

Abstract
Extracellular vesicles (EVs) are pivotal in intercellular communication, impacting diverse physiological and pathological processes. Current in vitro EV biogenesis studies often utilize pharmacological inhibitors, inducing off‐target effects and overlooking cell‐specific production nuances. Addressing these limitations, we utilized CRISPR/Cas9 to generate heterozygous full‐body and conditional sphingomyelin phosphodiesterase 3 (Smpd3) knockout (KO) transgenic mice. Smpd3, also known as neutral sphingomyelinase 2 (nSMase2), triggers membrane curvature through sphingomyelin hydrolysis to ceramide, thereby influencing exosome release. Intriguingly, Smpd3 deficiency demonstrated no impact on EV release both in vitro and in vivo, underscoring its potential cell‐type‐specific role in EV biogenesis. Notably, bone marrow derived macrophages (BMDMs) did exhibit reduced EV release upon Alix deletion. Our findings open avenues for subsequent inquiries, enriching our knowledge of EV biogenesis and illuminating intercellular communication in health and disease.
Keywords: ALG‐2‐interacting protein X, Alix, extracellular vesicles, neutral sphingomyelinase 2, nSMase2, Smpd3, sphingomyelin phosphodiesterase 3
1. INTRODUCTION
Extracellular vesicles (EVs) are nanosized membrane particles released by a broad variety of cell types, acting as important mediators of intercellular communication in both physiological and pathological conditions (EL Andaloussi et al., 2013; Gurung et al., 2021; van Niel et al., 2018). EV research is complicated due to the broad heterogeneity of EVs, as their size and composition rely on the cell of origin and the pathway(s) by which they are produced (EL Andaloussi et al., 2013; Gurung et al., 2021; van Niel et al., 2018). EVs are generally subdivided into two major subtypes, namely exosomes and ectosomes (or microvesicles), based on their way of biogenesis. On the one hand, exosomes are formed as a part of the endosomal pathway, initiated by the formation of intraluminal vesicles (ILVs) within multivesicular bodies (MVBs). Subsequently, ILVs are released as EVs into the extracellular environment via fusion of MVBs with the plasma membrane. The formation of ILVs can occur via different mechanisms, depending on the involvement of endosomal sorting complexes required for transport (ESCRT) (e.g., TSG101, STAM, and ALIX), or independent of ESCRT, which can be mediated by lipids (e.g., via neutral sphingomyelinases (nSMases)) or proteins of the tetraspanins family (e.g., CD63), amongst others (Abels & Breakefield, 2016; Gurung et al., 2021; Teng & Fussenegger, 2020; van Niel et al., 2018; Verderio et al., 2018). On the other hand, microvesicles originate directly from the plasma membrane via outwards membrane budding (EL Andaloussi et al., 2013; Gurung et al., 2021; van Niel et al., 2018).
EV studies often rely on the use of pharmacological EV biogenesis inhibitors to gather fundamental insights into the functional consequences of EVs and their applicability as disease biomarkers. Such inhibitors can affect one or more EV biogenesis pathways and therefore, the level of EVs produced by cells in vitro or present in biofluids in vivo can be targeted. For example, GW4869 is a pharmacological inhibitor that is widely used in EV research as it acts on neutral sphingomyelinase (nSMase) enzymes (Luberto et al., 2002; Shamseddine et al., 2015). These enzymes catalyse the conversion of membrane lipid sphingomyelin to ceramide, creating a local change of membrane fluidity and spontaneous membrane curvature (Jarsch et al., 2016; Verderio et al., 2018). More specifically, nSMases are reported as key players in ESCRT‐independent ILV formation (Trajkovic et al., 2008) and regulate fusion of MVBs with the plasma membrane (Choezom & Gross, 2022). Furthermore, inhibition of nSMases was shown to alter cargo and metabolite composition of EVs (Menck et al., 2017). Of all four mammalian nSMase enzymes that are described, nSMase2, encoded by the Smpd3 gene, is the most prominent and primarily studied nSMase (Airola & Hannun, 2013; Shamseddine et al., 2015). Hence, nSMase2 is believed to be the main nSMase that is predominantly involved in ESCRT‐independent exosome generation. This is supported by the observation that EV reduction can be achieved through general inhibition of nSMases with GW4869, as well as by specific silencing of Smpd3 expression via siRNA knockdown (Choezom & Gross, 2022; Menck et al., 2017; Mittelbrunn et al., 2011; Trajkovic et al., 2008; Yuyama et al., 2012). GW4869‐mediated nSMase inhibition was shown to block exosome generation and reduce EV release in vitro and in vivo. GW4869‐mediated EV reduction has been described in various cell types, including but not limited to astrocytes (Dinkins et al., 2014), neurons (Dinkins et al., 2014; Yuyama et al., 2012), oligodendrocytes (Trajkovic et al., 2008), T‐cells (Mittelbrunn et al., 2011), macrophages (Xu et al., 2016), HeLa cells (Choezom & Gross, 2022) and HEK cells (Kosaka et al., 2010). Also, systemic administration of GW4869 in mice was reported to lower EV levels in body fluids such as the blood and cerebrospinal fluid (CSF) (Balusu et al., 2016; Dinkins et al., 2014; Vandendriessche et al., 2021). Recently, a set of new nSMase inhibitors was discovered (i.e., DPTIP, PDDC, cambinol), showing improved pharmacokinetic and pharmacodynamic characteristics. These new pharmacological inhibitors are also reported to reduce EV levels in vitro and in vivo (Figuera et al., 2015; Rojas et al., 2018, 2019). Conclusively, the conventional wisdom in the research field is that nSMases are generally involved in EV biogenesis and that their inhibition reduces EV production.
The use of pharmacological inhibitors to influence EV production and/or release has a number of disadvantages. For example, pharmacological inhibitors embody off‐target effects that are not directly linked to EVs. Furthermore, they do not allow targeting EV processes in specific cell types when administered systemically for in vivo studies. To overcome the drawbacks of pharmacological inhibitors in EV research, we generated both a heterozygous full body Smpd3 knockout (KO) and conditional Smpd3 KO transgenic mouse line. Similar to other Smpd3 KO models, prenatal mortality was observed in our homozygous Smpd3 deficient mice. In our study, we used both Smpd3 +/− and Smpd3 fl /fl mice crossed with cell‐specific Cre recombinase expression mouse lines to characterize EV production and release in vivo and by specific cell types, namely MCC and BMDMs. The choice for MCC is based on the predominant expression of Smpd3 in the brain whereby neurons and glial cells are well‐known EV producing cell types (Ahmad et al., 2022), whereas BMDMs were selected as a cell type unrelated to the nervous system and with a potential impact on plasma EV levels. Indeed, immune cells including monocytes are reported as EV sources in the blood in both health and disease (Burgelman et al., 2021). With our study, we aimed to validate the applicability of these mouse lines as models to study the functional effects of EVs in physiological and pathological settings. In parallel, we evaluated the effect of Alix deficiency on released EV levels.
2. MATERIALS AND METHODS
2.1. Mice
Mice were housed with 14‐ to 10‐h light and dark cycles and free access to food and water in specific pathogen‐free (SPF) conditions. All experiments comply with the current laws of Belgium and were approved by the animal ethics committee of Ghent University. Smpd3 knockout (KO) (Smpd3 +/−; C57BL/6J background) and conditional Smpd3 KO mice (Smpd3 fl/fl; C57BL/6J background) were generated in house, as described below. Alix fl/fl mice (C57BL/6N background) were provided by Rémy Sadoul from the Grenoble Institut Neurosciences (Laporte et al., 2017). In all experiments age‐ and gender‐matched wildtype littermates (aged between 6 and 15 weeks) were used.
2.2. Generation of Smpd3 +/− and Smpd3 fl/fl mice using CRISPR/Cas9
Smpd3 KO (Smpd3 +/−) and conditional Smpd3 KO mice (Smpd3 fl/fl) were generated by injecting C57BL/6J zygotes with two RNP complexes and a ssDNA megamer (iDT) repair template. One RNP was loaded with a cr/tracrRNA duplex (iDT) with protospacer 5′‐GATTCCCAGGGAATAAGGGT‐3′ targeting intron 2 of the Smpd3 gene (ENSMUSG00000031906). The other RNP was loaded with a cr/tracrRNA duplex with protospacer 5′‐CCCAGGGGATCAGCTCCACA‐ 3′ targeting intron 3 of the Smpd3 gene. The 1974 nt long ssDNA Megamer repair template contains the following sequences: 123 nt 5′ homology, loxP, EcoRV site, 83 intronic nt, exon 3, 68 nt downstream of exon 3, EcoRI site, loxP, 98 nt 3′ homology. In the Smpd3 conditional KO mice, the LoxP sites are inserted between genomic base positions at chr8:106991154–106991155 and chr8: 106992826–106992827, respectively. The ssDNA Megamer also introduced a silent T/C point mutation at base (chr8:106991973).
crRNAs were designed using the Crispor algorithm (Haeussler et al., 2016). One‐cell C57BL/6JRj zygotes (Janvier Labs) were microinjected with the two RNP complexes (cr/tracr at 20 ng/µL each; Cas9 protein (Protein Service Facility, IRC‐VIB) at 50 ng/µL) and ssDNA Megamer (4 ng/µL) in DNase‐ and RNase‐free micro‐injection buffer (10 mM Tris and 1 mM EDTA, pH 7.5; iDT). Injected embryos were transferred into the oviducts of day 0.5 pseudo pregnant B6CBAF1 (Janvier Labs) recipient female mice.
In the Smpd3 KO mice, the ssDNA Megamer did not integrate. Instead, a 1788 bp deletion (chr8:106991037‐106992824) and a 2 bp insertion at the deletion site was created between both crRNA target sites. The deletion encompasses the complete exon3 (ENSMUSE00000222203) of the Smpd3 gene. This exon contains 205 nucleotides belonging to the 5′UTR and it codes for 439 of the in total 655 amino acids of the SMPD3 protein. All base annotations are according to C57BL/6J genome assembly GRCm39. When breeding the Smpd3 KO mice (Smpd3 +/− × Smpd3 +/−), no homozygous Smpd3 KO offspring was obtained, suggesting the appearance of prenatal mortality, similar to what has been reported to occur in previously generated full body Smpd3 KO models (Aubin et al., 2005; Sillence et al., 1993).
Genotyping of the Smpd3 KO mice and conditional Smpd3 mice was performed on crude DNA extracts from toe tissue samples. Primers used to genotype the mice are provided in Table 1. Confirmation of Smpd3 deletion in heterozygous Smpd3 KO mice and conditional Smpd3 KO mice, was validated in the organs and cell types of interest on DNA, RNA and protein level using deflox PCR, RT‐qPCR and western blot or protein activity assays, as described below.
TABLE 1.
