

Abstract

An adaptive catalytic system for selective hydrogenation
was developed
exploiting the H2 + CO2 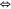 HCOOH equilibrium for reversible,
rapid, and robust on/off switch of the ketone hydrogenation activity
of ruthenium nanoparticles (Ru NPs). The catalyst design was based
on mechanistic studies and DFT calculations demonstrating that adsorption
of formic acid to Ru NPs on silica results in surface formate species
that prevent C=O hydrogenation. Ru NPs were immobilized on
readily accessible silica supports modified with guanidinium-based
ionic liquid phases (Ru@SILPGB) to generate in situ sufficient
amounts of HCOOH when CO2 was introduced into the H2 feed gas for switching off ketone hydrogenation while maintaining
the activity for hydrogenation of olefinic and aromatic C=C
bonds. Upon shutting down the CO2 supply, the C=O
hydrogenation activity was restored in real time due to the rapid
decarboxylation of the surface formate species without the need for
any changes in the reaction conditions. Thus, the newly developed
Ru@SILPGB catalysts allow controlled and alternating production
of either saturated alcohols or ketones from unsaturated substrates
depending on the use of H2 or H2/CO2 as feed gas. The major prerequisite for design of adaptive catalytic
systems based on CO2 as trigger is the ability to shift
the H2 + CO2
HCOOH equilibrium for reversible,
rapid, and robust on/off switch of the ketone hydrogenation activity
of ruthenium nanoparticles (Ru NPs). The catalyst design was based
on mechanistic studies and DFT calculations demonstrating that adsorption
of formic acid to Ru NPs on silica results in surface formate species
that prevent C=O hydrogenation. Ru NPs were immobilized on
readily accessible silica supports modified with guanidinium-based
ionic liquid phases (Ru@SILPGB) to generate in situ sufficient
amounts of HCOOH when CO2 was introduced into the H2 feed gas for switching off ketone hydrogenation while maintaining
the activity for hydrogenation of olefinic and aromatic C=C
bonds. Upon shutting down the CO2 supply, the C=O
hydrogenation activity was restored in real time due to the rapid
decarboxylation of the surface formate species without the need for
any changes in the reaction conditions. Thus, the newly developed
Ru@SILPGB catalysts allow controlled and alternating production
of either saturated alcohols or ketones from unsaturated substrates
depending on the use of H2 or H2/CO2 as feed gas. The major prerequisite for design of adaptive catalytic
systems based on CO2 as trigger is the ability to shift
the H2 + CO2 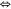 HCOOH equilibrium sufficiently to
exploit competing adsorption of surface formate and targeted functional
groups. Thus, the concept can be expected to be more generally applicable
beyond ruthenium as the active metal, paving the way for next-generation
adaptive catalytic systems in hydrogenation reactions more broadly.
HCOOH equilibrium sufficiently to
exploit competing adsorption of surface formate and targeted functional
groups. Thus, the concept can be expected to be more generally applicable
beyond ruthenium as the active metal, paving the way for next-generation
adaptive catalytic systems in hydrogenation reactions more broadly.
1. Introduction
As the production of fuels and chemicals is shifting from the use of fossil to renewable resources, challenges associated with the diversity and variability in energy and feedstock supplies are emerging.1−3 Consequently, catalysis research and development require innovative solutions to cope with the dynamics associated with the use of alternative energy resources to establish postfossil value chains.4,5 This is especially important when considering the use of “green” molecular hydrogen (H2) to convert renewable carbon feedstocks to essential value-added products (e.g., fuels, commodities, fine chemicals, agrochemicals, pharmaceuticals, etc.).5−7 A high degree of process flexibility or even adaptivity can be expected to be beneficial for the use of renewable resources, in particular to cope with quality variation in chemical feedstocks, and intermittent energy supply.8,9
In this context, the concept of adaptive catalysis takes inspiration from nature, where the reactivity of catalytic systems is modulated reversibly by various stimuli to promote countless series and parallel chemical transformations without interferences.10,11
The development of adaptive catalytic systems with the ability to control their performance through the application of external stimuli (e.g., temperature, chemical reactions, photochemical irradiation, redox switches, etc.) has attracted increasing attention in the past years.12−14 For example, following the pioneering work from Feringa et al. on light driven molecular rotors,15 the light-induced cis–trans isomerization of double bonds has been extensively studied as photoswitch to control the reactivity of organocatalysts, metal complexes, and metal nanoparticles (NPs).16−18 Most switchable catalysts developed so far attempt to control catalytic activity in response to fluctuating energy supply.19−23 Controlling product selectivity in a reversible manner can offer additional potential to enable customized production8,9 while coping with rapidly changing feedstock, market demand for products, or energy supply.17,18,24−26
Chemical modification of active sites on metal surfaces is a frequently applied tool to open or close individual pathways in complex catalytic networks.12 In order to be adaptive, the modification needs to be reversible, rapid and robust (“R3 rule”) allowing to switch between different modifications of a given catalyst material.14 A possible molecular process to fulfill this criteria is offered by the reversible reaction of H2 and CO2 to produce formic acid or formate.27−30 We have recently exploited this equilibrium to develop an adaptive catalytic system composed of ruthenium NPs immobilized on a tertiary amine-functionalized polymer grafted silica support (Ru@PGS).24 The selectivity switch in the hydrogenation of furan derivatives24 and bicyclic heteroaromatics26 was associated with the generation of ammonium formate species under H2/CO2, which decomposed upon heating under pure H2. The actual mechanism of action remained so far elusive, however. In addition, the elaborate synthesis of the polymeric structure of the surface molecular modifier and the heat treatment required to decompose the ammonium formate salt for regeneration of the original activity of Ru NPs limited the practical use of such systems.
In the present study, the influence of formic acid and formate species on the C=O hydrogenation activity of Ru NPs is investigated through mechanistic studies and DFT calculations. The acquired fundamental understanding serves as basis for the development of a new generation of simpler adaptive catalytic systems exploiting directly the reversible hydrogenation of CO2 to formic acid to control the selectivity of Ru NPs in aromatic ketone hydrogenation in real time (Figure 1).
Figure 1.
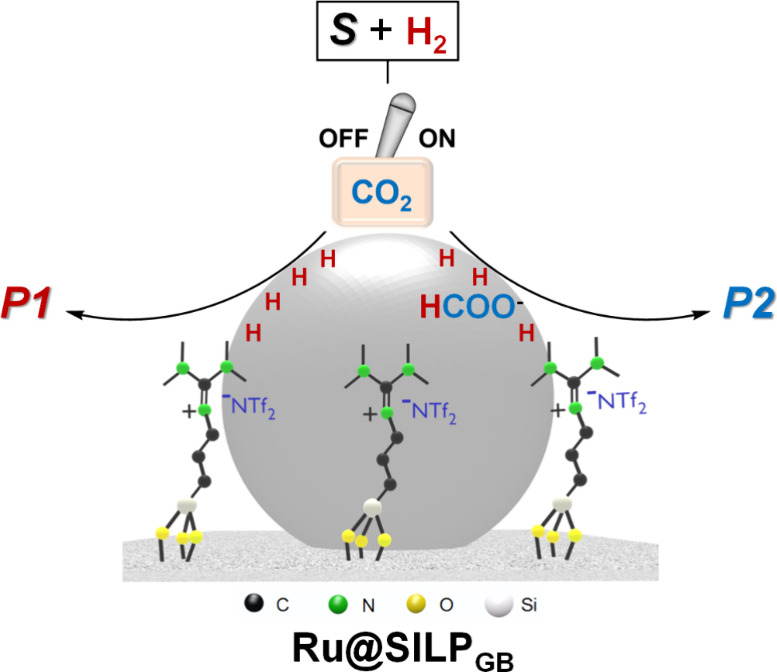
Adaptive catalytic system using in situ generated formic acid/formate as molecular trigger, illustrated for the Ru@SILPGB catalyst. S = substrate, P = product.
