFNγ production.502 IL-21 showed some benefit when used as monotherapy in the clinic but will probably need to be combined with other modalities.503 IL-21 is a B-cell growth factor that can potentially promote the growth of lymphomas.504 Therefore, its use as a soluble factor entails some risks. Acknowledging that strategies using single cytokines are less likely to succeed is also important. Instead, a rationale for efficient combinations of cytokines such as IL-2 or IL-15 with IL-21, IL-12, or IL-18 should be developed. For example, IL-15 may preserve telomeres better than IL-2. IL-21 may increase the metabolic fitness of NK cells, while IL-12 and IL-18 may reverse exhaustion. However, the best combination of cytokines may still require their use with other modalities.
Countering Immunosuppressive factors
TGFβ is one of the main driving forces in the TME to exhaust NK cells. It suppresses NK cells by the induction of miR-183, which binds and represses DAP12 transcription/translation, a common dysfunction in NK cells infiltrating lung cancers.505 TGFβ helps cancer immune evasion by converting NK cells into exhausted type1 innate lymphoid cells with reduced anticancer activity (Fig. 8) and sequestration in tissues due to overexpression of α1 integrin and CD103.183 NK cells exposed to TGFβ or to its relative Activin, acquire a gene signature and phenotype similar to the less cytotoxic ILCs, becoming unable to control tumor growth in mice,183–185 suggesting a possible mechanism of NK cells exhaustion by reverting to an ILCs state. Engineering efforts that effectively addressed this issue were the introduction of a dominant negative of the TGFβ1 receptor, which competes with the endogenous TGFβ1R506 and separately, in another study through the knockout of TGFβR2.507
Fig. 8.

Strategies for arming and deploying NK cells. Chimeric antigen receptor (CAR) comprises an extracellular domain with the single-chain variable fragment scFv region (H heavy and L light chain) connected by a flexible hinge region mainly derived from CD8 to the transmembrane domain region primarily derived from CD28. The intracellular domain evolved in multiple iterations from a first generation developed in 1993 with CD3ζ, DAP12, or DAP10. This first generation proved not to be very effective in the clinic. By 1998, a significant leap was achieved in the second generation by adding costimulatory domains such as CD28, introduced by the Sadelain group, and later 4-1BB and 2B4 for NK cells. This second generation showed persistence and utility in the clinic. The third generation built on adding multiple costimulatory domains, while the fourth added the cytokine component or cytokine receptors and STAT binding domains. Therapeutic antibodies can directly mediate ADCC by NK cells expressing CD16. Immune checkpoint blockade directed against PD-1, CTLA-4, and TIGIT is another strategy that removes the brakes on NK cells. Bi-specific engagers mainly target CD16 and link it with tumor antigens, while tri-specific links CD16 and an activating receptor on NK cells to an antigen on tumor cells. Another modality utilizes an antigen recognition domain that binds to inhibitory ligands coupled to intracellular domains of activating receptors. This strategy converts inhibition of the TME into an activation signal. Seven levels of intervention to enhance anti-tumor activity: 1- Improving collaboration between NK cells and other effector cells such as T-cells, DCs, Macrophages, and Neutrophils. 2-armoring NK cells with cytokine signaling, 3- Engineering NK cells to improve persistence, metabolism, and resistance to exhaustion, 4- Preventing immunosuppression by TGFβ1, IDO, PGE2, adenosine, 5- Improving NK cells homing into tumors, 6- Preventing tumor angiogenesis and immunosuppression, 7- Reducing immunosuppression by MDSCs, T-regs, and TAFs
Knockdown of IDO in cancer cells increased their sensitivity to NK cells in vitro and promoted their accumulation in the tumors in vivo.458 L-Kynuenie, generated through IDO, depletes tryptophan, starves immune cells, impairs NK cell viability, and inhibits the upregulation of NKp46 and NKG2D under IL-2 stimulation. IDO is upregulated in cancer cells, APCs, and endothelial cells by TGFβ, IFN-γ, PGE2, PD-1, CTLA-4, IL-6, and TNF-α (reviewed in ref. 508). Therefore, IDO inhibition must be coupled with other modalities, such as immune checkpoints inhibitor of PD-1/PDL-1.
PGE2 and IDO suppress NK cells.448 Specifically, PGE2 increases T-reg cell activity in lung cancer by signaling through PGE2 receptors, with receptor EP4 being the most potent in further antagonizing NK cells. Similarly, PGE2 impairs the NK cell and DCs interactions, reducing IL-12 secretion by DCs and CD4 T-cell polarization. Tumor PGE2 was also reported to inhibit chemokine receptors on cDC cells. Most importantly, PGE2 could induce PD-L1 expression.509 Therefore, PGE2 inhibition must be also coupled with other modalities, such as immune checkpoints PD-1/PDL-1. Inhibitors of the Cox2-PGE2 axis, such as Celecoxib, cause bleeding and cannot be used long-term. Therefore, targeting the EP4 receptor with antagonists in combination with PD-1/PDL-1 would be more efficacious (reviewed in ref. 510).
Extracellular ATP rises in pathological conditions such as inflammation, ischemia, tumorigenesis, and hypoxia. In tumors, this extracellular ATP is converted into immunosuppressive adenosine using two ectonucleotidases CD39 and CD73 expressed on cancer cells, T-cells, T-regs, macrophages, neutrophils, MDSCs, NK, and the vasculature, thereby enriching the TME with high levels of adenosine511 (Fig. 8). Adenosine binds to widely expressed adenosine receptor A2AR, including on NK cells, macrophages, and DCs. The immunosuppression of adenosine is illustrated by tumor rejection in more than 60% of A2AR -deficient mice407 and promoting the accumulation of highly cytotoxic CD56dim NK cells with upregulation of CX3CR1 transcription in NK cells, suggesting that adenosine prevents NK cells maturation and infiltration into tumors.512 Further, emphasizing the impact of adenosine inhibition of NK cells is the synergy between anti-PD1 and A2AR inhibitors, which inhibited metastatic melanoma and was primarily dependent on NK cells and IFNγ more than CD8+ T-cells.513 Blocking ATP hydrolysis using antibodies against CD39 and CD73 prevented adenosine accumulation, stimulated DCs and macrophages, and restored T-cell anti-tumor activity.514 Therefore, reducing adenosine accumulation in combination with ICIs is an efficacious strategy.
In another mechanism of tumor resistance to NK cells, tumor cells up-regulate collagen expression to enhance adhesive structures in the TME. This consolidation of collagen protects tumor cells by binding to soluble leukocyte-associated Ig-like receptor-1 (LAIR-1) expressing NK cells. Upon engagement with tumor collagen, human LAIR-1 associates with SHP-1 and SHP-2 and proceeds to dephosphorylate VAV1, thereby dampening NK activation (Fig. 6).
Restoring dysfunctional NK cells
The impact of immune checkpoints on lifting exhaustion in T-cells is well established. However, for NK cells, the state of exhaustion is not well defined, as NK cells require at least two activating signals in addition to cytokines to mature and operate. These conditions are challenging to produce in the TME, where NK cells are usually dysfunctional.515–518 However, even NK cells that are being created in the bone marrow could be affected by the growth of remote tumors in mice by a process involving the downregulation of IL-15Rα+ cells among bone marrow stromal cells and the interrupted maturation of NK cells,519 a dysfunction that is remedied by IL-15 administration. Immature NK cells found in tumors were also reported in humans, and their presence was correlated with poor survival.520 Overall, an NK cell could be rendered dysfunctional remotely, even before it encounters a cancer cell. A transcriptional profile for these dysfunctional/exhausted NK cells is the low expression of T-Bet and Eomes transcription factors521 necessary to sustain maturation, identity, and anticancer activity,522 as well as CD16 and KIRs expression.153 In the early TME, Cb11+ myeloid cells (Basophils, monocytes, macrophages, and DCs), express soluble IL-15, and this contributes to the inflammatory response that helps NK cells proliferate in the early tumor stage, making IL-15, an activating component of the early TME, but later, due to mounting immunosuppressive forces in established tumors, IL-15 production diminishes.523 Additionally, NK cells that engage cancer cells and interact with other cells in the TME could be exhausted due to an overwhelming multitude of immunosuppressive factors and lack of activating cytokines with increased expression of inhibitory receptors including, NKG2A, CD96, PD-1, and TIGIT,524–528 as well as an across the board downregulation of major activating receptors which include DNAM-1, NCRs, NKG2D, CD16.282,529–531 The function of NK cells in cancer patients could be restored by chemotherapy,532–534 through multiple mechanisms, or by surgical removal of the primary tumor.535 NK cell function will most likely also depends on the tumor burden. Therefore, these modalities may be necessary to complement new and emerging clinical interventions.
Immune checkpoints blockades
Since NK cell response integrates both inhibitory and activation signals, the blockade of any inhibitory receptor should enhance NK cell activation. Using ICI in the form of antibodies that bind these receptors and block the binding of their cognate ligands from cancer cells has shown a clear impact in T-cell immunotherapies, especially for PD-1 and CTLA-4. The participation of NK cells alongside T-cells in these therapies was noticeable,525,536–538 especially in MHC I-defective tumors. However, recent studies showed minimal PD-1 expression in NK cells from tumors, raising questions about its importance in PDL-1-expressing tumors.539 The same study suggested that TIGIT is markedly upregulated in these NK cells. CTLA-4 blockade with Ipilumab’s impact on NK cells is not clear. However, it was found to operate through the elimination of T-regs by NK cells mediated ADCC.540 CTLA4 engagement with ligands leads to its phosphorylation and recruitment of SHP-1 and SHP-2, leading to VAV1 dephosphorylation. After engagement with PDL-1, PD-1 ITSM domain phosphorylation at Tyrosine Y248 recruits SHP-2, suppressing NK cell activation (Fig. 6).
TIGIT is consistently upregulated in NK cells in human primary tumors and viral infection.539 TIGIT blockade reverses the exhaustion of NK cells from colon cancer patients and promotes their antitumor responses in mouse models.524 Additionally, NK cells expressing low TIGIT are resistant to MDSCs inhibition,432 suggesting the importance of this receptor in the crosstalk within the TME. In patients with metastatic melanoma, functionally impaired/exhausted, NK cells upregulated TIM-3 in NK cells compared to healthy subjects, and TIM-3 blockade in vitro reversed this exhausted phenotype.541 In T-cells, TIM-3 engagement with ligands leads to the phosphorylation of two tyrosines in its cytoplasmic tail (Y256 and Y263), leading to the dissociation of HLA-B associated transcript-3 (Bat-3). This dissociation disrupts LCK, ZAP70, and TCR activation.542 However, disengaged Bat-3 can also associate with P300, leading to transcription of MDM2, P21, BCL2, and the acetylation of P53, which may slow NK cell proliferation. Engagement of TIGIT with PVR and its phosphorylation through the Src- family kinases Fyn and Lck results in SHP-1 and SHP-2 recruitment, which in turn downregulates the PI3K, MAPK, and NF-KB signaling pathways and promotes VAV1 dephosphorylation. LAG-3 is highly expressed in T-cells from Hodgkin lymphoma and leukemia patients, and its synergy with anti-PD-1 was evident.543 In vitro, chronic stimulation of NK cells leads to epigenetic changes, upregulation of LAG-3 and PD-1, and NK cell dysfunction.544 LAG-3 mediated inhibition controls AKT phosphorylation and STAT5 activation leading to reduced mitochondria mass and quiescence.545 However, LAG-3 impact on NK cells remains obscure and needs more investigation. This might be due to the presence of many inhibiting receptors on NK cell surface that oppose any activation by ICIs. Additionally, a significant obstacle to ICI success is the simultaneous co-expression of many ICs in T-cells and probably NK cells, causing ICI failure.546
Eliminating NK cells fratricide
Acquisition of tumor antigens by NK cells through engagement with cancer cells in a membrane transfer process called trogocytosis could lead to the misidentification of these NK cells as targets and their death by other NK cells. Interaction between ligands and receptors leads to the loss of the membrane patch harboring the ligand to NK cells. The example of NKG2D interaction with ligand RaeI revealed that this process required clathrin-dependent internalization of NKG2D, leaving RaeI on the cell surface for a period of at least 24 h.547 RaeI-dressed NK cells do not kill each other, suggesting they lost NKG2D in this process and are only killed by cells that did not interact with cancer cells. The loss of NKG2D would render these trogocytic NK cells anergic. In mice, the interaction of receptor 2B4 on one NK cell with its ligand CD48 on another NK cell was reported to prevent fratricide. Blocking this interaction with antibodies led to fratricide.548 Because CD48 is expressed on all nucleated hematopoietic cells, it may provide a non-MHC mechanism of self-tolerance. Fratricide is worsened when using ADCC and CAR-NK mediated therapies that will increase the visibility of NK cells if they express the antigen naturally or acquire it by trogocytosis. Multiple myeloma expressing high levels of CD38 and targeted with anti-CD38 Daratumumab (Dara), reduces NK cell number due to ADCC-mediated fratricide.549 Knockout of CD38 in expanded primary NK cells prevented Dara-induced fratricide in NSG mice, which are devoid of NK cells.550 However, host NK cells would probably be targeted by ADCC if the patient is not lymphodepleted first. A similar strategy using iPSCs cells FT576, depleted of CD38 in combination with Dara, prevented fratricide and showed efficacy in preclinical models, opening a path to clinical translation.551
However, the fratricide caused by trogocytosis cannot be eliminated by knockout of the antigen on NK cells. Rezvani et al. cleverly put a brake on killing trogocytic NK cells by adding a self-inhibitory iCAR targeting co-receptor Cs1 which is expressed on all NK cells and transmits an inhibitory signal via the KIR2DL1 cytoplasmic domain in iCAR. CD19+ cells did not express Cs1 and were targeted by an additional CAR against CD19.552
NK cell engagers
Contact between immune cells and cancer cells can be encouraged by “engagers” that serve as a bridge by binding simultaneously to an activating receptor on the immune cell and a tumor-specific antigen on the cancer cell. Bite, trike, or tetraspecific engagers involve 2 or 3 or 4 receptors to strengthen the engagement and increase activation signals. The attractive aspect of engagers is the non-need to genetically modify immune cells and primarily target host immune cells to reactivate them. Early generations of engagers designed for T-cells and targeting CD3 have shown some success in hematological disease, with some toxicities that limited their efficacy. Among them, Blinatumomab, the first bite approved by the FDA, is a dual CD19 and CD3 engager. Blinatumomab is used to manage minimal residual disease after chemotherapy but is ineffective for certain patients who may relapse due to loss of CD19 or T-cell exhaustion. Several toxicities were reported for Blinatumomab, including neutropenia, neurotoxicity, infection, and cytokine release syndrome. Indeed, CD3-targeting engagers have been associated with severe toxicity, as in the case of Duvortuxizumab (anti-TAA+anti-CD3) and AFM11(anti-CD16a+anti-CD3). New generations of engagers seem to shift focus on the potential of innate immunity and enhancing its role in helping adaptive immunity. Three trifunctional NK cell engagers, targeting NKp46 and CD16 on NK cells and a tumor antigen on cancer cells (CD19, CD20, and EGFR) were shown to enhance cancer cell killing by human primary NK cells in vitro and in mice models.553 Another NK cell engager, AFM13 (CD16 + CD30), used with cord blood NK cells, exhibited enhanced killing of CD30+ leukemia and lymphoma targets.554 Several clinical trials evaluate multiple engagers, mainly targeting T-cells,555 with one ongoing trial NCT04074746 (Table 1) evaluating CB NK cells combined with AFM13 against R/R CD30+ Hodgkin lymphoma and non-Hodgkin lymphoma (reviewed in ref. 556).
Engineered NK cells
The therapeutic efficacy of non-engineered NK cells is suboptimal, and using autologous unmodified NK cells is not conducive to better antitumor activity.470–472 Similarly, autologous NK cells derived from cancer patients are particularly weak candidates as they may be already in exhausted and dysfunctional states516,528 that could even be beneficial to tumors.373,374 These states suggest NK cell’s plasticity and ability for re-education in the tumor microenvironment. Therefore, understanding how NK cells are co-opted in tumors may help design better strategies to engineer resilient and incorruptible NK cells.
Clonal cell lines a canvas still waiting for art
The first NK-based clinical trial was done using the cell line NK-92, which was originally derived from a patient’s blood with diffuse lymphadenopathy, B-symptoms and circulating LGL. Despite aggressive chemotherapy the patient passed of progressive lymphoma, roughly five weeks after admission. For this reason, the FDA requires NK-92 cell irradiation prior to administration. NK-92 is an obligate IL-2-dependent cell line with an anti-tumor activity superior to other NK cell lines and has a high safety profile despite its allogenic nature.465 NK-92 genome is aneuploid with a heterozygous stop mutation in the P53 gene. Clonal NK-92 cells are CD56bright, CD16neg, and KIRneg, a phenotype close to the recently identified NK2 population,42 making them a plausible descendent of the ILCP lineage. Whether it is possible to reprogram/reeducate NK-92 into a clonal cell line similar to an NK1 or an NK3 subset, is an intriguing question. Similar clonal NK cell lines were isolated (Table 2), but NK-92 unique characteristics, such as lack of KIRs and ease of genetic engineering are not yet fully exploited. This is probably due to the modest results using NK-92 in a phase I clinical trial, which showed minor responses in two patients out of twelve81 and another using CD33-CARNK-92 which showed safety but obvious clinical efficacy.557 The reported lack of efficacy in another phase I clinical trial for refractory and relapsed acute myeloid leukemia558 was attributed to circulating exosomes carrying an immunosuppressive cargo and disabling NK-92.559 Another clinical trial reported safety and some evidence of efficacy.560 A significant impediment to NK-92 use is the requirement for its irradiation. Thirty-one years after its first isolation,465 and despite widespread use, no reports of spontaneous IL-2-independent NK-92 clones exist. NK-92 does not cause tumors in ICR/scid mice even when supplemented with exogenous IL-2 or producing its IL-2.561,562 The risk that NK-92 cells could proliferate in vivo without a sustaining signaling has not been demonstrated in vivo. Still, it is speculated based on anecdotal findings with different tumor cell lines that caused subcutaneous nodules when implanted in terminal cancer patients despite failing in healthy volunteers.563 Additionally, numerous studies have shown no association between blood transfusion from precancerous blood donors and non-Hodgkin lymphoma risk,564–566 suggesting the unlikelihood of allogenic transfer of cancerous cells in healthy recipients. However, it cannot be excluded that NK-92 cells could proliferate if driven by self-sustaining IL-2 stimulation in severely immunodeficient patients. Therefore, combinations of tumor-suppressing signaling and suicide switches such as prodrug activating cytochrome P450 and HSV-TK enzymes or drug-activated iCasp9 switch, all inserted in multiple and separate chromosomal locations in the NK-92 cells genome could be a convincing step toward the potential use of this remarkable cell line in cancer patients, safely. This is possible through multiple rounds of infection and selection. However, more effort should be first deployed to improve the clonal cell lines’ efficacy by first determining the optimal signaling that drives cytotoxicity and resistance to exhaustion, followed by deciding what series of genes to add to improve tumor homing and strengthen activation signaling and what genes to eliminate, including ICs, to reinforce all these aspects and further enhance safety. Unfortunately, groups improving NK cell lines are rarely funded. Since its isolation, NK-92 and many other human NK cell lines have been instrumental in elucidating the biology of NK cells. However, except for NK-92 none of them advanced to clinical use in humans. The translational importance of NK cell lines will probably be more evident if they were to be used to treat animal cancers. A canine NK cell line (CN89)567 was reported as CD5+, CD8+, CD45+, CD56+, CD79a+ and NKp46+. However, its IL-2-independence and the B-cell marker CD79a+ as well as the absence of reports of cytolytic activity, cast doubt about its antitumor activity. While many canine clinical trials are ongoing (reviewed568), no canine NK cell line is being tested. However, the possibility of using human NK-92 for canine cancer by blocking xenoreaction with immunosuppressors is suggested by a phase I clinical trial in dogs, where a human T-cell line derived from child leukemia called TALL-104 was used safely.569 Unlike other T-cells, TALL-104 has lost its MHC I dependence and become an MHC I non-restricted T-cell line, much like NK cells. No toxicity to dogs was observed in the clinical trial. Seven dogs out of 19 showed a response, with one complete remission. Cyclosporin, an immunosuppressor was administered to dogs prior to TALL-104 infusion to prevent an anaphylactic reaction.