Sequences of the forward and reverse primers used for deflox PCR, RT‐qPCR and genotyping PCR.
| Deflox PCR | ||
|---|---|---|
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
| Smpd3 | CACCCTCAGATCCTAACGCA | GTCGCTCCCACTCTACAGAT |
| Alix | CAGTTGATTAGATGTGTGAACTTAG | GGATACTCACCATCATGGACAAACCACTCTATGCAAGCTTCAAAACTGACTGAG |
| RT‐qPCR | ||
|---|---|---|
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
| Rpl | CCTGCTGCTCTCAAGGTT | TGGTTGTCACTGCCTCGTACTT |
| Hprt | AGTGTTGGATACAGGCCAGAC | CGTGATTCAAATCCCTGAAGT |
| Ubc | AGGTCAAACAGGAAGACAGACGTA | TCACACCCAAGAACAAGCACA |
| Gapdh | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| Smpd1 | TGGGACTCCTTTGGATGGG | CGGCGCTATGGCACTGAAT |
| Smpd2 | TGGGACATCCCCTACCTGAG | TAGGTGAGCGATAGCCTTTGC |
| Smpd3 | ACACGACCCCCTTTCCTAATA | GGCGCTTCTCATAGGTGGTG |
| Alix | TAGTGTTTGCACGGAAGACAG | GGGAGGACTGATAGGCTGGA |
| Tsg101 | TCTAACCGTCCGTCAAACTGT | TTGTACCAGTGAGGTTCACCA |
| Stam1 | ACCCCTTCGACCAGGATGTT | CCACAGTTTGATACACATGCTCC |
| Genotyping PCR | ||
|---|---|---|
| Forward (5′–3′) | Reverse (5′–3′) | |
| Smpd3+/− | TATGAAGTGAAGCCAGGGG | CCCACACAACCCCAAGAAACGTGGCCTATCACTGTTAC |
| Smpd3fl/fl | CACCCTCAGATCCTAACGCA | TGTGACTCTGGATGATCAGT |
2.3. Mixed cortical cell (MCC) cultures
MCC culturing was performed as previously described with some adaptations (Balusu et al., 2016; Vandendriessche et al., 2021). Briefly, the cerebral cortex and hippocampus were obtained from neonatal P1‐P3 Smpd3 fl/fl and Alix fl/fl mice, from which the meninges were completely removed. Tissues were finely minced with a surgical scalpel in D‐PBS (14190‐169; Sigma). Next, the samples were spun at 400 g for 5 min at room temperature (RT), after which they were digested with 0.25% trypsin (T‐4424; Sigma) in 0.04% EDTA (Merck) for 10 min in a water bath at 37°C. After digestion, the cells were centrifuged at 400 g for 5 min and washed twice with complete culture medium (DMEM (41965‐062; Gibco) supplemented with 10% foetal calf serum (Gibco); 1X penicillin/streptomycin (P4333; Sigma); 1X non‐essential amino acids (BE13‐114E; Lonza), 0.4 mM sodium pyruvate (S8636; Sigma) and 2 mM L‐glutamine (BE17‐605F; Lonza)). Typically, 3–5 × 105–6 cells were obtained from each pup, which is a sufficient number of cells to plate one 24‐well plate. Cells were resuspended in complete culture medium and plated directly in 24‐well plates coated with 0.1 mg/mL poly‐l‐lysine (P6282; Sigma). Cells were maintained in standard tissue culture conditions and 50% of the medium was replaced every two days. The presence of three different cell types was confirmed previously by staining GFAP, IBA1 and TUBB3 for astrocytes, microglia, and neurons, respectively (Balusu et al., 2016). When the cells reached confluence, they were incubated for 24 h at 37°C and 5% CO2 with either TAT‐CRE (5 µg in Opti‐MEM; 9.23 mg/mL; > 95% purity; batch JL200827; Protein Core IRC‐VIB) or Opti‐MEM medium (11058‐021; Life Technologies Europe B.V.) alone. One week after TAT‐CRE mediated deletion, the culture medium was switched to Opti‐MEM (supplemented with 1X penicillin/streptomycin (P4333; Sigma)) and incubated for 8 h in standard culture conditions. EV‐containing culture medium was collected and centrifuged at 300 × g for 5 min at 4°C for removal of dead cells, followed by supernatant collection and an additional centrifugation of 2000 × g for 10 min at 4°C to remove debris. The final supernatant was stored into low adhesion tubes at −80°C until ExoView analysis. To lyse the cells for RNA extraction, TRIzol reagent (500 µL/well; 15596018; Life Technologies Europe) was added to the cells followed by storage at −80°C until RNA isolation.
2.4. Bone marrow‐derived macrophage (BMDM) cultures
Mice were euthanized in a CO2‐chamber followed by dissection of femur and tibia bones as described previously (Amend et al., 2016). After dissection, the bones were briefly rinsed in 70% ethanol and cold D‐PBS (14190‐169; Sigma) and femur and tibia of each limb were dislodged and opened at the knee side. Next, femur and tibia bones of one limb were combined into an 18‐G perforated 0.5 mL Eppendorf with the open knee side facing downwards. The 0.5 mL tube was placed into a 1.5 mL Eppendorf and centrifuged for 1 min at 1900 × g (RT) to collect the bone marrow (BM) into the 1.5 mL collection tubes. Subsequently, the resulting BM pellets were resuspended in ACK lysis buffer (1 mL/pellet, 10–548E; Lonza) and incubated at RT for 1 min, following filtration of the cell suspension over a 70 µm cell strainer (734‐0003; VWR International). Cell suspensions obtained from one mouse were pooled and spun for 5 min at 1500 rpm at 4°C to pellet the BM cells. After counting, the BM cells were resuspended in complete DMEM medium (41965‐062; Gibco) supplemented with 10% foetal calf serum (Gibco); 1X penicillin/streptomycin (P4333; Sigma); 1X non‐essential amino acids (BE13‐114E; Lonza), 0.4 mM sodium pyruvate (S8636; Sigma), 2 mM l‐glutamine (BE17‐605F; Lonza) and 20 ng/mL murine M‐CSF (Protein Service Facility, IRC‐VIB), and seeded into untreated 9 cm petridishes at 4 to 5 × 106 cells/dish. Cells were maintained in standard tissue culture conditions and the medium was replaced for 50% three days after seeding, with complete DMEM medium to which murine M‐CSF (40 ng/mL) was freshly added. Six days after seeding, the cells were detached using accutase solution (A‐6964; Sigma), counted and reseeded in 6‐well plates at 1 × 106 cells/well. One day after reseeding, the medium was switched to Opti‐MEM medium (11058‐021; Life Technologies Europe B.V.) supplemented with 20 ng/mL M‐CSF. The culture medium was collected for ExoView analysis 8 h after switching. To remove dead cells and debris, the medium samples were subjected to two subsequent centrifugations (5 min, 300 g, 4°C; 10 min, 2000 × g, 4°C). The resulting supernatant was collected and aliquoted into low adhesion tubes and stored at −80°C until ExoView analysis. The BMDM cells were lysed with TRIzol reagent (500 µL/well; 15596018; Life Technologies Europe) and stored at −80°C until RNA isolation. To check the purity of the BMDM culture, cells were detached and processed for flow cytometry, as described below.
2.5. BMDM purity assessment with flow cytometry
BMDMs were detached with accutase solution (A‐6964; Sigma), pelleted by centrifugation and resuspended in staining mix containing anti‐CD45‐BUV805 (1:400; 748370; Becton Dickinson Benelux NV), anti‐CD11b‐BUV395 (1:200; 563553; Becton Dickinson Benelux NV), anti‐F4/80‐BV785 (1:200; 123141; BioLegend) and fixable Viability Dye eFluor 506 (1:1000; 65‐0866‐18; Life Technologies Europe B.V.) in PBS. Cells were incubated for 30 min at 4°C and subsequently resuspended in FACS buffer (PBS with 5% FCS). Cells were then analysed using an BD FACSymphony A5 with FLow Jo software (Tree Star, Ashland, OR, USA). First, single cells were selected based on the forward and side scatter (FSC‐A and SSC‐A, respectively), followed by selecting the alive cell population based on the fixable live/dead staining signal. After selecting all CD45+ cells within the alive cell population, macrophages were selected based on the expression of CD11b and F4/80. Purity assessment following the described gating strategy showed >99% purity of BMDM cultures (Figure S1).
2.6. Blood collection and plasma preparation
Mice were anesthetized with 200 µL of ketamine/xylazine (100 mg/kg ketamine; 20 mg/kg xylazine) and blood was isolated via heart puncture and collected in EDTA tubes (Microvette 500, 20.1341; Sarstedt). On average, 500 µL of whole blood could be collected per mouse. To prepare blood plasma from whole blood samples, blood‐containing EDTA tubes were centrifuged at 1300 × g for 10 min at 4°C. The supernatant was transferred to a new Eppendorf and subjected to a second centrifugation at 2400 × g for 15 min at 4°C. Finally, the supernatant was transferred to a low adhesion Eppendorf (B74030; Biozym) and stored at −80°C until ExoView analysis.
2.7. Cerebrospinal fluid (CSF) collection
CSF was collected using the cisterna magna puncture method as described earlier (Van Hoecke et al., 2021; Vandendriessche et al., 2021). Briefly, capillaries to isolate CSF were made from borosilicate glass capillary tubes (B100‐75‐15; Sutter Instruments) using the Sutter P‐87 flaming micropipette puller (pressure 330 Pa, heat index 300). Just before CSF isolation, mice were anesthetized with 200 µL of ketamine/xylazine (100 mg/kg ketamine; 20 mg/kg xylazine). The occiput was disinfected with 70% ethanol and an incision was made inferior to the occiput. The dura mater was exposed by separating the muscle tissue on the dorsal side of the skull. Next, the animal was mounted at an angle of 135° and CSF was collected by puncturing the dura mater of the cisterna magna using the capillary needle. On average, 5 µL of CSF could be isolated per mouse. After collection, the CSF was centrifuged at 300 × g for 5 min at 4°C for removal of cells and debris, whereafter the supernatant was transferred to a low adhesion Eppendorf (B74029; Biozym) and stored at −80°C until ExoView analysis.
2.8. ExoView analysis
The ExoView Mouse Tetraspanin Kit (EV‐TETRA‐M2; Unchained Labs) was used to determine EV quantities in CSF, plasma and culture medium according to the manufacturer's instructions. Briefly, samples were diluted in 1X Incubation solution (CSF – 1:40; plasma – 1:50; MCC medium – 1:15; BMDM medium – 1:10) and 35–50 µL of diluted sample was spotted on a CD81, CD9 and IgG antibody‐coated Lunis chip and incubated overnight (O/N) at RT in a sealed Luni washer 24‐well plate (251‐1055; Unchained Labs). Next, the chips were washed with 1X Solution A using the CW100 ExoView Chip Washer (NanoView Biosciences). For secondary detection of CD81, CD9 and CD63 tetraspanins on the EV surface, a mixture of anti‐CD81 (CF® 555), anti‐CD9 (CF® 488A) and anti‐CD63 (CF® 647) (1:500 in blocking solution) was added to the chips. After 1 h of incubation at RT, the chips were washed with 1X Solution A, 1X Solution B and ultrapure MilliQ water in the plate washer. For chip drying, the chips were mounted at a 45° angle into the Luni plate wells and the remaining liquid was drained from the wells with the plate washer. Once dry, the chips were scanned with the ExoView R100 reader and the data were analysed using the NanoViewer analysis software version 3.2.
2.9. Tissue sample isolation for DNA, RNA and protein extraction
Following anaesthesia with ketamine/xylazine (100 mg/kg ketamine; 20 mg/kg xylazine), mice were transcardially perfused with a mixture of D‐PBS (14190‐169; Sigma) and 0.2% heparin sodium (5000 IU/mL; LEO Pharma B.V.). Next, the brain was isolated, and cortex and hippocampus tissue were microscopically dissected from the brain. In parallel, ileum and lung tissue were isolated for RNA extraction, and kidney, liver and spleen were dissected for DNA analysis. All tissues were snap‐frozen in liquid nitrogen and stored at −80°C until RNA or DNA extraction.
2.10. DNA extraction and Deflox PCR
DNA was extracted from the tissues and cells of interest using the ISOLATE II Genomic DNA Kit (BIO‐52066; GC Biotech BV) according to the manufacturer's instructions. DNA concentration was measured using the Nanodrop ND‐1000 (Nanodrop Technologies, Thermo Scientific). As input for the Deflox polymerase chain reaction (PCR), 30 ng of DNA was mixed with the correct primers (Table 1) and ALLIn HS Red Taq Mastermix (HSM0350; highQu). Next, amplified DNA samples were separated on a 2% agarose (15510‐027; Life Technologies) gel for 30−40 min at 160 V and visualized with Gel Doc XR+ (Biorad).
