

Abstract
Purpose of Review
The lack of adult human cardiomyocyte proliferative capacity impairs cardiac regeneration such as after myocardial injury. The sarcomere, a specialized actin cytoskeletal structure that is essential for twitch contraction in cardiomyocytes, has been considered a critical factor limiting adult human cardiomyocyte proliferation through incompletely understood mechanisms.
Recent Findings
This review summarizes known and emerging regulatory mechanisms connecting the human cardiomyocyte sarcomere to cell cycle regulation including structural and signaling mechanisms.
Summary
Cardiac regeneration could be augmented through targeting the inhibitory effects of the sarcomere on cardiomyocyte proliferation.
Keywords: Sarcomere, Cell cycle, Regeneration, Heart failure
Introduction
Heart failure (HF) is a rapidly growing cardiovascular condition with a prevalence of ~40 million individuals worldwide [1–3]. In the USA alone, HF affects over 6 million individuals and costs the healthcare system over $30 billion annually [4]. HF is a progressive condition that is frequently caused by cardiac injuries such as myocardial infarction, viral infection, and drug-related cardiotoxicity [5]. A shared consequence of many cardiac injuries is loss of cardiomyocyte number due to increased cell death, which promotes contractile dysfunction and maladaptive cardiac remodeling including cardiac chamber dilatation and fibrosis [6]. While in some contexts such as fetal cardiac development, it has been reported that cardiac regeneration can occur through cardiomyocyte proliferation (as defined by an increase in cell division and growth) [7], it is generally accepted to be insufficient in adult humans. The poor regenerative capacity of the adult human heart is also a consequence of the lack of a cardiac stem cell pool to replenish lost cardiomyocytes such that de novo cardiomyocyte generation must derive from proliferation of the existing cardiomyocyte pool [8]. While there are FDA-approved HF treatments such as adrenergic receptor blockers, neurohormonal antagonists, and implantable electrophysiological devices that have been shown to improve cardiac remodeling, HF symptoms, and even survival in human clinical studies [6], HF patients frequently develop advanced HF characterized by severe cardiac dysfunction that ultimately requires heart transplantation that is supply limited [5]. Therefore, the development of new HF therapeutics particularly those that target cardiac regeneration could be transformative.
Using development as a model to study cardiac regeneration (Figure 1), it has been observed that mammalian fetal cardiomyocytes utilize hyperplasia (as defined as cardiac growth by cell proliferation) for cardiac growth and in response to injury [8]; however, shortly after birth and distinct from other organisms such as the newt [9], cardiac growth and injury responses transition to cardiomyocyte hypertrophy (as defined by cardiac growth by cell enlargement) and fibrosis [10–12]. In parallel with the switch from hyperplasia to hypertrophy, adult mammalian cardiomyocytes also become progressively polyploid (defined by greater than 2 paired sets of chromosomes per cell) [13–17]. For example, the adult human cardiomyocyte is ~10% diploid, ~60% mononuclear polyploid, and ~30% multinuclear polyploid [12, 18]. While polyploidization can occur through several mechanisms [19], it is generally established that mammalian cardiomyocytes become polyploid through either endocycling (defined as genomic replication without mitosis) to generate mononuclear polyploid cells, or cytokinesis failure (defined as genomic replication and mitosis without cell division) to generate multinuclear cells [10]. Coincident with the shift from hyperplasia to hypertrophy and polyploidization, the mammalian adult heart also undergoes metabolic adaptations upregulating oxidative pathways [20] and biomechanical adaptations [21] including alterations in the expression of factors that sense and generate physical forces within the heart such as the force-producing sarcomere, a multiprotein machine evolved from the actin cytoskeleton that is responsible for cardiac pump function. Recent reviews of mammalian cardiac regeneration have focused on the therapeutic targeting of cardiac regeneration [22], biological relevance of polyploidy [10], and evolutionary perspectives [23], but the role of the sarcomere in cardiac regeneration has been less well summarized. Here, we address this gap in the literature by providing an overview of existing and emerging knowledge as well as potential future directions on the role of the cardiac sarcomere in mammalian cardiac regeneration focused on the cell cycle.
Fig. 1.