Table 2.
List of human clonal NK cell lines
| Cell line | Year established/published | Patient diagnosis | Age of donor | Sex | EBV status | Cytokine dependence | Clinical Trials |
|---|---|---|---|---|---|---|---|
| NK3.3 | 1982 | Cloning of primary MLC-activated cells in a medium containing interleukin-2 | N/A | N/A | EBV− | IL-2-dependent | NO |
| YT | 1983 | Acute lymphoblastic lymphoma (with thymoma) | 15 | Male | EBV+ | IL-2-independent | NO |
| NK-92 | 1992/1994 | LGL-NHL | 50 | Male | EBV− | IL-2-dependent | Yes (Table 1) |
| NKL | 1996 | NK-LGLL | 63 | Male | EBV− | IL-2-dependent | NO |
| NK-YS | 1996 | NK cell lymphoma, Nasal angiocentric, Leukemic state with systemic skin infiltration | 19 | Female | EBV+ | IL-2-dependent | NO |
| KHYG-1 | 1997 | Aggressive NK leukemia | 45 | Female | EBV− | IL-2-dependent | NO |
| HANK1 | 1998 | Nasal-like NK/T-cell lymphoma | 46 | Female | EBV+ | IL-2-dependent | NO |
| SNK-6 | 1998 | Nasal NK/T-cell lymphoma | 62 | Male | EBV+ | IL-2-dependent | NO |
| SNT-8 | 1998 | Nasal NK/T-cell lymphoma | 48 | Female | EBV+ | IL-2-dependent | NO |
| IMC-1 | 2004 | Aggressive NK cell leukemia | 42 | Male | EBV− | IL-2-dependent | NO |
LGL large granular lymphocyte, LGLL large granular lymphocyte leukemia, NHL non-Hodgkin’s lymphoma, EBV Epstein-Barr virus
Chimeric antigen receptors (CARs) advances in the clinic
CARs are synthetic constructs emulating the TCR function but without the HLA requirement developed first for T-cells (CAR-T). They comprised in their first iteration (first generation) an extracellular antigen recognition domain, which is the single-chain fragment variable (ScFv) derived from an antibody tethered to a transmembrane domain and the intracellular activation domain CD3ζ chain (Fig. 8). The binding of ScFv to a specific antigen triggers activation. However, this design allowed very short-term proliferation. The second-generation CARs added the CD28 activation domain, later reinforced by another costimulatory molecule, CD134 (OX40), or CD137 (4-1BB) for CAR-T570 and 2B4571 or CD137 for CAR-NK.572 The latest and fourth generation added cytokines IL-12 expression under the control of the NFAT6 minimal promoter that initiates IL-12 transcription upon CAR-T-cell activation.573 In another approach, cytokines such as IL-15 could be produced constitutively (reviewed in ref. 574).
Novel strategies to convert inhibition into activation are emerging. For example, TGFβ immunosuppression in the TME can be converted into activation by tethering TGFβR2 extracellular domain to NKG2D cytoplasmic domain.575 Another strategy converted IL-4 suppressive signals in the TME into proliferative signals from the ectodomain of the IL-4 receptor to the cytoplasmic domain of the IL-7 receptor.576 NKG2D, a specific NK activator and specifically its extracellular recognition domain, could replace the ScFv and be tethered to DAP10 and CD3ζ.577 These innovative strategies seem to work with NK cells and may improve immunotherapies. The mere expression of CAR in NK cells may enhance the baseline signaling through the interaction of endogenous cell components with the costimulatory domains, even without antigen stimulation. This phenomenon has been reported for CAR-T and is termed tonic signaling and has been explained by the heterodimerization of CAR’s CD28 with the endogenous CD28.578 We have seen a comparable effect in NK-92 expressing CD28-based CAR, leading to faster cell growth than naïve NK-92, without engagement with antigen (Chen et al. unpublished data). In T-cells, CARs induce cytotoxicity and proliferation by producing an autocrine loop of cytokines, whereas in NK cells they only induce cytotoxicity. Therefore, in contrast to CAR-T, CAR-NK cells need the addition of cytokines for survival and metabolic fitness.
Four representative clinical trials using different sources of NK cells (cord blood, iPSCs, and PB NK) are discussed here. Rezvani et al. in a clinical trial initiated in 2017 (NCT03056339), reported in 2020 phase I interim results.90 These showed that cord blood CAR-NK-CD19 cells armored with soluble IL-15 could persist in patients for over a year with a single infusion, with an overall response rate (ORR) of 73% and achieving complete remission (CR) for seven out of eleven patients, without any cytokine related syndrome (CRS), graft versus host disease (GVHD), Immune effector cell-associated neurotoxicity syndrome (ICANS) or NK-related toxicity as. The phase1/2 results were recently reported,579 with an ORR (d30)= 48.6%, ORR (d100)= 48.6%, 1 year: Overall survival (OS) = 86%, and progression-free survival (PFS) = 32%, with no CRS/ICANS/GVHD. Notably, the patients who achieved higher ORR had higher levels and longer persistence of CAR-NK cells. Most of these patients received a lymphodepleting nonmyeloablative preparative regimen of cyclophosphamide and fludarabine prior to CAR-NK infusion and received follow-up treatment 30 days post-infusion. Studies have shown that lymphodepletion enhances CAR-T efficacy by eliminating “cytokine sinks” competition by T-regs and other competing immune system elements.580 Lymphodepletion conditions the immune system to eliminate regulatory mechanisms that could hinder the functioning of infused CAR-T cells. Therefore, lymphodepletion prior to infusion most probably enhances CAR-NK efficacy. Furthermore, radiation treatment induced CXCL8 secretion from tumor cells and enhanced the directional migration of CD56dim NK cells to the tumor.581 Conditioning and multifactorial therapies will be even more necessary to give cell immunotherapies a winning chance against the more challenging solid tumors.
So far, the US Food and Drug Administration (FDA) has only approved CAR-T cell therapies against hematological cancers. Solid tumor heterogeneity and antigenic diversity, in addition to the TME immunosuppressive factors, are challenging. Therefore, better design of CARs incorporating other activation signals such as STAT3 and STAT5582 and other useful activators that may allow better survival of CAR-NK cells in the TME if combined with other modalities.
Another clinical trial (NCT04245722),583 used FT596, an off-the-shelf, CAR-NK-CD19 cell therapy derived from iPSCs, in patients with relapsed/refractory (R/R) B-cell lymphomas (BCLs) and chronic lymphocytic leukemia. FT596 employs three anti-tumor strategies: (1) a proprietary CD19-targeting CAR; (2) a high-affinity, non-cleavable CD16 Fc receptor that facilitates tumor targeting and enhances ADCC when paired with a therapeutic mAb; and (3) an IL-15/IL-15 receptor fusion that promotes cytokine-autonomous persistence. Preclinical in vivo models of leukemia and lymphoma have shown FT596 CAR-mediated effectiveness against CD19+ tumor cells. Additionally, when combined with the anti-CD20 agent rituximab, FT596 was effective against both CD19+ and CD19− tumor cells.
In the interim results reported in 2021, No dose-limiting toxicities, no ICANS/GVHD, but two cases of CRS were reported. After the first FT596 treatment cycle, ORR was observed in 5/8 patients receiving FT596 as monotherapy; when combined with Rituximab the ORR was obtained in 5/9 patients. At single-dose levels of at least 90 million FT596 cells as monotherapy, 8 of 11 patients achieved an OR, including 7 CRs. Among the 4 patients with prior CAR-T cell therapy treatment, a dose of at least 90 million FT596 cells, achieved CR in two patients. Most importantly, No B- or T-cell mediated anti-FT596 responses were seen. These results demonstrate the efficacy and safety of off-the-shelf NK cells derived from iPSCs. Noteworthy is the use of conditioning chemotherapy (cyclophosphamide and Fludarabine) prior to cell infusion.
In clinical trial NCT03415100, Xiao et al.584 used a novel chimeric antigen receptor (CAR) combining the extracellular domain of the natural killer (NK) cell receptor NKG2D with DAP12. Expression of the NKG2D-RNA-CAR significantly enhanced NK cell cytolytic activity in vitro, and in vivo in mice. The clinical trial interim results reported in 2019 showed that three patients with metastatic colorectal cancer were treated with local infusion of CAR-NK cells. Two patients experienced reduced ascites generation and a marked decrease in tumor cells in ascites samples (2/2, RECIST: SD), while one patient exhibited rapid tumor regression and a complete metabolic response in treated liver lesions (1/1, RECIST: SD). This small sample clinical trial shows the potency of PB NK cells when activated with hybrid chimeric antigen receptors based on NK cell receptors.
Another phase 1 clinical trial (NCT06325748) is currently enrolling adult patients with R/R CD33 and/or FLT3 expressing heme malignancies for allogenic treatment using SENTI-202, a Logic Gated off-the-shelf CAR-NK cell therapy candidate that selectively targets hematologic malignancies, using three technologies: 1) the OR GATE, which is an activating CAR that targets either or both CD33 and FLT3, 2) the NOT GATE, which recognize and protect healthy cells from being killed. And 3) a calibrated release of IL-15 to increase the persistence/expansion and activity of CAR-NK cells and potentially the host immune cells. This first-in-man trial will inform on the potential of this novel technology.
Engineered NK cells and the persistence problem
Infused allogenic haploidentical NK cells do not persist for more than 3 weeks and are eliminated by the recipient patient’s immune system.89,471,585,586 However, this rejection could be delayed by lymphodepletion,79 which, although not required in autologous NK cell transfer might improve NK cytolytic activity as mentioned earlier and especially when T-cells are weakened in the recipient.587 The most used lymphodepleting agents, cyclophosphamide or Fludarabine are given for one week. Lymphodepletion also eliminates “cytokine sinks” competition by T-regs and other competing immune system elements.580 The importance of lymphodepletion for NK cell expansion in the recipient is clearly demonstrated by the increased number of NK cells settling in the bone marrow when lymphodepletion intensity increases.588 Autologous NK cells would, in theory, persist more if the conditions that incapacitated/exhausted them in the cancer patient were removed. For this reason, we believe that engineering autologous NK cells, for example, by depletion of inhibiting receptors and increasing activating signaling, is a logical approach. Knockdown of HLA in haploidentical (half-identical) NK cells, assuming it is easily doable and does not lead to fratricide through missing self, may increase persistence and delay elimination by the recipient immune system. This approach was shown to work for human T-cells.589 A more practical solution is increasing the intensity of lymphodepletion, which may be an effective way to increase persistence, especially when combined with cytokine production by infused NK cells.590 Another possible therapeutic option could be the adaptive NK cells NKG2ChighCD57+ that expand in humans infected with HCMV.179,180 Interestingly, these cells downregulate PLZF, making them probably less susceptible to reverting to a less cytolytic ILC1 phenotype. These adaptive NK cells have significant persistence,591 pronounced ADCC, resistance to MDSCs and are intrinsically resistant to Treg cells.592
TCR-dressed NK cells
Engineering TCRs in allogeneic T-cells is a significant challenge since introduced TCRs will form mispaired non-specific TCRs.593,594 Therefore, expressing TCRs in NK cells that lack TCRs is a more sensible approach.595 TCRs that recognize tumor antigens presented by MHC can bind to all cellular antigens, including intracellular antigens unreachable by CAR-T and CAR-NK. CARs target only membrane proteins, which are encoded by one-fourth of the human genome,596 leaving 75% of proteins out of reach. This reduces the usefulness of the CARs approach, which also suffers from tumor antigen escape.597 Additionally, targeting normal overexpressed antigens by CARs can veer off-target or completely deplete normal tissue. This approach is exemplified by the anti-CD19 CAR strategy, which kills all B-cells,598 leaving cured survivors with a permanent need for antibody infusion and a lack of response to vaccination in pandemics.599 Tumor-infiltrating T-cells isolated from tumors often express tumor-specific TCRs.600–602 However, they may already be exhausted.603,604 NK cells naturally attack tumors in an MHC-independent manner, clearing tumors that antigen-specific T-cells cannot possibly target.38,605,606 NK cells are activated when MHC I expression is downregulated in transformed607 and virus-infected cells.170 The acquisition of resistance phenotype by tumor cells is often caused by the expression of inhibitory signals from MHC I.608 Indeed, HLA-E and HLA-G inhibit tumor cell lysis by NK cells.608,609 Therefore, combining the TCR-antigen-MHC-dependent recognition with innate MHC-independent tumor recognition will expand NK cells’ killing repertoire. TCR activation enables T-cells to manufacture IL-21 in an autocrine loop610 to activate Stat1611 and Stat3.612 Stat3 enhances telomeres maintenance.613 TCR activation also allows the production of IL-2,611 which promotes T-cell proliferation by activating STAT5. Finally, TCR activation prolongs cytokine signaling by downregulating CIS and Socs3.614 However, since CAR-NK and TCR-NK cells do not proliferate in response to antigens, as they require cytokines, adding a potent cytokine such as IL-15, IL-21, IL-2, or their combination will enhance the longevity and fitness of NK cells. The persistence of NK cells is another pressing problem, and lessons learned from CAR-T show that the shortening of telomeres depends on patients’ age and that the loss continues during the manufacturing process of CAR-T cells. Indeed, introducing hTERT mRNA in CD19-CAR-T led to more persistence in vivo,615 and the exhaustion of CAR-T directed against melanoma correlated with telomeres length.616 Similar strategies are probably needed to enhance NK cell persistence. Two clinical trials are currently testing TCR-NK. NCT06383572, a phase I/II trial evaluating the safety, and effectiveness of PRAME-T cell receptor-natural killer (PRAME-TCR-NK) cells against AML, MDS, and relapsed/refractory multiple myeloma. NCT06083883 is another phase 1/1b trial evaluating an Affinity-enhanced T-cell Receptor (TCR) Against the NY-ESO-1 in patients with advanced synovial sarcoma and myxoid/round cell liposarcoma. Both clinical trials use lymphodepleting chemotherapy.
Environmental factors
A series of studies evaluating the impact of the environment on human NK cells revealed that CD16pos NK cell cytolytic activity could be increased in males and females to last up to 7 days by a simple walk in the forest “forest bathing” while a walk in the city did not,617–620 with, nevertheless, a surprising decrease of CD4 cells. More recent studies confirmed the increase in CD56pos NK cells in a forest bathing group compared to an urban group.621,622 The earlier pioneering studies suspected phytoncides released from pine trees, which were confirmed to be the main factor in NK cell enhancement623 (Fig. 9). The relaxed feeling in the forest caused a decrease in the concentrations of cortisol in the blood and adrenaline in urine, suggesting the possibility of less immune inhibition from cortisol.619 A phytoncide (α-pinene) was shown to activate the ERK/AKT pathway in NK-92 cells and to increase their cytolytic activity,624 suggesting a direct effect on NK cells. Similar effects were noted for other phytochemicals, such as cymene and camphor. However, we hypothesize that the complex composition of phytoncide, which includes α-pinene, β-pinene, 1,8-Cineole, γ-Terpinene, Camphene, and Limonene, could have multiple targets that enhance innate immunity in particular NK cells. The Aryl Hydrocarbon Receptor (AhR) and aryl hydrocarbon receptor nuclear translocator (ARNT) are heterodimerizing transcription factors involved in sensing and responding to toxic xenobiotic chemicals by activating the transcription of CYP1A1, CYP1B1, IDO1, TDO, IL-22, GSTA and Aryl-Hydrocarbon Receptor Repressor AhRR. Recent work showed that in the pine beetle (Dendroctonus armandi), both AhR and ARNT were substantially induced by β-pinene and Limonene,625 leading to the induction of several phase I enzymes. IL-22 enhances Tryptophan synthesis in the gut and increases Trp hydroxylase (THP1), leading to Serotonin and endogenous tryptophan derivative, 6-formylindolo[3,2-b] carbazole (FICZ) production by deamination of Tryptamine.626 FICZ is also a potent ligand for AhR and can potentiate NK cell IFNγ production and cytolytic activity and control of tumors.627
Fig. 9.

