

Abstract
Polypyridine-ligated nickel complexes are widely used as privileged catalysts in a variety of cross-coupling reactions. The rapid adoption of these complexes is tentatively attributed to their ability to shuttle between different oxidation states and engage in electron-transfer reactions. However, these reactions are poorly understood in mechanistic terms. Here we investigate the reactivity of pseudohalide- and halide-ligated Ni(II) complexes, containing polypyridine ligands, in electron-transfer reactions. Specifically, Ni(II) halide complexes trigger comproportionation with Ni(0) with exceptional ease en route to Ni(I)Ln species, whereas the corresponding Ni(II) pseudohalide congeners are resistant to electron transfer, with Ni(I) pseudohalides being prone to disproportionation events. These observations are corroborated by electrochemical techniques and detailed quantum mechanical calculations. We also show that catalytically inactive Ni(II) pseudohalide complexes can be reactivated in the presence of exogeneous salts. From a broader perspective, this study provides rationalizations for overlooked and fundamental steps within the Ni-catalysed cross-coupling arena, thus offering blueprints for designing future Ni-catalysed reactions.
Recent years have witnessed the development of a myriad of Ni-catalysed reactions supported by polypyridine ligands as a new vehicle to forge sp2 or sp3 C–C(heteroatom) architectures. Unlike related studies based on phosphine (PR3) or N-heterocyclic carbene ligands, the exceptional ability of nickel complexes bearing polypyridine ligands to shuttle between odd and even oxidation states–including the merger of both in dual catalytic processes–represents a key contributory factor for their rapid adoption in both academic and industrial work1–5. Despite the advances realized, progress in Ni-catalysed reactions with polypyridine ligands is mainly based on empirical discoveries; indeed, these processes remain poorly understood in mechanistic terms, thus limiting innovation in this field of expertise. This observation is likely to be due to the fleeting nature of the intermediate nickel species, the ambiguity behind the exact role exerted by the polypyridine backbone and the inherent propensity for alternative, yet undesired, bimolecular pathways. A close inspection of the literature data tacitly indicates that electron-transfer events between nickel species in either comproportionation, disproportionation or reduction events supported by polypyridine backbones are poorly understood. This is somewhat surprising given that nearly all Ni-catalysed reactions based on polypyridine ligands are proposed to undergo redox changes at some stage within the catalytic cycle, thus reinforcing the notion that electron-transfer events might have a critical role in either speciation and/or catalytic turnover (Fig. 1a). Such lack of mechanistic information results in arduous screening efforts for successful reaction development, as it is difficult to substantiate or differentiate between additives that are beneficial or poisonous to productive Ni catalysis. Indeed, there is ample consensus that Ni-catalysed reactions with poor control over the speciation contribute to high catalyst loadings, low reaction rates and low catalytic turnovers, probably due to a reasonable uncertainty of the oxidation state at the nickel centre, an insufficient concentration of the key on-cycle species and the formation of unproductive off-cycle intermediates6–10. Investigations by our group have recently shown that catalytic C–O bond-functionalization reactions follow on-cycle Ni(II)/Ni(0) cycles with phosphine ancillary ligands, where generation of Ni(I) species significantly hinders catalysis11,12. However, note that the formation of Ni(I) complexes has been proven to be essential to promote catalytic carboxylation reactions with well-defined Ni(I) alkyl complexes containing polypyridine ligands, thus showing the subtleties exerted by the nature of the ligand backbone on reactivity13.
Fig. 1 |. Ambiguity in electron transfer.