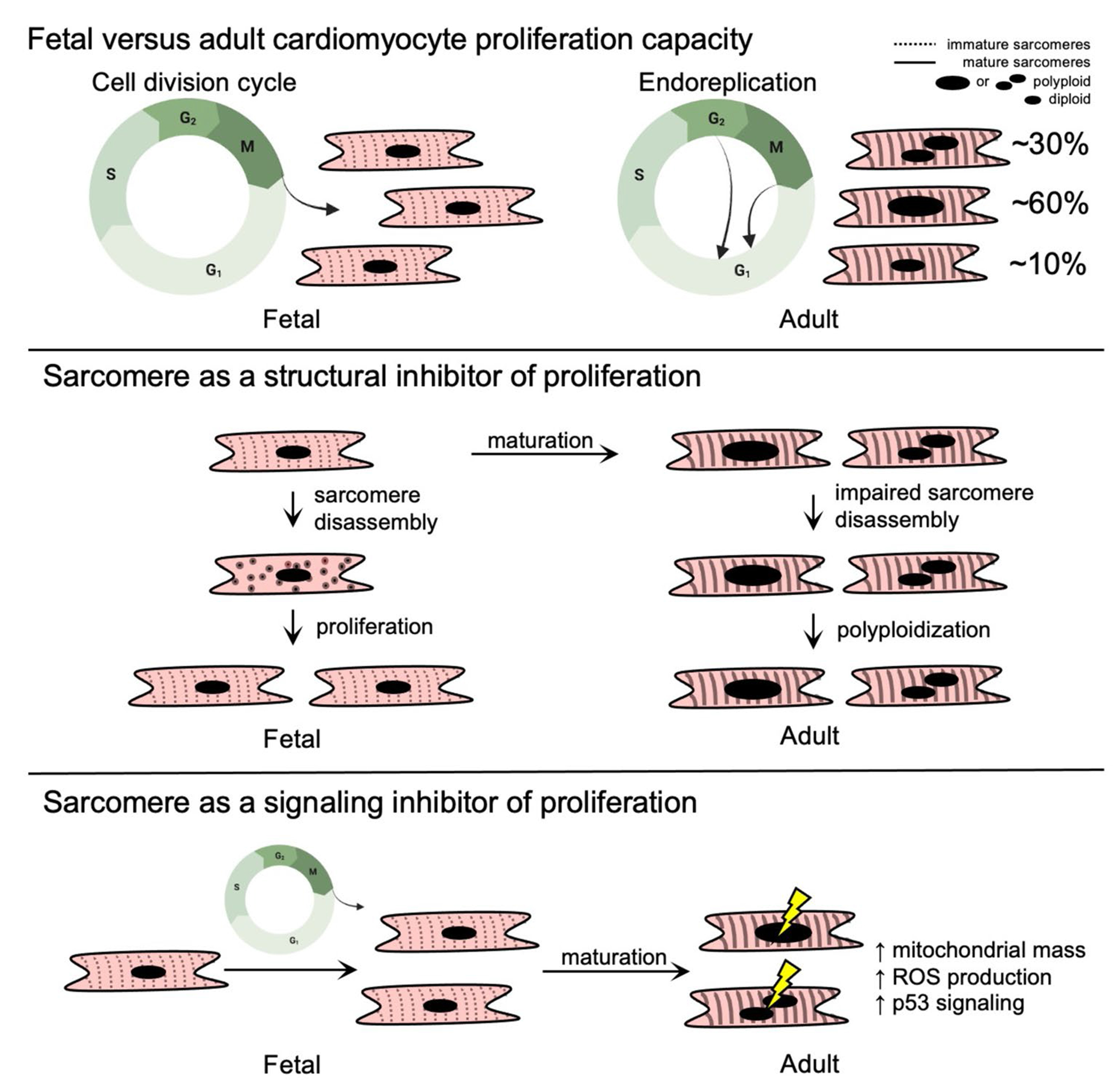
Fetal and adult human cardiomyocyte cell cycle patterns and sarcomere structure. While fetal human cardiomyocytes maintain their proliferative capacity as they progress through the stages of the cell cycle, adult cardiomyocytes lose their proliferative capacity and exit the cell cycle prior to completion of mitosis or cytokinesis (top panel). It has been proposed that the sarcomere inhibits proliferation because sarcomeres in the adult heart cannot be disassembled (middle panel). Alternative mechanisms may connect the sarcomere to cell cycle regulation such as contraction-induced metabolic and signaling alterations that promote cell cycle arrest (bottom panel)
Cardiac Sarcomere Structure
The sarcomere is the basic contractile unit of myocytes and is a unique specialization of the actin cytoskeleton found in higher organisms [24–26]. It is a cytosolic multiprotein complex that functions in mechanical twitch force production, as well as transcriptional regulation [27], cell signaling [28], and metabolic regulation [29]. The sarcomere is organized into parallel repeats of myosin-containing thick filaments, actin-containing thin filaments, and stabilizing titin filaments, all of which is laterally bounded by actinin-containing Z-disks that are crosslinked to the cytoskeleton by actinin. The area immediately surrounding the Z-disk lacks thick filament structures and is known as the I band, which is adjacent to a region that comprises the entire length of the thick filament known as the A band. Within the A band is a portion devoid of thin filament overlap called the H zone, containing the myomesin-rich M-line that corresponds to the center of the sarcomere [30–32]. In response to intracellular calcium, the troponin regulatory complex on the thin filament undergoes a series of conformational changes that expose myosin binding sites on actin, allowing the myosin head to bind actin and promote twitch contraction through an ATP-consuming power stroke that brings the Z-disk closer to the M-line, shortening the sarcomere [33]. When the sarcomere is not properly assembled, cardiac contractile dysfunction and heart failure can occur such as in the setting of sarcomere gene mutations [34–36]. Mechanosensory and signaling factors are also localized to multiple sarcomere sub-compartments including the Z-disk [37], titin filaments [38], and the M band [39]. The sarcomere also functions as a protein-protein interaction hub integrating mechanosensing with cell signaling pathways previously implicated in cell cycle regulation, in addition to a putative role in cell cycle inhibition through structural hindrance.