2. Results and Discussion
2.1. Mechanistic Studies and DFT Calculations
The hydrogenation of biomass-derived furfuralacetone (1) by 2.3 nm Ru NPs immobilized on SiO2 (Ru@SiO2, characterization described in the Figure S1 and Table S1) was used as a model reaction to investigate the potential impact of formic acid on the reactivity of Ru NPs. The hydrogenation of 1 can proceed via two reaction pathways possibly yielding four products (1a, 1b, 1c, 1d, Figure 2a). Catalytic reactions were conducted using 10 mL stainless steel high-pressure reactors equipped with a pressure gauge and heated in temperature-controlled aluminum blocks. Reaction conditions were set to 80 °C, 15 bar of H2, 16 h, and 1,4-dioxane as solvent.
Figure 2.

Hydrogenation of 1 using Ru@SiO2. (a) Reaction scheme; (b) hydrogenation under H2 with various amounts of formic acid as additive; (c) hydrogenation under H2 with various amounts of acetic acid as additive. Reaction conditions: Ru@SiO2 (19 mg, 0.007 mmol Ru), furfuralacetone (1, 0.25 mmol, 35 equiv), 1,4-dioxane (1 mL), 80 °C, 16 h, 500 rpm, H2 (15 bar), the red curve and blue curve are for products 1d and product 1b, respectively. HCOOH/Rusurface = molar ratio between HCOOH and Ru centers exposed at the surface of Ru NPs, see SI for details.
Under pure H2, full hydrogenation of 1 to the saturated alcohol 1d was observed with Ru@SiO2, which is the expected reactivity of Ru NPs under these conditions (Figure 2b,c).24 In sharp contrast, adding liquid formic acid to the solvent prior to the hydrogenation of 1 under pure H2 led to high yields of the saturated ketone 1b (Figure 2b). Initial addition of formic acid at a concentration of 22 mmol·L–1 (corresponding to a HCOOH/Ru molar ratio of 3, or HCOOH/Rusurface = 10 when considering only Ru centers exposed at the surface, see SI for details) was necessary to effectively suppress ketone hydrogenation (69% yield of 1b). It is worth noting that liquid formic acid partially decomposed under pure H2 as feed gas as evidenced by a decrease in HCOOH concentration with time under these reaction conditions (80 °C, 15 bar H2, Figure S2). Remarkably, introducing acetic acid did not show the same controlling effect as formic acid to suppress ketone hydrogenation. When acetic acid was used as an additive in the hydrogenation of 1 under pure H2, quantitative yields of the saturated alcohol 1d were formed even at high acetic acid/Rusurface ratios (up to 112, Figure 2c). These results indicate a specific action of formic acid on the catalytic performance of the Ru NPs resulting in the suppression of ketone hydrogenation activity. Notably, formic acid alone is capable of initiating the selectivity switch with Ru@SiO2, indicating that an amine functionality to generate ammonium formate species is not essential for the catalyst design.
Previous studies on the adsorption of formic acid on SiO2-supported metal NPs (e.g., Pd, Cu) evidenced the dissociative adsorption of formic acid at the metal surface to give bidentate formate species, alongside molecular adsorption on the SiO2 support.31−33 Interestingly, comparable studies performed using acetic acid evidenced a favored molecular adsorption at the SiO2 surface through H-bonding and silyl ester formation, and little to no formation of bidentate acetates species at the surface of supported metal NPs (e.g., Pd@SiO2).34−36
FTIR characterization of Ru@SiO2 after formic acid and acetic acid adsorption were found consistent with previous findings (Figure S3).31−36 In particular, formic acid adsorption resulted in a band at 1561 cm–1 characteristic of COO– in bidentate formate species, along with a C=O band at 1715 cm–1 corresponding to adsorbed molecular formic acid (Figure S3a). In contrast, acetic acid was found only in the molecularly adsorbed form (C=O band at 1730 cm–1) and none of the bands characteristic of acetate species (e.g., 1420, 1550, 1620 cm–1) was observed (Figure S3b), similar to what has been reported for Pd@SiO2 catalysts.35 This striking difference in the behavior of formic acid and acetic acid on Ru@SiO2 is possibly related to the difference in their O–H bond dissociation free energies (377 and 469 kJ/mol, respectively).37
These data suggest that the suppression of C=O hydrogenation activity induced by HCOOH is specifically linked to its dissociative adsorption at the Ru NPs surface in the presence of SiO2. To further investigate this hypothesis, the potential competitive adsorption of HCOOH and tetrahydrofuran ketones at the surface of Ru NPs was investigated by DFT calculations (details provided in SI).
The reactive surface of the Ru NPs was represented as extended surfaces of Ru, where the Ru(0001) plane was selected as the active surface, since it is thermodynamically stable and often used to represent catalytically active surfaces.38,39 The heteroaromatic 2-acetonylfuran (2) and saturated 2-acetonyltetrahydrofuran (2a) were selected as simple models to investigate their adsorption energies on the Ru surface in comparison to formic acid/formate species (Table 1). Different adsorption sites are available on Ru(0001) (Figure S4), and for each species investigated, several possible adsorption modes were considered. Heteroaromatic compound 2 can adsorb at the Ru(0001) surface via two dominant modes of similar adsorption energies (−2.08 and −2.09 eV, Table 1). The formation of a 2-acetonylfuran bridge is less likely, with a lower adsorption energy (−1.19 eV, Table 1). The saturated model 2a can adsorb at the Ru(0001) surface via two different modes of similar absorption energies (−1.13 and −1.21 eV, Table 1) involving a direct interaction of the carbonyl with the Ru surface. While the molecular adsorption of HCOOH on metal surfaces has been observed at low temperatures (e.g., 80 K),40,41 it is known to adsorb dissociatively through a proton transfer reaction on metal surfaces under conditions relevant to our study.34−36,42 The optimization of the dissociative adsorption of HCOOH affords a formate-like species interacting with the surface through the two oxygen atoms in a perpendicular fashion and a H atom in an hcp hollow site, exhibiting a typical bidentate formate species,40,41,43 with an overall adsorption energy of −1.8 eV (Table 1). Attempts to optimize this species on different starting adsorption modes converged toward the same optimized structure.
Table 1. Adsorption Modes of 2-Acetonylfuran (2), 2-Acetonyltetrahydrofuran (2a), and Formic Acid on a Ru(0001) Surface and Corresponding Adsorption Energiesa.

Details can be found in the Supporting Information file.
Dissociative adsorption.
The calculated adsorption energies indicate that the dissociative adsorption of HCOOH at the surface of Ru NPs is weaker than the adsorption of heteroaromatic 2, but stronger than the adsorption of saturated 2a. These data are in line with the observation that the C=C bond hydrogenation in furan rings is not inhibited upon addition of CO2 (vide infra). In turn, the strongly reduced activity of Ru@SiO2 for the hydrogenation of ketone-containing tetrahydrofuran derivatives under H2/CO2 can thus be rationalized by a competitive adsorption at the surface of Ru NPs between ketone intermediates and formate species resulting from the in situ hydrogenation of CO2 to formic acid. These new mechanistic insights indicate that generation of formic acid rather than ammonium formate should be sufficient to control the C=O hydrogenation ability of Ru NPs on silica support.