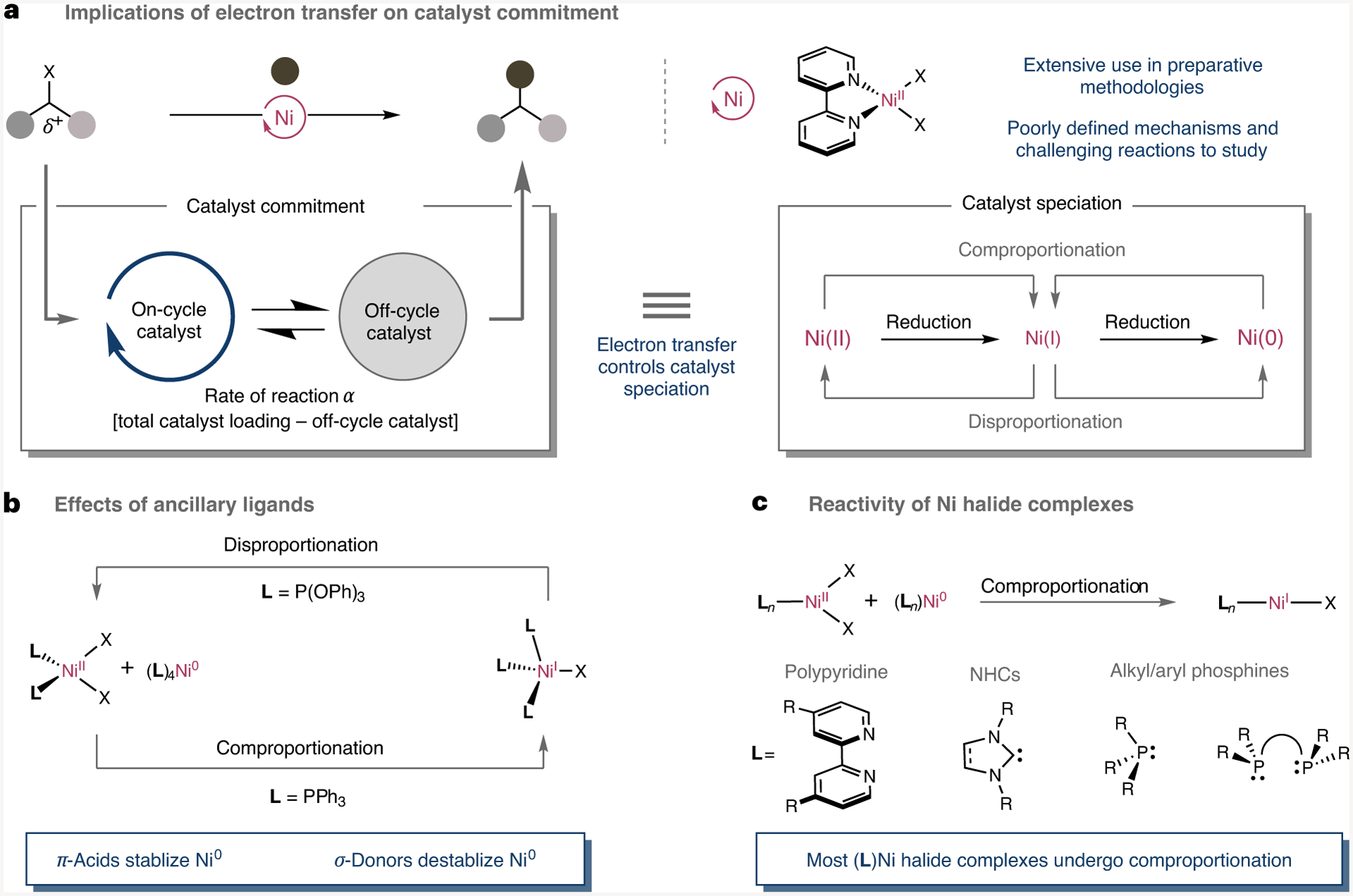
a, Implications of electron transfer on the formation of on-cycle or off-cycle nickel complexes. b, Impact of ancillary ligands on disproportionation or comproportionation. c, Comproportionation of Ni(II) halides supported by ancillary ligands. NHCs, N-heterocyclic carbenes.