Impact of the environment on NK cells: Phytoncide model. From the pine beetle to humans, a response to xenobiotics via the xenobiotic response element is mediated by the Aryl hydrocarbon receptor, which is induced by natural compounds and environmental pollutants that bind and activate AhR. Endogenous compound FICZ derived from Tryptophan metabolism can bind AhR with high affinity and trigger the expression of phase I enzymes, IDO, TDO, and IL-22 with wide-ranging physiological effects. AhR activation by FICZ affects NK cytolytic activity, and migration to tumors, with possibly a shift in Tryptophan metabolism by the microbiome from the Kynurenine pathway, which mainly degrades tryptophan in the liver by TDO, and IDO, to the beneficial 5-HT pathway by TPH, in the gut and brain and the Indole pathway catalyzed by the gut microbiota to generate indoles such FICZ which reinforces this signaling loop and enhances cancer control
More evidence is mounting to support AhR/ARNT role in mediating the transcription of genes involved in inflammation and control of the differentiation and activity of adaptive and innate immune cells.628 Additionally, NK cells stimulated by cytokine IL-2, IL-15, or IL-12 induce AhR expression, which can be activated by tryptophan derivative FICZ, the most potent ligand for the AhR produced endogenously and by gut microorganisms.629 Activated AhR also enhances the activation of the AKT serine/threonine kinase AKT pathway to promote cell survival.630 Activated AhR regulates NK cell migration through the Asb2 gene, which mediates the degradation of Filamin A via ubiquitination, leading to increased NK cell migration into tumors631 (Fig. 9). Therefore, we hypothesize that “forest bathing” might activate ERK/AKT by specific products such as α-pinene and induce AhR through β-pinene and Limonene. AhR is then activated by endogenous ligands, among which is tryptophan derivative FICZ, whose production by gut microbiota is promoted by phytoncide-AhR-induced IL-22. Interestingly, immunosuppressive tryptophan derivative Kynurenine, produced by IDO in tumors, can also activate AhR in T-cells to generate T-regs,632 inhibit CD4 and CD8 T cells,456 and reduce NK cytotoxicity.457 Therefore, tryptophan derivative FICZ and tryptophan metabolite Kynurenine have different impacts on AhR activation. However, FICZ binds to AhR in the nanomolar range and outcompetes kynurenine and its metabolites. Another metabolite of tryptophan is serotonin, which incidentally increases during forest bathing,633 suggesting that taking a walk in the forest may cause a shift in Tryptophan metabolism from the kynurenine pathway, which mostly degrades Tryptophan in the liver by Trp-2,3-dioxygenase (TDO), indoleamine-2,3-dioxygenase (IDO), to the 5-HT pathway by TPH, which takes place in both the gut and brain and the Indole pathway catalyzed by the gut microbiota to generate indoles such FICZ. Interestingly, the kynurenine pathway seems to dominate in tumors mostly through IDO, and consequently, tumors are depleted of Tryptophan. This suggests that supplementing indoles such as FICZ with its strong affinity to AhR or strengthening microbiota production of indoles, possibly by forest phytoncides, might counterbalance the kynurenine pathway inhibition of NK cells in tumors.
Concluding remarks
We attempted to review many aspects of NK biology in healthy and pathological conditions. How they are inhibited, and why their killing requires the synergy between multiple activation signals. NK cells can support angiogenesis in tumors and pregnancy and reduce T-cell overreaction to infection through IL-10 secretion to prevent tissue damage. They can also activate DCs, which in response, secrete IL-12 to enhance NK cell’s secretion of IFNγ, which is required for CD4 polarization. They help M0 and M2 macrophages transition to the pro-inflammatory M1 phenotype and induce monocyte conversion to DCs. They can detect and kill cancer cells lacking MHC I and senescent, stressed, and virus-infected cells. And can by delivering GNLY via nanotubes, specifically, kill bacteria in DCs, macrophages and trophoblasts, without harming host cells. We also examined the vast interactions with other immune cells of innate and adaptive immunity. It is clear at this point that NK cells are not created to do a unique specific task. This bewildering range of tasks will require many functionally adaptive or reprogramed NK cell states. This is probably achieved through origination from two lineages and egress at early stages from the bone marrow and subsequent wide variety of spatiotemporal education and maturation processes from which heterogenous NK cell populations emerge. Therefore, when using NK cells for therapeutic purposes, are these functionally adaptive/reprogrammed states of NK cells optimal? For example, the potential for reciprocal crosstalk between cytokine-producing CD56bright subset, which accumulates in draining lymph nodes634 with comigrating neutrophils,635 suggests an important role of this subset in the development of adaptive immune responses. Similarly, the CD56bright, CD16neg NK cell subset high antioxidative capacity and resistance to ROS produced by neutrophils636 suggests this subset more than the CD56dim, CD16pos is more suitable to resist inhibition by tumor resident neutrophils and participate in the cross-talk between neutrophil NK and DCs. Additionally, the downregulation of NKp46 and NKG2D expression, induced by phagocytes produced ROS was observed in the CD56dim but not the CD56bright subset of NK cells.637 On the other hand, a clear advantage of CD56dim and CD16pos is their potential combination with therapeutic antibodies. Additionally, CD56bright, like uneducated NK cells respond to inhibitory signals with strong production of phosphatase SHP-1, leading to rapid inactivation (Fig. 10), while CD56dim, educated, licensed NK cells produce less SHP-1 when encountering these inhibitory ligands, allowing them to better resist inhibition.145
Fig. 10.
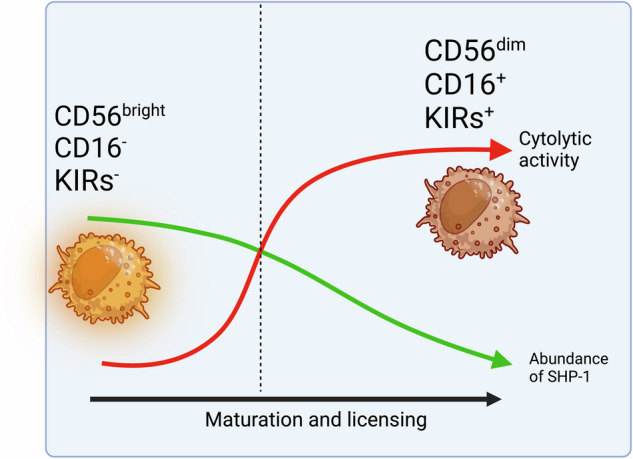
SHP-1 content is modulated by licensing and determines the degree of activation. CD56bright uneducated NK cells respond to inhibitory signals with strong production of phosphatase SHP-1, leading to their rapid inactivation, while educated, licensed CD56dim NK cells have reduced SHP-1 production when responding to inhibitory ligands, allowing them to resist deactivation
Similarly, clonal NK cell lines isolated from patients have distinct phenotypes reflecting NK cell diversity. They, however, provide a homogenous starting population that allows precise genetic engineering that is only limited by imagination. However, most genetic modifications on NK cell lines often implement progresses made in CAR-T for T-cells, not based on NK cell biology. For example, the NK-92 cell line, with its superior anticancer properties, has not been radically engineered to proliferate in vivo under suicide switches in animal models to test whether this proliferation can eradicate tumors. Studies showing that suicide switches can control NK-92 proliferation in vivo are also lacking. The fact that clinical trials using NK-92 showed little benefit suggests that NK-92 still needs further improvement by engineering. NK cells are among the early responders, and by lysing cancer cells, they expose tumor antigens to the care of DCs, which present them to T-cells in tumor-draining lymph nodes to induce polarization of CD4 T-cells into Th1 helper cells and the conversion of CD8 T-cells into cytotoxic T-cells (CTL). NK cells again intervene to allow the polarization of CD4 T-cells by secretion of IFNγ, which is also enhanced by IL-12 from DCs. This collaboration between DCs, NK, and T-cells is the prelude to establishing an effective adaptive anti-tumor immunity. The killing of immature DCs and activated T-cells by NK cells through the NKp30 and the modulation of MHC I HAL-E expression to prevent the killing or facilitate it, solidifies NK cell role in shaping adaptive immunity with significant consequences on CAR-T and NK-mediated immunotherapies. Therefore, strategies using NK cells will likely work if NK cells are engineered to enhance DCs and T-cell responses and to access and survive in the TME.
Immune checkpoint blockade has become a cornerstone in cancer immunotherapies. However, combinations with agents that block immunosuppression in the TME are necessary. Lymphodepletion and radiation therapy can help in this direction, but lessons learned suggest it will not be enough. An important direction is the genetic engineering of NK cells to create robust and versatile signaling incorporating several activation pathways found in NK cells without hyperactivating and exhausting NK cells. Another lesson learned is that NK cells can switch sides and play a supportive role in tumors and metastasis. Whether genetic engineering can preclude this is an important direction. Aside from cancer immunotherapy, NK cells can recognize stressed and senescent cells, and this developing area could prevent the development of cancers. This is an exciting direction for which genetically engineered NK cell lines might be more suitable.
CARs and TCRs not only enhance the killing of targeted cells but may also help the directional homing by collecting NK cells in antigen-rich tumors. Similarly, ectopic chemokine receptor expression could dramatically improve NK cells homing into tumors. Inhibition of IDO, PGE2, A2aR, and TGFβ, coupled with factors that enhance tumor perfusion, such as metronomic chemotherapy, which also increases NK cell recruitment into tumors, may enhance NK-mediated immunotherapies. Like CAR-T, the successes reported using CAR-NK immunotherapies are mostly for hematological cancers. Patients who respond can also relapse, suggesting either a lack of NK cells persistence, exhaustion, or possibly a conversion to a pro-tumorigenic function. While the two first possibilities could be remedied by genetic engineering, a re-education of NK cells in the cancer patient leading to a conversion into a less protumor phenotype would be a complex problem to solve. Such conversion could be determined by single-cell RNAseq analysis of CAR-NK cells from patients. For example, treatment with TFGβ1 converts NK cells to a less cytolytic state like ILC1s, presumably through the loss of EOMES. Blocking or reverting this conversion could restore NK cell cytolytic activity.
Another possible way to enhance NK function is by targeting intracellular checkpoints. VAV1 is the hub for various activating and inhibitory pathways, acting as a switch to turn off NK activation and prevent the downstream activation cascade268 (Fig. 5). Preventing VAV1 deactivation could offer a potent means to activate NK cells, with, however, the potential risk of higher toxicity to normal tissues. The dominant inhibitory signals originate from MHC I engagement with KIRs, leading to phosphorylation of their ITIMs domain, which creates a docking space for SHP-1 and SHP-2. The involvement of SHP-1 in VAV1, PIP3, and SLP76 dephosphorylation has been established,265,638 while SHP-2 dampens NK cells cytotoxicity, independently of KIR inhibition.639 Another regulator of NK cell function is CIS (cytokine-inducible SH2-containing protein). CIS is a member of the Socs family and targets STAT activation. The knockout of the CIS gene was reported to substantially improve CB-NK expressing soluble IL-15.488
The journey to enhance NK cell cytotoxicity and persistence is following in the footsteps of CAR-T development. However, there is a clear need to engineer activation signaling and co-stimulatory molecules specific to NK cells and their biology. The use of cytokine(s) and multiple activation signals will be mandatory. Incorporating elements to resist/modulate the TME will be necessary to allow NK cells to survive and engage in fruitful cross-talks with other effectors of immunity. The use of CARs, TCRs, ICI, engagers, radiation therapy, or bespoken chemotherapy regimens need to be optimized to increase the therapeutic efficacy of NK cells. All these improvements might benefit from immunomodulation by beneficial environmental factors and the strengthening of indoles-producing microbiota that enhance the immune response. These interventions could easily be incorporated into any treatment, and their beneficial impact merits further investigations.
Forward-looking perspective
Since the early 1970s, substantial research efforts have aimed to unravel NK cells’ functions and killing mechanisms. This effort is dwarfed by the extensive focus on adaptive immunity cells discovered earlier and which are fundamental to vaccine efficacy. As our understanding deepens, it becomes evident that NK cells are diverse populations engaged in various roles, using a complex balance of activating and inhibitory receptor signaling. Unfortunately, this balance does not always lead to desired outcomes, such as in cancer patients.
Two recent groundbreaking studies have identified the progenitor of NK cells, emphasizing that EOMES is crucial in differentiating NK cells from non-cytolytic ILC1s. Interestingly, NK cells exposed to TGFβ or Activin acquire a gene signature similar to ILCs, suggesting that NK cell exhaustion might involve reverting to an ILC state. Another significant study revealed that NK cells could kill activated T-cells, including CAR-CD19 T-cells, a capability they already possess against immature DCs. This finding raises important questions: Do patient NK cells hinder current CAR-T therapies? Could NK-mediated immunotherapies undermine the patient’s T-cell compartment, limiting adaptive immunity? And might combining CAR-T and CAR-NK therapies be counterproductive? Using high-dimensional single-cell analysis of human natural killer cells, an equally groundbreaking advance delineated three major populations of mature NK cells in PB.42 Two populations, NK1 and NK3, originated from ENKPs, and another NK2, originated from ILCPs. Which of these populations is a perfect fit for natural killer therapies remains to be confirmed.
Promising advances in NK-mediated immunotherapies are emerging, with numerous clinical trials underway90,487,590,640 (Table 1), making the full potential of NK cells increasingly attainable. These advances are bolstered by novel signal transduction engineering and precise gene editing using CRISPR/Cas9 technologies. Single-cell RNA sequencing (scRNA-seq) offers a detailed view of individual NK cell transcriptomes, enabling the identification of distinct NK cell subpopulations and their functional states.42 AI-driven clustering algorithms can categorize these cells based on gene expression, identifying subsets with enhanced cytotoxicity or resistance to tumor-induced immunosuppression and uncovering key regulatory genes and pathways. AI can design next-generation CARs for NK cells based on specific NK activating receptors, avoiding issues of exhaustion and overactivation. AI can optimize the ex vivo expansion and activation of NK cells, enhancing their potency and viability. Personalized data can guide the design of genetically engineered NK cells tailored to individual patient needs. Additionally, AI can identify synergistic combinations of NK cell therapies with other treatments, such as checkpoint inhibitors or cytokines.
Engineering autologous NK cells could address persistence issues more effectively than haploidentical transfers, especially if the KIR advantage of allogenic NK cells could be replicated through gene editing. AI can also predict which haplotype would persist more in a given patient. CAR-NK therapies may offer a cost-effective alternative to current prohibitively expensive CAR-T therapies, potentially improving access to treatment. Banking NK cells for multiple uses in multiple patients, particularly for cells from CB, could help bridge this gap. However, more clinical trials in large animal models, such as dogs with spontaneous tumors, are needed to validate therapies for tumors with similar signatures in humans and dogs, like osteosarcoma. Given recent reports of CAR-T lymphoma risks,641 this approach could also test the safety of new immunotherapies due to potential risks associated with gene editing strategies, with possible off-target effects, which might also be seen in CAR-NK therapies if used at larger scales. In this regard, more research is needed to examine the safety of combining elements that could increase the fitness and survival of NK cells but may lead to a gain of function. NK cell immunotherapies are extending to autoimmune diseases such as SLE with several clinical trials launched and recruiting (Table 1). These are targeting the B-cell compartment for elimination via CAR-CD19-NK. For other autoimmune disorders, NK cells expressing the extracellular domain of PDL-1 can target autoreactive T cells, which overexpress PD-1. This strategy has shown efficacy in the preclinical model and might be applicable to autoreactive follicular T-cells.642 Another encouraging direction is the bourgeoning of NK cell engagers as a safe way to enhance NK cells without genetic modifications. However, it appears they will not be sufficient as monotherapy and must be combined with other modalities. Engineering engagers with cytokines such as IL-15 provide better survival to NK cells. (reviewed41).
This is a time of great promise. As we gain deeper insights into NK cell signaling and the molecular mechanisms of their activation and inhibition, advances in NK-mediated immunotherapies will accelerate, leading us toward a brighter future.
Acknowledgements
Supported by Innovation Grants DAPCM (232129, 245130), and President and Fellows of Harvard College Myles shore fellowship (241317) to Y.J. S.C. was supported by Scientific Research Funds of Zhejiang Province Health Department 2024ky205, and Hangzhou Agricultural and Social Development Key Scientific Research Project 202004A19. H.Z. was supported by Guizhou Province Science Technology Project (2024)051. All figures were Created with BioRender.com.
Author contributions
Y.J. conceived the topics, wrote the review and drew figures. S.C. contributed to the research, writing, and editing of text and figures. H.Z. contributed to research and editing of text and figures. All authors have contributed, read, and approved the article.
Competing interests
The authors declare no competing interests.
Contributor Information
Sumei Chen, Email: sumeichen1986@outlook.com.
Youssef Jounaidi, Email: yjounaidi@mgh.harvard.edu.