2.2. Catalyst Design, Synthesis, and Characterization
The results of the mechanistic and DFT studies suggest that the
full reversibility of the H2 + CO2 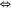 HCOOH equilibrium can be potentially
used as a rapid trigger to initiate selectivity switches in hydrogenation,
with clear benefits as compared to previously reported systems24 relying on complex polymeric surface molecular
modifiers and more stable ammonium formates species. However, its
effectiveness relies on the capacity of the considered catalytic system
to build up and stabilize significant concentrations of formic acid
under the reaction conditions considered.44 In this context, ionic liquids (ILs) are known for their potential
of strong solute–solvent interactions45 (e.g., Coulombic, H-bonding, van der Waals) and were found to shift
the endergonic CO2 hydrogenation equilibrium favorably
toward production of formic acid.46,47 In particular,
guanidinium-based ionic liquids have been reported to combine a high
affinity for CO248,49 with an exceptional
potential for stabilization of formic acid.50,51 Consequently, our catalyst design focused on a guanidinium-based
supported ionic liquid phase (SILPGB) as matrix for immobilization
of Ru NPs.52−54 The Ru NPs were chosen to act on one hand (under
H2) as hydrogenation catalysts for the substrate furfuralacetone 1, and on the other hand (under H2/CO2) also as CO2 hydrogenation catalysts producing the expected
molecular trigger formic acid stabilized by the guanidinium-based
surface molecular modifier.54
HCOOH equilibrium can be potentially
used as a rapid trigger to initiate selectivity switches in hydrogenation,
with clear benefits as compared to previously reported systems24 relying on complex polymeric surface molecular
modifiers and more stable ammonium formates species. However, its
effectiveness relies on the capacity of the considered catalytic system
to build up and stabilize significant concentrations of formic acid
under the reaction conditions considered.44 In this context, ionic liquids (ILs) are known for their potential
of strong solute–solvent interactions45 (e.g., Coulombic, H-bonding, van der Waals) and were found to shift
the endergonic CO2 hydrogenation equilibrium favorably
toward production of formic acid.46,47 In particular,
guanidinium-based ionic liquids have been reported to combine a high
affinity for CO248,49 with an exceptional
potential for stabilization of formic acid.50,51 Consequently, our catalyst design focused on a guanidinium-based
supported ionic liquid phase (SILPGB) as matrix for immobilization
of Ru NPs.52−54 The Ru NPs were chosen to act on one hand (under
H2) as hydrogenation catalysts for the substrate furfuralacetone 1, and on the other hand (under H2/CO2) also as CO2 hydrogenation catalysts producing the expected
molecular trigger formic acid stabilized by the guanidinium-based
surface molecular modifier.54
The silane-functionalized guanidinium-based ionic liquid [1,1,3,3-tetramethyl-2-[3-(triethoxysilyl)propyl] guanidium bis(trifluoromethylsulfonyl)imide] (ILGB) was prepared by adapting protocols from the literature,53 and its structure was confirmed by NMR spectroscopy (Figures S5 and S6). The silanization of ILGB on dehydroxylated SiO2 was achieved following a previously reported procedure52,53 and afforded the corresponding supported ionic liquid phase (SILPGB) with a loading of IL-like molecular modifiers of 0.58 mmol·g–1 (Figure 3a). Importantly, the preparation of SILPGB involves less and safer steps (4), better atom economy (AE = 24.5%) and cost efficiency (ca. 2.5 Euro/g) than that of the PGS support material (5 steps, AE = 4%, ca. 19 Euro/g) previously used24 for adaptive catalysis (see SI for detailed analysis, Figures S7–S8 and Tables S2–S3). For the immobilization of Ru NPs, SILPGB was subjected to wet impregnation with a solution of [Ru(2-methylallyl)2(cod)] (where cod = 1,5-cyclooctadiene) in tetrahydrofuran (THF). After removal of the solvent in vacuo, the dried powder was treated under an atmosphere of H2 (25 bar) at 100 °C for 18 h, giving the Ru@SILPGB catalyst as a fine black powder with a Ru loading of 3.5 wt % (or 0.35 mmol·g–1) as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Table S1).
Figure 3.

(a) Illustration of the design and preparation approach for Ru@SILPGB; (b) solid state 29Si CP-MAS of SiO2 (pink), SILPGB (green), and Ru@SILPGB (black); (c, d) HAADF-STEM images of Ru@SILPGB; (e) PDF G(r) of Ru NPs on Ru@SILPGB (after subtraction of SILPGB signal); (f) k2-weighted R-space FT-EXAFS spectra and (g) Ru K-edge XANES spectra (normalized) plot in Magnitude without phase correction) for Ru-foil, Ru@SILPGB and RuO2 (green, black and orange curves, respectively).
N2 physisorption analysis (Table S1) showed a decrease in the surface area of SILPGB and Ru@SILPGB (308 and 302 m2·g–1 respectively, calculated using Brunner–Emmett–Teller (BET) theory) in comparison to the starting SiO2 (453 m2·g–1), as expected due to the functionalization with ILGB. Solid-state 29Si CP-MAS NMR analysis of SILPGB and Ru@SILPGB (Figure 3b) showed the presence of two types of Si species: 1) tetra-functionalized (Q) Si with signals at −109 (Q4 = Si(OSi)4) and −100 ppm (Q3 = Si(OSi)3OH); and 2) trifunctionalized (T) Si with signals at −53 (T2 = R-Si(OSi)2OR’) and −62 ppm (T3 = R-Si(OSi)3). The T2 and T3 signals correspond to the Si atoms of ILGB covalently bound to the SiO2 surface, confirming the successful chemisorption of ILGB. Thermogravimetric analysis (TGA) performed under Ar showed that the SILPGB and Ru@SILPGB materials are thermally stable up to 280 °C (Figure S9). The transmission IR spectrum of Ru@SILPGB (Figure S10) exhibited several bands characteristic of the structure of ILGB, including N–H stretching at 3500–3100 cm–1, C–H stretching at 2950 cm–1, and C=N and C–N stretching at 1620 and 1420 cm–1, respectively.55,56 This indicates that the structure of ILGB was not affected by chemisorption nor by Ru NPs deposition. Bands at 1980 and 1873 cm–1 were attributed to silica overtone bands.56
High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM, Figure 3c,d) analysis confirmed the formation of small Ru NPs (diameter = 1.3 ± 0.4 nm, histogram provided in Figure S11) well-dispersed on the SILPGB support material. Lattice spacings (Table S4) of the Ru NPs were determined from the Fast Fourier Transformation (FFT, Figure S12) of the bright field STEM images (BF-STEM), which correspond well to hcp Ruthenium (P63/mmc).
Powder X-ray diffraction (PXRD) failed to provide insights into the structure of these very small Ru NPs, giving broad diffuse scattering rather than sharp Bragg peaks.57 Therefore, the atomic pair distribution function (PDF) technique was utilized, by applying a Fourier transformation on the total scattering data (Bragg peaks plus diffuse scattering). This method elucidates the nanostructures of the sample by representing them as a histogram of interatomic distances in real space (Figures 3e, S13 and S14). The PDF of Ru NPs in Ru@SILPGB (Figure 3e) is calculated from synchrotron PXRD data (Figure S13), and a refinement (see Figure S13f) confirms the presence of hcp Ru NPs with a P63/mmc space group, consistent with the FFT analysis. Potential interactions between Ru NPs and the IL layer of SILPGB cannot be concluded from the PDF data.
Ru K-edge X-ray absorption fine structure (XAFS) study of the Ru@SILPGB was performed to investigate the electronic and geometric structures of Ru NPs (Figure 3f,g). The X-ray absorption near edge structure (XANES) spectrum of Ru@SILPGB shows an absorption edge position (estimated by the energy position at 0.5 of normalized absorption) of 22119.3 eV, approximately 2.1 eV higher than that of Ru(0) in Ru metal foil (22117.2 eV), but 6.7 eV lower than that of Ru(IV) in RuO2 (22123.9 eV). This suggests that Ru is mainly in the metallic state with only traces of oxidation in Ru@SILPGB. Quantitative EXAFS fitting results of Ru@SILPGB (Table S5, Figure S15) showed Ru–Ru scattering with a coordination number (C.N.) of 9.1 ± 0.9 at 2.68 ± 0.01 Å, and an additional Ru–O scattering with C.N. of 1.3 ± 0.5 at 1.97 ± 0.03 Å, which is in good agreement with the slight oxidation observed from the XANES analysis.