The Actin Cytoskeleton and Cell Cycle
Because the sarcomere is a specialized version of the actin cytoskeleton, it is important to first consider how the actin cytoskeleton functions in the cell cycle. Cell division is a complex process [40], which is highly regulated and commonly organized into five sequential cell cycle phases—G0, G1, S, G2, and M. The G0 phase is a quiescent or resting state, which is followed by a gap/growth 1 (G1) phase in which RNA and protein synthesis occur in preparation for the subsequent phases. The synthesis (S) phase is when the genome is duplicated, which includes DNA replication, histone synthesis, and nucleosome formation. This is followed by a G2 phase in which rapid cell growth and protein synthesis occur in preparation for the mitotic (M) phase, in which the nucleus divides and the cell undergoes subsequent division or cytokinesis [40]. The final process of cytokinesis involves a contractile network composed of actin filaments, non-muscle myosin II, and other accessory factors that physically divide the parental cell into two daughter cells [41]. There are a number of key checkpoints that exist within and between phase transitions [42, 43]. Among these, the G1/S checkpoint is well-established to regulate the commitment to the cell cycle and has numerous stimuli that exert control, while the G2/M checkpoint regulates commitment to mitosis that is largely controlled by DNA damage or incomplete DNA replication, ensuring complete DNA replication in preparation for cytokinesis. While numerous factors control the cell cycle phases and checkpoints, key regulators are the cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors (CDKIs) [42]. It is known that cyclin-CDK complexes control specific steps of the cell cycle, and their respective protein levels and activities are tightly controlled through coordinated mechanisms such as by interactions with CDKIs [44–46]. For example, the cyclin D-CDK4/CDK6 complex plays a role in the progression through G1, while cyclin E-CDK2 is important for the G1/S transition. For S phase progression, the cyclin A-CDK2 complex is required, followed by cyclin A-CDK1 and cyclin B-CDK1 as critical for the progression through G2 and M phases [47, 48].
The actin cytoskeleton undergoes dynamic structural and functional changes that are essential for normal cell cycle progression [49–51]. For example, disruption of actin assembly using the toxin dihydrocytochalasin B [52] activates a G1/S checkpoint through the retinoblastoma pathway involving inhibition of cycle E/CDK2 [53], while the actin polymerization inhibitor cytochalasin D delays mitosis [54, 55]. The actin cytoskeleton also plays a role in cellular rounding and cortical stiffening to prepare for spindle assembly [56], as well as centrosome separation that is essential for mitosis as demonstrated by elegant studies using the actin polymerization toxin latrunculin [57, 58]. Additionally, the actin-binding protein, cortactin, has been found to be a key anchor between actin and the centrosome and is important in actin-facilitated centrosome separation during mitosis [59]. Cortactin is also a substrate of CDK1, which directly links this kinase to cortactin-mediated centrosome separation during mitosis [60]. In cytokinesis, the actin cytoskeleton plays a well-established role in contractile ring assembly at the cleavage furrow. Through force-generating interactions with myosin II, the cleavage furrow ingresses until a midbody structure is formed between daughter cells, after which abscission results in midbody cleavage and separation of the two daughter cells. This process requires actin filament disassembly in the cleavage furrow through the PKCε-14-3-3 complex and RhoA inactivation [61]. Moreover, cell cycle factors can also directly regulate the actin cytoskeleton such as has been shown for CDK1 that can directly phosphorylate the actin crosslinking protein filamin A at serine residues 1084, 1459, and 1533, which is essential for postmitotic daughter cell separation and migration [62]. It is important to note that most of these studies implicating the role of the actin cytoskeleton in cell cycle regulation utilize transformed cell lines and yeast models, and while these are powerful models to study these processes, their generalizability to cardiomyocyte cell cycle regulation may be limited. For example, unlike classical cell cycle model systems such as transformed cell lines, mammalian cardiomyocytes become progressively polyploid through incomplete cell cycle progression by incompletely understood mechanisms [10]. In summary, actin cytoskeletal functions are critical for general cell cycle progression particularly for mitosis and cytokinesis, but have not been sufficiently studied in mammalian cardiomyocyte models that proceed through complex cell cycle patterns.
Sarcomere Structure and Cell Cycle
Relative to adults, fetal mammalian cardiomyocytes that proliferate contain less organized and less abundant sarcomeres that are composed of distinct sarcomere gene and splice isoforms including fetal-enriched myosin heavy chain (MYH6 in humans), troponin I (TNNI1), and titin splice isoforms (TTN N2BA) [63]. With development, cardiomyocyte sarcomere content and longitudinal alignment both progressively increase (Figure 1) with a concomitant transition to adult-enriched sarcomere genes and splice isoforms including MYH7, TNNI3, and TTN N2B. It remains incompletely understood how the developmental transitions in sarcomere structure and gene expression contribute to the proliferative arrest observed in adult mammalian cardiomyocytes, which are considered essential to maintain normal cardiac contractile function in the adult mammalian heart.
During their proliferative phase, it has been observed that fetal mammalian cardiomyocytes in vivo undergo sarcomere disassembly prior to cytokinesis followed by reassembly in daughter cells [64]. This appears to be a coordinated process that involves marginalization of sarcomere structures to the cellular periphery during mitosis and cytokinesis [8, 65], which has been observed to occur by two sequential steps. The first step involves a collapse of the Z-disk and thin filament-associated proteins, followed by disassembly of titin, M band, and thick filament components including myosin heavy chain [64, 66, 67]. Hallmarks of this pattern of sarcomere disassembly have been observed in a number of studies investigating a broad scope of regulators of cardiomyocyte proliferation including overexpression of neuregulin1 (Nrg1) and its receptors (Erbb2 and Erbb4) [65, 68]; surgical resection of the left ventricular apex [8]; Meis1 gene knockout and Meis1-Hoxb13 double knockouts [69, 70•]; and treatment with the cyclin B1-CDK1 inhibitor Ro3306 [67]. In contrast, postnatal cardiomyocytes that express cell cycle markers but do not complete sarcomere disassembly have been observed to become multinucleated [71, 72]. Taken together, these studies provide evidence that sarcomere disassembly is a highly ordered process that is necessary for cell cycle completion, and the adult sarcomere may structurally impede cell cycle progression to promote cardiomyocyte polyploidization [73].