2.11. Real time quantitative PCR (RT‐qPCR)
RNA was isolated from cortex, hippocampus, ileum and lung tissue as well as from BMDM and MCC cells using the Aurum Total RNA Mini Kit (732‐6820; Bio‐Rad) according to the manufacturer's instructions. RNA concentration was measured using the Nanodrop ND‐1000 (Nanodrop Technologies, Thermo Scientific) and cDNA was prepared using the SensiFAST cDNA synthesis kit (BIO‐65054; Bioline). Real‐time quantitative polymerase chain reaction (RT‐qPCR) was performed with the Light Cycler 480 system (Roche) using the SensiFAST SYBR No‐ROX Kit (BIO‐98002; Bioline). Expression levels of the genes of interest were normalized to three to four most stable reference genes, as determined by the geNorm Housekeeping Gene Selection Software (Vandesompele et al., 2002). Housekeeping genes Gapdh, Hprt and/or Rpl were used in case of BMDM and MCC cells, while Gapdh, Hprt, Rpl and Ubc were used for normalizing expression values for all tissue samples. Expression values were scaled as relative expression to the wildtype condition (i.e., Smpd3 +/+, Smpd3 Mye‐WT and Alix Mye‐WT). The primer sequences of the forward and reverse primers for the different genes are provided in Table 1.
2.12. nSMase activity assay
To measure nSMase activity, BMDMs (1.5 × 106 cells/well) were lysed with ice‐cold buffer containing 1% Triton X‐100, 150 mM NaCl and 25 mM Tris‐HCl (pH 7.4) 24 h after switching to Opti‐MEM medium (11058‐021; Life Technologies Europe B.V.). Next, the lysates were transferred into eppendorfs and spun at 16,260 × g for 5 min at 4°C, the supernatant was collected, and the protein concentration was measured using the Pierce BCA Protein Assay Kit (23225; Thermo Fisher Scientific) according to the manufacturer's instructions. On average, 1.6 mg/mL of protein could be retrieved from the BMDM cell lysates. nSMase activity was assessed in BMDM cell lysates with the Neutral Sphingomyelinase Activity Assay kit (K‐1800; Echelon Biosciences) according to manufacturer's instructions. For each sample, 60 µg of protein was used. All appropriate controls recommended by the manufacturer, that is, no‐sphingomyelin controls of each sample and the choline chloride for positive control, were included in the assay. The assay was incubated O/N to increase sensitivity, as described by the manufacturer.
2.13. Western blot analysis
Tissues (cortex and hippocampus) or cells (BMDMs) were lysed with RIPA buffer containing 50 mM Tris HCl pH 8.0, 150 mM NaCl (1.064.045.000; Merck), 1% NP‐40 (I3021; Sigma), 0.5 mM EDTA (ED‐500 g; Sigma) pH 8.0, 1X complete protease inhibitor cocktail (11873580001; Sigma) in PBS and protein concentration was measured using the Pierce BCA Protein Assay Kit (23225; Thermo Fisher Scientific) according to the manufacturer's instructions. When protein extraction was intended for SMPD3 western blot, 0.5% sodium deoxycholate (D6750; Sigma) was added to the lysis buffer composition.
Equal protein amounts of each sample (i.e., 85 µg protein for tissue and 30 µg for cell lysate) were mixed with 6X loading buffer (0.35 M Tris HCl pH 6.8, 10% sodium dodecyl sulphate (442444H; VWR), 35% glycerol (7122301; Biosolve), 5% β‐mercaptoethanol (441433A; Sigma), 0.5% bromophenol blue (P0126; Sigma)) and boiled for 10 min at 95°C. Consequently, proteins were separated on a 10% SDS‐PAGE gel and transferred to a 0.45 µm nitrocellulose membrane (NBA085G001EA; Perkin Elmer) via wet blotting. Next, the membrane was blocked for 1 h at RT with blocking buffer, that is, 5% skimmed milk (232100; BD) in Tris‐buffered saline (10 mM Tris HCl pH 7.4, 140 mM NaCl in bidistilled water) for SMPD3 western blot, or 1:2 Intercept Blocking Buffer (927‐70001; Li‐Cor) in PBS in case of ALIX western blot, followed by O/N incubation with primary antibody mouse anti‐SMPD3 (1:700; sc‐166637; Bio‐Connect B.V.) in blocking buffer or rabbit anti‐ALIX (1:1000; ab76608; Abcam) in 1:4 Intercept Blocking Buffer (927‐70001; Li‐cor) in PBS. After washing, the membrane was incubated with secondary antibody goat anti‐mouse IgG Dylight 800 (1:10 000; SA5‐35521; Life Technologies Europe) in 0.1% Tween20 in Tris‐buffered saline or goat anti‐rabbit IgG Dylight 800 (1:10 000; SA5‐35571; Life Technologies Europe) in 0.1% Tween20 in Tris‐buffered saline. After washing the membrane, bands were visualized using the Odyssey Fc Imager (Li‐Cor, Germany). For the detection of ACTIN, the membrane was incubated with mouse anti‐β‐actin (1:1000; MA5‐15739; Life Technologies Europe B.V.) in 0.1% Tween20 in Tris‐buffered saline for 1 h at RT. After washing, the membrane was incubated with goat anti‐mouse IgG Dylight 680 (1:10 000; 35518; Life Technologies Europe) for 1 h at RT, followed by washing of the membrane and imaging with the Odyssey Fc Imager (Li‐Cor, Germany).
3. RESULTS
3.1. Validation of Smpd3 deletion in Smpd3 +/ − mice
To investigate the impact of Smpd3 deficiency in vivo, we aimed to generate conditional Smpd3 mice (Smpd3 fl/fl) using CRISPR/Cas9 technology. As shown in Figure 1a, this approach also resulted in the generation of heterozygous full body knockout (KO) mice (Smpd3 +/−). In agreement with literature, homozygous Smpd3 KO mice were not viable. Consequently, we used Smpd3 +/‐ mice in the next analyses. First, we validated Smpd3 deficiency on RNA and protein level in several brain regions and peripheral organs of Smpd3 +/− compared to Smpd3 +/+ mice. In cortex, hippocampus and ileum tissue, Smpd3 gene expression was significantly decreased to 50% or lower in Smpd3 +/− compared to wildtype Smpd3 +/+ littermates (Figure 1b–d). In the lung, a decreased trend for Smpd3 expression in Smpd3 +/− versus Smpd3 +/+ was detected (p = 0.0668) (Figure 1e). Additionally, western blot analysis on cortex and hippocampus tissue lysates showed significantly decreased SMPD3 protein expression in Smpd3 +/− versus Smpd3 +/+ mice (Figure 1f–i).
FIGURE 1.
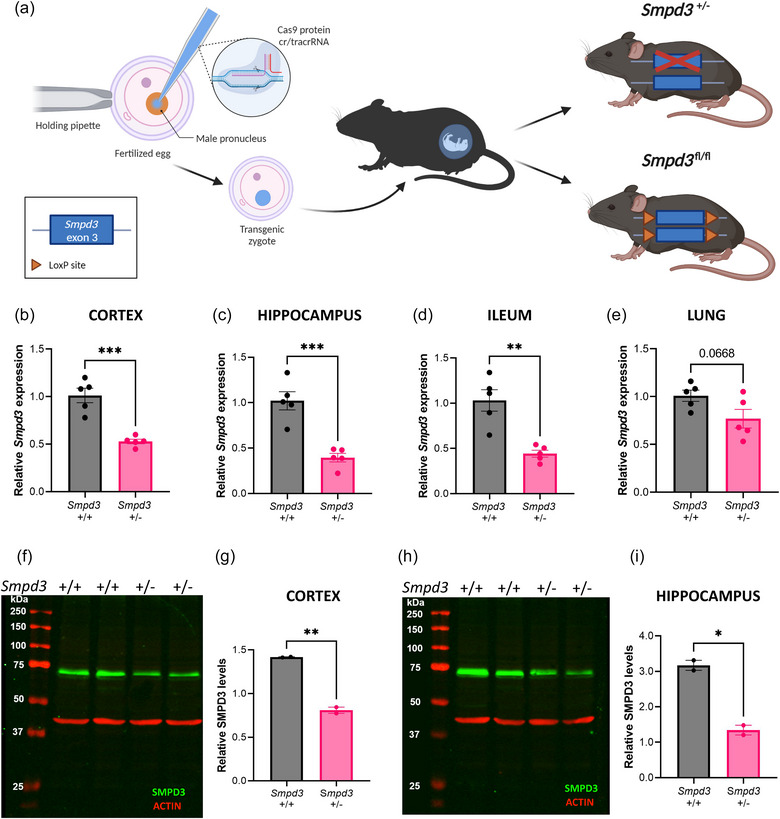
Validation of Smpd3 deficiency in heterozygous Smpd3 knockout mice (Smpd3 +/−). (a) Schematic overview of the generation of Smpd3 deficient (Smpd3 +/− and Smpd3 fl/fl) mice using CRISPR/Cas9 technology. (b–e) Smpd3 expression levels evaluated with RT‐qPCR on cortical (b), hippocampus (c), ileum (d) and lung (e) tissue samples from Smpd3 +/+ (n = 5) versus Smpd3 +/− mice (n = 5). Results are represented relative to Smpd3 +/+. (f–i) SMPD3 western blot analysis (f, h) and quantification of the SMPD3 expression (70 kDa) normalized to ACTIN (42 kDa) (g, i) in cortical (f, g) and hippocampal (h, i) brain tissue from Smpd3 +/+ (n = 2) versus Smpd3 +/− mice (n = 2). Data are represented as means ± SEM. Statistical analyses were performed by unpaired t‐test (*p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant). CRISPR, clustered regularly interspaced short palindromic repeats; cr/tracr RNA, crispr/trans‐activating crispr RNA; Smpd3, sphingomyelin phosphodiesterase 3.
3.2. EV levels in CSF and plasma of Smpd3 +/ − mice are not altered compared to Smpd3 +/ + mice
To assess the impact of SMPD3 deficiency on the amount of EVs in different biofluids, namely CSF and plasma, these biofluids were collected from Smpd3 +/− and Smpd3 +/+ mice and analysed using the ExoView technology (Figure 2). Detection of EVs with the scatter mode implies the quantification of captured EVs with a size between 50 and 200 nm, whereas EVs smaller than 50 nm can be captured with secondary fluorescent marker detection. According to the guidelines of the ExoView technology, detection of at least 500 EVs in fluorescent mode and at least 1000 EVs in label free scatter mode is stated as a reliable measurement. For both biofluids, total CD9 positive (+) EV levels, as well as CD9+/CD81+, CD9+/CD63+, CD9+/CD9+ and the label free scatter positive EVs (50–200 nm) detected on the CD9 capture spot were not significantly different between the two genotypes. Similarly, no altered EV levels were detected on the CD81 capture spot (Figure S2A–E, L–P), except for the significantly increased amount of scatter+ EVs in plasma of Smpd3 +/− mice (Figure S2P). However, the overall amount of scatter+ EVs captured on the CD81 capture was below the limit for reliable label free detection. To verify whether Smpd3 deficiency resulted in a different distribution of the tetraspanins on the released EVs, the colocalization of CD9, CD81 and CD63 was analysed in more detail (Figure S2F–K, Q–V), and in agreement with the other data, this did not reveal any significant alterations upon Smpd3 deficiency. Altogether, these data show that whole body heterozygous loss of Smpd3 expression does not affect EV levels in murine CSF and plasma.