Early organometallic studies dating back to the 1960s showed that comproportionation or disproportionation could be controlled by the ligand backbone, with σ-donating triphenylphosphine (PPh3) ligands promoting comproportionation reactions and π-accepting triphenyl phosphite (P(OPh)3) ligands favouring disproportionation reactions (Fig. 1b)14–18. However, in general, comproportionation reactions are favoured for Ni(II) halide complexes bearing differently substituted ligand backbones (Fig. 1c)8,14,15,17,19,20. The dichotomy exerted by the ligand backbone on catalysis has also been recognized by others in elegant work, highlighting the importance that the nickel speciation might have on reactivity by populating Ni(0), Ni(I), Ni(II), or even Ni(III) manifolds6–10,21–25. However, a close look at the literature data reveals that unravelling the mechanistic intricacies of catalytic cross-coupling reactions containing polypyridine Ni complexes represents a notoriously challenging task1,2,13,26–28. This is likely to be due to the apparent propensity of the latter to participate in one- or two-electron pathways with equal ease and the intermediacy of paramagnetic Ni(I) species and/or X-band electron paramagnetic resonance (EPR)-silent Ni(II) complexes. While elegant studies have demonstrated that polypyridyl LNi(I) and L2Ni(0) species can be accessed by either comproportionation or direct reduction with metallic reductants10,13,26,27,29–32, there still exists a lack of empirical evidence on disproportionation reactions28; indeed, speculation remains on how disproportionation, comproportionation and reduction are interconnected with polypyridine nickel complexes, an aspect of utmost relevance when designing future Ni-catalysed reactions operating via redox-transfer processes (Fig. 1b). In addition, further ambiguity exists surrounding the tendency of nickel pseudohalide complexes to share the same reactivity trends as the corresponding nickel halide species. Such an observation can hardly be underestimated given the recent popularity of C–O electrophiles as powerful alternatives to organic halides in the cross-coupling arena33–36, the enigmatic role that pseudohalide additives or base-sensitive functional groups such as alkoxides or carboxylates have in a wide variety of Ni-catalysed reactions of organic halides37–39 and the existence of Ni pseudohalide complexes as on-cycle intermediates in catalytic processes, including carboxylation reactions and reductive coupling techniques40–42. Putting all these observations into perspective, we anticipated that this ambiguity limits the full potential of Ni-catalysed reactions to maintain the propagating on-cycle intermediates while preventing the formation of alternative off-cycle species. Driven by these observations, together with the popularity exerted by polypyridine nickel complexes in medicinal chemistry programmes and their unique ability to facilitate redox processes, it was deemed necessary to study in detail the intricacies of electron-transfer events at the molecular level as it may provide the basis for future developments in the Ni-catalysed arena.
In this Article, we describe a combined experimental and computational study aimed at unravelling the fundamental factors contributing to electron-transfer events in comproportionation, disproportionation and/or reduction reactions that occur between well-defined polypyridine Ni(II), Ni(I) and Ni(0) complexes. Stochiometric comproportionation and disproportionation reactions of polypyridine-bound Ni(II) complexes reveal that complexes bearing halide ligands (Cl, Br) react via comproportionation while those bearing pseudohalide ligands (pivalate, benzoate, phenoxide) undergo spontaneous disproportionation. These findings are further supported by cyclic voltammetry studies and computational studies, which show that halide ligands stabilize the respective Ni(I) species through decreasing the spin density via π-back-donation compared to their pseudohalide counterparts. This in turn promotes comproportionation for halide-bearing complexes and disproportionation for pseudohalide-bearing complexes. We further show that reduction of Ni(II) halide complexes to Ni(0) readily occurs while their pseudohalide analogues are unreactive in reductive coupling reactions using Zn or Mn and that exogenous salt additives restore reduction to Ni(0). The implications of these findings are further extended to catalytic cross-electrophile coupling reactions in which blueprints for appropriate precatalyst and additive selection are described.
Results
Identification of model ligand systems
We began our investigations by synthesizing a series of (L)NiCl2 complexes containing either 1,10-phenanthroline (L1) or 2,2′-bipyridine (L2), the simplest ligands in the polypyridine series. It quickly became apparent that these complexes were not particularly soluble in conventional organic solvents (Fig. 2a). In addition, these species showed an inherent propensity to form mixtures of higher-order ligated nickel complexes of type (L)nNiCl2 (n = 0,1,2,3) or nickelate species such as [(L)3Ni][NiCl4] (ref.43) thus reinforcing the need to utilize a different ligand backbone (Fig. 2b). Taking these observations into consideration, we focused our attention on 1,10-phenanthrolines with substituents at the 2 and 9′ positions. The choice of these ligands is certainly not arbitrary given their rapid adoption in a myriad of Ni-catalysed site-selective cross-coupling reactions where success seems to be intimately attributed to the inclusion of such a substitution pattern at the polypyridine backbone (Fig. 2c). In sharp contrast to L1 and L2, single (L)NiCl2 species were invariably obtained for both neocuproine (L3) and bathocuproine (L4), thus indirectly highlighting the intriguing role that the substitution pattern on the ligand might have in the speciation within the catalytic cycle. The molecular structure of (L4)NiBr2 in the presence of additional L4 was characterized by X-ray crystallography, showing that the Ni atom is in a canonical tetrahedral geometry44. While (L3)NiX2 (X = Cl, Br) turned out to be insoluble in conventional solvents (L4)NiCl2 showed an improved solubility and well-defined speciation. In line with this notion, no change in the paramagnetic 1H NMR or UV-vis spectra was observed upon exposure of (L4)NiCl2 to excess amounts of L4, thus confirming both that (L4)NiCl2 exists as a single species and that these complexes meet the criteria for a well-behaved model system for studying redox-transfer events with catalytically relevant species (Fig. 2d).