Two reference catalysts comprising different surface modifiers were prepared using the same methods for Ru NP deposition and surface modification: Ru@Si-Dec (Ru NPs on SiO2 functionalized with decyl chains),58 and Ru@SILPIM (Ru NPs on an imidazolium-based SILP).54 Their structural and physicochemical properties were found very similar to that of Ru@SILPGB and Ru@SiO2 (Table S1, Figures S16 and S17) allowing for a direct comparison of the surface modification on the adaptivity of the catalyst materials.
2.3. Catalytic Study
2.3.1. Reactivity of Ru@SILPGB with H2/CO2–Hydrogenation of CO2 and Formic Acid Decomposition
The potential adaptivity toward the presence of carbon dioxide in the feed gas relies on the capacity of Ru@SILPGB to stabilize significant concentrations of formic acid upon adjustment of the equilibrium. Therefore, Ru@SILPGB was used as catalyst for the hydrogenation of CO2 (30 bar total pressure, H2/CO2 1/1) at 80 °C for 16 h, using deuterated tetrahydrofuran (THF-d8) as a solvent (Figure 4a). The amount of formic acid formed was quantified by 1H NMR using chloroform as a standard (see SI for detailed calculation). Under these conditions, a formic acid concentration in solution of 0.4 mmol·L–1 was detected (Table S6). While replacing THF-d8 by 1-butanol did not have a significant impact ([HCOOH] = 0.5 mmol·L–1), the use of 1,4-dioxane led to a 3-fold increase in the formic acid concentration (1.3 mmol·L–1). Such pronounced solvent effect in CO2 hydrogenation to formic acid reflects the impact of solvent properties on the solubility, diffusion, and interaction of reactants (H2 and CO2) with the catalyst surface, and is consistent with previous findings.59 Further optimization of the pressure and H2/CO2 ratio (Table S6) resulted in a noticeable enhancement up to 3.1 mmol·L–1 (1H and 13C NMR spectra provided in Figure 4b,c), corresponding to a HCOOH/Rusurface of 0.90. Thus, the following standard conditions were established for the rest of the study: 80 °C, 45 bar total pressure, H2/CO2 1/2, 16 h, 1,4-dioxane as a solvent. Interestingly, applying the reference Ru@SiO2, Ru@Si-Dec, and Ru@SILPIM catalysts under these conditions led to lower formic acid concentrations and HCOOH/Rusurface ratios, with the trend Ru@SILPGB > Ru@SILPIM > Ru@Si-Dec > Ru@SiO2 (Figure 4d). Since Ru loading, NPs size, and BET surface area are similar on all these catalysts, the enhanced formic acid concentration observed when using Ru@SILPGB can be associated with the capacity of the surface modifier to stabilize HCOOH.46,47 The enhanced formic acid stability around Ru@SILPGB presumably originates from electrostatic interactions with the strong Y-shaped delocalization of the cationic center,60 as well as from the higher basicity of guanidinium-based ILs as compared to imidazolium ones.61
Figure 4.

(a) Hydrogenation of CO2 using Ru@Support catalysts; (b) 1H and (c) 13C NMR spectra of the suspension solution after the hydrogenation of CO2, reaction conditions: Ru@SILPGB (20 mg, 0.007 mmol), 1,4-dioxane (1 mL), 80 °C, 16 h, 500 rpm, H2/CO2 (45 bar, 1:2); (d) the structure of Ru@Support catalysts: Ru@SiO2, Ru@Si-Dec, Ru@SILPIM, Ru@SILPGB; the corresponding concentration of HCOOH in the suspension solution and molar ratio of HCOOH/Rusurface, reaction conditions: Ru@Support catalyst (0.007 mmol Ru), 1,4-dioxane (1 mL), 80 °C, 16 h, 500 rpm, H2/CO2 (45 bar, 1:2), concentration of HCOOH determined by 1H NMR spectra using CHCl3 as an internal standard.
Since the rapid reversal of the trigger formation is an important aspect of the catalyst design for adaptivity, the reverse reaction of formic acid decomposition to CO2 and H2 was also investigated using the Ru@SILPGB. Solutions containing formic acid in the concentration formed under standard conditions were thus kept at 80 °C and 15 bar H2 in the absence of CO2 in the gas phase (see SI for detailed procedure). The formic acid was decomposed within 1 h to yield H2 and CO2 without any trace of CO as evidenced by headspace GC-TCD and FT-IR analyses (Figures S18 and S19, respectively). Similar observations were made with the reference catalysts (Figure S18). The high activity of Ru@SILPGB for the production and decomposition of formic acid from CO2 and H2 confirms its potential to show adaptivity toward the presence of carbon dioxide in the feed gas.
2.3.2. Adaptive Hydrogenation of Furanic Ketones Using Ru@SILPGB under H2 and H2/CO2
The hydrogenation of furfuralacetone (1) was selected to investigate the catalytic performance of Ru@SILPGB under H2 and H2/CO2 as feed gases, respectively (optimization provided in Table S7), and time profiles of product formation were recorded (Figure 5a,b). Under pure H2, the hydrogenation of the C=C double bond and the furan ring was fast (initial rate for the formation of 1b, r0(1b) = 102 mmol L–1 h–1), followed by the hydrogenation of the ketone group with an initial rate r0(1d) = 17 mmol L–1 h–1 (Figure 5a). The saturated alcohol 1d was obtained in excellent yield (91%), which is the expected reactivity of Ru NPs under these conditions (Figure 5a).24
Figure 5.

Hydrogenation of furfuralacetone (1) under H2 or H2/CO2 as feed gas. Time profiles of the hydrogenation of 1 using Ru@SILPGB under (a) H2 and (b) H2/CO2. (c) product distribution after hydrogenation of 1 under H2 or H2/CO2 using Ru@Suppost catalysts. Blue bars represent product 1b and red bars represent product 1d. (d) Relationship of yield of product 1b and HCOOH/Rusurface ratio. Reaction conditions: Ru@Support (0.007 mmol Ru), furfuralacetone (1, 0.25 mmol, 35 equiv), 1,4-dioxane (1 mL), 80 °C, 500 rpm, H2 (15 bar) or H2/CO2 (45 bar, 1:2). Product yield determined by GC-FID using tetradecane as the internal standard. Byproduct is 2,2′-(oxybis(butane-3,1-diyl))bis(tetrahydrofuran). Data points are average values of two to four experiments and error bars represent standard deviations. Red bars: product 1d; blue bars: product 1b.
In sharp contrast, the saturated ketone 1b was formed with high selectivity when H2/CO2 was used under otherwise identical conditions with the same partial pressure of H2 applied. The hydrogenation of the C=C double bond and the furan ring proceeded at a similar initial rate as under pure H2 (r0(1b) = 97 mmol L–1 h–1), but ketone hydrogenation was essentially shut down with an initial reaction rate r0(1d) = 6 mmol L–1 h–1 leading to less than 10% yield of 1d even after 24 h (Figure 5b). The progressive decline of C=O hydrogenation activity reflects the necessary time to build-up sufficient concentrations of formic acid to suppress ketone hydrogenation activity. This was confirmed by reference catalytic experiments started directly in the presence of suitable concentrations of formic acid, and for which the initial C=O hydrogenation rate r0(1d) was found much lower (1.7 mmol L–1 h–1, Figure S20).