FIGURE 2.
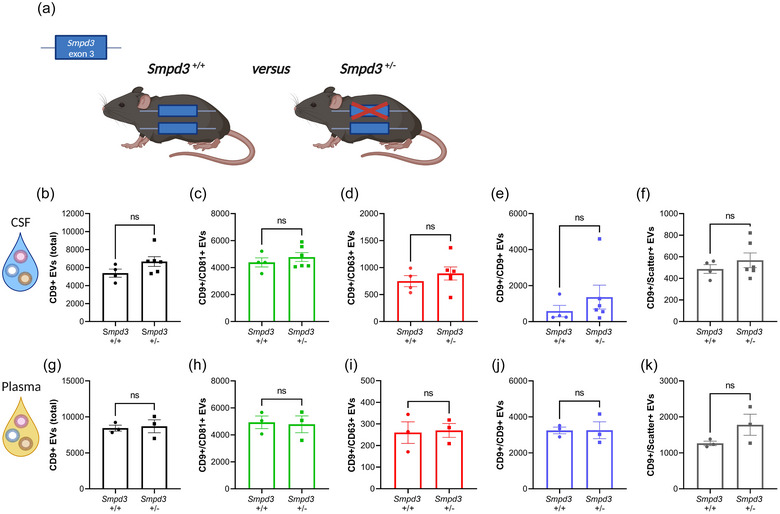
Heterozygous deletion of Smpd3 expression does not result in altered EV levels in biofluids. (a) Schematic representation of Smpd3 +/+ and Smpd3 +/− mice. (b–k) Quantification of CD9+ EVs in CSF (b–f) and plasma (g–k) from Smpd3 +/+ (n = 3–4) and Smpd3 +/− (n = 3–6) mice with the ExoView R100 platform, showing the total (b, g), CD81+ (c, h), CD63+ (d, i), CD9+ (e, j) and label‐free scatter+ (50–200 nm) (f, k) EVs, captured on the CD9 spot. Data are represented as means ± SEM. Statistical analyses were performed by unpaired t‐testing or Mann–Whitney testing (ns, not significant). CSF, cerebrospinal fluid; EV, extracellular vesicle; Smpd3, sphingomyelin phosphodiesterase 3.
3.3. Homozygous knock‐out of Smpd3 does not affect the amount of EVs released by mixed cortical cultures
Since the residual ∼50% Smpd3 expression in the Smpd3 +/ − mice might suffice to maintain ESCRT‐independent ILV production at a normal level, our next step was to achieve homozygous Smpd3 deletion in specific cell types. Thereto, we made use of our generated conditional Smpd3 mice (Smpd3 fl/fl) displayed in Figure 1a. Since Smpd3 is predominantly expressed in the brain (Hofmann et al., 2000; Sperker & Spence, 1983; Tan et al., 2018), we hypothesized that EV production by mixed cortical cells (MCCs) may be affected by Smpd3 deletion. To this end, we prepared MCCs starting from Smpd3 fl/fl pups, followed by TAT‐CRE recombinase treatment to induce deletion of the floxed Smpd3 gene fragment in the cell culture (Figure 3a). Verification of the deletion on both DNA and RNA level confirmed that Smpd3 expression was efficiently deleted upon TAT‐CRE incubation (Figure 3b,c). Next, the amount of EVs released over a time span of 8 h was compared between Smpd3 WT and Smpd3 KO MCC cultures. This showed that the total CD9+ EV levels, as well as CD9+/CD81+, CD9+/CD63+, CD9+/CD9+ and the label free scatter positive EVs (50–200 nm) detected on the CD9 capture spot were not significantly altered upon Smpd3 deletion in MCCs (Figure 3d–h). Similar effects were seen with EVs captured on the CD81 capture spot (Figure S3A–E). In addition, co‐expression of the different tetraspanins on the surface of EVs isolated from both genotypes was exactly the same (Figure S3F–K). Next, we checked whether Smpd3 deletion resulted in compensational upregulation of other genes that are known to be involved in EV biogenesis pathways and that could potentially explain the unaltered EV levels (Figure 3i–m). We selected Smpd1 and Smpd2 as members of the sphingomyelinase family together with targets that are more linked to ESCRT‐dependent EV pathways namely Alix, Tsg101 and Stam1. However, we could only observe a trend for downregulated Smpd2 expression in the Smpd3 KO MCCs. Taken together, the number of EVs released by MCCs in presence or absence of Smpd3 show that Smpd3 might not play a role in EV exosome biogenesis by cortical cells.
FIGURE 3.
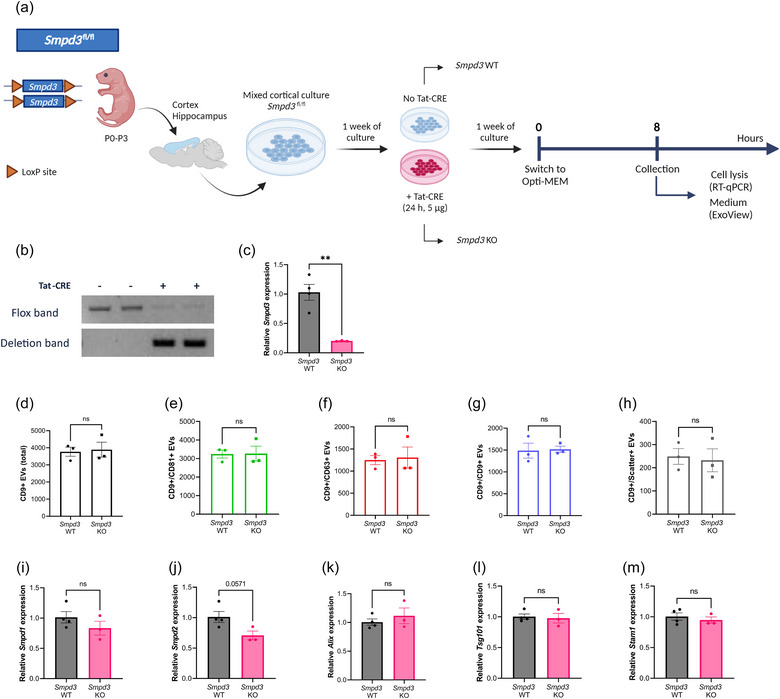
Smpd3 deletion in mixed cortical cell (MCC) cultures does not affect the level of EV production. (a) Schematic overview of the experimental set‐up for Smpd3 fl/fl MCC culture, TAT‐CRE mediated Smpd3 deletion and EV collection. (b) Deflox PCR on lysed MCC cells. The presence of the deletion band (402 bp) shows effective TAT‐CRE mediated ‘defloxing’. The flox band (2121 bp) is nearly absent in TAT‐CRE induced Smpd3 KO MCCs (Smpd3 KO) while still present in the Smpd3 WT MCCs (Smpd3 WT). (c) Smpd3 expression in Smpd3 KO MCCs analysed with RT‐qPCR, relative to Smpd3 WT MCCs. (d–h) ExoView quantification of CD9+ EVs in MCC culture medium from Smpd3 WT (n = 3) and Smpd3 KO (n = 3) MCC cultures, showing the total (d), CD81+ (e), CD63+ (f), CD9+ (g) and label‐free scatter+ (50–200 nm) (h) EVs on the CD9 capture spot. (I–M) Expression level of Smpd1 (i), Smpd2 (j), Alix (k), Tsg101 (l) and Stam1 (m) in MCCs from Smpd3 WT (n = 4) versus Smpd3 KO mice (n = 3). Results are represented relative to Smpd3 WT MCC expression values. Data are represented as means ± SEM. Statistical comparison of two groups was performed by unpaired t‐testing or a Mann–Whitney test (**p < 0.01; ns, not significant). Alix, apoptosis linked gene 2 interacting protein X; EV, extracellular vesicle; MCC, mixed cortical cell; Smpd, sphingomyelin phosphodiesterase.
3.4. Alix deficiency does not result in altered levels of EVs released by mixed cortical cells
Next, we investigated whether affecting a different EV biogenesis pathway may have more impact on EV release by these MCCs. Thereto, we analysed MCC‐derived EV production upon genetic Alix deletion (Figure 4a). Similar to Smpd3 deletion, also Alix deficiency (Figure 4b,c) had no impact on the number of released EVs (Figures 4d–h, S4A–E) or the tetraspanins colocalization of the released EVs (Figure S4F–K). We did observe a decreasing trend for the amount of scatter+ EVs on the CD9 capture spot (p = 0.07) (Figure 4h), but the amount of EVs detected was below the limit of reliable detection. In addition, expression analysis of EV‐related genes did not reveal any differences (Figure 4i–m).
FIGURE 4.
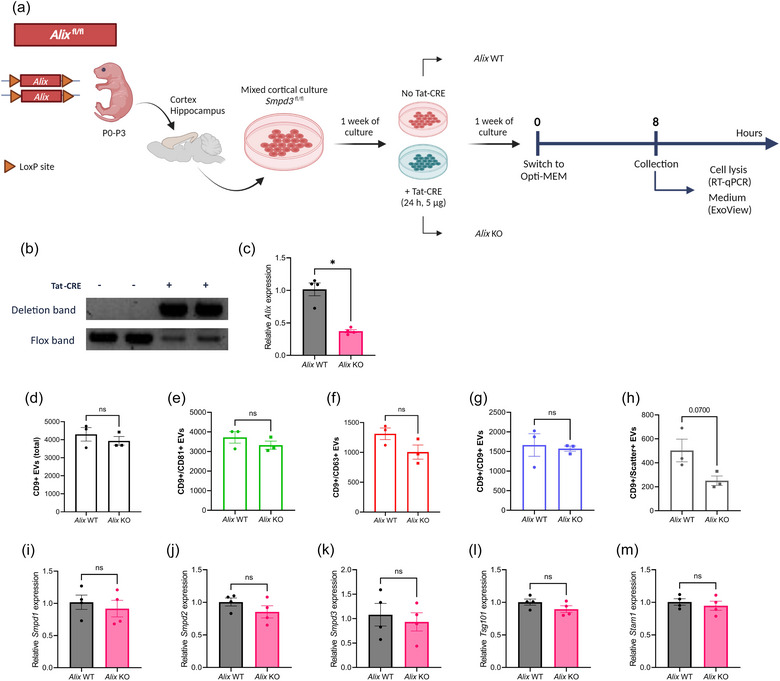
Alix deficiency in mixed cortical cell (MCC) cultures does not affect the level of EV production. (a) Schematic overview of the experimental set‐up for Alix fl/fl MCC culture, TAT‐CRE mediated Alix deletion and EV collection. (b) Deflox PCR on lysed MCC cells. The presence of the deletion band (473 bp) shows effective TAT‐CRE mediated ‘defloxing’. The flox band (368 bp) is less pronounced in Alix KO MCCs while clearly present in Alix WT MCCs. (c) Alix expression in Alix KO MCCs analyzed with RT‐qPCR, relative to Alix WT MCCs. (d–h) ExoView quantification of CD9+ EVs in MCC culture medium from Alix WT (n = 3) and Alix KO (n = 3) MCC cultures, showing the total (d), CD81+ (e), CD63+ (f), CD9+ (g) and label‐free scatter+ (50–200 nm) (h) EVs on the CD9 capture spot. (i–m) Expression level of Smpd1 (i), Smpd2 (j), Smpd3 (k), Tsg101 (l) and Stam1 (m) in MCCs from Alix WT (n = 4) versus Alix KO mice (n = 4). Results are represented relative to Alix WT MCC expression values. Data are represented as means ± SEM. Statistical comparison of two groups was performed by unpaired t‐testing or a Mann–Whitney test (*p < 0.05; ns, not significant). Alix, apoptosis linked gene 2 interacting protein X; EV, extracellular vesicle; MCC, mixed cortical cell; Smpd, sphingomyelin phosphodiesterase.