Fig. 2 |. Considerations for studying Ni polypyridine complexes.
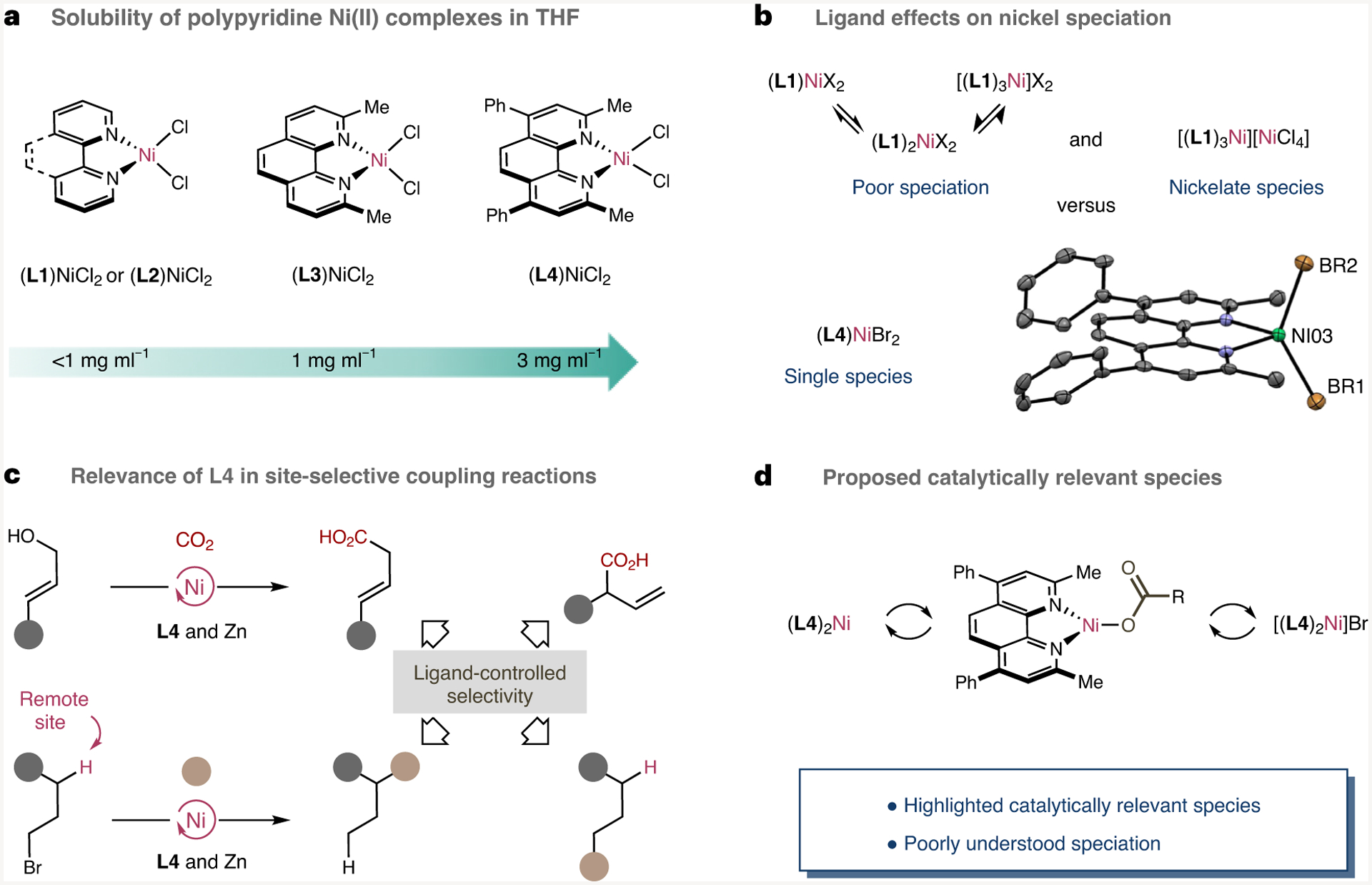
a, Effect of ligand substitution on complex solubility. b, Ligand effects on nickel speciation. c, Relevance of L4 in site-selective cross-coupling reactions. d, Catalytically relevant species in nickel-catalysed coupling reactions.
