

Abstract

Aqueous metal corrosion is a major economic concern in modern society. A phenomenon that has puzzled generations of scientists in this field is the so-called anomalous hydrogen evolution: the violent dissolution of magnesium under electron-deficient (anodic) conditions, accompanied by strong hydrogen evolution and a key mechanism hampering Mg technology. Experimental studies have indicated the presence of univalent Mg+ in solution, but these findings have been largely ignored because they defy our common chemical understanding and evaded direct experimental observation. Using recent advances in the ab initio description of solid–liquid electrochemical interfaces under controlled potential conditions, we describe the full reaction path of Mg atom dissolution from a kinked Mg surface under anodic conditions. Our study reveals the formation of a solvated [Mg2+(OH)−]+ ion complex, challenging the conventional assumption of Mg2+ ion formation. This insight provides an intuitive explanation for the postulated presence of (Coulombically) univalent Mg+ ions, and the absence of protective oxide/hydroxide layers normally formed under anodic/oxidizing conditions. The discovery of this unexpected and unconventional reaction mechanism is crucial for identifying new strategies for corrosion prevention and can be transferred to other metals.
Controlling material degradation in chemically harsh environments is an outstanding challenge for future sustainable technologies. Examples are electrochemical energy conversion and storage solutions,1−6 green metallurgy,7,8 and lightweight structural materials.9,10 The need to understand the fundamental corrosion mechanisms in such environments is highlighted by a number of deceptively simple, yet poorly understood degradation reactions such as the anomalous dissolution of metals under anodic conditions; their precise mechanistic details have remained elusive since their discovery more than 150 years ago.11
Magnesium is a prototypical example.12 Mg alloys are attractive materials for mechanical engineering or batteries,13 due to their lightweight, high-abundance, and low environmental impact. However, with Mg being one of the most reactive metals, a major technical weakness is its corrosion when it comes into contact with water. Many of the properties of magnesium in water are puzzling and not understood. For example, under anodic polarization, where magnesium dissolves, it simultaneously shows extreme rates of hydrogen evolution (HE), which would normally be expected exclusively for cathodic potentials. According to the Butler–Volmer equation, HE should decrease exponentially when increasing the potential toward the anodic direction. In marked contrast, however, magnesium and its alloys feature strongly enhanced HE with increasing anodic polarization. This effect is referred to in the literature as the “negative difference effect” or “anomalous hydrogen evolution reaction”.10,11,14 Multiple models have been proposed15,16 to explain the origin of the anomalous hydrogen evolution. The “enhanced catalytic activity mechanism”17 suggested the enrichment of impurities more noble than Mg or the formation of local active sites to catalyze the HE on the anode. However, the specific nature of the catalyst that drives the anodic HE has remained unknown.17,18
Even more puzzling, the amount of Mg dissolved was observed to be greater than coulometrically expected, assuming that Mg was oxidized to the dipositive Mg2+ ion,19 cf. Figure 1a. These findings were taken as evidence for the existence of a “unipositive Mg+ ion mechanism”.20 In a series of follow-up experiments, it was demonstrated that the unipositive Mg ions have a lifetime of several minutes in aqueous solutions and are able to reduce other species even macroscopic distances away from the oxidizing Mg anode.19,21 On the other hand, the unipositive Mg+ mechanism has been challenged on the grounds that such an ion should be extremely short-lived. So far, there is only indirect evidence for the existence of Mg+.10 Atomic emission spectroelectrochemical experiments,22,23 which clearly distinguish different oxidation states, found direct evidence only for the divalent Mg2+. Yet, this anomalous dissolution behavior is not limited to Mg, but has been observed, e.g., for Fe, Cr, and Zn as well.24 Despite their fundamental importance, however, the existence and exact chemical nature of the postulated unipositive metal ions as well as the precise atomistic reaction mechanisms responsible for anomalous dissolution have remained elusive.
Figure 1.