Regulators of sarcomere disassembly during cell cycle progression are not completely understood. This is in part due to the lack of robust mammalian cardiomyocyte model systems in which to study sarcomere disassembly through the cell cycle. In contrast, regulators of sarcomere assembly and maintenance have been better studied, such as members of the muscle chaperone [74] and E1-E3 ubiquitin proteasome systems [75]. For example, Hspb7 is a cardiac enriched heat shock protein that is essential for thin filament assembly in mice [76]. Disruption of the cardiomyocyte ubiquitin system also impairs sarcomere assembly as double knockout of E3 ubiquitin ligases MuRF1 and MuRF3 develops cardiac hypertrophy in vivo in association with accumulation of myosin heavy chains in the subsarcolemmal space [75]. While these studies have revealed factors regulating general sarcomere assembly and maintenance, how the sarcomere is regulated through the cell cycle remains largely unknown particularly the identity of the upstream regulatory factors as well as their sarcomere targets.
Sarcomere disassembly can be stimulated through activation of dedifferentiation programs that reprogram the sarcomere to its fetal-like structure, abundance, and gene expression pattern among other alterations in the state of the cardiomyocyte. For example, inhibition of miR-15 members or oncostatin M can induce dedifferentiation resembling a fetal-like cardiomyocyte state [77, 78], which results in enhanced capacity for proliferation and sarcomere disassembly. The dedifferentiated state also includes reduction in sarcomere content, re-expression of fetal-like sarcomere isoforms [78], upregulation of cell cycle promoting factors [79, 80], and downregulation of cell cycle checkpoint regulators including p21 and p53 [81]. Another method that has been demonstrated to activate a dedifferentiation program associated with enhanced cardiomyocyte proliferative capacity is transient expression of the Yamanaka reprogramming factors Oct3/4, Sox2, Klf4, and c-Myc [82], which has also been shown to improve cardiac stress responses following myocardial damage [83•]. Taken together, these dedifferentiation studies highlight the reversibility of adult cardiomyocyte cell cycle exit and sarcomere disassembly deficits as well as their intimate relationship with cellular differentiation state.
In addition to dedifferentiation, adult cardiomyocyte proliferation can also be stimulated using enhanced expression of cell cycle activators, which suggests both that the sarcomere may not be an irreversible barrier to cardiomyocyte proliferation and that cell cycle activators may be regulators of sarcomere disassembly. Intriguingly, CDK1 has been show to phosphorylate the actin crosslinking protein filamin A at residues that are highly conserved to other filamins including filamin C that is a major sarcomere component [62]. Disruption of filamin A phosphorylation at CDK1 sites impairs post-mitotic daughter cell separation and cell migration in non-cardiomyocytes [84], but this process has not been studied in cardiomyocytes or filamins enriched in the sarcomere. Coincident with the loss of proliferative capacity of adult cardiomyocytes, cell cycle activators are downregulated including cyclins and CDKs [85–87•]. To test the functional impact of restoration of candidate cell cycle activators, rodent studies have evaluated the role of enhanced cyclin A2, cyclin B1, CDK1, cyclin D1, cyclin D2, and CDK4, among others [85, 87•–92]. With enhancement of a single activation factor, these studies have reported limited and variable proliferative rates including the promotion of polyploidization. For example, transgenic overexpression of cardiomyocyte G1/S-specific cyclins including cyclin D1 [91] and cyclin D2 [92] promotes polyploidization. In addition to enhancing a single cell cycle activator, recent studies have assessed the combinatorial overexpression of cyclin B1, CDK1, cyclin D1, and CDK4 that can induce adult cardiomyocyte cell cycle reactivation [85]. This combination of factors was observed to promote higher mitosis marker expression (histone H3 phosphorylation) relative to single factors in vitro. Interestingly, cyclin B1, CDK1, cyclin D1, and CDK4 overexpression did not promote cardiomyocyte multinucleation, though ploidy was not measured [85].
In association with the loss of cell cycle activator expression, adult mammalian cardiomyocytes also increase expression and activity for cell cycle inhibitors such as the CDKIs including p21 and p27 [93, 94] and tumor suppressors such as p53 and Rb [95]. While knockout of these factors such as p27 [94] and double knockout of p27 and p21 [96] can prolong the rodent cardiomyocyte proliferative window, targeting these repressive pathways exclusively in adult cardiomyocytes towards promoting cardiac regeneration has not been well studied, particularly how and whether these cell cycle inhibitory factors regulate sarcomere structure and function. In summary, while modulation of mammalian cardiomyocyte cell cycle regulators has had variable success in enhancing rodent cardiac regeneration, it remains incompletely known how these pathways converge on the regulation of the sarcomere particularly in the adult cardiomyocyte.