References
- 1.Kiessling, R., Klein, E. & Wigzell, H. “Natural” killer cells in the mouse. I. cytotoxic cells with specificity for mouse Moloney leukemia cells. specificity and distribution according to genotype. Eur. J. Immunol.5, 112–117 (1975). [DOI] [PubMed] [Google Scholar]
- 2.Herberman, R. B., Nunn, M. E. & Lavrin, D. H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. distribution of reactivity and specificity. Int. J. Cancer16, 216–229 (1975). [DOI] [PubMed] [Google Scholar]
- 3.Aktas, E., Erten, G., Kucuksezer, U. C. & Deniz, G. Natural killer cells: versatile roles in autoimmune and infectious diseases. Expert Rev. Clin. Immunol.5, 405–420 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Liu, M., Liang, S. & Zhang, C. NK cells in autoimmune diseases: protective or pathogenic? Front. Immunol.12, 624687 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kucuksezer, U. C. et al. The role of natural killer cells in autoimmune diseases. Front. Immunol.12, 622306 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radomska-Lesniewska, D. M., Bialoszewska, A. & Kaminski, P. Angiogenic properties of NK cells in cancer and other angiogenesis-dependent diseases. Cells. 10,1621 (2021). [DOI] [PMC free article] [PubMed]
- 7.Liu, Q. et al. NK cells modulate the inflammatory response to corneal epithelial abrasion and thereby support wound healing. Am. J. Pathol.181, 452–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobecki, M. et al. NK cells in hypoxic skin mediate a trade-off between wound healing and antibacterial defence. Nat. Commun.12, 4700 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavalcante-Silva, J. & Koh, T. J. Role of NK cells in skin wound healing of mice. J. Immunol.210, 981–990 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Von Woon, E. et al. Number and function of uterine natural killer cells in recurrent miscarriage and implantation failure: a systematic review and meta-analysis. Hum. Reprod. Update28, 548–582 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camous, X., Pera, A., Solana, R. & Larbi, A. NK cells in healthy aging and age-associated diseases. J. Biomed. Biotechnol.2012, 195956 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonangeli, F., Zingoni, A., Soriani, A. & Santoni, A. Senescent cells: living or dying is a matter of NK cells. J. Leukoc. Biol.105, 1275–1283 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Chelyapov, N., Nguyen, T. T. & Gonzalez, R. Autologous NK cells propagated and activated ex vivo decrease senescence markers in human PBMCs. Biochem. Biophys. Rep.32, 101380 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Podack, E. R. et al. Structure, function, and expression of murine and human perforin 1 (P1). Immunol. Rev.103, 203–211 (1988). [DOI] [PubMed] [Google Scholar]
- 15.Podack, E. R. & Dennert, G. Assembly of two types of tubules with putative cytolytic function by cloned natural killer cells. Nature302, 442–445 (1983). [DOI] [PubMed] [Google Scholar]
- 16.Jenne, D. et al. Identification and sequencing of cDNA clones encoding the granule-associated serine proteases granzymes D, E, and F of cytolytic T lymphocytes. Proc. Natl. Acad. Sci. USA85, 4814–4818 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson, D. H. et al. Granulysin crystal structure and a structure-derived lytic mechanism. J. Mol. Biol.325, 355–365 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Stenger, S. et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science282, 121–125 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Crespo, A. C. et al. Decidual NK cells transfer granulysin to selectively kill bacteria in trophoblasts. Cell182, 1125–1139 e1118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bykovskaja, S. N., Rytenko, A. N., Rauschenbach, M. O. & Bykovsky, A. F. Ultrastructural alteration of cytolytic T lymphocytes following their interaction with target cells. I. Hypertrophy and change of orientation of the Golgi apparatus. Cell Immunol.40, 164–174 (1978). [DOI] [PubMed] [Google Scholar]
- 21.Bykovskaja, S. N., Rytenko, A. N., Rauschenbach, M. O. & Bykovsky, A. F. Ultrastructural alteration of cytolytic T lymphocytes following their interaction with target cells. II. morphogenesis of secretory granules and intracellular vacuoles. Cell Immunol.40, 175–185 (1978). [DOI] [PubMed] [Google Scholar]
- 22.Boehm, U., Klamp, T., Groot, M. & Howard, J. C. Cellular responses to interferon-gamma. Annu. Rev. Immunol.15, 749–795 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Perussia, B. Lymphokine-activated killer cells, natural killer cells, and cytokines. Curr. Opin. Immunol.3, 49–55 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Cassatella, M. A. et al. Molecular basis of interferon-gamma and lipopolysaccharide enhancement of phagocyte respiratory burst capability. studies on the gene expression of several NADPH oxidase components. J. Biol. Chem.265, 20241–20246 (1990). [PubMed] [Google Scholar]
- 25.Bradley, L. M., Dalton, D. K. & Croft, M. A direct role for IFN-gamma in regulation of Th1 cell development. J. Immunol.157, 1350–1358 (1996). [PubMed] [Google Scholar]
- 26.Snapper, C. M. & Paul, W. E. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science236, 944–947 (1987). [DOI] [PubMed] [Google Scholar]
- 27.Gronberg, A. et al. IFN-gamma treatment of K562 cells inhibits natural killer cell triggering and decreases the susceptibility to lysis by cytoplasmic granules from large granular lymphocytes. J. Immunol.140, 4397–4402 (1988). [PubMed] [Google Scholar]
- 28.Gray, J. D., Hirokawa, M., Ohtsuka, K. & Horwitz, D. A. Generation of an inhibitory circuit involving CD8+ T cells, IL-2, and NK cell-derived TGF-beta: contrasting effects of anti-CD2 and anti-CD3. J. Immunol.160, 2248–2254 (1998). [PubMed] [Google Scholar]
- 29.Wahl, S. M. Transforming growth factor beta: the good, the bad, and the ugly. J. Exp. Med.180, 1587–1590 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sporn, M. B., Roberts, A. B., Wakefield, L. M. & de Crombrugghe, B. Some recent advances in the chemistry and biology of transforming growth factor-beta. J. Cell Biol.105, 1039–1045 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostapchuk, Y. O. et al. Peripheral blood NK cells expressing HLA-G, IL-10, and TGF-beta in healthy donors and breast cancer patients. Cell Immunol.298, 37–46 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Perona-Wright, G. et al. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe6, 503–512 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vivier, E. & Ugolini, S. Regulatory natural killer cells: new players in the IL-10 anti-inflammatory response. Cell Host Microbe6, 493–495 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Moore, K. W., de Waal Malefyt, R., Coffman, R. L. & O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol.19, 683–765 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Lee, S. H. et al. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J. Exp. Med.206, 2235–2251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks, D. G. et al. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med.12, 1301–1309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, Z. et al. IL-10 enhances human natural killer cell effector functions via metabolic reprogramming regulated by mTORC1 signaling. Front. Immunol.12, 619195 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caligiuri, M. A. Human natural killer cells. Blood112, 461–469 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper, M. A. et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood97, 3146–3151 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Cooper, M. A., Fehniger, T. A. & Caligiuri, M. A. The biology of human natural killer-cell subsets. Trends Immunol.22, 633–640 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Vivier, E. et al. Natural killer cell therapies. Nature626, 727–736 (2024). [DOI] [PubMed] [Google Scholar]
- 42.Rebuffet, L. et al. High-dimensional single-cell analysis of human natural killer cell heterogeneity. Nat. Immunol. 25,1474–1488 (2024). [DOI] [PMC free article] [PubMed]
- 43.Horowitz, A. et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med.5, 208ra145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cudkowicz, G. & Bennett, M. Peculiar immunobiology of bone marrow allografts. II. rejection of parental grafts by resistant F 1 hybrid mice. J. Exp. Med.134, 1513–1528 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herberman, R. B., Nunn, M. E., Lavrin, D. H. & Asofsky, R. Effect of antibody to theta antigen on cell-mediated immunity induced in syngeneic mice by murine sarcoma virus. J. Natl. Cancer Inst.51, 1509–1512 (1973). [DOI] [PubMed] [Google Scholar]
- 46.Morgan, D. A., Ruscetti, F. W. & Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science193, 1007–1008 (1976). [DOI] [PubMed] [Google Scholar]
- 47.Karre, K., Ljunggren, H. G., Piontek, G. & Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature319, 675–678 (1986). [DOI] [PubMed] [Google Scholar]
- 48.Bonavida, B., Katz, J. & Gottlieb, M. Mechanism of defective NK cell activity in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. I. Defective trigger on NK cells for NKCF production by target cells, and partial restoration by IL 2. J. Immunol.137, 1157–1163 (1986). [PubMed] [Google Scholar]
- 49.Lanier, L. L., Ruitenberg, J. J. & Phillips, J. H. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol.141, 3478–3485 (1988). [PubMed] [Google Scholar]
- 50.Nagler, A., Lanier, L. L., Cwirla, S. & Phillips, J. H. Comparative studies of human FcRIII-positive and negative natural killer cells. J. Immunol.143, 3183–3191 (1989). [PubMed] [Google Scholar]
- 51.Kobayashi, M. et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med.170, 827–845 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson, P., Caligiuri, M., Ritz, J. & Schlossman, S. F. CD3-negative natural killer cells express zeta TCR as part of a novel molecular complex. Nature341, 159–162 (1989). [DOI] [PubMed] [Google Scholar]
- 53.Lanier, L. L., Yu, G. & Phillips, J. H. Co-association of CD3 zeta with a receptor (CD16) for IgG Fc on human natural killer cells. Nature342, 803–805 (1989). [DOI] [PubMed] [Google Scholar]
- 54.Moretta, A. et al. A novel surface antigen expressed by a subset of human CD3- CD16+ natural killer cells. role in cell activation and regulation of cytolytic function. J. Exp. Med.171, 695–714 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moretta, A. et al. Novel surface molecules involved in human NK cell activation and triggering of the lytic machinery. Int. J. Cancer Suppl.7, 6–10 (1992). [PubMed] [Google Scholar]
- 56.Karlhofer, F. M., Ribaudo, R. K. & Yokoyama, W. M. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature358, 66–70 (1992). [DOI] [PubMed] [Google Scholar]
- 57.Colonna, M. & Samaridis, J. Cloning of immunoglobulin-superfamily members associated with HLA-C and HLA-B recognition by human natural killer cells. Science268, 405–408 (1995). [DOI] [PubMed] [Google Scholar]
- 58.D’Andrea, A. et al. Molecular cloning of NKB1. a natural killer cell receptor for HLA-B allotypes. J. Immunol.155, 2306–2310 (1995). [PubMed] [Google Scholar]
- 59.Wagtmann, N. et al. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity2, 439–449 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Gong, J. H., Maki, G. & Klingemann, H. G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia8, 652–658 (1994). [PubMed] [Google Scholar]
- 61.Burton, J. D. et al. A lymphokine, provisionally designated interleukin T and produced by a human adult T-cell leukemia line, stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc. Natl. Acad. Sci. USA91, 4935–4939 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grabstein, K. H. et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science264, 965–968 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Shibuya, A. et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity4, 573–581 (1996). [DOI] [PubMed] [Google Scholar]
- 64.Sivori, S. et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J. Exp. Med.186, 1129–1136 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vitale, M. et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med.187, 2065–2072 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pende, D. et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med.190, 1505–1516 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lanier, L. L. et al. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature391, 703–707 (1998). [DOI] [PubMed] [Google Scholar]
- 68.Braud, V. M. et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature391, 795–799 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Bauer, S. et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science285, 727–729 (1999). [PubMed] [Google Scholar]
- 70.Kubin, M. et al. ULBP1, 2, 3: novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur. J. Immunol.31, 1428–1437 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Cerwenka, A. et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity12, 721–727 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Parrish-Novak, J. et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature408, 57–63 (2000). [DOI] [PubMed] [Google Scholar]
- 73.Ruggeri, L. et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood94, 333–339 (1999). [PubMed] [Google Scholar]
- 74.Piccioli, D., Sbrana, S., Melandri, E. & Valiante, N. M. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J. Exp. Med.195, 335–341 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gerosa, F. et al. Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med.195, 327–333 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferlazzo, G. et al. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med.195, 343–351 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castriconi, R. et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA100, 4120–4125 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bottino, C. et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med.198, 557–567 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller, J. S. et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood105, 3051–3057 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Della Chiesa, M. et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood108, 4118–4125 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Arai, S. et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy10, 625–632 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Cella, M. et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature457, 722–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bellora, F. et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc. Natl. Acad. Sci. USA107, 21659–21664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thoren, F. B. et al. Human NK cells induce neutrophil apoptosis via an NKp46- and fas-dependent mechanism. J. Immunol.188, 1668–1674 (2012). [DOI] [PubMed] [Google Scholar]
- 85.Foley, B. et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol.189, 5082–5088 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee, J. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity42, 431–442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schlums, H. et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity42, 443–456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Szmania, S. et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J. Immunother.38, 24–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Romee, R. et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med.8, 357ra123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med.382, 545–553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Osman, M. et al. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv.4, 5035–5039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ruckert, T. et al. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol.23, 1551–1563 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liang, Z. et al. Eomes expression identifies the early bone marrow precursor to classical NK cells. Nat. Immunol.25, 1172–1182 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ding, Y. et al. Distinct developmental pathways generate functionally distinct populations of natural killer cells. Nat. Immunol.25, 1183–1192 (2024). [DOI] [PubMed] [Google Scholar]
- 95.Kilian, M. et al. The immunoglobulin superfamily ligand B7H6 subjects T cell responses to NK cell surveillance. Sci. Immunol.9, eadj7970 (2024). [DOI] [PubMed] [Google Scholar]
- 96.Karre, K. Natural killer cell recognition of missing self. Nat. Immunol.9, 477–480 (2008). [DOI] [PubMed] [Google Scholar]
- 97.Moretta, A. & Moretta, L. HLA class I specific inhibitory receptors. Curr. Opin. Immunol.9, 694–701 (1997). [DOI] [PubMed] [Google Scholar]
- 98.Lanier, L. L. NK cell receptors. Annu. Rev. Immunol.16, 359–393 (1998). [DOI] [PubMed] [Google Scholar]
- 99.Benichou, G. et al. Limited T cell response to donor MHC peptides during allograft rejection. Implications for selective immune therapy in transplantation. J. Immunol.153, 938–945 (1994). [PubMed] [Google Scholar]
- 100.Liu, Z. et al. Limited usage of T cell receptor V beta genes by allopeptide-specific T cells. J. Immunol.150, 3180–3186 (1993). [PubMed] [Google Scholar]
- 101.Ljunggren, H. G. & Karre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today11, 237–244 (1990). [DOI] [PubMed] [Google Scholar]
- 102.Ljunggren, H. G. & Karre, K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J. Exp. Med.162, 1745–1759 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Piontek, G. E. et al. YAC-1 MHC class I variants reveal an association between decreased NK sensitivity and increased H-2 expression after interferon treatment or in vivo passage. J. Immunol.135, 4281–4288 (1985). [PubMed] [Google Scholar]
- 104.Zappacosta, F. et al. Peptides isolated from HLA-Cw*0304 confer different degrees of protection from natural killer cell-mediated lysis. Proc. Natl. Acad. Sci. USA94, 6313–6318 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mandelboim, O. et al. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J. Exp. Med.184, 913–922 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blum, J. S., Wearsch, P. A. & Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol.31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ljunggren, H. G. et al. Empty MHC class I molecules come out in the cold. Nature346, 476–480 (1990). [DOI] [PubMed] [Google Scholar]
- 108.Peruzzi, M., Parker, K. C., Long, E. O. & Malnati, M. S. Peptide sequence requirements for the recognition of HLA-B*2705 by specific natural killer cells. J. Immunol.157, 3350–3356 (1996). [PubMed] [Google Scholar]
- 109.Peruzzi, M., Wagtmann, N. & Long, E. O. A p70 killer cell inhibitory receptor specific for several HLA-B allotypes discriminates among peptides bound to HLA-B*2705. J. Exp. Med.184, 1585–1590 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rajagopalan, S. & Long, E. O. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J. Exp. Med.185, 1523–1528 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim, S. et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature436, 709–713 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Garcia-Lora, A., Algarra, I. & Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell Physiol.195, 346–355 (2003). [DOI] [PubMed] [Google Scholar]
- 113.Garrido, F. & Algarra, I. MHC antigens and tumor escape from immune surveillance. Adv. Cancer Res.83, 117–158 (2001). [DOI] [PubMed] [Google Scholar]
- 114.Ardolino, M. et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J. Clin. Investig.124, 4781–4794 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Azumi, K. et al. Genomic analysis of immunity in a Urochordate and the emergence of the vertebrate immune system: “waiting for Godot”. Immunogenetics55, 570–581 (2003). [DOI] [PubMed] [Google Scholar]
- 116.Flajnik, M. F. A cold-blooded view of adaptive immunity. Nat. Rev. Immunol.18, 438–453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hirano, M., Das, S., Guo, P. & Cooper, M. D. The evolution of adaptive immunity in vertebrates. Adv. Immunol.109, 125–157 (2011). [DOI] [PubMed] [Google Scholar]
- 118.Scofield, V. L., Schlumpberger, J. M., West, L. A. & Weissman, I. L. Protochordate allorecognition is controlled by a MHC-like gene system. Nature295, 499–502 (1982). [DOI] [PubMed] [Google Scholar]
- 119.Stoner, D. S., Quattro, J. M. & Weissman, I. L. Highly polymorphic microsatellite loci in the colonial ascidian Botryllus schlosseri. Mol. Mar. Biol. Biotechnol.6, 163–171 (1997). [PubMed] [Google Scholar]
- 120.Stoner, D. S. & Weissman, I. L. Somatic and germ cell parasitism in a colonial ascidian: possible role for a highly polymorphic allorecognition system. Proc. Natl. Acad. Sci. USA93, 15254–15259 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khalturin, K., Becker, M., Rinkevich, B. & Bosch, T. C. Urochordates and the origin of natural killer cells: identification of a CD94/NKR-P1-related receptor in blood cells of Botryllus. Proc. Natl. Acad. Sci. USA100, 622–627 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boyington, J. C. et al. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature405, 537–543 (2000). [DOI] [PubMed] [Google Scholar]
- 123.Fan, Q. R., Long, E. O. & Wiley, D. C. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat. Immunol.2, 452–460 (2001). [DOI] [PubMed] [Google Scholar]
- 124.Maenaka, K., Juji, T., Stuart, D. I. & Jones, E. Y. Crystal structure of the human p58 killer cell inhibitory receptor (KIR2DL3) specific for HLA-Cw3-related MHC class I. Structure7, 391–398 (1999). [DOI] [PubMed] [Google Scholar]
- 125.Moesta, A. K. et al. Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J. Immunol.180, 3969–3979 (2008). [DOI] [PubMed] [Google Scholar]