3.5. Smpd3 deletion in macrophages does not affect the amount of released EVs
To evaluate the effect of Smpd3 deficiency in a peripheral cell type, we created Smpd3 fl/fl × LysM‐Cre Tg/+ mice, thereby establishing a myeloid cell specific Smpd3 KO (Smpd3 Mye‐KO). As controls, age‐ and gender‐matched Smpd3 fl/fl × LysM‐Cre +/+ (Smpd3 Mye‐WT) littermates were used. First, we confirmed myeloid Smpd3 deficiency on isolated macrophages followed by DNA, RNA and protein analysis, using deflox PCR, RT‐qPCR and nSMase activity assay, respectively. On DNA level, the deletion band of 402 bp could be detected in BMDMs derived from Smpd3 Mye‐KO mice, while there was no detection of LoxP site(s) by means of the flox band of 2121 bp (Figure 5b). All peripheral organs from Smpd3 Mye‐KO mice showed both the deletion and flox band due to the local presence of myeloid cells (Figure 5b). RNA analysis confirmed that Smpd3 expression in Smpd3 Mye‐KO BMDMs is almost completely absent compared to Smpd3 Mye‐WT BMDMs (Figure 5c). To confirm Smpd3 deficiency on protein level, we assessed nSMase activity in BMDM cell lysate. Importantly, the performed nSMase activity assay does not exclusively measure nSMase2 activity, but also activity of other nSMase enzymes such as nSMase1 and nSMase3. In BMDM cell lysate from Smpd3 Mye‐KO mice, we observed a lower nSMase activity, illustrating that the genomic Smpd3 deletion is accompanied with a decrease in nSMase activity (Figure 5d).
FIGURE 5.
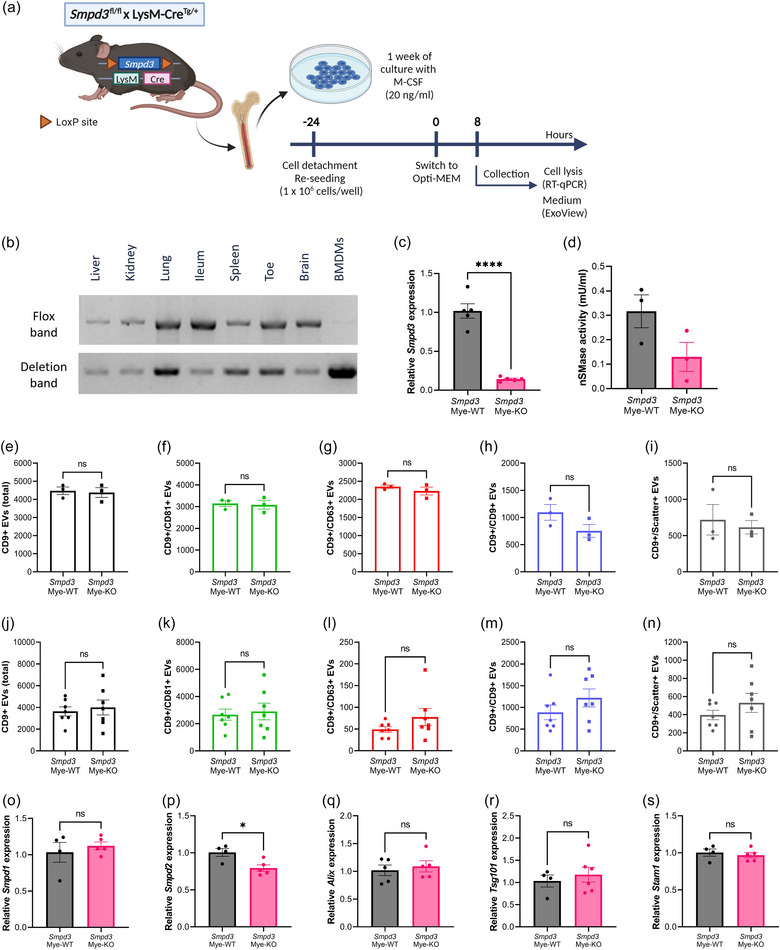
Smpd3 deletion in bone marrow‐derived macrophages (BMDMs) does not affect the amount of EV release. (a) BMDMs were differentiated from whole bone marrow from Smpd3 fl/fl × LysM‐CreTg/+ (Smpd3 Mye‐KO) mice and Smpd3 fl/fl × LysM‐Cre+/+ (Smpd3 Mye‐WT). Twenty four hours before the start of the assays, BMDMs were re‐seeded at equal cell densities. At the start of incubation, the culture medium was switched to Opti‐MEM. Cells were lysed and culture medium was collected for analyses 8 h after switching to Opti‐MEM medium. (b) Deflox PCR on several myeloid cell‐containing organs and BMDMs derived from Smpd3Mye‐KO mice. The presence of the deletion band (402 bp) shows effective Cre‐mediated ‘defloxing’. The flox band (2121 bp) is absent in BMDMs while still present in the organs. (c) Smpd3 expression in BMDMs analysed with RT‐qPCR, relative to Smpd3Mye‐WT, showing efficient KO of Smpd3 on RNA level. (d) nSMase activity assay illustrating reduced nSMase enzyme activity in Smpd3Mye‐KO mice. (e–n) ExoView quantification of CD9+ EVs in BMDM culture medium (e–i) and plasma (j–n) from Smpd3Mye‐WT (n = 3–7) and Smpd3Mye‐KO (n = 3–7) mice, showing the total (e, j), CD81+ (f, k), CD63+ (g, l), CD9+ (h, m) and label‐free scatter+ (50–200 nm) (i, n) EVs captured on the CD9 spot. (O–S) Expression level of Smpd1 (o), Smpd2 (p), Alix (p), Tsg101 (r) and Stam1 (s) in BMDMs from Smpd3Mye‐WT (n = 4–5) versus Smpd3Mye‐KO mice (n = 5–6). Results are represented relative to Smpd3Mye‐WT. Data are represented as means ± SEM. Statistical analyses were performed by unpaired t‐testing (*p < 0.05, ****p < 0.0001; ns, not significant). Alix, apoptosis linked gene 2 interacting protein X; BMDM, bone marrow‐derived macrophage; EV, extracellular vesicle; Smpd, sphingomyelin phosphodiesterase.
After confirming specific deletion of Smpd3 in BMDMs from Smpd3 Mye‐KO mice, we compared the amount of EVs released by these cells. Thereto, the medium of BMDM cells (Figure 5a) was collected after culturing for 8 h followed by ExoView analysis (Figures 5e–I, S5A–K). This revealed no significant changes in total CD9+ EV levels, as well as CD9+/CD81+, CD9+/CD63+, CD9+/CD9+ and the label free scatter positive EVs (50–200 nm) detected on the CD9 capture spot. Moreover, also plasma EV levels from the Smpd3 Mye‐KO mice did not show significant differences compared to Smpd3 Mye‐WT (Figures 5j–n, S5L–V). Because no changes in EV levels could be detected upon Smpd3 Mye‐KO, we investigated whether Smpd3 deletion mediated a compensational upregulation of other EV‐biogenesis related‐genes. There were no significant differences observed in the expression levels of Smpd1, Alix, Tsg101 and Stam1 in Smpd3 Mye‐KO compared to Smpd3 Mye‐WT (Figure 5o–s), which may suggest the absence of compensatory upregulation of the analysed alternative EV biogenesis pathways at gene expression level. However, we did detect significant Smpd2 downregulation in Smpd3 Mye‐KO derived BMDMs. Taken together, these results indicate that Smpd3 deletion does not impact the amount of EVs released by macrophages.
3.6. Alix deletion significantly decreases EV release by macrophages
Next, we investigated whether alternative EV biogenesis pathways may be more important for the overall EV production by macrophages. Hence, Alix fl/fl mice were crossed with LysM‐CreTg/+ mice to generate myeloid cell specific Alix KO mice (Alix Mye‐KO) (Figure 6a). First, we verified the specificity and efficiency of Alix deletion in the macrophages. As shown in Figure 6b,c, we could show defloxing at DNA level, as well as strongly downregulated Alix gene expression in the BMDMs from Alix Mye‐KO mice. Moreover, western blot analysis on BMDM cell lysate showed that ALIX protein expression was completely absent in Alix Mye‐KO derived BMDMs (Figure 6d). Next, we performed ExoView analysis on BMDM culture medium from Alix Mye‐WT and Alix Mye‐KO cultures 8 h after incubation (Figure 6a,e–i). This analysis revealed that the total amount of CD9‐captured EVs, as well as CD9+/CD81+, CD9+/CD63+ and CD9+/CD9+ EV numbers were significantly decreased in Alix Mye‐KO BMDM medium compared to Alix Mye‐WT. In addition, also the level of label free, scatter positive EVs (50–200 nm) captured on the CD9 spot showed a decreased trend for Alix Mye‐KO versus Alix Mye‐WT (p = 0.0588). The same trends could be observed on the EVs captured by the CD81 capture spot (Figure S6A–E). Moreover, Alix deficiency resulted in a significant decrease of CD9+/CD63+ double positive EVs, CD9+/CD81+/CD63+ triple positive EVs as well as a small but significant decrease in EVs that were only positive for tetraspanin CD9 (Figure S6F–K). In contrast, ExoView analysis of plasma of Alix Mye‐WT and Alix Mye‐KO mice did not show differences in EV numbers (Figures 6j–n, S6L–P) or tetraspanin colocalization (Figure S6Q–V). Furthermore, we investigated whether other EV‐biogenesis related genes (i.e. Smpd1, Smpd2, Smpd3, Tsg101 and Stam1) were differentially expressed as a response to Alix deletion (Figure 6o–s). No compensatory expression of other EV‐related genes upon Alix deletion was detected. Collectively, these results indicate that Alix deletion in macrophages results in a decrease in EV release, while this has no impact on the overall plasma EV levels in vivo.
FIGURE 6.