Emerging Human Cardiomyocyte Models to Study the Sarcomere and Cell Cycle
To study how the sarcomere regulates the cell cycle, human cardiomyocyte models that lack sarcomeres have been recently developed [36]. While in vivo rodent models that lack cardiac sarcomeres are embryonic lethal, in vitro models are viable such as human fetal-like cardiomyocytes (iCMs) differentiated from pluripotent stem cells using robust direct differentiation methods [97]. While iCMs may not fully recapitulate in vivo biology, iCMs have been observed to undergo differentiation time-dependent proliferative arrest and polyploidization resembling in vivo human cardiomyocyte patterns (Figure 2) [10]. After producing iCMs that lack cardiac sarcomeres using genetic knockout of cardiac troponin, enhanced cell cycle marker activation and reduced polyploidization rates could be observed [87•]. When sarcomere assembly-deficient iCMs were transplanted into myocardial infarction rodent models, graft size was increased ~4× relative to wildtype iCMs, which was related to enhanced iCM proliferation rates supporting an inhibitory role for the sarcomere. Moreover, sarcomere assembly-deficient iCMs did not result in a highly proliferative state 3-month post-engraftment as only 0.50% of iCMs demonstrated expression of the cell cycle marker Ki67 relative to 0.14% in wildtype iCMs. These proliferation levels were much lower than what was observed prior to transplantation. Also, no tumor-like intramyocardial growths were observed in grafts, confirming that additional mechanisms beyond sarcomere assembly may be responsible for promoting proliferative arrest and polyploidization in human cardiomyocytes. Recently, one such mechanism identified was cell-cell contact, which was shown in iCMs to promote inhibition of canonical Wnt signaling and promote early cell cycle exit [98]. Reducing cell-cell contact in 2D tissue culture via sparse seeding of iCMs suppressed maturation and stimulated cell cycle activation, promoting massive cellular proliferation without subsequent contractility deficits once assembled into 3D engineered heart tissues. Going forward, the iCM model system could be a powerful new tool to assist with the identification of additional mechanisms of human cardiomyocyte cell cycle control such as by the sarcomere and beyond.
Fig. 2.
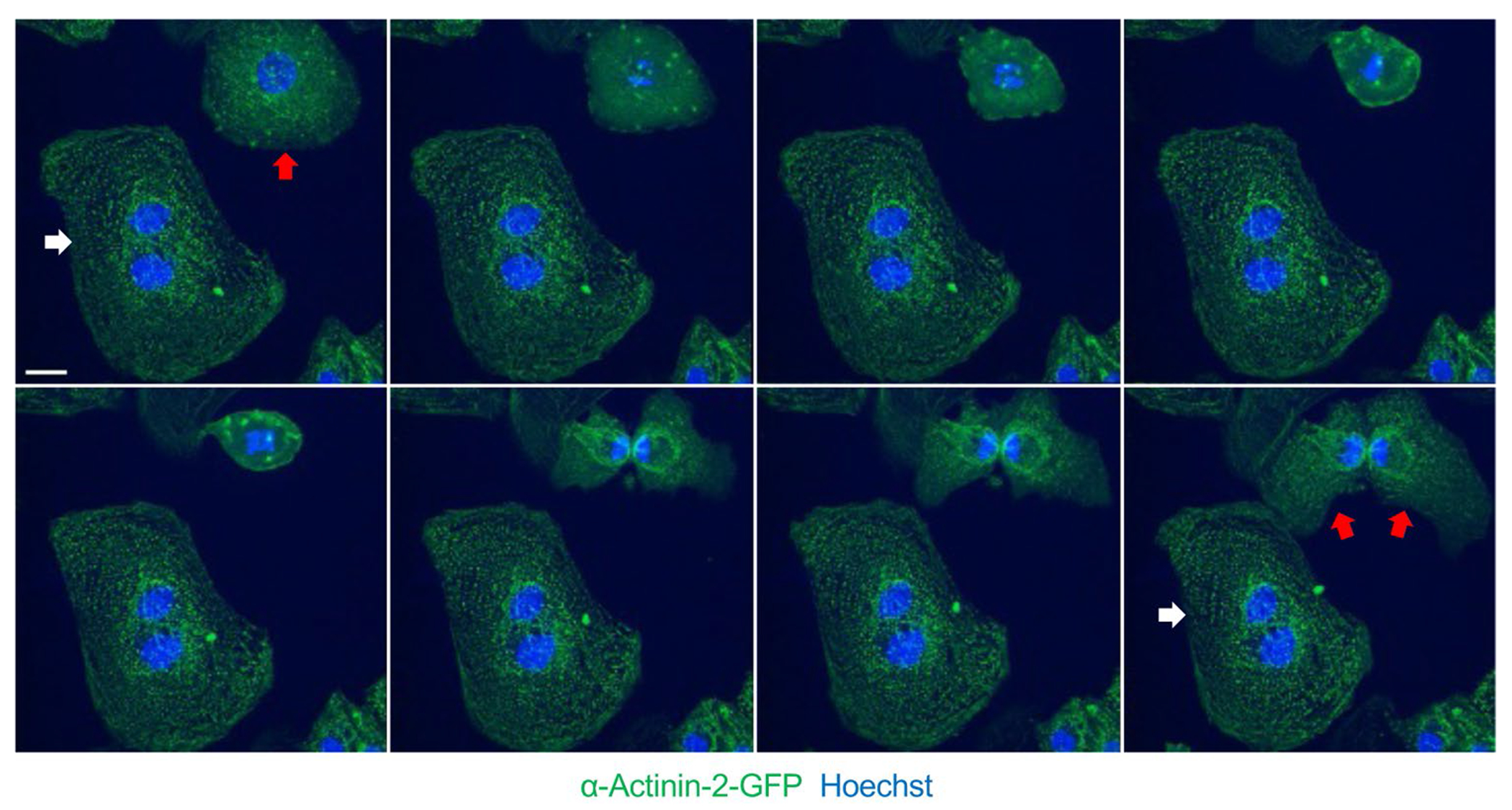
Human iPS–derived cardiomyocyte (iCM) mitosis and cytokinesis visualized using time-lapse live cell confocal imaging (micrographs at ~15-min intervals). Sarcomere Z-disk structures as demonstrated by alpha actinin 2-GFP signal disassemble during early stages of mitosis and reassemble after mitosis. White arrow denotes multinuclear iCM, while red arrow denotes a mononuclear iCM (top left image) that successfully completes cytokinesis (bottom right image). Scale bar = 10 μ
ICM models have been recently engineered to harbor cell cycle reporters such as by fusing green fluorescent protein (GFP) to cyclin B1 within the endogenous genomic locus using CRISPR technology [99]. As cyclin B1 is expressed in S, G2, and M phases, GFP expression status can provide a live cell readout on iCM cell cycle status. Using this cyclin B1-GFP iCM model combined with a genome-wide CRISPR knockout library, genetic levers of iCM proliferation and polyploidization have been studied [87•]. Among hits, the well-studied tumor suppressor p53 was found to be a strong activator of polyploidization and inhibitor of iCM proliferation, which was found to also be dependent on sarcomere function through a p53-dependent DNA damage response [87•]. Intriguingly, sarcomere gene mutations that activate contractile function such as in hypertrophic cardiomyopathy individuals have also been shown to induce a DNA damage response [100]. Taken together, these emerging studies implicate sarcomere function as regulatory factor promoting proliferative arrest and polyploidization through a DNA damage response involving p53. Future studies utilizing iCM models could reveal additional mechanisms connecting the sarcomere to cardiomyocyte cell cycle regulation.