(a) Conventional picture of Mg corrosion: dissolution proceeds via formation of divalent Mg2+ ions, resulting in the transfer of 2e– per dissolved Mg ion. In solution, Mg2+ reacts with OH– and precipitates. (b) Under basic conditions, OH– may be consumed, forming a hydroxide layer that partially passivates the Mg surface. (c) In the present work, we reveal a low barrier pathway where Mg dissolves as an effectively monovalent [Mg2+(OH)−]+ ion complex.
First-principles techniques could be the method of choice to reveal the origin of these anomalous dissolution reactions. However, studies that explore the corrosion process taking into account the full complexity of the realistic surface water interface are still lacking.
Via ab initio thermopotentiostat molecular dynamics simulations, we demonstrate that the hypothesized unipositive metal ions are in fact ion complexes, consisting of a divalent metal ion and an OH– group. In the conventional picture, cf. Figure 1a, Mg dissolves via the formation of divalent Mg2+ ions, transferring 2e– to the metal surface per dissolved Mg ion. Under basic conditions, cf. Figure 1b, water adsorbs dissociatively, partially passivating the metal surface via formation of a Mg(OH)2 surface hydroxide. In the present work, we identified an energetically and kinetically favorable reaction pathway where a surface metal atom becomes solvated in conjunction with an attached surface hydroxyl group (Figure 1c). This pathway completely circumvents the passivating nature of the hydroxide film. In turn, this reaction process supports a continued dissociative water adsorption, explaining the anomalous hydrogen evolution at the anodically polarized surface. The effectively unipositive [Mg2+(OH)−]+ ion complex is responsible for the observed amount of metal dissolved being larger than coulometrically expected. The [Mg2+(OH)−]+ ion complex may subsequently decay into the divalent metal ion via the reduction of another species. We expect it to be long-lived due to a strong Coulomb barrier separating the effectively unipositive ion complex from prospective reactants such as H+.
To reveal the dissolution mechanism and the nature of the unipositive Mg+ ion, we described the solvated Mg-surface by a supercell containing a 6-layer slab oriented in the (1 2 3̅ 15) direction and 64 explicit water molecules. Dissolution is generally understood to proceed via kink atoms due to their weaker bonds and the greater exposure of kink-sites to both adsorbing molecules and the electric field. We therefore induced a miscut in the Mg-slab, resulting in a surface with two kink-sites in the supercell. Already during equilibration under open-circuit conditions, a water molecule adsorbs dissociatively at one of the kink-sites according to
| 1 |
Two further H2O adsorbed subsequently at the same kink-site as intact molecules, leading to the configuration shown schematically in Figure 2i. For clarity, only the participating water molecules are shown. Under open-circuit conditions, this configuration remained stable on the time scale of our simulations.
Figure 2.

Solvation shell formation at kink-site Mg: (a) normal distance between the dissolving kink-site Mg-atom and its Mg bonding partner located directly underneath. The top panel shows schematic representations of the structural evolution along the AIMD trajectory, at times marked by red arrows. For clarity, only water molecules belonging to the solvation shell of the Mg2+ ion are shown explicitly. (b) Local system potential (green curve) and counter charge (red curve) used to balance the surface charge on the working electrode. Until turning on potential control at time t = 5.5 ps, the green dashed line marks the mean open circuit potential of ⟨ϕ(t)⟩ = −0.65 V, obtained in our simulations. Afterward the green dashed line indicates the targeted potential of ⟨ϕ(t)⟩ = −2 V. The Mg2+ ion remains attached to the surface via a hydroxyl bridge, see text.