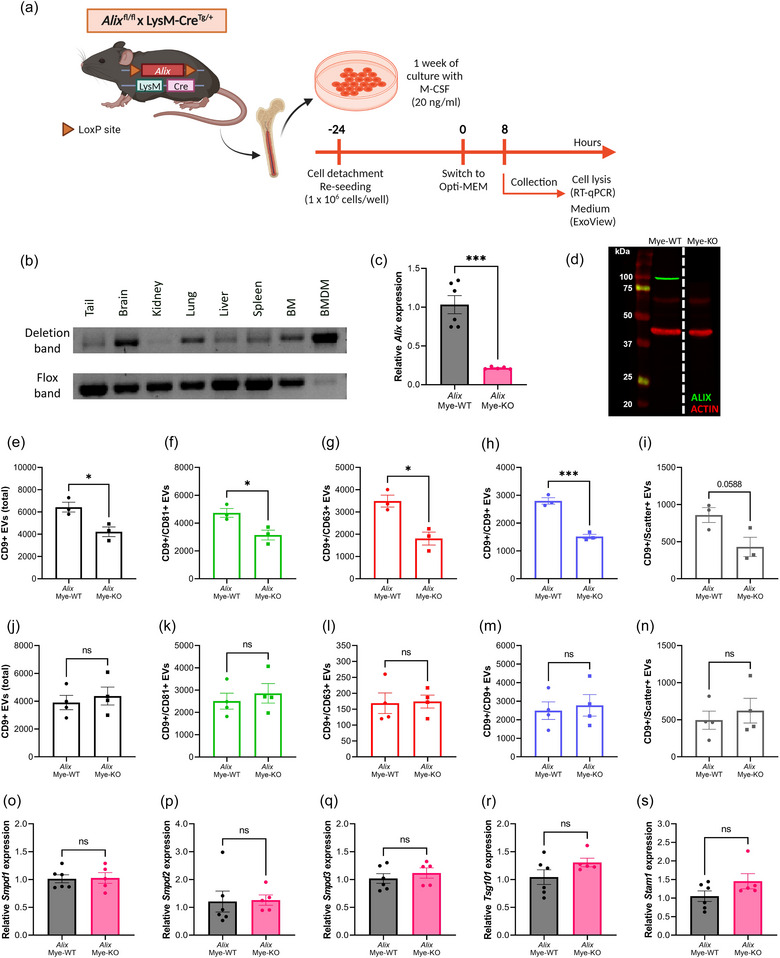
Alix deletion in bone marrow‐derived macrophages (BMDMs) results in decreased EV production. (a) BMDMs were differentiated from whole bone marrow from Alix fl/fl × LysM‐CreTg/+ (Alix Mye‐KO) mice and Alix fl/fl × LysM‐Cre+/+ (Alix Mye‐WT). At 24 h before the start of the assays, BMDMs were re‐seeded at equal cell densities. At the start of incubation, the culture medium was switched to Opti‐MEM. Cells were lysed and culture medium was collected for analyses 8 h after switching to Opti‐MEM medium. (b) Deflox PCR on several myeloid cell‐containing organs and BMDMs derived from AlixMye‐KO mice. The presence of the deletion band (473 bp) shows effective cre‐mediated ‘defloxing’. The flox band (368 bp) is merely absent in BMDMs while still present in the organs. (c) Alix expression in BMDMs analysed with RT‐qPCR, relative to AlixMye‐WT, showing efficient KO of Alix on RNA level. (d) ALIX western blot, visualizing the absence of ALIX (96 kDa) in BMDMs from AlixMye‐KO BMDM lysates. Red bands represent ACTIN protein (42 kDa) expression. (e–n) ExoView quantification of CD9+ EVs in BMDM culture medium (e–i) and plasma (j–n) from AlixMye‐WT (n = 3–4) and AlixMye‐KO (n = 3–4) mice, showing the total (e, j), CD81+ (f, k), CD63+ (g, l), CD9+ (h, m) and label‐free scatter+ (50–200 nm) (i, n) EVs captured on the CD9 spot. (O–S) Expression level of Smpd1 (o), Smpd2 (p), Smpd3 (q), Tsg101 (r) and Stam1 (s) in BMDMs from AlixMye‐WT (n = 6) versus AlixMye‐KO mice (n = 5). Results are represented relative to AlixMye‐WT. Data are represented as means ± SEM. Statistical analyses were performed by unpaired t‐testing or Mann–Whitney testing (*p < 0.05, ***p < 0.001, ns, not significant). Alix, apoptosis linked gene 2 interacting protein X; BMDM, bone marrow‐derived macrophage; EV, extracellular vesicle; Smpd, sphingomyelin phosphodiesterase.
4. DISCUSSION
Due to the drawbacks of using pharmacological inhibitors of EV production, more specifically non‐EV related off‐target effects and the inability to target specific cell types or tissues, we aimed to generate a transgenic mouse model that would allow to study the effect of EV inhibition in several physiological and pathological conditions. Given the number of studies available showing GW4869‐mediated nSMase inhibition and siRNA‐mediated Smpd3 silencing, Smpd3 was selected as suitable candidate and we generated a classical and conditional Smpd3 KO transgenic mouse line to study the effect of nSMase2‐specific deletion on EV release. However, as CSF and plasma of Smpd3 +/− mice both did not show a reduction in EV levels, we hypothesized that complete Smpd3 deficiency might be necessary to affect ESCRT‐independent EV production. Moreover, we considered the fact that the importance of the ESCRT‐independent, Smpd3‐mediated EV generation may be more relevant in certain cell types compared to others. As a consequence, while in some cell types EV production could be affected upon Smpd3 deletion, this might be masked or compensated by EV release by unaffected cell types. Therefore, we used the conditional Smpd3 fl/fl mice to achieve complete Smpd3 deletion in a mix of cortical brain cells, as well as in bone marrow‐derived macrophages (BMDMs). Unexpectedly, Smpd3 deletion had no effect on the amount of released EVs in any of the investigated cell types.
The results of our study are intriguing and raise questions regarding the generalization of nSMase2's role as regulator of exosome biogenesis. It is essential to consider that much of the pioneering research on nSMase2‐dependent effects on EV production was conducted on a limited amount of cell types or cell lines. For instance, Trajkovic et al. initially described nSMase(2)‐mediated ceramide production as involved in ESCRT‐independent biogenesis of intraluminal vesicles (ILVs) inside multivesicular bodies (MVBs) (Trajkovic et al., 2008). In this study, both pharmacological nSMase inhibition as well as siRNA‐mediated Smpd3 silencing could reduce the amount of proteolipid protein (PLP) enriched EVs, but this was specifically described for an oligodendrocyte cell line. Similarly, Menck et al. observed decreased levels of small EVs, defined as EVs obtained via ultracentrifugation at 100,000 × g, upon GW4869 treatment or Smpd3 siRNA delivery in human SKBR3 cells and mouse L‐cells, while EVs pelleted by 14,000 × g ultracentrifugation increased upon nSMase inhibition (Menck et al., 2017). A potential mechanism for participation of nSMase2 in controlling MVB fusion with the plasma membrane was recently provided by Choezom et al., demonstrating that nSMase2 regulates V‐ATPase‐mediated endosomal acidification, which in term determines MVB fate (Choezom & Gross, 2022). However, this mechanism was proposed based on results obtained by only studying HeLa cells. In addition, it is important to note that, when nSMase2 indeed controls MVB‐plasma membrane fusion, this might not exclusively affect MVBs containing ILVs formed through the nSMase2‐mediated, ESCRT‐independent pathway but may also influence the fate of MVBs with ILVs resulting from ESCRT‐dependent pathways. In our study, neither mixed cortical cells (MCCs) nor BMDMs showed decreased EV release upon Smpd3 deletion, whereas in BMDMs an EV reduction was observed upon Alix deletion. This finding may suggest that BMDM‐mediated ILV production in basal conditions is mainly dependent on ESCRT‐dependent machinery rather than ESCRT‐independent mechanisms, but this remains to be investigated further. Notably, a few studies also reported that nSMase2 blockage in some specific cell types (melanoma cells, PC‐3 cells) did not result in altered MVB biogenesis or EV release (Niel et al., 2011; Phuyal et al., 2014).
As mentioned above, there are many additional studies reporting reduced EV production by several other cell types upon nSMase(2) blockage (Balusu et al., 2016; Cheng et al., 2018; Kosaka et al., 2010; Mittelbrunn et al., 2011; Vandendriessche et al., 2021; Xu et al., 2016; Yuyama et al., 2012). However, the EV analysis methods differ strongly from our approach, as the ExoView platform has never been used before to quantify the amount of EVs present in biofluids or culture medium in the context of nSMase inhibition or deletion. Indeed, previous studies mainly use Nanoparticle Tracking Analysis (NTA) and western blot on purified EV fractions. Quantifying EVs through a limited set of EV markers using western blot is not always reliable because of potential shifts in marker abundance under different conditions and the lack of suitable reference proteins for normalization. Moreover, the technique used to isolate EVs can introduce bias towards certain EV subpopulations or co‐isolation of particular contaminants (Clos‐Sansalvador et al., 2022; Monguió‐Tortajada et al., 2019; Veerman et al., 2021). ExoView encounters the advantage to be able to analyse EV counts without prior EV isolation, since the technique is based on marker‐specific capturing of the EVs. For mouse studies, tetraspanins CD81 and CD9 are standardly used to capture EVs, while secondary detection for CD81, CD9 and CD63 is possible. Since these tetraspanins are believed to be present on all EV populations, ExoView offers efficient inspection of EVs. However, the lack of CD63 capture spots entails a risk for losing EVs which are only CD63+ or harbor very limited levels of CD81 and CD9. Indeed, there are examples illustrating that the distribution or abundance of general tetraspanin EV markers on EV populations can differ depending on the EV‐producing cell type. For example, a study on MHC‐II‐expressing HeLa cells noted that CD63 was most enriched in small EVs, while CD63 negative EVs were also present (Colombo et al., 2013). Furthermore, it is important to note that, according to the MISEV2018 guidelines, CD81 and CD63 are recognized as non‐tissue specific markers, whereas CD9 is absent on NK‐, B‐ and some MSC‐cells (Théry et al., 2018). Another ExoView‐related limitation is its restricted size range, detecting EVs between 50 and 200 nm in the label‐free scatter mode and <50–200 nm in fluorescent mode (Deng et al., 2022), thus excluding larger EVs. Therefore, we can't exclude that SMPD3 deficiency causes a potential shift in EV size distribution. Moreover, sample dilutions implemented in this study were optimized for quantification of fluorescently labelled EVs while it was not appropriate for reliable detection of EVs with label‐free quantification.
While the amount of EVs released by cells was not changed upon absence of Smpd3, we did not check intracellular effects of Smpd3 deletion on MVB and ILV formation. Since the role of Smpd3 in EV biogenesis is related to ILV formation within MVBs as well as MVB fusion with the plasma membrane (Choezom & Gross, 2022; Trajkovic et al., 2008), Smpd3 deficiency could potentially lead to intracellular MVB accumulation and/or reduced ILV generation. In addition, we did not analyse the effect of Smpd3 deficiency on the levels of its substrate sphingomyelin and its released product ceramide. Nevertheless, irrespective of potential MVB accumulation or decreased ILV formation, the results of this study indicate that this does not influence the number of EVs that are eventually released. The latter observation may also suggest that the proposed role of nSMase in favouring MVB‐plasma membrane fusion, which potentially affects MVBs containing ESCRT‐dependent and ‐independent formed ILVs, is not as important in all cell types or environmental contexts.
Importantly, we did not distinguish exosomes from ectosomes during our analyses. Up to date, there is no good discriminative marker that allows to differentiate between both EV subtypes, thus we cannot eliminate the possibility that Smpd3 deletion did affect specific EV subtypes. As mentioned above, previous research by Menck et al. showed that nSMase inhibition can differentially affect EV populations (Menck et al., 2017). They defined exosomes and ectosomes based on pelleting at differential centrifugation forces of 100,000 g and 14,000 × g, respectively, which were referred to as P100 and P14 EV fractions. Here, nSMase inhibition reduced the release of P100 ‘exosomes’ while increasing the release of P14 ‘ectosomes’ (Menck et al., 2017). While pelleted EV fractions showed overlap in size, there was more enrichment for EV markers CD81 and ALIX, in the P100 “exosome” fraction compared to the P14 ‘ectosome’ fraction. Moreover, reduced levels of CD81, ALIX and syntenin in the P100 fraction and enrichment for Wnt proteins in the P14 EV fraction were reported upon nSMase inhibition via GW4869 as well as siRNA‐mediated silencing of Smpd2 and/or Smpd3 (Menck et al., 2017). These findings illustrate the ability of nSMases to potentially mediate EV content alterations. This statement is further supported by the study of Trajkovic et al. describing that a specific subset of PLP‐containing EVs, produced by Oli‐neu cells was three times enriched for ceramide (Trajkovic et al., 2008). The production of this specific EV population was shown to depend on nSMase. While we checked whether Smpd3 deletion could mediate a shift in EV tetraspanin distribution, we did not address other potential content alterations of EVs, so it is still a possibility that, despite the lack of EV reduction upon Smpd3 deletion, EV populations show shifts into content. Furthermore, our ExoView protocol was only able to detect surface and not internal tetraspanins, which could be evaluated by adding a fixation and permeabilization step to the ExoView protocol.