In order to evaluate the influence of the molecular modifier on the adaptivity of the catalytic system, the performance of Ru@SILPGB was compared to reference catalysts Ru@SiO2, Ru@Si-Dec, and Ru@SILPIM. The presence of CO2 together with H2 in the feed gas did not impact the performance of Ru@SiO2 leading to full hydrogenation to product 1d under both gas mixtures in agreement with previous observations (Figure S21).24 However, mixtures of products 1b and 1d were obtained with the three other catalysts once CO2 was introduced. The selectivity toward the saturated ketone 1b followed the order Ru@SILPGB (90%) > Ru@SILPIM (63%) > Ru@Si-Dec (25%, Figure 5c,d). Interestingly, this trend correlates directly with the HCOOH/Rusurface ratio determined for each catalyst (Figure 5d), indicating that the C=O hydrogenation activity of Ru NPs can be effectively suppressed provided that a sufficient amount of HCOOH is adjusted by the surface molecular modifier via the CO2 hydrogenation equilibrium. This is consistent with reference experiments involving the hydrogenation of 1 under H2 with Ru@SiO2 and Ru@SILPGB in the presence of various amount of formic acid as additive (Figure S22). The required starting HCOOH/Rusurface ratio to suppress C=O hydrogenation is higher for Ru@SiO2 (11) than for Ru@SILPGB (3), reflecting important differences in the catalysts’ abilities to stabilize a certain level of formic acid concentration under reaction conditions.
Satisfyingly, the selectivity switch was found fully reversible, and alternating between H2 or H2/CO2 as feed gas allowed producing product 1d (86–88%) or product 1b (88–91%) in high yields and selectivity in six consecutive cycles without catalyst regeneration (Figure 6a). The direct correlation of the selectivity switch with the reversible formation of HCOOH was demonstrated by concomitant 1H NMR monitoring of the reaction mixture under H2 and H2/CO2 (Figure 6b). Importantly, no workup (e.g., heat treatment or washing step, Figures 6a,b and S23) was necessary between the cycles to regenerate rapidly the activity of pristine Ru NPs, demonstrating the real time reversibility of the selectivity switch as a noticeable improvement over previously reported Ru@PGS systems.24,26 The robustness of Ru@SILPGB was further investigated through recycling experiments under H2 and H2/CO2. The conditions were slightly modified to obtain product mixtures to probe changes in performance directly. Five consecutive cycles were performed with H2 (Figure 6c) and with H2/CO2 (Figure 6e). Product distributions remained constant within experimental error under both sets of conditions without obvious signs of deactivation.
Figure 6.

(a) Product distributions for consecutive cycles of the hydrogenation of 1 using Ru@SILPGB while alternating the feed gas between H2 and H2/CO2 and (b) 1H NMR monitoring of the HCOOH content of the reaction solutions as a function of the feed gas composition. (c, e) Recycling experiments for the hydrogenation of furfuralacetone (1) using Ru@SILPGB under (c) H2 and (e) H2/CO2; corresponding (d) k2-weighted R-space FT-EXAFS spectra and (f) Ru K-edge XANES spectra (normalized) plot in Magnitude without phase correction) for Ru@SILPGB after reaction under H2 and H2/CO2. Reaction conditions: Ru@SILPGB (20 mg, 0.007 mmol Ru), furfuralacetone (1, 0.25 mmol, 35 equiv), 1,4-dioxane (1 mL), H2 (15 bar) or H2/CO2 (45 bar, 1:2), 80 °C, 16 h for (a, b), 2 h for (c,e). Product yield determined by GC-FID using tetradecane as the internal standard. Conversion >99% and the byproduct is 2,2′-(oxybis(butane-3,1-diyl))bis(tetrahydrofuran). Data points are average values of three experiments and error bars represent standard deviations. Red bars: product 1d; blue bars: product 1b.
Independent of the used feed gas, T2 and T3 signals were unchanged in the solid-state 29Si CP-MAS NMR analysis of Ru@SILPGB after catalysis indicating the presence of the chemisorbed IL-like molecular modifier (Figure S24). BET surface areas (Table S8) increased slightly to 330 m2·g–1 after reaction under H2 and 354 m2·g–1 after reaction under H2/CO2 as compared to that of fresh catalyst (302 m2·g–1). This results presumably from loss of small quantities of the guanidinium modifier physisorbed rather than chemisorbed to the SiO2 surface during preparation. Ru loadings after 5 cycles under H2 or H2/CO2 were determined to 3.7 and 4.0 wt %, respectively (Table S8). The slight increase relative to the fresh catalyst (3.5 wt %) also reflects probably the removal of the nonchemisorbed modifier. The heterogeneous nature of the catalysis was confirmed as the Ru content in the reaction solution was below 2 ppm (Table S9) and the reaction solution after hot filtration of the catalyst did not show any activity under both H2 and H2/CO2 (Figure S25). The used Ru@SILPGB catalysts exhibited slight aggregation of Ru NPs as compared to the fresh catalyst, again independently of the used gas composition (Figure S26). XANES and EXAFS spectra of Ru@SILPGB after catalysis were nearly identical to the fresh Ru@SILPGB, indicating good Ru NPs stability as no noticeable change in oxidation state nor coordination structure were observed (Figure 6d,f). These results demonstrate the robustness of the Ru@SILPGB catalyst and the absence of irreversible structural or electronic modifications arising from its use under different feed gases.
The adaptivity of Ru@SILPGB was further explored by expanding the substrate scope to a variety of ketone-containing furan derivatives. Satisfyingly, the hydrogenation selectivity could be controlled using CO2 as the molecular trigger for these substrates as well under optimized conditions, yielding either the saturated alcohol or ketone as products in high yields (optimization steps in Table S10). Together with the previous findings,24 this further substantiates the generalization of the concept for selective production of different hydrogenation products under H2 or under H2/CO2 with introduction of carbon dioxide into the feed gas as the only change in conditions (Table 2).
Table 2. Hydrogenation of Ketone Derivatives using Ru@SILPGB under H2 or H2/CO2a.
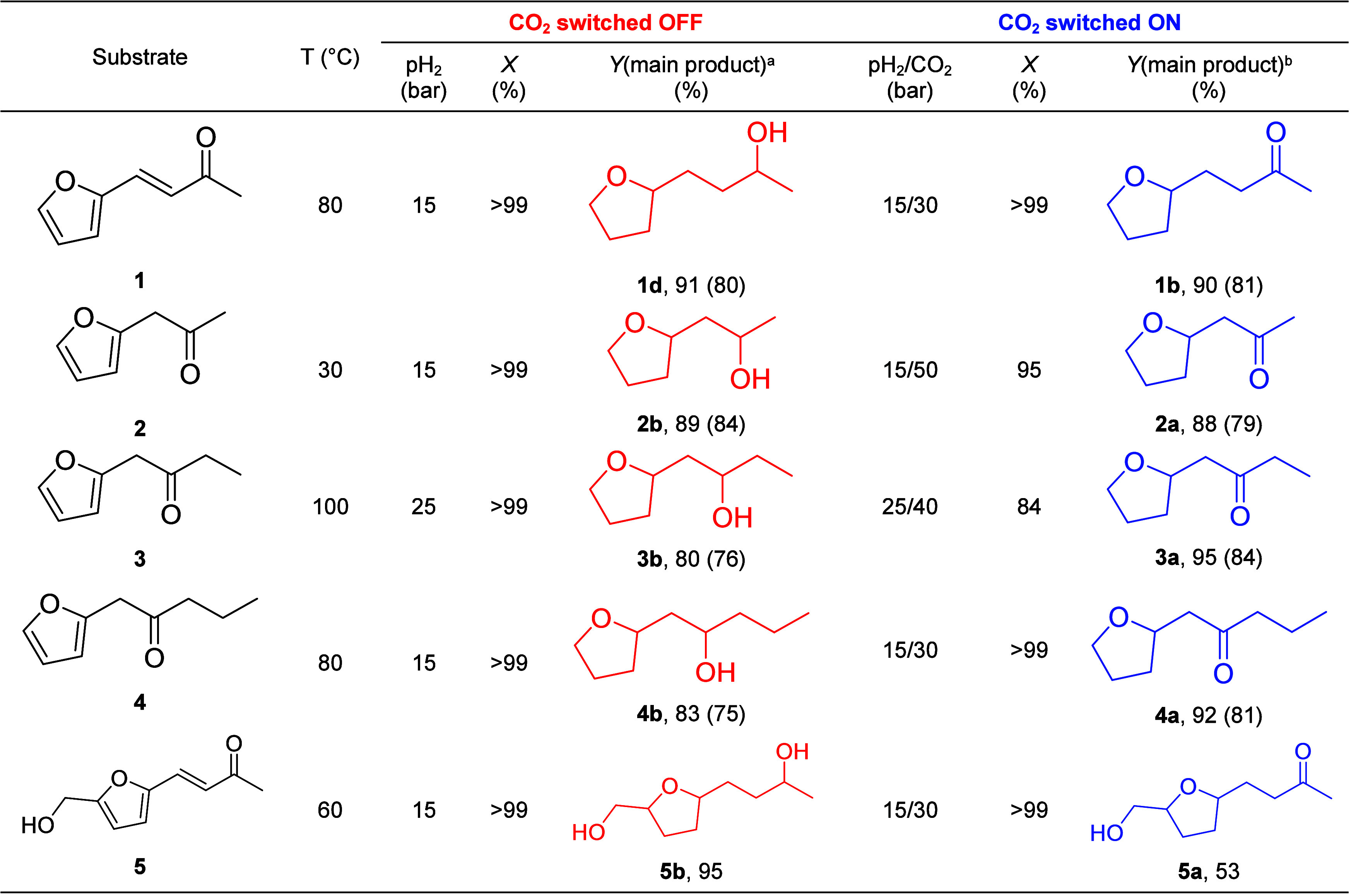
Reaction conditions: Ru@SILPGB (20 mg, 0.007 mmol Ru), substrate (0.25 mmol, 35 equiv), 1,4-dioxane (1 mL), 16 h, X = conversion, Y = yield. Conversions and product yields determined by GC-FID using tetradecane as the internal standard.