In order to drive a dissolution reaction, we subsequently polarized the Mg slab anodically. The electrode charge is controlled by our recently introduced thermopotentiostat.25,26 Within 1.6 ps after switching on the thermopotentiostat with a target potential of ⟨ϕ⟩ = −2 V, a fourth water molecule approached the kink atom (cf. Figure 2ii) and adsorbed (Figure 2iii), starting to form a solvation shell. The kink-atom is then increasingly being lifted out of the surface. Figure 2a shows the distance parallel to the surface normal between the kink-atom and its Mg bonding partner underneath. A maximum extension of 3.6 Å is reached after the solvation shell is completed (Figure 2iv,v). Although the surface is charged with 2 additional electrons (cf. Figure 2b), indicating that the kink atom is now fully ionized, the solvated Mg2+ ion remains firmly bound to the surface: in conjunction with the hydroxyl group created in the reaction (eq 1), the solvated ion forms an [Mg2+(OH)–]+ad complex, where the hydroxyl group connects the kink atom to its nearest Mg neighbor (cf. Figure 2v). We speculate that this process is common to anodically polarized metal surfaces. A “place-exchange mechanism”, where a surface-adsorbed oxygen and a metal atom underneath exchange their position, was first proposed by Lanyon et al.,27 later observed by Vetter and Schultze28 for Pt surfaces and reexamined more recently by Rost et al.29
This hydroxyl bridge bond is highly stable. Removing any water molecules except the ones constituting the solvation shell and separating the [Mg2+(OH)–]+ad ion complex from the surface in a vacuum calculation, we estimated the binding energy to be ∼2 eV. Such a large binding energy is inconsistent with the experimentally observed high dissolution rates.10,11,14 In order for the dissolution to proceed, we therefore expect the breaking of the hydroxyl bridge bond to be catalyzed by its surrounding environment. Indeed, our simulations showed a possible candidate: a concerted double proton transfer from a neighboring adsorbed water molecule via a solvated H2O molecule that is hydrogen bridge bonded to the hydroxyl group (cf. Figure 3i) relocates the hydroxyl group laterally to a neighboring site. Thereby, the [Mg2+OH–]+ ion complex is oxidized to Mg2+ and left with a complete solvation shell consisting of 6 H2O molecules (Figure 3ii)
| 2 |
As a result, the Mg2+ ion quickly moved into the liquid water region (dashed blue line in Figure 2), leaving the hydroxyl group behind on the surface.
Figure 3.

Two distinct pathways for Mg-dissolution. (1) A concerted double proton transfer from a surface adsorbed H2O molecule to the hydroxyl bridge releases the Mg2+(ad) ion into solution. The corresponding normal distance between the dissolving kink-site Mg-atom and its Mg bonding partner underneath is indicated by the blue dashed line. The hydroxyl group remained on the surface. (2) Alternatively, an intra solvation shell single proton transfer to the hydroxyl bridge equally detaches the Mg2+(ad) ion from the surface (solid blue line). The green line denotes the distance between the transferring proton and the hydroxyl bridge. The hydroxyl remains attached to the Mg2+(aq) ion, resulting in an effectively +1 charged [Mg2+(OH)–]+(aq) ion complex.
This double proton transfer proceeding on the surface is, however, not the only conceivable reaction to catalyze the dissolution. In order to search for alternative processes, we moved the Mg2+(ad) ion with a constant velocity of v = 2/3 Å/ps parallel to the surface normal into the solution. In response, one of the H2O molecules forming the solvation shell turned one of its OH-bonds toward the hydroxyl group. In Figure 3, we show the distance between the hydrogen in the corresponding OH-bond and the oxygen atom of the hydroxyl group (green solid curve). In the time frame from 10.8 to 12.5 ps, multiple transfer attempts are visible, until at ∼13 ps an intra solvation shell single proton transfer occurs to the hydroxyl group (Figure 3a,b). Thereby, the [Mg2+(OH)–]+(ad) ion complex as a whole becomes fully solvated and moves into the liquid water region (solid blue curve, Figure 3c)
| 3 |
We emphasize that the outcome of these two competing processes is fundamentally different. For reaction (eq 2), the hydroxyl remains on the surface. Therefore, the surface will be quickly hydroxylated and becomes electrochemically passive. No more dissociative H2O adsorption is possible, preventing any further anomalous hydrogen evolution. Reaction (eq 3), on the other hand, removes the hydroxyl group from the surface of the metal into the solution, leaving the next kink site exposed to further dissociative H2O adsorption. It is therefore only the reaction (eq 3) that is associated with ongoing anomalous hydrogen evolution. Consistent with the experimental observation that only the unipositive Mg ion is associated with the anomalous anodic hydrogen evolution,19,20 the [Mg2+(OH)–]+(aq) ion complex created in reaction (eq 3) is effectively charged +1. We therefore propose that the elusive unipositive Mg+ ion is in fact an [Mg2+(OH)–]+ ion complex. This interpretation is supported by the fact that the reaction Mg2+(OH)− ⇌ Mg2+ + OH– is known to have a pKb value of 2.56.30 Hence, for pH > 11.44 the [Mg2+(OH)–]+(aq) ion complex becomes the dominant species. Due to the hydroxylation of the surface, we speculate that the local pH becomes sufficiently large so that the ion complex may even be thermodynamically stabilized.