In our study, we carefully examined the expression levels of other genes involved in both ESCRT‐dependent and ‐independent ILV biogenesis pathways, ruling out compensatory upregulation. Interestingly, we observed a decrease in the expression of Smpd2 in Smpd3 deficient cells. We examined whether this could be due to unspecific binding of the Smpd2 primers with Smpd3 cDNA, but BLAST analysis rules this out. Hence, the reduction in Smpd2 expression appears to be a genuine biological consequence of Smpd3 deficiency. In contrast to Smpd3, the functions of Smpd2 are less well understood, and its physiological role remains unknown. Neither Smpd2 KO nor overexpression significantly affects lipid storage or metabolism (Tallon et al., 2021; Wu et al., 2010). Importantly, its role in EV biogenesis is poorly investigated. In fact, only a few studies compared the effect of Smpd3 versus Smpd2 knock‐down on EV release. For example, the study of Menck et al. (2017) observed that siRNA‐mediated Smpd2 silencing showed similar effects to Smpd3 knock‐down in terms of EV production by SKBR3 cells, whereas Yuyama et al. (2012) reported that released EV levels were only reduced in case of Smpd3 knock‐down in neuronal N2a cells. Regarding studies using siRNA‐mediated Smpd3 silencing, it is worth noting that not all studies describe siRNA specificity for Smpd3 compared to other nSMases. Overall, further research is needed to fully understand the contribution of Smpd2 to ESCRT‐independent EV biogenesis.
Lastly, it is noteworthy that we are not the first to generate Smpd3 deficient or KO mice. For example, a spontaneous recessive Smpd3 mutation referred to as fragilitas ossium (fro) in mice significantly attenuated nSMase activity and displays a phenotype similar to a skeletal disorder namely osteogenesis imperfecta (Aubin et al., 2005; Muriel et al., 1991; Sillence et al., 1993). These so‐called fro/fro mice suffer from perinatal mortality in approximately 90% of the offspring (Sillence et al., 1993). Similarly, conditional Smpd3 KO mice were previously generated by crossing Smpd3 fl/fl mice with chondrocyte‐ and osteoblast‐specific Cre lines, successfully mimicking the skeletal phenotype of fro/fro mice (Li et al., 2016). Additionally, Stoffel et al. (2005) described Smpd3 −/− mice that exhibit severe retardation of late embryonic and postnatal growth, described as a dwarfism phenotype with skeletal defects and growth hormone deficits (Stoffel et al., 2005). However, these studies did not investigate the effect of Smpd3 deficiency on EV biogenesis. One study described that Smpd3 KO mediates impaired vesicular protein transport at the Golgi membrane in neurons, leading to the accumulation of neurotoxic proteins such as amyloid precursor protein (APP), amyloid‐beta and phosphorylated Tau, consequently leading to neuronal cell death and cognitive decline (Stoffel et al., 2018). Nonetheless, this study did not quantify EV production by neurons or in CSF of these mice.
In conclusion, our findings reveal no significant impact of Smpd3 deficiency on EV release in the cell types tested. These intriguing results suggest that the involvement of Smpd3 in EV biogenesis may be cell type or species dependent, indicating that its effects might not be universally applicable across all cellular contexts. These findings open new avenues for further research aimed at understanding the intricate interplay between EVs and their parent cells, providing valuable insights into the fundamental processes governing intercellular communication in both healthy and diseased conditions.
AUTHOR CONTRIBUTIONS
Marlies Burgelman: Conceptualization (equal); formal analysis (equal); investigation (equal); methodology (equal); visualization (lead); writing—original draft (lead). Pieter Dujardin: Conceptualization (equal); formal analysis (equal); investigation (equal); methodology (equal); writing—original draft (equal). Anthony Willems: Formal analysis (equal); investigation (equal). Tino Hochepied: Methodology (equal); resources (equal). Griet van Imschoot: Investigation (equal). Elien van Wonterghem: Investigation (equal); Lien van Hoecke: Formal analysis (equal); investigation (equal); methodology (equal). Charysse Vandendriessche: Conceptualization (equal); formal analysis (equal); methodology (equal); supervision (equal); writing—review and editing (equal). Roosmarijn E. Vandenbroucke: Conceptualization (equal); formal analysis (equal); funding acquisition (lead); methodology (equal); resources (equal); supervision (equal); writing‐review and editing (equal)
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Figure S1. Gating strategy for BMDM purity testing with flow cytometry.
Figure S2. Supplementary data of ExoView analysis of CSF and plasma of heterozygous Smpd3 KO mice.
Figure S3. Supplementary data of ExoView analysis of mixed cortical cell (MCC) culture medium of Smpd3 WT and Smpd3 KO MCC cultures.
Figure S4. Supplementary data of ExoView analysis of mixed cortical cell (MCC) culture medium of Alix WT and Alix KO MCC cultures.
Figure S5. Supplementary data of ExoView analysis of BMDM culture medium and plasma of myeloid Smpd3 KO mice.
Figure S6. Supplementary data of ExoView analysis of BMDM culture medium and plasma of myeloid Alix KO mice.
ACKNOWLEDGEMENTS
We wish to thank the VIB Transgenic Core Facility (Ghent, Belgium) for generating the Smpd3 mice and Rémy Sadoul (Grenoble Institut Neurosciences, France) for providing the Alix fl/fl mice. We also want to thank the VIB Flow Core and BioImaging Core (Ghent, Belgium) for training, support and access to the instrument park. The illustrations in this manuscript were partially created with BioRender.com. This work was supported by the Research Foundation‐Flanders (FWO Vlaanderen; 3F003118; 3F013720; 1268823N; 1295223N) and the Alzheimer Research Foundation (SAO‐FRA; 20220031).
Burgelman, M. , Dujardin, P. , Willems, A. , Hochepied, T. , Van Imschoot, G. , Van Wonterghem, E. , Van Hoecke, L. , Vandendriessche, C. , & Vandenbroucke, R. E. (2024). Challenging the conventional wisdom: Re‐evaluating Smpd3's role in extracellular vesicle biogenesis. Journal of Extracellular Biology, 3, e70015. 10.1002/jex2.70015
REFERENCES
- Abels, E. R. , & Breakefield, X. O. (2016). Introduction to extracellular vesicles: Biogenesis, RNA cargo selection, content, release, and uptake. Cellular and Molecular Neurobiology, 36(3), 301–312. 10.1007/s10571-016-0366-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, S. , Srivastava, R. K. , Singh, P. , Naik, U. P. , & Srivastava, A. K. (2022). Role of extracellular vesicles in glia‐neuron intercellular communication. Frontiers in Molecular Neuroscience, 15, 844194. 10.3389/fnmol.2022.844194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airola, M. V. , & Hannun, Y. A. (2013). Sphingolipid metabolism and neutral sphingomyelinases. Handbook of Experimental Pharmacology, 215, 57–76. 10.1007/978-3-7091-1368-4_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amend, S. R. , Valkenburg, K. C. , & Pienta, K. J. (2016). Murine hind limb long bone dissection and bone marrow isolation. Journal of Visualized Experiments: JoVE, 110, 53936. 10.3791/53936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin, I. , Adams, C. P. , Opsahl, S. , Septier, D. , Bishop, C. E. , Auge, N. , Salvayre, R. , Negre‐Salvayre, A. , Goldberg, M. , Guénet, J. L. , & Poirier, C. (2005). A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nature Genetics, 37(8), 803–805. 10.1038/ng1603 [DOI] [PubMed] [Google Scholar]
- Balusu, S. , Van Wonterghem, E. , De Rycke, R. , Raemdonck, K. , Stremersch, S. , Gevaert, K. , Brkic, M. , Demeestere, D. , Vanhooren, V. , Hendrix, A. , Libert, C. , & Vandenbroucke, R. E. (2016). Identification of a novel mechanism of blood‐brain communication during peripheral inflammation via choroid plexus‐derived extracellular vesicles. EMBO Molecular Medicine, 8(10), 1162–1183. 10.15252/emmm.201606271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgelman, M. , Vandendriessche, C. , & Vandenbroucke, R. E. (2021). Extracellular vesicles: A double‐edged sword in sepsis. Pharmaceuticals (Basel, Switzerland), 14(8), 829. 10.3390/ph14080829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Q. , Li, X. , Wang, Y. , Dong, M. , Zhan, F. H. , & Liu, J. (2018). The ceramide pathway is involved in the survival, apoptosis and exosome functions of human multiple myeloma cells in vitro. Acta Pharmacologica Sinica, 39(4), 561–568. 10.1038/aps.2017.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choezom, D. , & Gross, J. C. (2022). Neutral sphingomyelinase 2 controls exosome secretion by counteracting V‐ATPase‐mediated endosome acidification. Journal of Cell Science, 135(5), jcs259324. 10.1242/jcs.259324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clos‐Sansalvador, M. , Monguió‐Tortajada, M. , Roura, S. , Franquesa, M. , & Borràs, F. E. (2022). Commonly used methods for extracellular vesicles’ enrichment: Implications in downstream analyses and use. European Journal of Cell Biology, 101(3), 151227. 10.1016/j.ejcb.2022.151227 [DOI] [PubMed] [Google Scholar]
- Colombo, M. , Moita, C. , van Niel, G. , Kowal, J. , Vigneron, J. , Benaroch, P. , Manel, N. , Moita, L. F. , Théry, C. , & Raposo, G. (2013). Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. Journal of Cell Science, 126(Pt 24), 5553–5565. 10.1242/jcs.128868 [DOI] [PubMed] [Google Scholar]
- Deng, F. , Ratri, A. , Deighan, C. , Daaboul, G. , Geiger, P. C. , & Christenson, L. K. (2022). Single‐particle interferometric reflectance imaging characterization of individual extracellular vesicles and population dynamics. Journal of Visualized Experiments : JoVE, 179, 10.3791/62988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins, M. B. , Dasgupta, S. , Wang, G. , Zhu, G. , & Bieberich, E. (2014). Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiology of Aging, 35(8), 1792–1800. 10.1016/j.neurobiolaging.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL Andaloussi, S. , Mäger, I. , Breakefield, X. O. , & Wood, M. J. (2013). Extracellular vesicles: Biology and emerging therapeutic opportunities. Nature Reviews. Drug Discovery, 12(5), 347–357. 10.1038/nrd3978 [DOI] [PubMed] [Google Scholar]