Rest = corresponding byproduct formed by the dehydration of saturated alcohol.
Rest = saturated alcohol products. Isolated yields are given in parentheses and the NMR spectra of isolated products are provided in Figures S28–S43.
3. Conclusions
In conclusion, the adaptive control of ketone hydrogenation using Ru-based catalysts employing H2 or H2/CO2 feed gas was elucidated by mechanistic studies supported by DFT calculations, revealing formate coverage at the Ru surface as the key control factor for the reversible selectivity switch. The fundamental understanding served as basis for the development of a new adaptive catalytic system exploiting the reversible hydrogenation of CO2 to formic acid as a trigger to control product formation upon hydrogenation of furanic ketones. In particular, Ru NPs were immobilized on a new guanidinium-based supported ionic liquid phase (SILPGB). The resulting Ru@SILPGB was characterized in pristine form as well as after exposure to catalytic relevant conditions using various techniques including N2 physisorption, electron microscopy, solid state NMR, pair distribution function analysis, and X-ray absorption spectroscopy. The hydrogenation of biomass-derived furfuralacetone and related ketone substrates confirmed the practically instantaneous on/off switching of the C=O hydrogenation with Ru@SILPGB under H2 or H2/CO2. The guanidinium-based surface molecular modifier was found essential to stabilize concentrations of formic acid sufficient to observe a selectivity switch (HCOOH/Rusurface ratio close to 1), which was not observed using imidazolium-based Ru@SILPIM, C10-chain modified Ru@Si-Dec, and Ru@SiO2 catalysts. The selectivity switch was found fully reversible, rapid, and robust, providing either full hydrogenation under H2 or partial hydrogenation under H2/CO2.
The elucidation
of the control mechanism from formic acid/formate
adsorption on metal surfaces may help to pave the way toward the development
of next-generation adaptive catalytic systems exploiting more generally
formic acid from CO2 as molecular trigger in NPs-catalyzed
reactions. As evidenced in this study, the major prerequisite for
catalyst design is the ability to shift the H2 + CO2 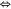 HCOOH equilibrium sufficiently to
exploit a competing adsorption of surface formate and targeted functional
groups. Consequently, the concept is not restricted to ruthenium nanoparticles
as the active component nor to ketone vs furan hydrogenation and further
studies to explore this potential seem promising.
HCOOH equilibrium sufficiently to
exploit a competing adsorption of surface formate and targeted functional
groups. Consequently, the concept is not restricted to ruthenium nanoparticles
as the active component nor to ketone vs furan hydrogenation and further
studies to explore this potential seem promising.
Acknowledgments
The authors acknowledge financial support by the Max Planck Society and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832. The authors would like to thank Norbert Pfänder (MPI CEC) for STEM analysis, and Dr. Meike Emondts (DWI, RWTH Aachen) for solid state 29Si CP-MAS NMR measurements. The authors are also thankful to Annika Gurowski, Alina Jakubowski, and Justus Werkmeister (MPI CEC) for GC and GC-MS measurements. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III (proposal No. I-20220137), and we would like to thank Dr. Edmund Welter for assistance in using P65 Applied XAFS Beamline. We acknowledge Diamond Light Source (UK) for the experiment time on Beamline B18 for XAFS measurements (proposal No. SP35401). We would like to thank Dr. Diego Gianolio, Dr. Iuliia Mikulska and Dr. Veronica Celorrio for their assistance during the beamtime. We acknowledge the electron Physical Science Imaging Centre (ePSIC) in Diamond Light Source for experiment time in lab E02 for STEM characterizations (proposal No. MG33118). We would like to thank Dr. David Hopkinson, Dr. Chris Allen and Dr. Mohsen Danaie for their assistance during the experiment time. The authors would like to thank the Max Planck Computing & Data Facility (MPCDF) for the access to their high computing facility and their user support. L.K. acknowledges Alexander von Humboldt Foundation for a postdoctoral fellowship and funding support. The authors would like to thank Marius Heise-Podleska for TGA experiments and Johanna Taing for N2 physisorption measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c06765.
Experimental details, data processing and evaluation, materials, theoretical calculations, additional data including reference catalyst characterization, IR spectra, data fitting details, spent catalyst characterization, parameter optimization, and NMR spectra (PDF)
Open access funded by Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- Schlögl R. Chemistry‘s Role in Regenerative Energy. Ale. Chem. Int. Ed. 2011, 50, 6424–6426. 10.1002/anie.201103415. [DOI] [PubMed] [Google Scholar]
- Zimmerman J. B.; Anastas P. T.; Erythropel H. C.; Leitner W. Designing for a Green Chemistry Future. Science 2020, 367, 397–400. 10.1126/science.aay3060. [DOI] [PubMed] [Google Scholar]
- Horvath I. T. Introduction: Sustainable Chemistry. Chem. Rev. 2018, 118, 369–371. 10.1021/acs.chemrev.7b00721. [DOI] [PubMed] [Google Scholar]
- Vogt C.; Weckhuysen B. M. The Concept of Active Site in Heterogeneous Catalysis. Nat. Rev. Chem. 2022, 6, 89–111. 10.1038/s41570-021-00340-y. [DOI] [PubMed] [Google Scholar]
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: from Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck C. O.; Pérez E.; Horváth I. T.; Sheldon R. A.; Poliakoff M. Valorization of Biomass: Deriving More Value from Waste. Science 2012, 337, 695–699. 10.1126/science.1218930. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhou M.; Wang A.; Zhang T. Selective Hydrogenation over Supported Metal Catalysts: from Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. 10.1021/acs.chemrev.9b00230. [DOI] [PubMed] [Google Scholar]
- European Commission. Luxembourg. Publications Office of the European Union EFFRA Factories of the Future. Multi-annual Roadmap for the Contractual PPP under Horizon 2020, 2013.
- The US Chemical Industry. Technology Vision 2020, 1996.