Moreover, refs (19) and (21) pointed
out that the unipositive Mg+ ion remains stable for several
minutes in aqueous solutions and is able to reduce other species even
at macroscopic distances away from the Mg anode. Since the  ion complex is positively charged and requires,
e.g., an H+(aq) ion to oxidize to
ion complex is positively charged and requires,
e.g., an H+(aq) ion to oxidize to 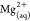 , we expect the complex to be able to reduce
other species. In addition, due to the Coulomb barrier between
, we expect the complex to be able to reduce
other species. In addition, due to the Coulomb barrier between  and
and 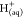 , which both
are positively charged, the
ion complex will be rather long-lived. An alternative reaction to
obtain
, which both
are positively charged, the
ion complex will be rather long-lived. An alternative reaction to
obtain 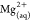 is the dissociation of the
is the dissociation of the  ion complex into its constituents
ion complex into its constituents 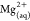 and
and 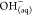 . Analogous to the
reduction via other species,
this reaction is kinetically hindered by the large Coulomb attraction
between the positive
. Analogous to the
reduction via other species,
this reaction is kinetically hindered by the large Coulomb attraction
between the positive 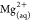 and the negative
and the negative 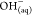 .
.
Next to applying a potential of Ũ = −1.35 V, we also performed simulations at weak anodic conditions (Ũ = −0.35 V), as well as cathodic conditions (Ũ = +1.15 V). Ũ corresponds to the voltage measured against open circuit conditions.
The reaction steps observed in our thermopotentiostat AIMD simulations imply the following model for Mg dissolution and the anomalous HER via unipositive Mg: starting from the hydroxylated surface (Figure 4a), two distinct reaction pathways are available. On the one hand, with a rate of (1 – k) the hydroxylated kink atom is first ionized as
| 4 |
and subsequently solvated according to (Figure 4c)
| 5 |
Figure 4.

Reaction model for Mg dissolution and anomalous hydrogen evolution via unipositive Mg; see text.
After that, the process can start over at the next exposed kink atom (see Figure 4a). We note that summing eqs 4 and 5 results in
| 6 |
On the other hand,
with a rate of k the  ion complex
is solvated as a whole (Figure 4b). Since the hydroxyl
group becomes detached from the surface, the surface is thereby left
exposed to another dissociative adsorption event (Figure 4d)
ion complex
is solvated as a whole (Figure 4b). Since the hydroxyl
group becomes detached from the surface, the surface is thereby left
exposed to another dissociative adsorption event (Figure 4d)
| 7 |
This is the step that triggers the anomalous anodic hydrogen evolution reaction (HER), explaining why only the effectively unipositive Mg ion complex contributes to the anodic HER.
Eventually, the  complexes
in the anolyte reduce other species—such
as,
complexes
in the anolyte reduce other species—such
as, 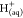 —and
oxidize in the process according
to
—and
oxidize in the process according
to
| 8 |
| 9 |
We now see that eqs 6 and 9 are balanced by the cathodic half-reaction
| 10 |
so that the sum of eqs 6, 9, and 10 yields the well-known total balance equation
| 11 |
We emphasize, that the key steps eqs 4, 5, and 7 are directly observable in our AIMD simulations. Only steps eqs 8 and 10 have been inferred.