Studying comproportionation and disproportionation
Aiming at investigating in detail electron transfer in polypyridyl nickel species, a representative set of (L4)NiX2 complexes was synthesized by systematically varying the corresponding anionic ligand, with the corresponding Ni(II) alkoxide complexes being easily within reach upon exposure of the Ni(II) halide analogues to either KOPiv, PhCO2K or NaOPh (Fig. 3a). Interestingly, attempts at accessing Ni(II) alkoxide complexes by reacting LiOtBu with (L4)NiCl2 resulted in LiOtBu Ni clusters of the formal composition [NiLi3Cl2(OtBu)3]2·4THF with loss of L4, thus showing the influence that the alkoxide residue might have on Ni speciation (for X-ray diffraction see Supplementary Fig. 68). As expected, comproportionation between (L4)NiX2 (X = Cl (3a-Cl), Br (3a-Br)) and (L4)Ni(COD) (COD = cyclooctadiene) provided a rapid and reliable access to well-defined Ni(I) halide complexes [(L4)2NiI] X (X = Cl (4a-Cl), Br (4a-Br)), in which 4a-Br could be unambiguously determined by X-ray crystallography (Fig. 3b). Kinetic studies by UV-vis spectroscopy showed that the reaction occurred within seconds (Supplementary Fig. 14), consistent with rapid electron transfer where 5a formally acts as a reductant and 3a-Cl/Br acts as an oxidant to form stable Ni(I) species 4a-Cl/Br. Interestingly, a strikingly different reactivity trend was observed for (L4)Ni(OR)2 complexes (3b, R = Ph; 3c, R = COPh; 3d, R = Piv). Indeed, none of these complexes underwent reaction with (L4)2Ni(0) as judged by both EPR spectroscopy and paramagnetic 1H NMR, thus ruling out the intervention of Ni(I) species (Supplementary Figs. 1–8). To validate whether these results were specific to L4 or not, we synthesized (L3)Ni(OPiv)2 (3e), the structure of which was determined by X-ray diffraction. In line with our expectations, no reaction was observed upon exposure of 3e to (L3)2Ni (5b), thus contributing to the perception that the nature of the halide backbone exerts a much more profound impact on both speciation and reactivity than initially anticipated.