- 126.Manser, A. R., Weinhold, S. & Uhrberg, M. Human KIR repertoires: shaped by genetic diversity and evolution. Immunol. Rev.267, 178–196 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Sakaue, S. et al. Decoding the diversity of killer immunoglobulin-like receptors by deep sequencing and a high-resolution imputation method. Cell Genom.2, 100101 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Djaoud, Z. & Parham, P. HLAs, TCRs, and KIRs, a triumvirate of human cell-mediated immunity. Annu. Rev. Biochem.89, 717–739 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Kikuchi-Maki, A., Catina, T. L. & Campbell, K. S. Cutting edge: KIR2DL4 transduces signals into human NK cells through association with the Fc receptor gamma protein. J. Immunol.174, 3859–3863 (2005). [DOI] [PubMed] [Google Scholar]
- 130.Rajagopalan, S., Fu, J. & Long, E. O. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J. Immunol.167, 1877–1881 (2001). [DOI] [PubMed] [Google Scholar]
- 131.Goodridge, J. P., Witt, C. S., Christiansen, F. T. & Warren, H. S. KIR2DL4 (CD158d) genotype influences expression and function in NK cells. J. Immunol.171, 1768–1774 (2003). [DOI] [PubMed] [Google Scholar]
- 132.Ferreira, L. M. R., Meissner, T. B., Tilburgs, T. & Strominger, J. L. HLA-G: at the interface of maternal-fetal tolerance. Trends Immunol.38, 272–286 (2017). [DOI] [PubMed] [Google Scholar]
- 133.Rajagopalan, S., Moyle, M. W., Joosten, I. & Long, E. O. DNA-PKcs controls an endosomal signaling pathway for a proinflammatory response by natural killer cells. Sci. Signal.3, ra14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rajagopalan, S. et al. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol.4, e9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tilburgs, T., Evans, J. H., Crespo, A. C. & Strominger, J. L. The HLA-G cycle provides for both NK tolerance and immunity at the maternal-fetal interface. Proc. Natl. Acad. Sci. USA112, 13312–13317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zheng, G. et al. Interaction between HLA-G and NK cell receptor KIR2DL4 orchestrates HER2-positive breast cancer resistance to trastuzumab. Signal Transduct. Target Ther.6, 236 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Blunt, M. D. & Khakoo, S. I. Activating killer cell immunoglobulin-like receptors: detection, function and therapeutic use. Int. J. Immunogenet47, 1–12 (2020). [DOI] [PubMed] [Google Scholar]
- 138.Burian, A. et al. HLA-F and MHC-I open conformers bind natural killer cell ig-like receptor KIR3DS1. PLoS ONE11, e0163297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garcia-Beltran, W. F. et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol.17, 1067–1074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Alter, G. et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J. Virol.83, 6798–6805 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Aranda-Romo, S. et al. Killer-cell immunoglobulin-like receptors (KIR) in severe A (H1N1) 2009 influenza infections. Immunogenetics64, 653–662 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Zhi-ming, L. et al. Polymorphisms of killer cell immunoglobulin-like receptor gene: possible association with susceptibility to or clearance of hepatitis B virus infection in Chinese Han population. Croat. Med. J.48, 800–806 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Carotta, S., Pang, S. H., Nutt, S. L. & Belz, G. T. Identification of the earliest NK-cell precursor in the mouse BM. Blood117, 5449–5452 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Brodin, P. et al. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood113, 2434–2441 (2009). [DOI] [PubMed] [Google Scholar]
- 145.Wu, Z. et al. Dynamic variability in SHP-1 abundance determines natural killer cell responsiveness. Sci. Signal.14, eabe5380 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Boos, M. D., Ramirez, K. & Kee, B. L. Extrinsic and intrinsic regulation of early natural killer cell development. Immunol. Res.40, 193–207 (2008). [DOI] [PubMed] [Google Scholar]
- 147.Yu, J., Freud, A. G. & Caligiuri, M. A. Location and cellular stages of natural killer cell development. Trends Immunol.34, 573–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ouyang, Q., Baerlocher, G., Vulto, I. & Lansdorp, P. M. Telomere length in human natural killer cell subsets. Ann. N.Y. Acad. Sci.1106, 240–252 (2007). [DOI] [PubMed] [Google Scholar]
- 149.Romagnani, C. et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J. Immunol.178, 4947–4955 (2007). [DOI] [PubMed] [Google Scholar]
- 150.Freud, A. G. et al. Evidence for discrete stages of human natural killer cell differentiation in vivo. J. Exp. Med.203, 1033–1043 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grzywacz, B. et al. Coordinated acquisition of inhibitory and activating receptors and functional properties by developing human natural killer cells. Blood108, 3824–3833 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Perussia, B., Chen, Y. & Loza, M. J. Peripheral NK cell phenotypes: multiple changing of faces of an adapting, developing cell. Mol. Immunol.42, 385–395 (2005). [DOI] [PubMed] [Google Scholar]
- 153.Kiekens, L. et al. T-BET and EOMES accelerate and enhance functional differentiation of human natural killer cells. Front. Immunol.12, 732511 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Molla, V. C. et al. Natural killer cells 56(bright)16(-) have higher counts in the umbilical cord blood than in the adult peripheral blood. Hematol. Transfus. Cell Ther.45, 419–427 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Almeida-Oliveira, A. et al. Age-related changes in natural killer cell receptors from childhood through old age. Hum. Immunol.72, 319–329 (2011). [DOI] [PubMed] [Google Scholar]
- 156.Mocchegiani, E. & Malavolta, M. NK and NKT cell functions in immunosenescence. Aging Cell3, 177–184 (2004). [DOI] [PubMed] [Google Scholar]
- 157.Murakami, G. & Taniguchi, I. Histologic heterogeneity and intranodal shunt flow in lymph nodes from elderly subjects: a cadaveric study. Ann. Surg. Oncol.11, 279S–284S (2004). [DOI] [PubMed] [Google Scholar]
- 158.Turner, V. M. & Mabbott, N. A. Influence of ageing on the microarchitecture of the spleen and lymph nodes. Biogerontology18, 723–738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Miller, J. S., Alley, K. A. & McGlave, P. Differentiation of natural killer (NK) cells from human primitive marrow progenitors in a stroma-based long-term culture system: identification of a CD34+7+ NK progenitor. Blood83, 2594–2601 (1994). [PubMed] [Google Scholar]
- 160.Briard, D., Brouty-Boye, D., Azzarone, B. & Jasmin, C. Fibroblasts from human spleen regulate NK cell differentiation from blood CD34(+) progenitors via cell surface IL-15. J. Immunol.168, 4326–4332 (2002). [DOI] [PubMed] [Google Scholar]
- 161.Carson, W. E. et al. Endogenous production of interleukin 15 by activated human monocytes is critical for optimal production of interferon-gamma by natural killer cells in vitro. J. Clin. Investig.96, 2578–2582 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Freud, A. G. et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity22, 295–304 (2005). [DOI] [PubMed] [Google Scholar]
- 163.de Rham, C. et al. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Res. Ther.9, R125 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wagner, J. A. et al. Cytokine-induced memory-like differentiation enhances unlicensed natural killer cell antileukemia and fcgammaRIIIa-triggered responses. Biol. Blood Marrow Transpl.23, 398–404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cooley, S. et al. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood110, 578–586 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Juelke, K. et al. Education of hyporesponsive NK cells by cytokines. Eur. J. Immunol.39, 2548–2555 (2009). [DOI] [PubMed] [Google Scholar]
- 167.Levi, I. et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget6, 13835–13843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Orr, M. T., Murphy, W. J. & Lanier, L. L. Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat. Immunol.11, 321–327 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Benson, D. M. Jr et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood120, 4324–4333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tortorella, D. et al. Viral subversion of the immune system. Annu. Rev. Immunol.18, 861–926 (2000). [DOI] [PubMed] [Google Scholar]
- 171.Smith, H. R. et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA99, 8826–8831 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Eriksson, M., Ryan, J. C., Nakamura, M. C. & Sentman, C. L. Ly49A inhibitory receptors redistribute on natural killer cells during target cell interaction. Immunology97, 341–347 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell174, 1054–1066 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Cortez, V. S. & Colonna, M. Diversity and function of group 1 innate lymphoid cells. Immunol. Lett.179, 19–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Constantinides, M. G., McDonald, B. D., Verhoef, P. A. & Bendelac, A. A committed precursor to innate lymphoid cells. Nature508, 397–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xu, W. et al. An Id2(RFP)-reporter mouse redefines innate lymphoid cell precursor potentials. Immunity50, 1054–1068.e1053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gordon, S. M. et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity36, 55–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Flommersfeld, S. et al. Fate mapping of single NK cells identifies a type 1 innate lymphoid-like lineage that bridges innate and adaptive recognition of viral infection. Immunity54, 2288–2304 e2287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Paust, S., Blish, C. A. & Reeves, R. K. Redefining memory: building the case for adaptive NK cells. J. Virol.91, e00169–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Myers, J. A. & Miller, J. S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol.18, 85–100 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dowell, A. C. et al. Long-term proliferation of functional human NK cells, with conversion of CD56(dim) NK cells to a CD56 (bright) phenotype, induced by carcinoma cells co-expressing 4-1BBL and IL-12. Cancer Immunol. Immunother.61, 615–628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Keskin, D. B. et al. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc. Natl. Acad. Sci. USA104, 3378–3383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gao, Y. et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol.18, 1004–1015 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Cortez, V. S. et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat. Immunol.18, 995–1003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rautela, J. et al. Therapeutic blockade of activin-A improves NK cell function and antitumor immunity. Sci. Signal.12, eaat7527 (2019). [DOI] [PubMed] [Google Scholar]
- 186.Lopez-Verges, S. et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood116, 3865–3874 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Krishnaraj, R. & Svanborg, A. Preferential accumulation of mature NK cells during human immunosenescence. J. Cell Biochem.50, 386–391 (1992). [DOI] [PubMed] [Google Scholar]
- 188.Brenchley, J. M. et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood101, 2711–2720 (2003). [DOI] [PubMed] [Google Scholar]
- 189.Focosi, D., Bestagno, M., Burrone, O. & Petrini, M. CD57+ T lymphocytes and functional immune deficiency. J. Leukoc. Biol.87, 107–116 (2010). [DOI] [PubMed] [Google Scholar]
- 190.Andrade, F. et al. Granzyme H destroys the function of critical adenoviral proteins required for viral DNA replication and granzyme B inhibition. EMBO J.26, 2148–2157 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Romero, V., Fellows, E., Jenne, D. E. & Andrade, F. Cleavage of La protein by granzyme H induces cytoplasmic translocation and interferes with La-mediated HCV-IRES translational activity. Cell Death Differ.16, 340–348 (2009). [DOI] [PubMed] [Google Scholar]
- 192.Silva, T. et al. The chemokine CCL5 inhibits the replication of influenza A virus through SAMHD1 modulation. Front. Cell Infect. Microbiol.11, 549020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zepp, J. A., Nold-Petry, C. A., Dinarello, C. A. & Nold, M. F. Protection from RNA and DNA viruses by IL-32. J. Immunol.186, 4110–4118 (2011). [DOI] [PubMed] [Google Scholar]
- 194.Vietzen, H. et al. NKG2C deletion is a risk factor for human cytomegalovirus viremia and disease after lung transplantation. J. Infect. Dis.217, 802–806 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Liu, L. L. et al. Critical Role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep.15, 1088–1099 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sato, Y. et al. CXCL10 expression status is prognostic in patients with advanced thoracic esophageal squamous cell carcinoma. Ann. Surg. Oncol.23, 936–942 (2016). [DOI] [PubMed] [Google Scholar]
- 197.Campbell, J. J. et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J. Immunol.166, 6477–6482 (2001). [DOI] [PubMed] [Google Scholar]
- 198.Farag, S. S. et al. Natural killer cell receptors: new biology and insights into the graft-versus-leukemia effect. Blood100, 1935–1947 (2002). [DOI] [PubMed] [Google Scholar]
- 199.Bryceson, Y. T., March, M. E., Ljunggren, H. G. & Long, E. O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood107, 159–166 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bryceson, Y. T., Ljunggren, H. G. & Long, E. O. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood114, 2657–2666 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Anderson, P. et al. Fc gamma receptor type III (CD16) is included in the zeta NK receptor complex expressed by human natural killer cells. Proc. Natl. Acad. Sci. USA87, 2274–2278 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lanier, L. L. NK cell recognition. Annu. Rev. Immunol.23, 225–274 (2005). [DOI] [PubMed] [Google Scholar]
- 203.Campbell, K. S., Yusa, S., Kikuchi-Maki, A. & Catina, T. L. NKp44 triggers NK cell activation through DAP12 association that is not influenced by a putative cytoplasmic inhibitory sequence. J. Immunol.172, 899–906 (2004). [DOI] [PubMed] [Google Scholar]
- 204.Bottino, C., Castriconi, R., Moretta, L. & Moretta, A. Cellular ligands of activating NK receptors. Trends Immunol.26, 221–226 (2005). [DOI] [PubMed] [Google Scholar]
- 205.Koch, J., Steinle, A., Watzl, C. & Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol.34, 182–191 (2013). [DOI] [PubMed] [Google Scholar]
- 206.Pessino, A. et al. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med.188, 953–960 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sen Santara, S. et al. The NK cell receptor NKp46 recognizes ecto-calreticulin on ER-stressed cells. Nature616, 348–356 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Glasner, A. et al. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J. Immunol.188, 2509–2515 (2012). [DOI] [PubMed] [Google Scholar]
- 209.Cagnano, E. et al. Expression of ligands to NKp46 in benign and malignant melanocytes. J. Investig. Dermatol.128, 972–979 (2008). [DOI] [PubMed] [Google Scholar]
- 210.Ziblat, A. et al. IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur. J. Immunol.45, 192–202 (2015). [DOI] [PubMed] [Google Scholar]
- 211.Cantoni, C. et al. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med.189, 787–796 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Baychelier, F. et al. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood122, 2935–2942 (2013). [DOI] [PubMed] [Google Scholar]
- 213.Shemesh, A. et al. Survival in acute myeloid leukemia is associated with NKp44 splice variants. Oncotarget7, 32933–32945 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Siewiera, J. et al. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat. Commun.6, 10183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Korgun, E. T. et al. Location of cell cycle regulators cyclin B1, cyclin A, PCNA, Ki67 and cell cycle inhibitors p21, p27 and p57 in human first trimester placenta and deciduas. Histochem. Cell Biol.125, 615–624 (2006). [DOI] [PubMed] [Google Scholar]
- 216.Delahaye, N. F. et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med.17, 700–707 (2011). [DOI] [PubMed] [Google Scholar]
- 217.Pesce, S. et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology4, e1001224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Binici, J. et al. A soluble fragment of the tumor antigen BCL2-associated athanogene 6 (BAG-6) is essential and sufficient for inhibition of NKp30 receptor-dependent cytotoxicity of natural killer cells. J. Biol. Chem.288, 34295–34303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Reiners, K. S. et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood121, 3658–3665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Poggi, A. et al. Tumor-induced apoptosis of human IL-2-activated NK cells: role of natural cytotoxicity receptors. J. Immunol.174, 2653–2660 (2005). [DOI] [PubMed] [Google Scholar]
- 221.Rosental, B. et al. A novel mechanism for cancer cells to evade immune attack by NK cells: the interaction between NKp44 and proliferating cell nuclear antigen. Oncoimmunology1, 572–574 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rosental, B. et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol.187, 5693–5702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lanier, L. L. Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol.9, 495–502 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Watzl, C. & Long, E. O. Signal transduction during activation and inhibition of natural killer cells. Curr. Protoc. Immunol. 90, 11–9, (2010). [DOI] [PMC free article] [PubMed]
- 225.Franke, T. F., Kaplan, D. R., Cantley, L. C. & Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science275, 665–668 (1997). [DOI] [PubMed] [Google Scholar]
- 226.Marcenaro, E. et al. CD59 is physically and functionally associated with natural cytotoxicity receptors and activates human NK cell-mediated cytotoxicity. Eur. J. Immunol.33, 3367–3376 (2003). [DOI] [PubMed] [Google Scholar]
- 227.Diefenbach, A. et al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol.3, 1142–1149 (2002). [DOI] [PubMed] [Google Scholar]
- 228.Diefenbach, A. et al. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol.1, 119–126 (2000). [DOI] [PubMed] [Google Scholar]
- 229.Venkataraman, G. M. et al. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J. Immunol.178, 961–969 (2007). [DOI] [PubMed] [Google Scholar]
- 230.Weiss, T. et al. NKG2D-dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin. Cancer Res.24, 882–895 (2018). [DOI] [PubMed] [Google Scholar]
- 231.Cai, X. et al. Control of tumor initiation by NKG2D naturally expressed on ovarian cancer cells.Neoplasia19, 471–482 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.McGilvray, R. W. et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin. Cancer Res.15, 6993–7002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cerwenka, A., Baron, J. L. & Lanier, L. L. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA98, 11521–11526 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Guerra, N. et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity28, 571–580 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Vyas, M. et al. Soluble NKG2D ligands in the ovarian cancer microenvironment are associated with an adverse clinical outcome and decreased memory effector T cells independent of NKG2D downregulation. Oncoimmunology6, e1339854 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Salih, H. R., Rammensee, H. G. & Steinle, A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol.169, 4098–4102 (2002). [DOI] [PubMed] [Google Scholar]
- 237.Zhang, J. et al. Antibody-mediated neutralization of soluble MIC significantly enhances CTLA4 blockade therapy. Sci. Adv.3, e1602133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang, J. et al. Antibody targeting tumor-derived soluble NKG2D ligand sMIC provides dual co-stimulation of CD8 T cells and enables sMIC(+) tumors respond to PD1/PD-L1 blockade therapy. J. Immunother. Cancer7, 223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tamaki, S. et al. Soluble MICB serum levels correlate with disease stage and survival rate in patients with oral squamous cell carcinoma. Anticancer Res.30, 4097–4101 (2010). [PubMed] [Google Scholar]
- 240.Wu, B. J. et al. Serum soluble MICB (sMICB) correlates with disease progression and survival in melanoma patients. Tumour Biol.34, 565–569 (2013). [DOI] [PubMed] [Google Scholar]
- 241.Yamaguchi, K. et al. Diagnostic and prognostic impact of serum-soluble UL16-binding protein 2 in lung cancer patients. Cancer Sci.103, 1405–1413 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Nakajima, H. et al. Activating interactions in human NK cell recognition: the role of 2B4-CD48. Eur. J. Immunol.29, 1676–1683 (1999). [DOI] [PubMed] [Google Scholar]
- 243.Sivori, S. et al. 2B4 functions as a co-receptor in human NK cell activation. Eur. J. Immunol.30, 787–793 (2000). [DOI] [PubMed] [Google Scholar]
- 244.Eissmann, P. & Watzl, C. Molecular analysis of NTB-A signaling: a role for EAT-2 in NTB-A-mediated activation of human NK cells. J. Immunol.177, 3170–3177 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Bottino, C. et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J. Exp. Med. 194, 235–235 (2001). [DOI] [PMC free article] [PubMed]
- 246.Dong, Z. et al. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity36, 974–985 (2012). [DOI] [PubMed] [Google Scholar]
- 247.Tangye, S. G. et al. Cutting edge: human 2B4, an activating NK cell receptor, recruits the protein tyrosine phosphatase SHP-2 and the adaptor signaling protein SAP. J. Immunol.162, 6981–6985 (1999). [PubMed] [Google Scholar]
- 248.Bloch-Queyrat, C. et al. Regulation of natural cytotoxicity by the adaptor SAP and the Src-related kinase Fyn. J. Exp. Med202, 181–192 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tassi, I. & Colonna, M. The cytotoxicity receptor CRACC (CS-1) recruits EAT-2 and activates the PI3K and phospholipase Cgamma signaling pathways in human NK cells. J. Immunol.175, 7996–8002 (2005). [DOI] [PubMed] [Google Scholar]