The proposed model allows us to explain experimental observations15 showing that the amount of hydrogen produced
per unit charge depends on the current density. We speculate that
at higher current densities an increasingly larger fraction of the
current will be carried by bivalent 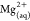 ions, as opposed to the effectively monovalent
ion complexes. As only the ion complexes contribute to the anodic
HER, the amount of H2 produced per unit charge also decreases.
Since the ion complexes efficiently remove hydroxyl groups from the
surface while
ions, as opposed to the effectively monovalent
ion complexes. As only the ion complexes contribute to the anodic
HER, the amount of H2 produced per unit charge also decreases.
Since the ion complexes efficiently remove hydroxyl groups from the
surface while 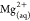 does not, we expect the formation of distinctly
different surface structures, depending on which pathway dominates.
does not, we expect the formation of distinctly
different surface structures, depending on which pathway dominates.
Further approaches to test the proposed reaction mechanism include
detection of the ion complexes themselves. We propose to extend the
experiments by Petty et al.19 where fresh
electrolyte flowing past an anodically polarized magnesium electrode
was collected in another vessel. Hydroxide precipitation, induced,
e.g., via titration by NaOH or KOH, may be able to determine the ratio
between  and
and 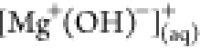 present in the electrolyte. Alternatively,
THz solvation shell spectroscopy, UV–vis and 1H
or 17O NMR may be sensitive to the presence of
present in the electrolyte. Alternatively,
THz solvation shell spectroscopy, UV–vis and 1H
or 17O NMR may be sensitive to the presence of 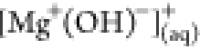 via changes in the
via changes in the 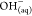 signals due to complexation.
signals due to complexation.
In summary, by using ab initio molecular dynamics
simulations of aqueous magnesium interfaces under potential control,
taking into account the full complexity of the realistic metal–water
interface, we have discovered a novel and completely unexpected reaction
pathway. The identified dissolution product—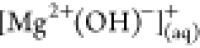 —naturally explains one
of the most
studied and debated corrosion mechanisms—the anomalous anodic
hydrogen evolution, which has puzzled scientists since it was first
reported more than 150 years ago. Our results clearly show that water
is not just a spectator but an active reactant. Under anodic conditions,
water dissociatively adsorbs to form a surface hydroxide. Subsequently,
the interfacial water provides a low-barrier pathway for proton transfer
reactions that allow the surface hydroxide to dissolve via the formation
of
—naturally explains one
of the most
studied and debated corrosion mechanisms—the anomalous anodic
hydrogen evolution, which has puzzled scientists since it was first
reported more than 150 years ago. Our results clearly show that water
is not just a spectator but an active reactant. Under anodic conditions,
water dissociatively adsorbs to form a surface hydroxide. Subsequently,
the interfacial water provides a low-barrier pathway for proton transfer
reactions that allow the surface hydroxide to dissolve via the formation
of 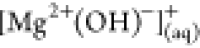 ion complexes. This pathway bypasses
the
usual passivation effect of surface films and explains the unusually
high anodic corrosion rates and the chemical nature of the hypothesized
solvated
ion complexes. This pathway bypasses
the
usual passivation effect of surface films and explains the unusually
high anodic corrosion rates and the chemical nature of the hypothesized
solvated  ions. The discovery of such an
unexpected
reaction pathway also demonstrates the level and potential that ab
initio molecular dynamics simulations have reached, thanks to recent
methodological advances in the description of electrochemical interfaces.
ions. The discovery of such an
unexpected
reaction pathway also demonstrates the level and potential that ab
initio molecular dynamics simulations have reached, thanks to recent
methodological advances in the description of electrochemical interfaces.
Acknowledgments
The authors acknowledge the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for funding through Project No. 409476157 (SFB1394), Project No. 506711657 (SFB1625) and support under the Germany’s Excellence Strategy, EXC 2033-390677874-RESOLV.