Fig. 3 |. Effect of anonic ligands on electron transfer.
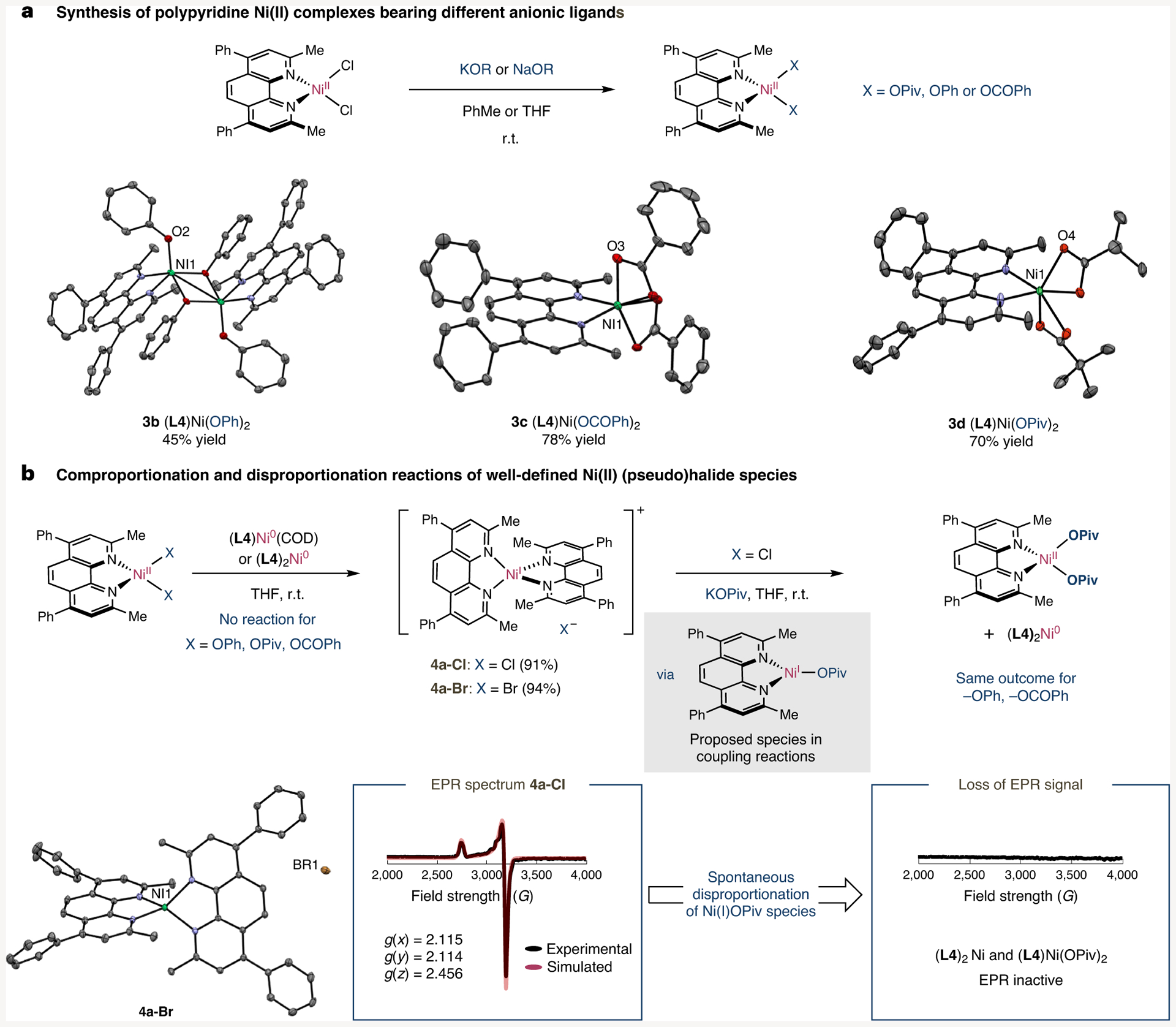
a, Synthesis of Ni(II) pseudohalide complexes [KOPiv (3 equiv.), PhMe, 16 h, r.t.; PhCO2K (2 equiv.), THF, 16 h, r.t.; NaOPh (3 equiv.), PhMe, 16 h, r.t.]. b, Investigation of the nature of the anionic ligand on comproportionation and disproportionation. COD, cyclooctadiene; OPiv, pivalate; r.t., room temperature.
Taking into consideration that reactions between Ni(0) and Ni(II) formally constitute electron-transfer events, it is reasonable to assume that they are under thermodynamic control. Therefore, if comproportionation between Ni(0) and Ni(II) carboxylate complexes does not occur, we tentatively speculated that the principle of microscopic reversibility should apply and disproportionation of Ni(I) alkoxides en route to both Ni(0) and Ni(II) should occur spontaneously. As shown in Fig. 3b, this hypothesis was indirectly confirmed by salt metathesis of 4a-Cl with KOPiv. Interestingly, not even traces of (L4)NiI(OPiv) were observed by in situ monitoring of the reaction by EPR spectroscopy. Instead, formation of (L4)Ni(OPiv)2 (3d) and (L4)2Ni(0) was observed in quantitative yields, thus contributing to the perception that rapid disproportionation of transiently generated (L4)NiI(OPiv) comes into play. An otherwise identical outcome was obtained by either utilizing solvents with higher dielectric constants such as MeCN (ε = 37.5) or by replacing KOPiv with either PhCO2K or NaOPh (Supplementary Figs. 15–22). These findings could be extended with Ni complexes containing L3 instead, forming statistical mixtures of (L3)Ni(OPiv)2 (3d) and (L3)2Ni (5b) in quantitative yields upon reaction of [(L3)2Ni] Cl (4b-Cl) with KOPiv.