- Krauss G.Biochemistry of Signal Transduction and Regulation; Wiley-VCH: Weinheim, 2003. [Google Scholar]
- Traut T.Allosteric Regulatory Enzymes; Springer: New York, 2008. [Google Scholar]
- Blanco V.; Leigh D. A.; Marcos V. Artificial Switchable Catalysts. Chem. Soc. Rev. 2015, 44, 5341–5370. 10.1039/C5CS00096C. [DOI] [PubMed] [Google Scholar]
- Teator A. J.; Lastovickova D. N.; Bielawski C. W. Switchable Polymerization Catalysts. Chem. Rev. 2016, 116, 1969–1992. 10.1021/acs.chemrev.5b00426. [DOI] [PubMed] [Google Scholar]
- Bordet A.; Leitner W. Adaptive Catalytic Systems for Chemical Energy Conversion. Angew. Chem., Int. Ed. 2023, 62, e202301956 10.1002/anie.202301956. [DOI] [PubMed] [Google Scholar]
- Koumura N.; Zijlstra R. W. J.; van Delden R. A.; Harada N.; Feringa B. L. Light-Driven Monodirectional Molecular Rotor. Nature 1999, 401, 153–155. 10.1038/43646. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Han S.; Kim J.; Soh S.; Grzybowski B. A. Photoswitchable Catalysis Mediated by Dynamic Aggregation of Nanoparticles. J. Am. Chem. Soc. 2010, 132, 11018–11020. 10.1021/ja104260n. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Sen S.; Udayabhaskararao T.; Sawczyk M.; Kucanda K.; Manna D.; Kundu P. K.; Lee J.-W.; Král P.; Klajn R. Reversible Trapping and Reaction Acceleration within Dynamically Self-Assembling Nanoflasks. Nat. Nanotechnol. 2016, 11, 82–88. 10.1038/nnano.2015.256. [DOI] [PubMed] [Google Scholar]
- Wang J.; Feringa B. L. Dynamic Control of Chiral Space in a Catalytic Asymmetric Reaction using a Molecular Motor. Science 2011, 331, 1429–1432. 10.1126/science.1199844. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Han S.; Kim J.; Soh S.; Grzybowski B. A. Photoswitchable Catalysis Mediated by Dynamic Aggregation of Nanoparticles. J. Am. Chem. Soc. 2010, 132, 11018–11020. 10.1021/ja104260n. [DOI] [PubMed] [Google Scholar]
- Li Y.; Hao J.; Song H.; Zhang F.; Bai X.; Meng X.; Zhang H.; Wang S.; Hu Y.; Ye J. Selective Light Absorber-Assisted Single Nickel Atom Catalysts for Ambient Sunlight-Driven CO2 Methanation. Nat. Commun. 2019, 10, 2359–2368. 10.1038/s41467-019-10304-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Zambrana C.; Gutiérrez-Blanco A.; Gonell S.; Poyatos M.; Peris E. Redox-Switchable Cycloisomerization of Alkynoic Acids with Napthalenediimide-Derived N-Heterocyclic Carbene Complexes. Angew. Chem., Int. Ed. 2021, 60, 20003–20011. 10.1002/anie.202107973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreissl H.; Jin J.; Lin S.-H.; Schuette D.; Stortte S.; Levin N.; Chaudret B.; Vorholt A. J.; Bordet A.; Leitner W. Commercial Cu2Cr2O5 Decorated with Iron Carbide Nanoparticles as a Multifunctional Catalyst for Magnetically Induced Continuous-Flow Hydrogenation of Aromatic Ketones. Angew. Chem., Int. Ed. 2021, 60, 26639–26646. 10.1002/anie.202107916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.-H.; Hetaba W.; Chaudret B.; Leitner W.; Bordet A. Copper-Decorated Iron Carbide Nanoparticles Heated by Magnetic Induction as Adaptive Multifunctional Catalysts for the Selective Hydrodeoxygenation of Aldehydes. Adv. Energy Mater. 2022, 12, 2201783 10.1002/aenm.202201783. [DOI] [Google Scholar]
- Bordet A.; El Sayed S.; Sanger M.; Boniface K. J.; Kalsi D.; Luska K. L.; Jessop P. G.; Leitner W. Selectivity Control in Hydrogenation through Adaptive Catalysis using Ruthenium Nanoparticles on a CO2-Responsive Support. Nat. Chem. 2021, 13, 916–922. 10.1038/s41557-021-00735-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh V.; Chatterjee B.; Chang W. C.; Cramer H. H.; Hindemith C.; Randel H.; Weyhermuller T.; Fares C.; Werle C. An Adaptive Rhodium Catalyst to Control the Hydrogenation Network of Nitroarenes. Angew. Chem., Int. Ed. 2022, 61, e202205515 10.1002/anie.202205515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; El Sayed S.; Kang L.; Sanger M.; Wiegand T.; Jessop P. G.; DeBeer S.; Bordet A.; Leitner W. Adaptive Catalysts for the Selective Hydrogenation of Bicyclic Heteroaromatics using Ruthenium Nanoparticles on a CO2-Responsive Support. Angew. Chem., Int. Ed. 2023, 62, e202311427 10.1002/anie.202311427. [DOI] [PubMed] [Google Scholar]
- Leitner W.; Dinjus E.; Gaßner F. Activation of Carbon Dioxide: IV. Rhodium-Catalysed Hydrogenation of Carbon Dioxide to Formic Acid. J. Organomet. Chem. 1994, 475, 257–266. 10.1016/0022-328X(94)84030-X. [DOI] [Google Scholar]
- Klankermayer J.; Wesselbaum S.; Beydoun K.; Leitner W. Selective Catalytic Synthesis using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem., Int. Ed. 2016, 55, 7296–7343. 10.1002/anie.201507458. [DOI] [PubMed] [Google Scholar]
- Mellmann D.; Sponholz P.; Junge H.; Beller M. Formic Acid as A Hydrogen Storage Material–Development of Homogeneous Catalysts for Selective Hydrogen Release. Chem. Soc. Rev. 2016, 45, 3954–3988. 10.1039/C5CS00618J. [DOI] [PubMed] [Google Scholar]
- Eppinger J.; Huang K.-W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. 10.1021/acsenergylett.6b00574. [DOI] [Google Scholar]
- Hirota K.; Fueki K.; Shindo K.; Nakai Y. Studies on the State of Formic Acid Adsorbed on Silica and Alumina by a Combined Method of Nuclear Magnetic Resonance and Infrared Absorption. Bull. Chem. Soc. Jpn. 1959, 32, 1261–1263. 10.1246/bcsj.32.1261. [DOI] [Google Scholar]
- Millar G. J.; Rochester C. H. Infrared Study of the Adsorption of Formic Acid on Silica-Supported Copper and Oxidised Copper Catalysts. J. Chem. Soc., Faraday Trans. 1991, 87, 1491–1496. 10.1039/ft9918701491. [DOI] [Google Scholar]
- Cabilla G. C.; Bonivardi A. L.; Baltanàs M. A. Infrared Study of the Adsorption of Formic Acid on Clean and Ca-Promoted Pd/SiO2 Catalysts. Appl. Catal. A: General 2003, 255, 181–195. 10.1016/S0926-860X(03)00546-5. [DOI] [Google Scholar]
- Rachmady W.; Vannice M. A. Acetic Acid Reduction by H2 over Supported Pt Catalysts: A DRIFTS and TPD/TPR Study. J. Catal. 2002, 207, 317–330. 10.1006/jcat.2002.3556. [DOI] [Google Scholar]
- Brijaldo M. H.; Rojas H. A.; Martínez J. J.; Passos F. B. Effect of Support on Acetic Acid Decomposition over Palladium Catalysts. J. Catal. 2015, 331, 63–75. 10.1016/j.jcat.2015.08.019. [DOI] [Google Scholar]