Open access funded by Max Planck Society. Open access funded by Max Planck Society.
The authors declare no competing financial interest.
References
- Ciamician G. The photochemistry of the future. Science 1912, 36, 385–394. 10.1126/science.36.926.385. [DOI] [PubMed] [Google Scholar]
- Seh Z. W.; Kibsgaard J.; Dickens C. F.; Chorkendorff I.; Norskov J. K.; Jaramillo T. F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Song Y.; Xia Y. Electrochemical capacitors: Mechanism, materials, systems, characterization and applications. Chem. Soc. Rev. 2016, 45, 5925–5950. 10.1039/C5CS00580A. [DOI] [PubMed] [Google Scholar]
- Suntivich J.; Gasteiger H. A.; Yabuuchi N.; Nakanishi H.; Goodenough J. B.; Shao-Horn Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 2011, 3, 546–550. 10.1038/nchem.1069. [DOI] [PubMed] [Google Scholar]
- Hwang S.-W.; et al. A Physically Transient Form of Silicon Electronics. Science 2012, 337, 1640–1644. 10.1126/science.1226325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K. J.; Kuzum D.; Hwang S.-W.; Kim B. H.; Juul H.; Kim N. H.; Won S. M.; Chiang K.; Trumpis M.; Richardson A. G.; et al. Bioresorbable silicon electronics for transient spatiotemporal mapping of electrical activity from the cerebral cortex. Nat. Mater. 2016, 15, 782–791. 10.1038/nmat4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabat K. C.; Rajput P.; Paramguru R. K.; Bhoi B.; Mishra B. K. Reduction of Oxide Minerals by Hydrogen Plasma: An Overview. Plasma Chem. Plasma Process. 2014, 34, 1–23. 10.1007/s11090-013-9484-2. [DOI] [Google Scholar]
- Cavaliere P.Hydrogen Assisted Direct Reduction of Iron Oxides; Springer Cham, 2022. [Google Scholar]
- Makar G. L.; Kruger J. Corrosion of magnesium. Int. Mater. Rev. 1993, 38, 138–153. 10.1179/imr.1993.38.3.138. [DOI] [Google Scholar]
- Esmaily M.; Svensson J.; Fajardo S.; Birbilis N.; Frankel G.; Virtanen S.; Arrabal R.; Thomas S.; Johansson L. Fundamentals and advances in magnesium alloy corrosion. Prog. Mater. Sci. 2017, 89, 92–193. 10.1016/j.pmatsci.2017.04.011. [DOI] [Google Scholar]
- Beetz W. XXXIV. On the development of hydrogen from the anode. Lond. Edinb. Dubl. Phil. Mag. J. Sci. 1866, 32, 269. 10.1080/14786446608644179. [DOI] [Google Scholar]
- Song G.; Atrens A. Understanding Magnesium Corrosion–A Framework for Improved Alloy Performance. Adv. Eng. Mater. 2003, 5, 837–858. 10.1002/adem.200310405. [DOI] [Google Scholar]
- Abbott T. B. Magnesium: Industrial and research developments over the last 15 years. Corrosion 2015, 71, 120–127. 10.5006/1474. [DOI] [Google Scholar]
- Glicksman R. Anodic dissolution of Magnesium alloys in aqueous salt solutions. J. Electrochem. Soc. 1959, 106, 83. 10.1149/1.2427299. [DOI] [Google Scholar]
- Frankel G.; Samaniego A.; Birbilis N. Evolution of hydrogen at dissolving magnesium surfaces. Corros. Sci. 2013, 70, 104–111. 10.1016/j.corsci.2013.01.017. [DOI] [Google Scholar]
- Liao X.; Sun C.; Huang J.; Xiao B.; Ying T.; Cao F. The positive difference effect and anomalous hydrogen evolution on the anodically polarized Mg surface in alkaline NH4+ containing solutions. J. Mater. Sci. 2024, 59, 8535–8555. 10.1007/s10853-024-09716-z. [DOI] [Google Scholar]