Density functional theory studies on disproportionation and comproportionation
To gain insights into the distinct reactivity of nickel complexes and their ability to undergo comproportionation or disproportionation, we next turned to dispersion-corrected density functional theory (DFT) and domain-based local pair natural orbital coupled cluster method with single and double and perturbative triple excitations (DLPNO-CCSD(T)) calculations (Fig. 4 and see Supplementary Figs. 70–75 for additional information). First, we focused on the energy coordinate of comproportionation between (L4)2Ni(0) (5a) and (L4)NiCl2 (3a-Cl). As shown in Fig. 4a, an energetically viable pathway for comproportionation was found via ligand dissociation from (L4)2Ni(0) (5a) and solvent (L4) NiCl2 (3a-Cl) coordination en route to a bridging Ni(I) dimer (via 3TS1) (see Supplementary Fig. 71 for full energy diagrams). In comparison to previously isolated [(dtbbpy)Ni(I)Cl]2 dimeric species (dtbbpy = 4,4′-Di-tert-butyl-2,2′-dipyridyl), [(L4)Ni(I)Cl]2 has longer Ni–Cl and Ni–Ni distances (2.41 versus ~2.33 Å and 3.35 versus 2.68 Å) presumably due to the increased steric effects associated with methyl substituents in the bathocuproine versus bipyridine scaffold27. The corresponding [(L4)Ni(I)Cl]2 undergoes rapid dissociation (~2 kcal mol−1 barrier) to form the more thermodynamically favourable L4Ni(I)Cl monomeric species. In line with our experimental findings, comproportionation of all Ni(II) pseudohalide series was computed to be thermodynamically disfavoured by 6–8 kcal mol−1 (Fig. 4b). Despite exhaustive attempts we were not able to identify the comproportionation transition state (akin to TS1) for pseudohalide analogues. Nonetheless, these results strongly suggest that even if the barrier to undergo comproportionation is low, monomeric Ni(I) pseudohalide complexes might favour the reverse disproportionation pathway (Supplementary Fig. 72). Presumably, halide ligands decrease the spin density (via π-back-donation)45 of the monomeric LNi(I)X complexes that, in turn, stabilizes these species when compared to their parent Ni(II) and Ni(0) precursors45. This hypothesis was assessed by examining a range of