- 250.Saborit-Villarroya, I. et al. The adaptor protein 3BP2 binds human CD244 and links this receptor to Vav signaling, ERK activation, and NK cell killing. J. Immunol.175, 4226–4235 (2005). [DOI] [PubMed] [Google Scholar]
- 251.Zhang, Z. et al. DNAM-1 controls NK cell activation via an ITT-like motif. J. Exp. Med.212, 2165–2182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shibuya, K. et al. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity11, 615–623 (1999). [DOI] [PubMed] [Google Scholar]
- 253.Shirakawa, J. et al. LFA-1-dependent lipid raft recruitment of DNAM-1 (CD226) in CD4+ T cell. Int. Immunol.18, 951–957 (2006). [DOI] [PubMed] [Google Scholar]
- 254.Ralston, K. J. et al. The LFA-1-associated molecule PTA-1 (CD226) on T cells forms a dynamic molecular complex with protein 4.1G and human discs large. J. Biol. Chem.279, 33816–33828 (2004). [DOI] [PubMed] [Google Scholar]
- 255.Masson, D. et al. Overexpression of the CD155 gene in human colorectal carcinoma. Gut49, 236–240 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bevelacqua, V. et al. Nectin-like-5 overexpression correlates with the malignant phenotype in cutaneous melanoma. Oncotarget3, 882–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Nakai, R. et al. Overexpression of Necl-5 correlates with unfavorable prognosis in patients with lung adenocarcinoma. Cancer Sci.101, 1326–1330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kim, H. S. et al. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity32, 175–186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kim, H. S. & Long, E. O. Complementary phosphorylation sites in the adaptor protein SLP-76 promote synergistic activation of natural killer cells. Sci. Signal.5, ra49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Milito, N. D. et al. NKG2D engagement on human NK cells leads to DNAM-1 hypo-responsiveness through different converging mechanisms. Eur. J. Immunol.53, e2250198 (2023). [DOI] [PubMed] [Google Scholar]
- 261.Sayitoglu, E. C. et al. Boosting natural killer cell-mediated targeting of sarcoma through DNAM-1 and NKG2D. Front. Immunol.11, 40 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Banta, K. L. et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity55, 512–526.e519 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Weulersse, M. et al. Eomes-dependent loss of the co-activating receptor CD226 restrains CD8(+) T cell anti-tumor functions and limits the efficacy of cancer immunotherapy. Immunity53, 824–839.e810 (2020). [DOI] [PubMed] [Google Scholar]
- 264.Braun, M. et al. CD155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor CD226 in CD8(+) T cells. Immunity53, 805–823.e815 (2020). [DOI] [PubMed] [Google Scholar]
- 265.Stebbins, C. C. et al. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol. Cell Biol.23, 6291–6299 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Liu, D., Peterson, M. E. & Long, E. O. The adaptor protein Crk controls activation and inhibition of natural killer cells. Immunity36, 600–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Chirino, L. M. et al. TAM receptors attenuate murine NK-cell responses via E3 ubiquitin ligase Cbl-b. Eur. J. Immunol.50, 48–55 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Mesecke, S. et al. Integration of activating and inhibitory receptor signaling by regulated phosphorylation of Vav1 in immune cells. Sci. Signal.4, ra36 (2011). [DOI] [PubMed] [Google Scholar]
- 269.Kaiser, B. K., Pizarro, J. C., Kerns, J. & Strong, R. K. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc. Natl. Acad. Sci. USA105, 6696–6701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.van der Merwe, P. A. et al. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med.185, 393–403 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yu, X. et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol.10, 48–57 (2009). [DOI] [PubMed] [Google Scholar]
- 272.Tahara-Hanaoka, S. et al. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int. Immunol.16, 533–538 (2004). [DOI] [PubMed] [Google Scholar]
- 273.Reches, A. et al. Nectin4 is a novel TIGIT ligand which combines checkpoint inhibition and tumor specificity. J. Immunother. Cancer8, e000266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Zhu, Y. et al. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med.213, 167–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chan, C. J. et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol.15, 431–438 (2014). [DOI] [PubMed] [Google Scholar]
- 276.Ren, X. et al. Blockade of the immunosuppressive KIR2DL5/PVR pathway elicits potent human NK cell-mediated antitumor immunity. J. Clin. Investig.132, e163620 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Johnston, R. J. et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell26, 923–937 (2014). [DOI] [PubMed] [Google Scholar]
- 278.Deng, Y. et al. Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity42, 457–470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Du, X. et al. CD226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc. Natl. Acad. Sci. USA115, E11731–E11740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Carlsten, M. et al. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol.183, 4921–4930 (2009). [DOI] [PubMed] [Google Scholar]
- 281.Carlsten, M. et al. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia24, 1607–1616 (2010). [DOI] [PubMed] [Google Scholar]
- 282.Mamessier, E. et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig.121, 3609–3622 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Oda, T., Ohka, S. & Nomoto, A. Ligand stimulation of CD155alpha inhibits cell adhesion and enhances cell migration in fibroblasts. Biochem. Biophys. Res. Commun.319, 1253–1264 (2004). [DOI] [PubMed] [Google Scholar]
- 284.Kakunaga, S. et al. Enhancement of serum- and platelet-derived growth factor-induced cell proliferation by Necl-5/Tage4/poliovirus receptor/CD155 through the Ras-Raf-MEK-ERK signaling. J. Biol. Chem.279, 36419–36425 (2004). [DOI] [PubMed] [Google Scholar]
- 285.Reymond, N. et al. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J. Exp. Med.199, 1331–1341 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Cerdeira, A. S. et al. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J. Immunol.190, 3939–3948 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Choi, P. J. & Mitchison, T. J. Imaging burst kinetics and spatial coordination during serial killing by single natural killer cells. Proc. Natl. Acad. Sci. USA110, 6488–6493 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Jounaidi, Y., Cotten, J. F., Miller, K. W. & Forman, S. A. Tethering IL2 to its receptor IL2Rbeta enhances antitumor activity and expansion of natural killer NK92 cells. Cancer Res.77, 5938–5951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zagury, D. et al. Studies on the mechanism of T cell-mediated lysis at the single effector cell level. I. Kinetic analysis of lethal hits and target cell lysis in multicellular conjugates. J. Immunol.123, 1604–1609 (1979). [PubMed] [Google Scholar]
- 290.Wiedemann, A., Depoil, D., Faroudi, M. & Valitutti, S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. USA103, 10985–10990 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Bhat, R. & Watzl, C. Serial killing of tumor cells by human natural killer cells–enhancement by therapeutic antibodies. PLoS ONE2, e326 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Shi, F. D., Ljunggren, H. G., La Cava, A. & Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol.11, 658–671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Frey, M. et al. Differential expression and function of L-selectin on CD56bright and CD56dim natural killer cell subsets. J. Immunol.161, 400–408 (1998). [PubMed] [Google Scholar]
- 294.Casilli, F. et al. Inhibition of interleukin-8 (CXCL8/IL-8) responses by repertaxin, a new inhibitor of the chemokine receptors CXCR1 and CXCR2. Biochem. Pharm.69, 385–394 (2005). [DOI] [PubMed] [Google Scholar]
- 295.Chuntharapai, A., Lee, J., Hebert, C. A. & Kim, K. J. Monoclonal antibodies detect different distribution patterns of IL-8 receptor A and IL-8 receptor B on human peripheral blood leukocytes. J. Immunol.153, 5682–5688 (1994). [PubMed] [Google Scholar]
- 296.Morohashi, H. et al. Expression of both types of human interleukin-8 receptors on mature neutrophils, monocytes, and natural killer cells. J. Leukoc. Biol.57, 180–187 (1995). [DOI] [PubMed] [Google Scholar]
- 297.Imai, T. et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell91, 521–530 (1997). [DOI] [PubMed] [Google Scholar]
- 298.Yoneda, O. et al. Fractalkine-mediated endothelial cell injury by NK cells. J. Immunol.164, 4055–4062 (2000). [DOI] [PubMed] [Google Scholar]
- 299.Bernardini, G. et al. CCL3 and CXCL12 regulate trafficking of mouse bone marrow NK cell subsets. Blood111, 3626–3634 (2008). [DOI] [PubMed] [Google Scholar]
- 300.Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell121, 335–348 (2005). [DOI] [PubMed] [Google Scholar]
- 301.Zhang, S. et al. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS ONE11, e0164514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Mailloux, A. W. & Young, M. R. Regulatory T-cell trafficking: from thymic development to tumor-induced immune suppression. Crit. Rev. Immunol.30, 435–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Zhang, Q. W. et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE7, e50946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Templeton, A. J. et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Natl. Cancer Inst.106, dju124 (2014). [DOI] [PubMed] [Google Scholar]
- 305.Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol. Immunol.15, 335–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Sarvaiya, P. J. et al. Chemokines in tumor progression and metastasis. Oncotarget4, 2171–2185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Muller, N. et al. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-secreting Glioblastoma. J. Immunother.38, 197–210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Biasci, D. et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA117, 28960–28970 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Chheda, Z. S. et al. Chemoattractant receptors BLT1 and CXCR3 regulate antitumor immunity by facilitating CD8+ T cell migration into tumors. J. Immunol.197, 2016–2026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Taki, M. et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun.9, 1685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Berahovich, R. D. et al. Evidence for NK cell subsets based on chemokine receptor expression. J. Immunol.177, 7833–7840 (2006). [DOI] [PubMed] [Google Scholar]
- 312.Lima, M. et al. Chemokine receptor expression on normal blood CD56(+) NK-cells elucidates cell partners that comigrate during the innate and adaptive immune responses and identifies a transitional NK-cell population. J. Immunol. Res.2015, 839684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Zabel, B. A. et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med.205, 2207–2220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Parolini, S. et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood109, 3625–3632 (2007). [DOI] [PubMed] [Google Scholar]
- 315.Vermi, W. et al. Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med.201, 509–515 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Wittamer, V. et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med.198, 977–985 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kaur, J. et al. Identification of chemerin receptor (ChemR23) in human endothelial cells: chemerin-induced endothelial angiogenesis. Biochem. Biophys. Res. Commun.391, 1762–1768 (2010). [DOI] [PubMed] [Google Scholar]
- 318.Kveberg, L. et al. Sphingosine 1 phosphate induces the chemotaxis of human natural killer cells. Role for heterotrimeric G proteins and phosphoinositide 3 kinases. Eur. J. Immunol.32, 1856–1864 (2002). [DOI] [PubMed] [Google Scholar]
- 319.Maghazachi, A. A. G protein-coupled receptors in natural killer cells. J. Leukoc. Biol.74, 16–24 (2003). [DOI] [PubMed] [Google Scholar]
- 320.Walzer, T. et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol.8, 1337–1344 (2007). [DOI] [PubMed] [Google Scholar]
- 321.Ledgerwood, L. G. et al. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat. Immunol.9, 42–53 (2008). [DOI] [PubMed] [Google Scholar]
- 322.Bottcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell172, 1022–1037.e1014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Wennerberg, E., Kremer, V., Childs, R. & Lundqvist, A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol. Immunother.64, 225–235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Miao, H. et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS ONE9, e94281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Kremer, V. et al. Correction to: genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer5, 88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Liblau, R. et al. T cell response to myelin basic protein epitopes in multiple sclerosis patients and healthy subjects. Eur. J. Immunol.21, 1391–1395 (1991). [DOI] [PubMed] [Google Scholar]
- 327.Ota, K. et al. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature346, 183–187 (1990). [DOI] [PubMed] [Google Scholar]
- 328.Wardemann, H. et al. Predominant autoantibody production by early human B cell precursors. Science301, 1374–1377 (2003). [DOI] [PubMed] [Google Scholar]
- 329.Ganguly, D., Haak, S., Sisirak, V. & Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol.13, 566–577 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Zhang, A. L. et al. Natural killer cells trigger differentiation of monocytes into dendritic cells. Blood110, 2484–2493 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Vitale, M. et al. The small subset of CD56brightCD16- natural killer cells is selectively responsible for both cell proliferation and interferon-gamma production upon interaction with dendritic cells. Eur. J. Immunol.34, 1715–1722 (2004). [DOI] [PubMed] [Google Scholar]
- 332.Lin, S. J. et al. Phenotypic and functional characterization of natural killer cells in rheumatoid arthritis-regulation with interleukin-15. Sci. Rep.10, 5858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Aramaki, T. et al. A significantly impaired natural killer cell activity due to a low activity on a per-cell basis in rheumatoid arthritis. Mod. Rheumatol.19, 245–252 (2009). [DOI] [PubMed] [Google Scholar]
- 334.Louis, C. et al. NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J. Exp. Med.217, e20191421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Laroni, A. et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J. Autoimmun.72, 8–18 (2016). [DOI] [PubMed] [Google Scholar]
- 336.Gross, C. C. et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA113, E2973–E2982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Park, Y. W. et al. Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum.60, 1753–1763 (2009). [DOI] [PubMed] [Google Scholar]
- 338.Henriques, A. et al. NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin. Rheumatol.32, 805–813 (2013). [DOI] [PubMed] [Google Scholar]
- 339.Hagberg, N. et al. IFN-alpha production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1beta and LFA-1. J. Immunol.186, 5085–5094 (2011). [DOI] [PubMed] [Google Scholar]
- 340.Huang, Z. et al. Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J. Immunol.186, 3421–3431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lin, S. J. et al. Activating and inhibitory receptors on natural killer cells in patients with systemic lupus erythematosis-regulation with interleukin-15. PLoS ONE12, e0186223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Rodacki, M. et al. Altered natural killer cells in type 1 diabetic patients. Diabetes56, 177–185 (2007). [DOI] [PubMed] [Google Scholar]
- 343.Qin, H. et al. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes60, 857–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Kim, J. H. et al. Relationship between natural killer cell activity and glucose control in patients with type 2 diabetes and prediabetes. J. Diabetes Investig.10, 1223–1228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Peters, L., Posgai, A. & Brusko, T. M. Islet-immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clin. Exp. Immunol.198, 326–340 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Dotta, F. et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA104, 5115–5120 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Traherne, J. A. et al. KIR haplotypes are associated with late-onset type 1 diabetes in European-American families. Genes Immun.17, 8–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Tirpe, A. A. et al. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int. J. Mol. Sci.20, 6140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Guan, Y. et al. Renal cell tumors convert natural killer cells to a proangiogenic phenotype. Oncotarget11, 2571–2585 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Tabiasco, J. et al. Human decidual NK cells: unique phenotype and functional properties–a review. Placenta27, S34–S39 (2006). [DOI] [PubMed] [Google Scholar]
- 351.Kalkunte, S. S. et al. Vascular endothelial growth factor C facilitates immune tolerance and endovascular activity of human uterine NK cells at the maternal-fetal interface. J. Immunol.182, 4085–4092 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Kusumi, M. et al. Expression patterns of lectin-like natural killer receptors, inhibitory CD94/NKG2A, and activating CD94/NKG2C on decidual CD56bright natural killer cells differ from those on peripheral CD56dim natural killer cells. J. Reprod. Immunol.70, 33–42 (2006). [DOI] [PubMed] [Google Scholar]
- 353.Crespo, A. C., Strominger, J. L. & Tilburgs, T. Expression of KIR2DS1 by decidual natural killer cells increases their ability to control placental HCMV infection. Proc. Natl. Acad. Sci. USA113, 15072–15077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Male, V. et al. The effect of pregnancy on the uterine NK cell KIR repertoire. Eur. J. Immunol.41, 3017–3027 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Mor, G., Cardenas, I., Abrahams, V. & Guller, S. Inflammation and pregnancy: the role of the immune system at the implantation site. Ann. N. Y Acad. Sci.1221, 80–87 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.King, A. et al. HLA-E is expressed on trophoblast and interacts with CD94/NKG2 receptors on decidual NK cells. Eur. J. Immunol.30, 1623–1631 (2000). [DOI] [PubMed] [Google Scholar]
- 357.Shreeve, N. et al. The CD94/NKG2A inhibitory receptor educates uterine NK cells to optimize pregnancy outcomes in humans and mice. Immunity54, 1231–1244 e1234 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Hanna, J. et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med.12, 1065–1074 (2006). [DOI] [PubMed] [Google Scholar]
- 359.Chakraborty, D., Rumi, M. A. & Soares, M. J. NK cells, hypoxia and trophoblast cell differentiation. Cell Cycle11, 2427–2430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Trowsdale, J. & Moffett, A. NK receptor interactions with MHC class I molecules in pregnancy. Semin. Immunol.20, 317–320 (2008). [DOI] [PubMed] [Google Scholar]
- 361.Chazara, O., Xiong, S. & Moffett, A. Maternal KIR and fetal HLA-C: a fine balance. J. Leukoc. Biol.90, 703–716 (2011). [DOI] [PubMed] [Google Scholar]
- 362.Krzywinska, E. et al. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun.8, 1597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Ni, J. et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1alpha unleashes NK cell activity. Immunity52, 1075–1087 e1078 (2020). [DOI] [PubMed] [Google Scholar]
- 364.Kim, J. et al. Hypoxia-induced IL-18 increases hypoxia-inducible factor-1alpha expression through a Rac1-dependent NF-kappaB pathway. Mol. Biol. Cell19, 433–444 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Lob, S. et al. The role of Interleukin-18 in recurrent early pregnancy loss. J. Reprod. Immunol.148, 103432 (2021). [DOI] [PubMed] [Google Scholar]
- 366.Cui, A. et al. Dictionary of immune responses to cytokines at single-cell resolution. Nature625, 377–384 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Mailliard, R. B. et al. IL-18-induced CD83+CCR7+ NK helper cells. J. Exp. Med.202, 941–953 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Williams, P. J. et al. Altered decidual leucocyte populations in the placental bed in pre-eclampsia and foetal growth restriction: a comparison with late normal pregnancy. Reproduction138, 177–184 (2009). [DOI] [PubMed] [Google Scholar]
- 369.Carosella, E. D. et al. HLA-G: An Immune Checkpoint Molecule. Adv. Immunol.127, 33–144 (2015). [DOI] [PubMed] [Google Scholar]
- 370.Hackmon, R. et al. Definitive class I human leukocyte antigen expression in gestational placentation: HLA-F, HLA-E, HLA-C, and HLA-G in extravillous trophoblast invasion on placentation, pregnancy, and parturition. Am. J. Reprod. Immunol. 77,e12643 (2017). [DOI] [PubMed]
- 371.Wang, S. et al. Harnessing the potential of HLA-G in cancer therapy: advances, challenges, and prospects. J. Transl. Med.22, 130 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Yang, Y., Chu, W., Geraghty, D. E. & Hunt, J. S. Expression of HLA-G in human mononuclear phagocytes and selective induction by IFN-gamma. J. Immunol.156, 4224–4231 (1996). [PubMed] [Google Scholar]
- 373.Chan, I. S. et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol.219, e202001134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.de Jonge, K. et al. Circulating CD56(bright) NK cells inversely correlate with survival of melanoma patients. Sci. Rep.9, 4487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Sender, R. et al. The total mass, number, and distribution of immune cells in the human body. Proc. Natl. Acad. Sci. USA120, e2308511120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov.12, 31–46 (2022). [DOI] [PubMed] [Google Scholar]