Conclusion
The sarcomere is an essential multiprotein machine that generates the pumping force necessary for the heart to deliver oxygen and nutrients to maintain organismal viability and functions. It is not surprising that this complex structure interfaces with nearly all functions of the cardiomyocyte including the cell cycle. It has been postulated that adult mammalian cardiomyocytes do not proliferate because sarcomere disassembly would promote HF and death. Yet, emerging studies particularly in rodent models have demonstrated that reactivation of cardiomyocyte proliferative capacity can be protective against cardiac injuries such as myocardial infarctions [85]. Future studies particularly utilizing in vitro human iCM and in vivo murine models to delineate new levers to promote cardiac regeneration such as by targeting the sarcomere could be transformative for not only HF patients, but other conditions characterized by insufficient cardiomyocyte numbers such as congenital heart disorders like hypoplastic left heart syndrome [101].
Funding
J.T.H. is supported by the National Institutes of Health (HL142787 and EB028898). A.M.P. and F.A.L. are supported by the American Heart Association (PRE34381021 and PRE35110005).
Footnotes
This article is part of the Topical Collection on Regenerative Medicine
Conflict of Interest The authors declare no competing interests.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):368–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roth GA, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol. ‘2017;70(1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moran AE, et al. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: the Global Burden of Disease 2010 study. Circulation. 2014;129(14):1483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Virani SS, et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143(8):e254–743. [DOI] [PubMed] [Google Scholar]
- 5.Yancy CW, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):1810–52. [DOI] [PubMed] [Google Scholar]
- 6.Metra M, Teerlink JR. Heart failure. Lancet. 2017;390(10106):1981–95. [DOI] [PubMed] [Google Scholar]
- 7.Patterson M, et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet. 2017;49(9):1346–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porrello ER, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187(2):249–53. [DOI] [PubMed] [Google Scholar]
- 10.Derks W, Bergmann O. Polyploidy in Cardiomyocytes: Road-block to Heart Regeneration? Circ Res. 2020;126(4):552–65. [DOI] [PubMed] [Google Scholar]
- 11.Alkass K, et al. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell. 2015;163(4):1026–36. [DOI] [PubMed] [Google Scholar]
- 12.Bergmann O, et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell. 2015;161(7):1566–75. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Rosa JM, Burns CE, Burns CG. Zebrafish heart regeneration: 15 years of discoveries. Regeneration (Oxf). 2017;4(3):105–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bettencourt-Dias M, Mittnacht S, Brockes JP. Heterogeneous proliferative potential in regenerative adult newt cardiomyocytes. J Cell Sci. 2003;116(Pt 19):4001–9. [DOI] [PubMed] [Google Scholar]
- 15.Hirose K, et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science. 2019;364(6436):184–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298(5601):2188–90. [DOI] [PubMed] [Google Scholar]
- 17.Becker RO, Chapin S, Sherry R. Regeneration of the ventricular myocardium in amphibians. Nature. 1974;248(5444):145–7. [DOI] [PubMed] [Google Scholar]
- 18.Mollova M, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110(4):1446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ovrebo JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development. 2018;145(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puente BN, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157(3):565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Granados-Riveron JT, Brook JD. The impact of mechanical forces in heart morphogenesis. Circ Cardiovasc Genet. 2012;5(1):132–42. [DOI] [PubMed] [Google Scholar]
- 22.Sadek H, Olson EN. Toward the Goal of Human Heart Regeneration. Cell Stem Cell. 2020;26(1):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vivien CJ, Hudson JE, Porrello ER. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen Med. 2016;1:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gautel M, Djinovic-Carugo K. The sarcomeric cytoskeleton: from molecules to motion. J Exp Biol. 2016;219(Pt 2):135–45. [DOI] [PubMed] [Google Scholar]
- 25.Sequeira V, et al. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta. 2014;1838(2):700–22. [DOI] [PubMed] [Google Scholar]
- 26.Virel A, Backman L. Molecular evolution and structure of alpha-actinin. Mol Biol Evol. 2004;21(6):1024–31. [DOI] [PubMed] [Google Scholar]
- 27.Heineke J, et al. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci U S A. 2005;102(5):1655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheikh F, et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest. 2008;118(12):3870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ladha FA, et al. Actinin BioID reveals sarcomere crosstalk with oxidative metabolism through interactions with IGF2BP2. Cell Rep. 2021;36(6):109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henderson CA, et al. Overview of the Muscle Cytoskeleton. Compr Physiol. 2017;7(3):891–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woodcock EA, Matkovich SJ. Cardiomyocytes structure, function and associated pathologies. Int J Biochem Cell Biol. 2005;37(9):1746–51. [DOI] [PubMed] [Google Scholar]
- 32.Knoll R, Buyandelger B, Lab M. The sarcomeric Z-disc and Z-discopathies. J Biomed Biotechnol. 2011;2011:569628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda). 2007;22:73–80. [DOI] [PubMed] [Google Scholar]