- Figuera‐Losada, M. , Stathis, M. , Dorskind, J. M. , Thomas, A. G. , Bandaru, V. V. , Yoo, S. W. , Westwood, N. J. , Rogers, G. W. , McArthur, J. C. , Haughey, N. J. , Slusher, B. S. , & Rojas, C. (2015). Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS ONE, 10(5), e0124481. 10.1371/journal.pone.0124481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung, S. , Perocheau, D. , Touramanidou, L. , & Baruteau, J. (2021). The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Communication and Signaling: CCS, 19(1), 47. 10.1186/s12964-021-00730-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler, M. , Schönig, K. , Eckert, H. , Eschstruth, A. , Mianné, J. , Renaud, J. B. , Schneider‐Maunoury, S. , Shkumatava, A. , Teboul, L. , Kent, J. , Joly, J. S. , & Concordet, J. P. (2016). Evaluation of off‐target and on‐target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology, 17(1), 148. 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, K. , Tomiuk, S. , Wolff, G. , & Stoffel, W. (2000). Cloning and characterization of the mammalian brain‐specific, Mg2+‐dependent neutral sphingomyelinase. Proceedings of the National Academy of Sciences of the United States of America, 97(11), 5895–5900. 10.1073/pnas.97.11.5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsch, I. K. , Daste, F. , & Gallop, J. L. (2016). Membrane curvature in cell biology: An integration of molecular mechanisms. The Journal of Cell Biology, 214(4), 375–387. 10.1083/jcb.201604003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka, N. , Iguchi, H. , Yoshioka, Y. , Takeshita, F. , Matsuki, Y. , & Ochiya, T. (2010). Secretory mechanisms and intercellular transfer of microRNAs in living cells. The Journal of Biological Chemistry, 285(23), 17442–17452. 10.1074/jbc.M110.107821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte, M. H. , Chatellard, C. , Vauchez, V. , Hemming, F. J. , Deloulme, J. C. , Vossier, F. , Blot, B. , Fraboulet, S. , & Sadoul, R. (2017). Alix is required during development for normal growth of the mouse brain. Scientific Reports, 7, 44767. 10.1038/srep44767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Manickam, G. , Ray, S. , Oh, C. D. , Yasuda, H. , Moffatt, P. , & Murshed, M. (2016). Smpd3 expression in both chondrocytes and osteoblasts is required for normal endochondral bone development. Molecular and Cellular Biology, 36(17), 2282–2299. 10.1128/MCB.01077-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luberto, C. , Hassler, D. F. , Signorelli, P. , Okamoto, Y. , Sawai, H. , Boros, E. , Hazen‐Martin, D. J. , Obeid, L. M. , Hannun, Y. A. , & Smith, G. K. (2002). Inhibition of tumor necrosis factor‐induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. The Journal of Biological Chemistry, 277(43), 41128–41139. 10.1074/jbc.M206747200 [DOI] [PubMed] [Google Scholar]
- Menck, K. , Sönmezer, C. , Worst, T. S. , Schulz, M. , Dihazi, G. H. , Streit, F. , Erdmann, G. , Kling, S. , Boutros, M. , Binder, C. , & Gross, J. C. (2017). Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. Journal of Extracellular Vesicles, 6(1), 1378056. 10.1080/20013078.2017.1378056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbrunn, M. , Gutiérrez‐Vázquez, C. , Villarroya‐Beltri, C. , González, S. , Sánchez‐Cabo, F. , González, M. Á. , Bernad, A. , & Sánchez‐Madrid, F. (2011). Unidirectional transfer of microRNA‐loaded exosomes from T cells to antigen‐presenting cells. Nature Communications, 2, 282. 10.1038/ncomms1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monguió‐Tortajada, M. , Gálvez‐Montón, C. , Bayes‐Genis, A. , Roura, S. , & Borràs, F. E. (2019). Extracellular vesicle isolation methods: Rising impact of size‐exclusion chromatography. Cellular and Molecular Life Sciences: CMLS, 76(12), 2369–2382. 10.1007/s00018-019-03071-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriel, M. P. , Bonaventure, J. , Stanescu, R. , Maroteaux, P. , Guénet, J. L. , & Stanescu, V. (1991). Morphological and biochemical studies of a mouse mutant (fro/fro) with bone fragility. Bone, 12(4), 241–248. 10.1016/8756-3282(91)90070-y [DOI] [PubMed] [Google Scholar]
- van Niel , G. , Charrin, S. , Simoes, S. , Romao, M. , Rochin, L. , Saftig, P. , Marks, M. S. , Rubinstein, E. , & Raposo, G. (2011). The tetraspanin CD63 regulates ESCRT‐independent and ‐dependent endosomal sorting during melanogenesis. Developmental Cell, 21(4), 708–721. 10.1016/j.devcel.2011.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuyal, S. , Hessvik, N. P. , Skotland, T. , Sandvig, K. , & Llorente, A. (2014). Regulation of exosome release by glycosphingolipids and flotillins. The FEBS Journal, 281(9), 2214–2227. 10.1111/febs.12775 [DOI] [PubMed] [Google Scholar]
- Rojas, C. , Barnaeva, E. , Thomas, A. G. , Hu, X. , Southall, N. , Marugan, J. , Chaudhuri, A. D. , Yoo, S. W. , Hin, N. , Stepanek, O. , Wu, Y. , Zimmermann, S. C. , Gadiano, A. G. , Tsukamoto, T. , Rais, R. , Haughey, N. , Ferrer, M. , & Slusher, B. S. (2018). DPTIP, a newly identified potent brain penetrant neutral sphingomyelinase 2 inhibitor, regulates astrocyte‐peripheral immune communication following brain inflammation. Scientific Reports, 8(1), 17715. 10.1038/s41598-018-36144-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas, C. , Sala, M. , Thomas, A. G. , Datta Chaudhuri, A. , Yoo, S. W. , Li, Z. , Dash, R. P. , Rais, R. , Haughey, N. J. , Nencka, R. , & Slusher, B. (2019). A novel and potent brain penetrant inhibitor of extracellular vesicle release. British Journal of Pharmacology, 176(19), 3857–3870. 10.1111/bph.14789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamseddine, A. A. , Airola, M. V. , & Hannun, Y. A. (2015). Roles and regulation of neutral sphingomyelinase‐2 in cellular and pathological processes. Advances in Biological Regulation, 57, 24–41. 10.1016/j.jbior.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence, D. O. , Ritchie, H. E. , Dibbayawan, T. , Eteson, D. , & Brown, K. (1993). Fragilitas ossium (fro/fro) in the mouse: A model for a recessively inherited type of osteogenesis imperfecta. American Journal of Medical Genetics, 45(2), 276–283. 10.1002/ajmg.1320450227 [DOI] [PubMed] [Google Scholar]
- Sperker, E. R. , & Spence, M. W. (1983). Neutral and acid sphingomyelinases of rat brain: Somatotopographical distribution and activity following experimental manipulation of the dopaminergic system in vivo. Journal of Neurochemistry, 40(4), 1182–1184. 10.1111/j.1471-4159.1983.tb08112.x [DOI] [PubMed] [Google Scholar]
- Stoffel, W. , Jenke, B. , Blöck, B. , Zumbansen, M. , & Koebke, J. (2005). Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proceedings of the National Academy of Sciences of the United States of America, 102(12), 4554–4559. 10.1073/pnas.0406380102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel, W. , Jenke, B. , Schmidt‐Soltau, I. , Binczek, E. , Brodesser, S. , & Hammels, I. (2018). SMPD3 deficiency perturbs neuronal proteostasis and causes progressive cognitive impairment. Cell Death & Disease, 9(5), 507. 10.1038/s41419-018-0560-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallon, C. , Hollinger, K. R. , Pal, A. , Bell, B. J. , Rais, R. , Tsukamoto, T. , Witwer, K. W. , Haughey, N. J. , & Slusher, B. S. (2021). Nipping disease in the bud: NSMase2 inhibitors as therapeutics in extracellular vesicle‐mediated diseases. Drug Discovery Today, 26(7), 1656–1668. 10.1016/j.drudis.2021.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, L. H. , Tan, A. J. , Ng, Y. Y. , Chua, J. J. , Chew, W. S. , Muralidharan, S. , Torta, F. , Dutta, B. , Sze, S. K. , Herr, D. R. , & Ong, W. Y. (2018). Enriched expression of neutral sphingomyelinase 2 in the striatum is essential for regulation of lipid raft content and motor coordination. Molecular Neurobiology, 55(7), 5741–5756. 10.1007/s12035-017-0784-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, F. , & Fussenegger, M. (2020). Shedding light on extracellular vesicle biogenesis and bioengineering. Advanced Science (Weinheim, Baden‐Wurttemberg, Germany), 8(1), 2003505. 10.1002/advs.202003505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … & Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7(1), 1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic, K. , Hsu, C. , Chiantia, S. , Rajendran, L. , Wenzel, D. , Wieland, F. , Schwille, P. , Brügger, B. , & Simons, M. (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (New York, N.Y.), 319(5867), 1244–1247. 10.1126/science.1153124 [DOI] [PubMed] [Google Scholar]
- Vandendriessche, C. , Balusu, S. , Van Cauwenberghe, C. , Brkic, M. , Pauwels, M. , Plehiers, N. , Bruggeman, A. , Dujardin, P. , Van Imschoot, G. , Van Wonterghem, E. , Hendrix, A. , Baeke, F. , De Rycke, R. , Gevaert, K. , & Vandenbroucke, R. E. (2021). Importance of extracellular vesicle secretion at the blood‐cerebrospinal fluid interface in the pathogenesis of Alzheimer's disease. Acta Neuropathologica Communications, 9(1), 143. 10.1186/s40478-021-01245-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele, J. , De Preter, K. , Pattyn, F. , Poppe, B. , Van Roy, N. , De Paepe, A. , & Speleman, F. (2002). Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biology, 3(7), RESEARCH0034. 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoecke, L. , Van Cauwenberghe, C. , Dominko, K. , Van Imschoot, G. , Van Wonterghem, E. , Castelein, J. , Xie, J. , Claeys, W. , Vandendriessche, C. , Kremer, A. , Borghgraef, P. , De Rycke, R. , Hecimovic, S. , & Vandenbroucke, R. E. (2021). Involvement of the choroid plexus in the pathogenesis of niemann‐pick disease type C. Frontiers in Cellular Neuroscience, 15, 757482. 10.3389/fncel.2021.757482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel, G. , D'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nature Reviews. Molecular Cell Biology, 19(4), 213–228. 10.1038/nrm.2017.125 [DOI] [PubMed] [Google Scholar]
- Veerman, R. E. , Teeuwen, L. , Czarnewski, P. , Güclüler Akpinar, G. , Sandberg, A. , Cao, X. , Pernemalm, M. , Orre, L. M. , Gabrielsson, S. , & Eldh, M. (2021). Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. Journal of Extracellular Vesicles, 10(9), e12128. 10.1002/jev2.12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio, C. , Gabrielli, M. , & Giussani, P. (2018). Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. Journal of Lipid Research, 59(8), 1325–1340. 10.1194/jlr.R083915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, B. X. , Clarke, C. J. , & Hannun, Y. A. (2010). Mammalian neutral sphingomyelinases: Regulation and roles in cell signaling responses. Neuromolecular Medicine, 12(4), 320–330. 10.1007/s12017-010-8120-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Liu, Y. , Yang, C. , Kang, L. , Wang, M. , Hu, J. , He, H. , Song, W. , & Tang, H. (2016). Macrophages transfer antigens to dendritic cells by releasing exosomes containing dead‐cell‐associated antigens partially through a ceramide‐dependent pathway to enhance CD4(+) T‐cell responses. Immunology, 149(2), 157–171. 10.1111/imm.12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuyama, K. , Sun, H. , Mitsutake, S. , & Igarashi, Y. (2012). Sphingolipid‐modulated exosome secretion promotes clearance of amyloid‐β by microglia. The Journal of Biological Chemistry, 287(14), 10977–10989. 10.1074/jbc.M111.324616 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gating strategy for BMDM purity testing with flow cytometry.
Figure S2. Supplementary data of ExoView analysis of CSF and plasma of heterozygous Smpd3 KO mice.
Figure S3. Supplementary data of ExoView analysis of mixed cortical cell (MCC) culture medium of Smpd3 WT and Smpd3 KO MCC cultures.
Figure S4. Supplementary data of ExoView analysis of mixed cortical cell (MCC) culture medium of Alix WT and Alix KO MCC cultures.
Figure S5. Supplementary data of ExoView analysis of BMDM culture medium and plasma of myeloid Smpd3 KO mice.
Figure S6. Supplementary data of ExoView analysis of BMDM culture medium and plasma of myeloid Alix KO mice.