- 377.Marin, I. et al. Cellular senescence is immunogenic and promotes antitumor immunity. Cancer Discov.13, 410–431 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Hernandez-Mercado, E. et al. Increased CD47 and MHC class I inhibitory signals expression in senescent CD1 primary mouse lung fibroblasts. Int. J. Mol. Sci.22, 10215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Wang, C. et al. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell8, 311–323 (2009). [DOI] [PubMed] [Google Scholar]
- 380.Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Stout, R. D. & Suttles, J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol. Rev.205, 60–71 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Uyemura, K., Castle, S. C. & Makinodan, T. The frail elderly: role of dendritic cells in the susceptibility of infection. Mech. Ageing Dev.123, 955–962 (2002). [DOI] [PubMed] [Google Scholar]
- 383.Han, S. et al. Enhanced differentiation of splenic plasma cells but diminished long-lived high-affinity bone marrow plasma cells in aged mice. J. Immunol.170, 1267–1273 (2003). [DOI] [PubMed] [Google Scholar]
- 384.Voehringer, D., Koschella, M. & Pircher, H. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood100, 3698–3702 (2002). [DOI] [PubMed] [Google Scholar]
- 385.Naylor, K. et al. The influence of age on T cell generation and TCR diversity. J. Immunol.174, 7446–7452 (2005). [DOI] [PubMed] [Google Scholar]
- 386.Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer15, 397–408 (2015). [DOI] [PubMed] [Google Scholar]
- 387.Pereira, B. I. et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun.10, 2387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Baker, D. J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Lutz, C. T. et al. Reciprocal age related change in natural killer cell receptors for MHC class I. Mech. Ageing Dev.126, 722–731 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Walzer, T. et al. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood106, 2252–2258 (2005). [DOI] [PubMed] [Google Scholar]
- 391.Borrego, F. et al. NK phenotypic markers and IL2 response in NK cells from elderly people. Exp. Gerontol.34, 253–265 (1999). [DOI] [PubMed] [Google Scholar]
- 392.Severino, V. et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis.4, e911 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Coppe, J. P., Kauser, K., Campisi, J. & Beausejour, C. M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem.281, 29568–29574 (2006). [DOI] [PubMed] [Google Scholar]
- 394.Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 396.Borrelli, C. et al. Drug-induced senescent multiple myeloma cells elicit nk cell proliferation by direct or exosome-mediated IL15 trans-presentation. Cancer Immunol. Res.6, 860–869 (2018). [DOI] [PubMed] [Google Scholar]
- 397.Lv, L. H. et al. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem.287, 15874–15885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Nelson, G. et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell11, 345–349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Lee, S. & Schmitt, C. A. The dynamic nature of senescence in cancer. Nat. Cell Biol.21, 94–101 (2019). [DOI] [PubMed] [Google Scholar]
- 400.Soriani, A. et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood113, 3503–3511 (2009). [DOI] [PubMed] [Google Scholar]
- 401.Sagiv, A. et al. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene32, 1971–1977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Antonangeli, F. et al. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. Oncoimmunology5, e1218105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Atochina, O. & Harn, D. LNFPIII/LeX-stimulated macrophages activate natural killer cells via CD40-CD40L interaction. Clin. Diagn. Lab Immunol.12, 1041–1049 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.O’Sullivan, T. et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med.209, 1869–1882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Nedvetzki, S. et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood109, 3776–3785 (2007). [DOI] [PubMed] [Google Scholar]
- 406.Romo, N. et al. Natural killer cell-mediated response to human cytomegalovirus-infected macrophages is modulated by their functional polarization. J. Leukoc. Biol.90, 717–726 (2011). [DOI] [PubMed] [Google Scholar]
- 407.Elhaik-Goldman, S. et al. The natural cytotoxicity receptor 1 contribution to early clearance of Streptococcus pneumoniae and to natural killer-macrophage cross talk. PLoS ONE6, e23472 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Siren, J. et al. Cytokine and contact-dependent activation of natural killer cells by influenza A or Sendai virus-infected macrophages. J. Gen. Virol.85, 2357–2364 (2004). [DOI] [PubMed] [Google Scholar]
- 409.Hamerman, J. A., Ogasawara, K. & Lanier, L. L. Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J. Immunol.172, 2001–2005 (2004). [DOI] [PubMed] [Google Scholar]
- 410.Kloss, M. et al. Interaction of monocytes with NK cells upon toll-like receptor-induced expression of the NKG2D ligand MICA. J. Immunol.181, 6711–6719 (2008). [DOI] [PubMed] [Google Scholar]
- 411.Krneta, T. et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol.101, 285–295 (2017). [DOI] [PubMed] [Google Scholar]
- 412.Funes, S. C., Rios, M., Escobar-Vera, J. & Kalergis, A. M. Implications of macrophage polarization in autoimmunity. Immunology154, 186–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Soudja, S. M., Ruiz, A. L., Marie, J. C. & Lauvau, G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity37, 549–562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Jaeger, B. N. et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J. Exp. Med.209, 565–580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Sohlberg, E. et al. Perturbed NK-cell homeostasis associated with disease severity in chronic neutropenia. Blood139, 704–716 (2022). [DOI] [PubMed] [Google Scholar]
- 416.Koga, Y. et al. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): a novel mechanism of antitumor effect by neutrophils. Cancer Res.64, 1037–1043 (2004). [DOI] [PubMed] [Google Scholar]
- 417.Gaggero, S., Witt, K., Carlsten, M. & Mitra, S. Cytokines orchestrating the natural killer-myeloid cell crosstalk in the tumor microenvironment: implications for natural killer cell-based cancer immunotherapy. Front Immunol.11, 621225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Sun, R. et al. Tumor-associated neutrophils suppress antitumor immunity of NK cells through the PD-L1/PD-1 axis. Transl. Oncol.13, 100825 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Elinav, E. et al. Inflammation-induced cancer: crosstalk between tumours, immune cells, and microorganisms. Nat. Rev. Cancer13, 759–771 (2013). [DOI] [PubMed] [Google Scholar]
- 420.Jaillon, S. et al. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer20, 485–503 (2020). [DOI] [PubMed] [Google Scholar]
- 421.Costantini, C. et al. Neutrophil activation and survival are modulated by interaction with NK cells. Int. Immunol.22, 827–838 (2010). [DOI] [PubMed] [Google Scholar]
- 422.Bhatnagar, N. et al. Cytokine-activated NK cells inhibit PMN apoptosis and preserve their functional capacity. Blood116, 1308–1316 (2010). [DOI] [PubMed] [Google Scholar]
- 423.Bertin, F. R. et al. Natural killer cells induce neutrophil extracellular trap formation in venous thrombosis. J. Thromb. Haemost.17, 403–414 (2019). [DOI] [PubMed] [Google Scholar]
- 424.Valayer, A. et al. Neutrophils can disarm NK cell response through cleavage of NKp46. J. Leukoc. Biol.101, 253–259 (2017). [DOI] [PubMed] [Google Scholar]
- 425.Zhang, H. et al. Annexin A2/TLR2/MYD88 pathway induces arginase 1 expression in tumor-associated neutrophils. J. Clin. Investig.132, e153643 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Munder, M. Arginase: an emerging key player in the mammalian immune system. Br. J. Pharm.158, 638–651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Mellqvist, U. H. et al. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood96, 1961–1968 (2000). [PubMed] [Google Scholar]
- 428.Mao, Y. et al. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res.20, 4096–4106 (2014). [DOI] [PubMed] [Google Scholar]
- 429.Bluestone, J. A. & Abbas, A. K. Natural versus adaptive regulatory T cells. Nat. Rev. Immunol.3, 253–257 (2003). [DOI] [PubMed] [Google Scholar]
- 430.Dahlberg, C. I. et al. Natural killer cell-based therapies targeting cancer: possible strategies to gain and sustain anti-tumor activity. Front. Immunol.6, 605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Littwitz-Salomon, E. et al. Activated regulatory T cells suppress effector NK cell responses by an IL-2-mediated mechanism during an acute retroviral infection. Retrovirology12, 66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Sarhan, D. et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res.76, 5696–5706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Vaknin, I. et al. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood111, 1437–1447 (2008). [DOI] [PubMed] [Google Scholar]
- 434.Yue, J. et al. Myeloid-derived suppressor cells inhibit natural killer cells in myelodysplastic syndromes through the TIGIT/CD155 pathway. Hematology28, 2166333 (2023). [DOI] [PubMed] [Google Scholar]
- 435.Nausch, N., Galani, I. E., Schlecker, E. & Cerwenka, A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood112, 4080–4089 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Hoechst, B. et al. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood117, 6532–6541 (2011). [DOI] [PubMed] [Google Scholar]
- 437.Raskovalova, T. et al. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J. Immunol.175, 4383–4391 (2005). [DOI] [PubMed] [Google Scholar]
- 438.Horowitz, A. et al. Cross-talk between T cells and NK cells generates rapid effector responses to Plasmodium falciparum-infected erythrocytes. J. Immunol.184, 6043–6052 (2010). [DOI] [PubMed] [Google Scholar]
- 439.Fehniger, T. A. et al. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood101, 3052–3057 (2003). [DOI] [PubMed] [Google Scholar]
- 440.Agaugue, S. et al. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood112, 1776–1783 (2008). [DOI] [PubMed] [Google Scholar]
- 441.Adam, C. et al. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood106, 338–344 (2005). [DOI] [PubMed] [Google Scholar]
- 442.Srivastava, R. M. et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin. Cancer Res.19, 1858–1872 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Zingoni, A. et al. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J. Immunol.173, 3716–3724 (2004). [DOI] [PubMed] [Google Scholar]
- 444.Sconocchia, G. et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology3, e952197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Ing, R. & Stevenson, M. M. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun.77, 770–782 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Della Chiesa, M. et al. The natural killer cell-mediated killing of autologous dendritic cells is confined to a cell subset expressing CD94/NKG2A, but lacking inhibitory killer Ig-like receptors. Eur. J. Immunol.33, 1657–1666 (2003). [DOI] [PubMed] [Google Scholar]
- 447.Ferlazzo, G., Semino, C. & Melioli, G. HLA class I molecule expression is up-regulated during maturation of dendritic cells, protecting them from natural killer cell-mediated lysis. Immunol. Lett.76, 37–41 (2001). [DOI] [PubMed] [Google Scholar]
- 448.Li, T. et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett.318, 154–161 (2012). [DOI] [PubMed] [Google Scholar]
- 449.Linnemeyer, P. A. & Pollack, S. B. Prostaglandin E2-induced changes in the phenotype, morphology, and lytic activity of IL-2-activated natural killer cells. J. Immunol.150, 3747–3754 (1993). [PubMed] [Google Scholar]
- 450.Joshi, P. C., Zhou, X., Cuchens, M. & Jones, Q. Prostaglandin E2 suppressed IL-15-mediated human NK cell function through down-regulation of common gamma-chain. J. Immunol.166, 885–891 (2001). [DOI] [PubMed] [Google Scholar]
- 451.Yakar, I. et al. Prostaglandin e(2) suppresses NK activity in vivo and promotes postoperative tumor metastasis in rats. Ann. Surg. Oncol.10, 469–479 (2003). [DOI] [PubMed] [Google Scholar]
- 452.Holt, D., Ma, X., Kundu, N. & Fulton, A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol. Immunother.60, 1577–1586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Sharma, S. et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res.65, 5211–5220 (2005). [DOI] [PubMed] [Google Scholar]
- 454.Van Elssen, C. H. et al. Inflammation-restraining effects of prostaglandin E2 on natural killer-dendritic cell (NK-DC) interaction are imprinted during DC maturation. Blood118, 2473–2482 (2011). [DOI] [PubMed] [Google Scholar]
- 455.Uyttenhove, C. et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med.9, 1269–1274 (2003). [DOI] [PubMed] [Google Scholar]
- 456.Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature478, 197–203 (2011). [DOI] [PubMed] [Google Scholar]
- 457.Trikha, P. et al. Defining the AHR-regulated transcriptome in NK cells reveals gene expression programs relevant to development and function. Blood Adv.5, 4605–4618 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Wang, D. et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int. J. Oncol.40, 929–934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Palumbo, J. S. et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood105, 178–185 (2005). [DOI] [PubMed] [Google Scholar]
- 460.Placke, T. et al. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res.72, 440–448 (2012). [DOI] [PubMed] [Google Scholar]
- 461.Kopp, H. G., Placke, T. & Salih, H. R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res.69, 7775–7783 (2009). [DOI] [PubMed] [Google Scholar]
- 462.Romee, R. et al. Cytokine activation induces human memory-like NK cells. Blood120, 4751–4760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Spanholtz, J. et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS ONE5, e9221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Knorr, D. A. et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med.2, 274–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Klingemann, H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy25, 451–457 (2023). [DOI] [PubMed] [Google Scholar]
- 466.Toffoli, E. C. et al. Allogeneic NK cells induce monocyte-to-dendritic cell conversion, control tumor growth, and trigger a pro-inflammatory shift in patient-derived cultures of primary and metastatic colorectal cancer. J. Immunother. Cancer11, e007554 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Condiotti, R., Zakai, Y. B., Barak, V. & Nagler, A. Ex vivo expansion of CD56+ cytotoxic cells from human umbilical cord blood. Exp. Hematol.29, 104–113 (2001). [DOI] [PubMed] [Google Scholar]
- 468.Shah, N. et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS ONE8, e76781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Ojo, E. O. et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci. Rep.9, 14916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Parkhurst, M. R., Riley, J. P., Dudley, M. E. & Rosenberg, S. A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res.17, 6287–6297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Rubnitz, J. E. et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol.28, 955–959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Vales-Gomez, M., Reyburn, H. T., Mandelboim, M. & Strominger, J. L. Kinetics of interaction of HLA-C ligands with natural killer cell inhibitory receptors. Immunity9, 337–344 (1998). [DOI] [PubMed] [Google Scholar]
- 473.Ruggeri, L. et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science295, 2097–2100 (2002). [DOI] [PubMed] [Google Scholar]
- 474.Asai, O. et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J. Clin. Investig.101, 1835–1842 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Huber, C. M., Doisne, J. M. & Colucci, F. IL-12/15/18-preactivated NK cells suppress GvHD in a mouse model of mismatched hematopoietic cell transplantation. Eur. J. Immunol.45, 1727–1735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Geller, M. A. et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy13, 98–107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Sotillo, E. et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov.5, 1282–1295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Fujisaki, H. et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res.69, 4010–4017 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Sim, G. C. et al. IL2 variant circumvents ICOS+ regulatory T-cell expansion and promotes NK cell activation. Cancer Immunol. Res.4, 983–994 (2016). [DOI] [PubMed] [Google Scholar]
- 480.Lopes, J. E. et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J. Immunother. Cancer8, e000673 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Levin, A. M. et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine. Nature484, 529–533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Di Pilato, M. et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell184, 4512–4530 e4522 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Guimond, M. et al. In vivo role of Flt3 ligand and dendritic cells in NK cell homeostasis. J. Immunol.184, 2769–2775 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Watkinson, F. et al. IL-15 upregulates telomerase expression and potently increases proliferative capacity of NK, NKT-like, and CD8 T cells. Front. Immunol.11, 594620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Rufer, N. et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J. Exp. Med.190, 157–167 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Akbar, A. N., Beverley, P. C. & Salmon, M. Will telomere erosion lead to a loss of T-cell memory? Nat. Rev. Immunol.4, 737–743 (2004). [DOI] [PubMed] [Google Scholar]
- 487.Liu, E. et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia32, 520–531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Daher, M. et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood137, 624–636 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Christodoulou, I. et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J. Immunother. Cancer9, e003894 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Felices, M. et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight3, e96219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Lasek, W., Zagozdzon, R. & Jakobisiak, M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother.63, 419–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Sun, J. C. et al. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J. Exp. Med.209, 947–954 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Leong, J. W. et al. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol. Blood Marrow Transpl.20, 463–473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Konjevic, G. M. et al. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine117, 30–40 (2019). [DOI] [PubMed] [Google Scholar]
- 495.Zhou, T. et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature583, 609–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Spolski, R. & Leonard, W. J. Interleukin-21: a double-edged sword with therapeutic potential. Nat. Rev. Drug Discov.13, 379–395 (2014). [DOI] [PubMed] [Google Scholar]
- 497.Endo, T. A. et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature387, 921–924 (1997). [DOI] [PubMed] [Google Scholar]
- 498.Cohney, S. J. et al. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol. Cell Biol.19, 4980–4988 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Asao, H. et al. Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J. Immunol.167, 1–5 (2001). [DOI] [PubMed] [Google Scholar]
- 500.Strengell, M. et al. IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J. Immunol.169, 3600–3605 (2002). [DOI] [PubMed] [Google Scholar]
- 501.Zeng, R. et al. The molecular basis of IL-21-mediated proliferation. Blood109, 4135–4142 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Strengell, M. et al. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J. Immunol.170, 5464–5469 (2003). [DOI] [PubMed] [Google Scholar]
- 503.Petrella, T. M. et al. Interleukin-21 has activity in patients with metastatic melanoma: a phase II study. J. Clin. Oncol.30, 3396–3401 (2012). [DOI] [PubMed] [Google Scholar]
- 504.Bhatt, S., Sarosiek, K. A. & Lossos, I. S. Interleukin 21 - its potential role in the therapy of B-cell lymphomas. Leuk. Lymphoma58, 17–29 (2017). [DOI] [PubMed] [Google Scholar]
- 505.Donatelli, S. S. et al. TGF-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc. Natl. Acad. Sci. USA111, 4203–4208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Yvon, E. S. et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy19, 408–418 (2017). [DOI] [PubMed] [Google Scholar]
- 507.Shaim, H. et al. Targeting the alphav integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig.131, e142116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Tang, K., Wu, Y. H., Song, Y. & Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J. Hematol. Oncol.14, 68 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Goto, S. et al. Upregulation of PD-L1 expression by prostaglandin E(2) and the enhancement of IFN-gamma by anti-PD-L1 antibody combined with a COX-2 inhibitor in mycoplasma bovis Infection. Front. Vet. Sci.7, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Jin, K., Qian, C., Lin, J. & Liu, B. Cyclooxygenase-2-Prostaglandin E2 pathway: a key player in tumor-associated immune cells. Front. Oncol.13, 1099811 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Blay, J., White, T. D. & Hoskin, D. W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res.57, 2602–2605 (1997). [PubMed] [Google Scholar]