- Tang M.; Larish W. A.; Fang Y.; Gankanda A.; Grassian V. H. Heterogeneous Reactions of Acetic Acid with Oxide Surfaces: Effects of Mineralogy and Relative Humidity. J. Phys. Chem. C 2016, 120, 5609–5616. 10.1021/acs.jpca.6b05395. [DOI] [PubMed] [Google Scholar]
- Kerr J. A. Bond Dissociation Energies by Kinetic Methods. Chem. Rev. 1966, 66, 465–500. 10.1021/cr60243a001. [DOI] [Google Scholar]
- Lu X.; Wang W.; Deng Z.; Zhu H.; Wei S.; Ng S.-P.; Guo W.; Wu C.-M. L. Methanol Oxidation on Ru(0001) for Direct Methanol Fuel Cells: Analysis of the Competitive Reaction Mechanism. RSC Adv. 2016, 6, 1729–1737. 10.1039/C5RA21793H. [DOI] [Google Scholar]
- Zhao P.; He Y.; Cao D. B.; Wen X.; Xiang H.; Li Y.-W.; Wang J.; Jiao H. High Coverage Adsorption and Co-Adsorption of CO and H2 on Ru(0001) from DFT and Thermodynamics. Phys. Chem. Chem. Phys. 2015, 17, 19446–19456. 10.1039/C5CP02486B. [DOI] [PubMed] [Google Scholar]
- Sun Y.-K.; Weinberg W. H. Catalytic Decomposition of Formic Acid on Ru(001): Transient Measurements. J. Chem. Phys. 1991, 94, 4587–4599. 10.1063/1.460587. [DOI] [Google Scholar]
- Toby B. H.; Avery N. R.; Anton A. B.; Weinberg W. H. Electron Energy Loss Spectroscopy of the Decomposition of Formic Acid on Ru(111). Stud. Surf. Sci. Catal. 1983, 14, 317–321. 10.1016/S0167-2991(09)61071-5. [DOI] [Google Scholar]
- Podrojková N.; Sans V.; Oriňak A.; Oriňaková R. Recent Developments in the Modelling of Heterogeneous Catalysts for CO2 Conversion to Chemicals. ChemCatChem. 2020, 12, 1802–1825. 10.1002/cctc.201901879. [DOI] [Google Scholar]
- Columbia M. R.; Thiel P. A. The Interaction of Formic Acid with Transition Metal Surfaces, Studied in Ultrahigh Vacuum. J. Electroanal 1994, 369, 1–14. 10.1016/0022-0728(94)87077-2. [DOI] [Google Scholar]
- Leitner W.; Dinjus E.; Gaßner F. Activation of Carbon Dioxide: IV. Rhodium-Catalysed Hydrogenation of Carbon Dioxide to Formic Acid. J. Organomet. Chem. 1994, 475, 257–266. 10.1016/0022-328X(94)84030-X. [DOI] [Google Scholar]
- Anderson J. L.; Ding J.; Welton T.; Armstrong D. W. Characterizing Ionic Liquids On the Basis of Multiple Solvation Interactions. J. Am. Chem. Soc. 2002, 124, 14247–14254. 10.1021/ja028156h. [DOI] [PubMed] [Google Scholar]
- Yasaka Y.; Wakai C.; Matubayasi N.; Nakahara M. Controlling the Equilibrium of Formic Acid with Hydrogen and Carbon Dioxide Using Ionic Liquid. J. Phys. Chem. A 2010, 114, 3510–3515. 10.1021/jp908174s. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Mu T. Conversion of CO2 to Value-Added Products Mediated by Ionic Liquids. Green Chem. 2019, 21, 2544–2574. 10.1039/C9GC00827F. [DOI] [Google Scholar]
- Zhang S.; He L.-N. Capture and Fixation of CO2 Promoted by Guanidine Derivatives. Aust. J. Chem. 2014, 67, 980–988. 10.1071/CH14125. [DOI] [Google Scholar]
- Shukla S. K.; Khokarale S. G.; Bui T. Q.; Mikkola J.-P. T. Ionic Liquids: Potential Materials for Carbon Dioxide Capture and Utilization. Front. Mater. 2019, 6, 42. 10.3389/fmats.2019.00042. [DOI] [Google Scholar]
- Brar N. K.; Brown R. T.; Shahbaz K.; Hunt P. A.; Weber C. C. Guanidinium Solvents with Exceptional Hydrogen Bond Donating Abilities. Chem. Commun. 2022, 58, 3505. 10.1039/D1CC06938A. [DOI] [PubMed] [Google Scholar]
- Rauber D.; Philippi F.; Becker J.; Zapp J.; Morgenstern B.; Kuttich B.; Kraus T.; Hempelmann R.; Hunt P.; Welton T.; Kay C. W. M. Anion and Ether Group Influence in Protic Guanidinium Ionic Liquids. Phys. Chem. Chem. Phys. 2023, 25, 6436–6453. 10.1039/D2CP05724G. [DOI] [PubMed] [Google Scholar]
- Bordet A.; Leitner W. Metal Nanoparticles Immobilized on Molecularly Modified Surfaces: Versatile Catalytic Systems for Controlled Hydrogenation and Hydrogenolysis. Acc. Chem. Res. 2021, 54, 2144–2157. 10.1021/acs.accounts.1c00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordet A.; Moos G.; Welsh C.; License P.; Luska K. L.; Leitner W. Molecular Control of the Catalytic Properties of Rhodium Nanoparticles in Supported Ionic Liquid Phase (SILP) Systems. ACS Catal. 2020, 10, 13904–13912. 10.1021/acscatal.0c03559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis Anandaraj S. J.; Kang L.; DeBeer S.; Bordet A.; Leitner W. Catalytic Hydrogenation of CO2 to Formate Using Ruthenium Nanoparticles Immobilized on Supported Ionic Liquid Phases. Small 2023, 19, e2206806 10.1002/smll.202206806. [DOI] [PubMed] [Google Scholar]
- Chipurici P.; Vlaicu A.; Calinescu I.; Vinatoru M.; Busuioc C.; Dinescu A.; Ghebaur A.; Rusen E.; Voicu G.; Ignat M.; Diacon A. Magnetic Silica Particles Functionalized with Guanidine Derivatives for Microwave-Assisted Transesterification of Waste Oil. Sci. Rep. 2021, 11, 17518. 10.1038/s41598-021-97097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbino J. M.; de Menezes E. W.; Benvenutti E. V.; Cataluña R.; Ebeling G.; Dupont J. Silica-Supported Guanidine Catalyst for Continuous Flow Biodiesel Production. Green Chem. 2011, 13, 3111–3116. 10.1039/c1gc15727b. [DOI] [Google Scholar]
- Lipp J.; Banerjee R.; Patwary M. F.; Patra N.; Dong A.; Girgsdies F.; Bare S. R.; Regalbuto J. R. Extension of Rietveld Refinement for Benchtop Powder XRD Analysis of Ultrasmall Supported Nanoparticles. Chem. Mater. 2022, 34, 8091–8111. 10.1021/acs.chemmater.2c00101. [DOI] [Google Scholar]
- Kacem S.; Emondts M.; Bordet A.; Leitner W. Selective Hydrogenation of Fluorinated Arenes using Rhodium Nanoparticles on Molecularly Modified Silica. Catal. Sci. Technol. 2020, 10, 8120–8126. 10.1039/D0CY01716G. [DOI] [Google Scholar]
- Rohmann K.; Kothe J.; Haenel M. W.; Englert U.; Hölscher M.; Leitner W. Hydrogenation of CO2 to Formic Acid with a Highly Active Ruthenium Acriphos Complex in DMSO and DMSO/Water. Angew. Chem., Int. Ed. 2016, 55, 8966–8969. 10.1002/anie.201603878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos A. R.; Blundell R. K.; Licence P. XPS of Guanidinium Ionic Liquids: a Comparison of Charge Distribution in Nitrogenous Cations. Phys. Chem. Chem. Phys. 2015, 17, 11839–11847. 10.1039/C5CP01069A. [DOI] [PubMed] [Google Scholar]
- Drozdov F. V.; Kotov V. M. Guanidine: a Simple Molecule with Great Potential: from Catalysts to Biocides and Molecular Glues. INEOS Open 2020, 3, 200–213. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.