- Fajardo S.; Glover C.; Williams G.; Frankel G. The Source of Anodic Hydrogen Evolution on Ultra High Purity Magnesium. Electrochim. Acta 2016, 212, 510–521. 10.1016/j.electacta.2016.07.018. [DOI] [Google Scholar]
- Mercier D.; Światowska J.; Zanna S.; Seyeux A.; Marcus P. Role of Segregated Iron at Grain Boundaries on Mg Corrosion. J. Electrochem. Soc. 2018, 165, C42. 10.1149/2.0621802jes. [DOI] [Google Scholar]
- Petty R. L.; Davidson A. W.; Kleinberg J. The Anodic Oxidation of Magnesium Metal: Evidence for the Existence of Unipositive Magnesium1,2. J. Am. Chem. Soc. 1954, 76, 363–366. 10.1021/ja01631a013. [DOI] [Google Scholar]
- Atrens A.; Dietzel W. The Negative Difference Effect and Unipositive Mg+. Adv. Eng. Mater. 2007, 9, 292–297. 10.1002/adem.200600275. [DOI] [Google Scholar]
- Rausch M. D.; McEwen W. E.; Kleinberg J. Reductions Involving Unipositive Magnesium. Chem. Rev. 1957, 57, 417–437. 10.1021/cr50015a001. [DOI] [Google Scholar]
- Światowska J.; Volovitch P.; Ogle K. The anodic dissolution of Mg in NaCl and Na2SO4 electrolytes by atomic emission spectroelectrochemistry. Corros. Sci. 2010, 52, 2372–2378. 10.1016/j.corsci.2010.02.038. [DOI] [Google Scholar]
- Lebouil S.; Gharbi O.; Volovitch P.; Ogle K. Mg Dissolution in Phosphate and Chloride Electrolytes: Insight into the Mechanism of the Negative Difference Effect. Corrosion 2015, 71, 234–241. 10.5006/1459. [DOI] [Google Scholar]
- Drazic D. M.; Popic J. P. Anomalous dissolution of metals and chemical corrosion. J. Serb. Chem. Soc. 2005, 70, 489–511. 10.2298/JSC0503489D. [DOI] [Google Scholar]
- Deissenbeck F.; Freysoldt C.; Todorova M.; Neugebauer J.; Wippermann S. Dielectric Properties of Nanoconfined Water: A Canonical Thermopotentiostat Approach. Phys. Rev. Lett. 2021, 126, 136803. 10.1103/PhysRevLett.126.136803. [DOI] [PubMed] [Google Scholar]
- Deißenbeck F.; Wippermann S. Dielectric Properties of Nanoconfined Water from Ab Initio Thermopotentiostat Molecular Dynamics. J. Chem. Theory Comput. 2023, 19, 1035–1043. 10.1021/acs.jctc.2c00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanyon M. A. H.; Trapnell B. M. W.; Hinshelwood C. N. The interaction of oxygen with clean metal surfaces. Proc. R. Soc. London, Ser. A 1955, 227, 387–399. 10.1098/rspa.1955.0018. [DOI] [Google Scholar]
- Vetter K. J.; Schultze J. W. The kinetics of the electrochemical formation and reduction of monomolecular oxide layers on platinum in 0.5 M H2SO4: Part I. Potentiostatic pulse measurements. J. Electroanal. Chem. Interfacial Electrochem. 1972, 34, 131–139. 10.1016/S0022-0728(72)80509-6. [DOI] [Google Scholar]
- Rost M. J.; Jacobse L.; Koper M. T. M. Non-Random Island Nucleation in the Electrochemical Roughening on Pt(111). Angew. Chem., Int. Ed. 2023, 62, e202216376 10.1002/anie.202216376. [DOI] [PubMed] [Google Scholar]
- Haynes W. M.; Lide D. R.; Bruni T. J.. CRC Handbook of Chemistry and Physics; Taylor and Francis, 2017. [Google Scholar]