- 512.Young, A. et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res.78, 1003–1016 (2018). [DOI] [PubMed] [Google Scholar]
- 513.Mittal, D. et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res.74, 3652–3658 (2014). [DOI] [PubMed] [Google Scholar]
- 514.Perrot, I. et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep.27, 2411–2425.e2419 (2019). [DOI] [PubMed] [Google Scholar]
- 515.Paul, S., Kulkarni, N., Shilpi & Lal, G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. Oncoimmunology5, e1235106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Platonova, S. et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res.71, 5412–5422 (2011). [DOI] [PubMed] [Google Scholar]
- 517.Zhang, Q. F. et al. Liver-infiltrating CD11b(-)CD27(-) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol. Immunol.14, 819–829 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Jovic, V. et al. Impaired perforin-dependent NK cell cytotoxicity and proliferative activity of peripheral blood T cells is associated with metastatic melanoma. Tumori87, 324–329 (2001). [DOI] [PubMed] [Google Scholar]
- 519.Richards, J. O. et al. Tumor growth impedes natural-killer-cell maturation in the bone marrow. Blood108, 246–252 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Chretien, A. S. et al. Natural killer defective maturation is associated with adverse clinical outcome in patients with acute myeloid leukemia. Front. Immunol.8, 573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Gill, S. et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood119, 5758–5768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Wong, P. et al. T-BET and EOMES sustain mature human NK cell identity and antitumor function. J. Clin. Investig.133, e162530 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Santana Carrero, R. M. et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc. Natl. Acad. Sci. USA116, 599–608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Zhang, Q. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol.19, 723–732 (2018). [DOI] [PubMed] [Google Scholar]
- 525.Beldi-Ferchiou, A. et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget7, 72961–72977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Benson, D. M. Jr. et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood116, 2286–2294 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Sun, H. et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology70, 168–183 (2019). [DOI] [PubMed] [Google Scholar]
- 528.Sun, C. et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology6, e1264562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Guillerey, C. et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Investig.125, 2904 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Peng, Y. P. et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J. Transl. Med.11, 262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Parry, H. M. et al. NK cell function is markedly impaired in patients with chronic lymphocytic leukaemia but is preserved in patients with small lymphocytic lymphoma. Oncotarget7, 68513–68526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Acebes-Huerta, A. et al. Drug-induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM-1 receptors. Oncoimmunology5, e1074378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Zhang, X. et al. Low-dose gemcitabine treatment enhances immunogenicity and natural killer cell-driven tumor immunity in lung cancer. Front. Immunol.11, 331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Ghiringhelli, F. et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother.56, 641–648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.MacFarlane, A. W. T. et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol. Res.2, 320–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig.128, 4654–4668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Barry, K. C. et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med.24, 1178–1191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Zemek, R. M. et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med.11, eaav7816 (2019). [DOI] [PubMed] [Google Scholar]
- 539.Judge, S. J. et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig.130, 3051–3068 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Sanseviero, E. et al. Anti-CTLA-4 activates intratumoral NK cells and combined with IL15/IL15Ralpha complexes enhances tumor control. Cancer Immunol. Res.7, 1371–1380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.da Silva, I. P. et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res2, 410–422 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Acharya, N., Sabatos-Peyton, C. & Anderson, A. C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer8, e000911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Andrews, L. P., Marciscano, A. E., Drake, C. G. & Vignali, D. A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev.276, 80–96 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Merino, A. et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig.129, 3770–3785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Previte, D. M. et al. Lymphocyte activation gene-3 maintains mitochondrial and metabolic quiescence in naive CD4(+) T cells. Cell Rep.27, 129–141 e124 (2019). [DOI] [PubMed] [Google Scholar]
- 546.Thommen, D. S. et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol. Res.3, 1344–1355 (2015). [DOI] [PubMed] [Google Scholar]
- 547.Nakamura, K. et al. Fratricide of natural killer cells dressed with tumor-derived NKG2D ligand. Proc. Natl. Acad. Sci. USA110, 9421–9426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Taniguchi, R. T., Guzior, D. & Kumar, V. 2B4 inhibits NK-cell fratricide. Blood110, 2020–2023 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Casneuf, T. et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv.1, 2105–2114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Nagai, Y. et al. CD38 knockout primary NK cells to prevent “fratricide” and boost daratumumab activity. Blood134, 870–870 (2019). [Google Scholar]
- 551.Goodridge, J. P. et al. Abstract 1550: FT576 path to first-of-kind clinical trial: translation of a versatile multi-antigen specific off-the-shelf NK cell for treatment of multiple myeloma. Cancer Res.81, 1550–1550 (2021). [Google Scholar]
- 552.Li, Y. et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat. Med. 28, 2133–2144 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Gauthier, L. et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell177, 1701–1713.e1716 (2019). [DOI] [PubMed] [Google Scholar]
- 554.Kerbauy, L. N. et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30(+) malignancies. Clin. Cancer Res.27, 3744–3756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Fenis, A. et al. New immune cell engagers for cancer immunotherapy. Nat. Rev. Immunol. 24, 471-486(2024). [DOI] [PubMed]
- 556.Maskalenko, N. A., Zhigarev, D. & Campbell, K. S. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat. Rev. Drug Discov.21, 559–577 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Tang, X. et al. Erratum: First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res.8, 1899 (2018). [PMC free article] [PubMed] [Google Scholar]
- 558.Boyiadzis, M. et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy19, 1225–1232 (2017). [DOI] [PubMed] [Google Scholar]
- 559.Hong, C. S. et al. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci. Rep.7, 14684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Williams, B. A. et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget8, 89256–89268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Tam, Y. K., Miyagawa, B., Ho, V. C. & Klingemann, H. G. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J. Hematother.8, 281–290 (1999). [DOI] [PubMed] [Google Scholar]
- 562.Yan, Y. et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res.4, 2859–2868 (1998). [PubMed] [Google Scholar]
- 563.Southam, C. M. Homotransplantation of human cell lines. Bull. N. Y Acad. Med.34, 416–423 (1958). [PMC free article] [PubMed] [Google Scholar]
- 564.Edgren, G. et al. Risk of cancer after blood transfusion from donors with subclinical cancer: a retrospective cohort study. Lancet369, 1724–1730 (2007). [DOI] [PubMed] [Google Scholar]
- 565.Adami, J. et al. Blood transfusion and non-Hodgkin lymphoma: lack of association. Ann. Intern. Med.127, 365–371 (1997). [DOI] [PubMed] [Google Scholar]
- 566.Cerhan, J. R. et al. Blood transfusion history and risk of non-Hodgkin lymphoma: an InterLymph pooled analysis. Cancer Causes Control30, 889–900 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Grudzien, M. et al. A newly established canine NK-type cell line and its cytotoxic properties. Vet. Comp. Oncol.19, 567–577 (2021). [DOI] [PubMed] [Google Scholar]
- 568.Razmara, A. M. et al. Improved characterization and translation of NK cells for canine immunotherapy. Front. Vet. Sci.11, 1336158 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Cesano, A. et al. Phase I clinical trial with a human major histocompatibility complex nonrestricted cytotoxic T-cell line (TALL-104) in dogs with advanced tumors. Cancer Res.56, 3021–3029 (1996). [PubMed] [Google Scholar]
- 570.Wang, J. et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum. Gene Ther.18, 712–725 (2007). [DOI] [PubMed] [Google Scholar]
- 571.Altvater, B. et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res.15, 4857–4866 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Tassev, D. V., Cheng, M. & Cheung, N. K. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther.19, 84–100 (2012). [DOI] [PubMed] [Google Scholar]
- 573.Chmielewski, M., Kopecky, C., Hombach, A. A. & Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res.71, 5697–5706 (2011). [DOI] [PubMed] [Google Scholar]
- 574.Zhang, B. et al. Chimeric antigen receptor-based natural killer cell immunotherapy in cancer: from bench to bedside. Cell Death Dis.15, 50 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Wang, Z. et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-betaR II and NKG2D. Cancer Immunol. Immunother.66, 537–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Mohammed, S. et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther.25, 249–258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Chang, Y. H. et al. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res.73, 1777–1786 (2013). [DOI] [PubMed] [Google Scholar]
- 578.Muller, Y. D. et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front. Immunol.12, 639818 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Marin, D. et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat. Med.30, 772–784 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Klebanoff, C. A. et al. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol.26, 111–117 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Walle, T. et al. Radiotherapy orchestrates natural killer cell dependent antitumor immune responses through CXCL8. Sci. Adv.8, eabh4050 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Kagoya, Y. et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med.24, 352–359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Bachanova, V. et al. Safety and efficacy of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood138, 823–823 (2021). [Google Scholar]
- 584.Xiao, L. et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol. Ther.27, 1114–1125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Dolstra, H. et al. Successful transfer of umbilical cord blood CD34(+) hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin. Cancer Res.23, 4107–4118 (2017). [DOI] [PubMed] [Google Scholar]
- 586.Shimasaki, N., Coustan-Smith, E., Kamiya, T. & Campana, D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy18, 1422–1434 (2016). [DOI] [PubMed] [Google Scholar]
- 587.Williams, R. L. et al. Recipient T cell exhaustion and successful adoptive transfer of haploidentical natural killer cells. Biol. Blood Marrow Transpl.24, 618–622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Grzywacz, B. et al. Natural killer cell homing and persistence in the bone marrow after adoptive immunotherapy correlates with better leukemia control. J. Immunother.42, 65–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Torikai, H. et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood122, 1341–1349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Shimasaki, N., Jain, A. & Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov.19, 200–218 (2020). [DOI] [PubMed] [Google Scholar]
- 591.Cichocki, F. et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia30, 456–463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Sarhan, D. et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol. Res.6, 766–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Kershaw, M. H., Westwood, J. A. & Darcy, P. K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer13, 525–541 (2013). [DOI] [PubMed] [Google Scholar]
- 594.Met, O. et al. Principles of adoptive T cell therapy in cancer. Semin Immunopathol.41, 49–58 (2019). [DOI] [PubMed] [Google Scholar]
- 595.Parlar, A. et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur. J. Immunol.49, 1278–1290 (2019). [DOI] [PubMed] [Google Scholar]
- 596.Fagerberg, L. et al. Prediction of the human membrane proteome. Proteomics10, 1141–1149 (2010). [DOI] [PubMed] [Google Scholar]
- 597.Majzner, R. G. & Mackall, C. L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov.8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 598.June, C. H. & Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med.379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Jarisch, A. et al. Immune responses to SARS-CoV-2 vaccination in young patients with anti-CD19 chimeric antigen receptor T cell-induced B cell aplasia. Transpl. Cell Ther.28, 366.e361–366.e367 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Rosenberg, S. A. et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. a preliminary report. N. Engl. J. Med.319, 1676–1680 (1988). [DOI] [PubMed] [Google Scholar]
- 601.Wolfel, T. et al. Lysis of human melanoma cells by autologous cytolytic T cell clones. identification of human histocompatibility leukocyte antigen A2 as a restriction element for three different antigens. J. Exp. Med.170, 797–810 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Van, den & Eynde, B. et al. Presence on a human melanoma of multiple antigens recognized by autologous CTL. Int. J. Cancer44, 634–640 (1989). [DOI] [PubMed] [Google Scholar]
- 603.Lin, Y. & Okada, H. Cellular immunotherapy for malignant gliomas. Expert Opin. Biol. Ther.16, 1265–1275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Vacchelli, E. et al. Trial watch: adoptive cell transfer for anticancer immunotherapy. Oncoimmunology2, e24238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Vidal, S. M., Khakoo, S. I. & Biron, C. A. Natural killer cell responses during viral infections: flexibility and conditioning of innate immunity by experience. Curr. Opin. Virol.1, 497–512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Orr, M. T. & Lanier, L. L. Natural killer cell education and tolerance. Cell142, 847–856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Algarra, I., Cabrera, T. & Garrido, F. The HLA crossroad in tumor immunology. Hum. Immunol.61, 65–73 (2000). [DOI] [PubMed] [Google Scholar]
- 608.Kochan, G., Escors, D., Breckpot, K. & Guerrero-Setas, D. Role of non-classical MHC class I molecules in cancer immunosuppression. Oncoimmunology2, e26491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Lin, A. et al. HLA-G expression in human ovarian carcinoma counteracts NK cell function. Ann. Oncol.18, 1804–1809 (2007). [DOI] [PubMed] [Google Scholar]
- 610.Suto, A. et al. Development and characterization of IL-21-producing CD4+ T cells. J. Exp. Med.205, 1369–1379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Spolski, R., Li, P. & Leonard, W. J. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat. Rev. Immunol.18, 648–659 (2018). [DOI] [PubMed] [Google Scholar]
- 612.Cui, W. et al. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity35, 792–805 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Denman, C. J. et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE7, e30264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Chueh, F. Y. & Yu, C. L. Engagement of T-cell antigen receptor and CD4/CD8 co-receptors induces prolonged STAT activation through autocrine/paracrine stimulation in human primary T cells. Biochem. Biophys. Res. Commun.426, 242–246 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Bai, Y. et al. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discov.1, 15040 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Zhou, J. et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J. Immunol.175, 7046–7052 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Li, Q. et al. Visiting a forest, but not a city, increases human natural killer activity and expression of anti-cancer proteins. Int. J. Immunopathol. Pharm.21, 117–127 (2008). [DOI] [PubMed] [Google Scholar]
- 618.Li, Q. et al. A forest bathing trip increases human natural killer activity and expression of anti-cancer proteins in female subjects. J. Biol. Regul. Homeost. Agents22, 45–55 (2008). [PubMed] [Google Scholar]
- 619.Li, Q. et al. A day trip to a forest park increases human natural killer activity and the expression of anti-cancer proteins in male subjects. J. Biol. Regul. Homeost. Agents24, 157–165 (2010). [PubMed] [Google Scholar]
- 620.Li, Q. Effect of forest bathing trips on human immune function. Environ. Health Prev. Med.15, 9–17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Tsao, T. M. et al. Health effects of a forest environment on natural killer cells in humans: an observational pilot study. Oncotarget9, 16501–16511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Lyu, B. et al. Benefits of a three-day bamboo forest therapy session on the psychophysiology and immune system responses of male college students. Int. J. Environ. Res. Public Health16, 4991 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Li, Q. et al. Effect of phytoncide from trees on human natural killer cell function. Int. J. Immunopathol. Pharm.22, 951–959 (2009). [DOI] [PubMed] [Google Scholar]
- 624.Jo, H. et al. alpha-pinene enhances the anticancer activity of natural killer cells via ERK/AKT pathway. Int. J. Mol. Sci.22, 656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Liu, B. & Chen, H. Identification and functional characterization of the transcription factors AhR/ARNT in Dendroctonus Armandi. Cells11, 3856 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Smirnova, A. et al. Evidence for new light-independent pathways for generation of the endogenous aryl hydrocarbon receptor agonist FICZ. Chem. Res. Toxicol.29, 75–86 (2016). [DOI] [PubMed] [Google Scholar]
- 627.Shin, J. H. et al. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA110, 12391–12396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Helm, E. Y. & Zhou, L. Transcriptional regulation of innate lymphoid cells and T cells by aryl hydrocarbon receptor. Front. Immunol.14, 1056267 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Rannug, A. 6-formylindolo[3,2-b]carbazole, a potent ligand for the aryl hydrocarbon receptor produced both endogenously and by microorganisms, can either promote or restrain inflammatory responses. Front. Toxicol.4, 775010 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Shi, F. et al. Endogenous regulation of the Akt pathway by the aryl hydrocarbon receptor (AhR) in lung fibroblasts. Sci. Rep.11, 23189 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Shin, J. H. et al. AHR regulates NK cell migration via ASB2-mediated ubiquitination of filamin A. Front. Immunol.12, 624284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol.185, 3190–3198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Li, Q. et al. Effects of forest bathing (shinrin-yoku) on serotonin in serum, depressive symptoms and subjective sleep quality in middle-aged males. Environ. Health Prev. Med.27, 44 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Vivier, E. et al. Functions of natural killer cells. Nat. Immunol.9, 503–510 (2008). [DOI] [PubMed] [Google Scholar]
- 635.Boudaly, S. Activation of dendritic cells by polymorphonuclear neutrophils. Front. Biosci.14, 1589–1595 (2009). [DOI] [PubMed] [Google Scholar]
- 636.Harlin, H. et al. The CD16- CD56(bright) NK cell subset is resistant to reactive oxygen species produced by activated granulocytes and has higher antioxidative capacity than the CD16+ CD56(dim) subset. J. Immunol.179, 4513–4519 (2007). [DOI] [PubMed] [Google Scholar]
- 637.Romero, A. I., Thoren, F. B., Brune, M. & Hellstrand, K. NKp46 and NKG2D receptor expression in NK cells with CD56dim and CD56bright phenotype: regulation by histamine and reactive oxygen species. Br. J. Haematol.132, 91–98 (2006). [DOI] [PubMed] [Google Scholar]
- 638.Binstadt, B. A. et al. SLP-76 is a direct substrate of SHP-1 recruited to killer cell inhibitory receptors. J. Biol. Chem.273, 27518–27523 (1998). [DOI] [PubMed] [Google Scholar]
- 639.Purdy, A. K. & Campbell, K. S. SHP-2 expression negatively regulates NK cell function. J. Immunol.183, 7234–7243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Laskowski, T. J., Biederstadt, A. & Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer22, 557–575 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Micklethwaite, K. P. et al. Investigation of product-derived lymphoma following infusion of piggyBac-modified CD19 chimeric antigen receptor T cells. Blood138, 1391–1405 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Reighard, S. D. et al. Therapeutic targeting of follicular T cells with chimeric antigen receptor-expressing natural killer cells. Cell Rep. Med.1, 100003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]