- 34.Hinson JT, et al. Heart disease. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349(6251):982–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts AM, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7(270):270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettinato AM, et al. Development of a Cardiac Sarcomere Functional Genomics Platform to Enable Scalable Interrogation of Human TNNT2 Variants. Circulation. 2020;142(23):2262–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luther PK. The vertebrate muscle Z-disc: sarcomere anchor for structure and signalling. J Muscle Res Cell Motil. 2009;30(5–6):171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayans O, et al. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395(6705):863–9. [DOI] [PubMed] [Google Scholar]
- 39.Li HL, et al. Overexpression of myofibrillogenesis regulator-1 aggravates cardiac hypertrophy induced by angiotensin II in mice. Hypertension. 2007;49(6):1399–408. [DOI] [PubMed] [Google Scholar]
- 40.Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000;100(1):71–8. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen LTS, Robinson DN. The Unusual Suspects in Cytokinesis: Fitting the Pieces Together. Front Cell Dev Biol. 2020;8:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barnum KJ, O’Connell MJ. Cell cycle regulation by checkpoints. Methods Mol Biol. 2014;1170:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst). 2004;3(8–9):997–1007. [DOI] [PubMed] [Google Scholar]
- 44.Gerard C, Goldbeter A. From quiescence to proliferation: Cdk oscillations drive the mammalian cell cycle. Front Physiol. 2012;3:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan DO. Principles of CDK regulation. Nature. 1995;374(6518):131–4. [DOI] [PubMed] [Google Scholar]
- 46.Murray AW, Kirschner MW. Dominoes and clocks: the union of two views of the cell cycle. Science. 1989;246(4930):614–21. [DOI] [PubMed] [Google Scholar]
- 47.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925–39. [DOI] [PubMed] [Google Scholar]
- 48.Koff A, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257(5077):1689–94. [DOI] [PubMed] [Google Scholar]
- 49.Thery M, et al. The extracellular matrix guides the orientation of the cell division axis. Nat Cell Biol. 2005;7(10):947–53. [DOI] [PubMed] [Google Scholar]
- 50.Pollard TD, O’Shaughnessy B. Molecular Mechanism of Cytokinesis. Annu Rev Biochem. 2019;88:661–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones MC, Zha J, Humphries MJ. Connections between the cell cycle, cell adhesion and the cytoskeleton. Philos Trans R Soc Lond B Biol Sci. 2019;374(1779):20180227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin DC, et al. Cytochalasins inhibit nuclei-induced actin polymerization by blocking filament elongation. J Cell Biol. 1980;84(2):455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reshetnikova G, et al. Disruption of the actin cytoskeleton leads to inhibition of mitogen-induced cyclin E expression, Cdk2 phosphorylation, and nuclear accumulation of the retinoblastoma protein-related p107 protein. Exp Cell Res. 2000;259(1):35–53. [DOI] [PubMed] [Google Scholar]
- 54.Lee K, Song K. Actin dysfunction activates ERK1/2 and delays entry into mitosis in mammalian cells. Cell Cycle. 2007;6(12):1487–95. [PubMed] [Google Scholar]
- 55.Gachet Y, et al. A MAP kinase-dependent actin checkpoint ensures proper spindle orientation in fission yeast. Nature. 2001;412(6844):352–5. [DOI] [PubMed] [Google Scholar]
- 56.Uzbekov R, Kireyev I, Prigent C. Centrosome separation: respective role of microtubules and actin filaments. Biol Cell. 2002;94(4–5):275–88. [DOI] [PubMed] [Google Scholar]
- 57.Rosenblatt J, et al. Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly. Cell. 2004;117(3):361–72. [DOI] [PubMed] [Google Scholar]
- 58.Coue M, et al. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213(2):316–8. [DOI] [PubMed] [Google Scholar]
- 59.Blethrow JD, et al. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci U S A. 2008;105(5):1442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang W, et al. Centrosome separation driven by actin-microfilaments during mitosis is mediated by centrosome-associated tyrosine-phosphorylated cortactin. J Cell Sci. 2008;121(Pt 8):1334–43. [DOI] [PubMed] [Google Scholar]
- 61.Saurin AT, et al. The regulated assembly of a PKCepsilon complex controls the completion of cytokinesis. Nat Cell Biol. 2008;10(8):891–901. [DOI] [PubMed] [Google Scholar]
- 62.Cukier IH, Li Y, Lee JM. Cyclin B1/Cdk1 binds and phosphorylates Filamin A and regulates its ability to cross-link actin. FEBS Lett. 2007;581(8):1661–72. [DOI] [PubMed] [Google Scholar]
- 63.Guo Y, Pu WT. Cardiomyocyte Maturation: New Phase in Development. Circ Res. 2020;126(8):1086–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahuja P, et al. Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J Cell Sci. 2004;117(Pt 15):3295–306. [DOI] [PubMed] [Google Scholar]
- 65.Bersell K, et al. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138(2):257–70. [DOI] [PubMed] [Google Scholar]
- 66.Engel FB, Schebesta M, Keating MT. Anillin localization defect in cardiomyocyte binucleation. J Mol Cell Cardiol. 2006;41(4):601–12. [DOI] [PubMed] [Google Scholar]
- 67.Fan X, et al. Dynamic Alterations to alpha-Actinin Accompanying Sarcomere Disassembly and Reassembly during Cardiomyocyte Mitosis. PLoS One. 2015;10(6):e0129176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.D’Uva G, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17(5):627–38. [DOI] [PubMed] [Google Scholar]
- 69.Mahmoud AI, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497(7448):249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.•.Nguyen NUN, et al. A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature. 2020;582(7811):271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; Findings from this study implicate a previously uncrecognized signaling axis that regulates mouse cardiomyocyte replication.
- 71.Li F, Wang X, Gerdes AM. Formation of binucleated cardiac myocytes in rat heart: II Cytoskeletal organisation. J Mol Cell Cardiol. 1997;29(6):1553–65. [DOI] [PubMed] [Google Scholar]
- 72.Li F, et al. Formation of binucleated cardiac myocytes in rat heart: I. Role of actin-myosin contractile ring. J Mol Cell Cardiol. 1997;29(6):1541–51. [DOI] [PubMed] [Google Scholar]
- 73.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87(2):521–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith DA, et al. Getting folded: chaperone proteins in muscle development, maintenance and disease. Anat Rec (Hoboken). 2014;297(9):1637–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fielitz J, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest. 2007;117(9):2486–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu T, et al. HSPB7 is indispensable for heart development by modulating actin filament assembly. Proc Natl Acad Sci U S A. 2017;114(45):11956–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Porrello ER, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110(1):187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kubin T, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011;9(5):420–32. [DOI] [PubMed] [Google Scholar]
- 79.O’Meara CC, et al. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ Res. 2015;116(5):804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li G, et al. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development. 2019;146(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y, et al. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS One. 2010;5(9):e12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. [DOI] [PubMed] [Google Scholar]
- 83.•.Chen Y, et al. Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science. 2021;373(6562):1537–40. [DOI] [PubMed] [Google Scholar]; Findings from this study implicate reversibility of adult mouse cardiomyocyte cell cycle arrest by transient activation of a dedifferentiation program using four transcription factors.
- 84.Szeto SGY, et al. Phosphorylation of filamin A by Cdk1 regulates filamin A localization and daughter cell separation. Exp Cell Res. 2015;330(2):248–66. [DOI] [PubMed] [Google Scholar]
- 85.Mohamed TMA, et al. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell. 2018;173(1):104–116 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gilsbach R, et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun. 2018;9(1):391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.•.Pettinato AM, et al. Sarcomere function activates a p53-dependent DNA damage response that promotes polyploidization and limits in vivo cell engraftment. Cell Rep. 2021;35(5):109088. [DOI] [PMC free article] [PubMed] [Google Scholar]; Findings from this study characterize a human cardiomyocyte model system to study human cardiomyocyte replication and polyploidization mechanisms.
- 88.Chaudhry HW, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem. 2004;279(34):35858–66. [DOI] [PubMed] [Google Scholar]
- 89.Shapiro SD, et al. Cyclin A2 induces cardiac regeneration after myocardial infarction through cytokinesis of adult cardiomyocytes. Sci Transl Med. 2014;6(224):224ra27. [DOI] [PubMed] [Google Scholar]
- 90.Bicknell KA, Coxon CH, Brooks G. Forced expression of the cyclin B1-CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem J. 2004;382(Pt 2):411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soonpaa MH, et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99(11):2644–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pasumarthi KB, et al. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96(1):110–8. [DOI] [PubMed] [Google Scholar]
- 93.Koh KN, et al. Persistent and heterogenous expression of the cyclin-dependent kinase inhibitor, p27KIP1, in rat hearts during development. J Mol Cell Cardiol. 1998;30(3):463–74. [DOI] [PubMed] [Google Scholar]
- 94.Poolman RA, et al. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85(2):117–27. [DOI] [PubMed] [Google Scholar]
- 95.Flink IL, et al. Changes in E2F complexes containing retinoblastoma protein family members and increased cyclin-dependent kinase inhibitor activities during terminal differentiation of cardiomyocytes. J Mol Cell Cardiol. 1998;30(3):563–78. [DOI] [PubMed] [Google Scholar]
- 96.Tane S, et al. CDK inhibitors, p21(Cip1) and p27(Kip1), participate in cell cycle exit of mammalian cardiomyocytes. Biochem Biophys Res Commun. 2014;443(3):1105–9. [DOI] [PubMed] [Google Scholar]
- 97.Lian X, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buikema JW, et al. Wnt Activation and Reduced Cell-Cell Contact Synergistically Induce Massive Expansion of Functional Human iPSC-Derived Cardiomyocytes. Cell Stem Cell. 2020;27(1):50–63 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koch B, et al. Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing. Nat Protoc. 2018;13(6):1465–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sharifi-Sanjani M, et al. Cardiomyocyte-Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. J Am Heart Assoc. 2017;6(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bittle GJ, et al. Stem Cell Therapy for Hypoplastic Left Heart Syndrome: Mechanism, Clinical Application, and Future Directions. Circ Res. 2018;123(2):288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]