pseudohalide ligands computationally (see Supplementary Fig. 74 for details). Interestingly, calculations revealed that subtle modulation of the electron properties of the pseudohalide ligand could decrease the electron density at the metal centre, thus making comproportionation thermodynamically favourable (Supplementary Figs. 74 and 75). These findings are particularly important, as they might lead to the foundation of methods for rational modulation of nickel complexes to control comproportionation and disproportionation pathways.
Fig. 4 |. Computational study of comproportionation.
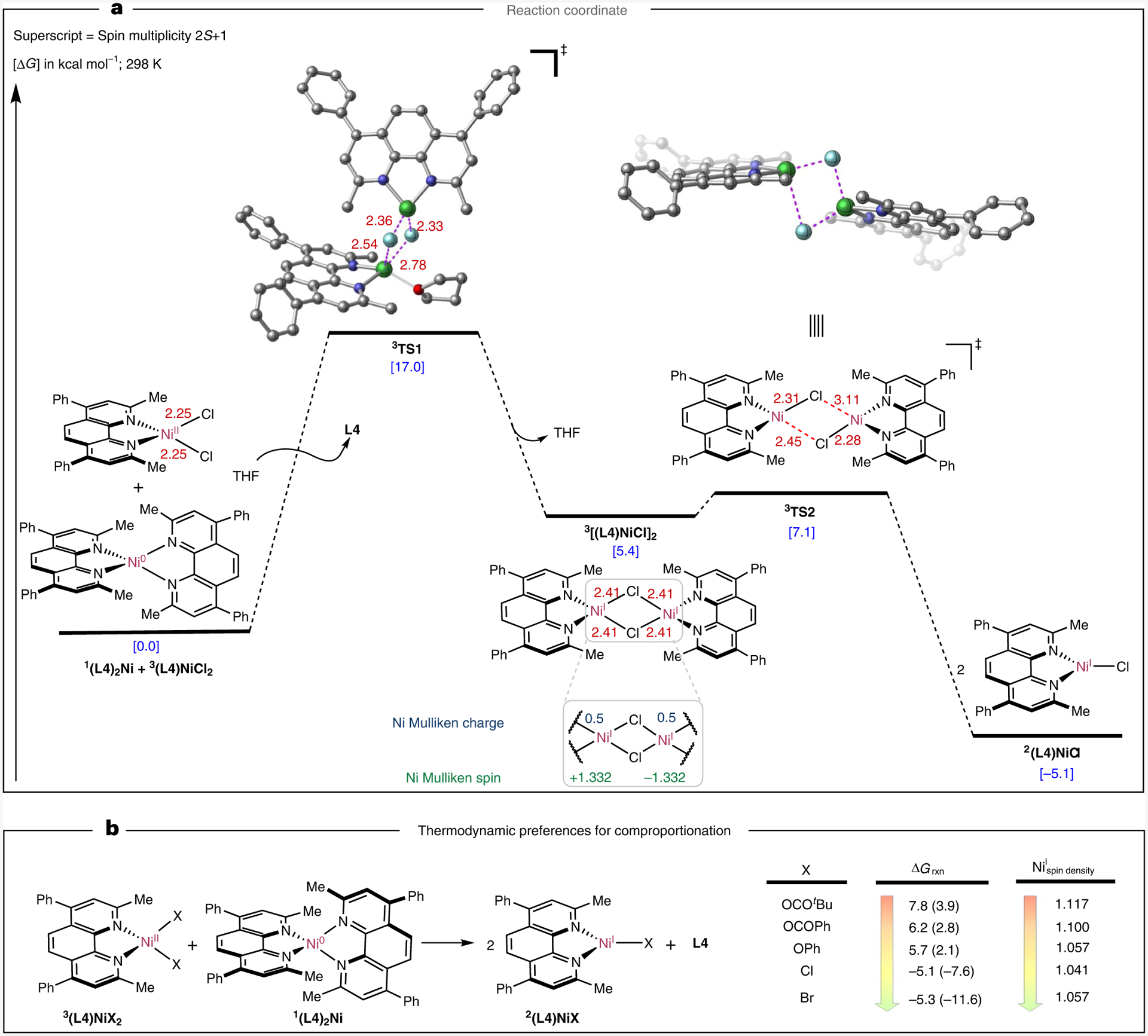
a, Lowest energy pathway computed at the UB3LYP-D3/def2tzvpp-CPCM(THF)//UB3LYP-D3/def2svp-CPCM(THF) level for comproportionation of (L4)2NiCl2. b, Computed free energies for the formation of monomeric (L4)Ni(I)-X species from the corresponding Ni(II) and Ni(0) complexes calculated at the UB3LYP-D3/def2tzvpp-CPCM(THF)//UB3LYP-D3/def2svp-CPCM(THF) and DLPNO-CCSD(T)/def2svp-CPCM(THF)//UB3LYP-D3/def2svp-CPCM(THF) (in parenthesis) levels of theory.
Electrochemical and stochiometric investigations
Having identified a strikingly divergent reactivity depending on the anionic ligand at Ni(II) and Ni(I) centres, we wondered whether these electron-transfer events could be indirectly assessed by comparing the cyclic voltammograms (CVs) of Ni(II) halide complexes to their corresponding Ni(II) carboxylate analogues. As shown in Fig. 5a, this turned out to be the case. Specifically, the CVs of (L4)NiCl2 (3a-Cl) and (L4)NiBr2 (3a-Br) resulted in two distinct, separated redox potentials of the Ni(II)/Ni(I) (Cl = −0.86 V, Br = −0.71 V versus saturated calomel electrode (SCE) in MeCN) and Ni(I)/Ni(0) (Cl = −1.24 V, Br = −1.10 V versus SCE in MeCN) couple. The separation of redox potentials and oxidation states is consistent with the ability to generate stable Ni(I) oxidation states, thus confirming that electron transfer and comproportionation between (L4)NiX2 and (L4)2Ni is particularly downhill. In striking contrast, the CV of (L4)Ni(OPiv)2 (3d) revealed that only two electron redox events occur, in which Ni(II) is directly reduced to Ni(0) (Ep/2 = −1.43 V versus SCE in MeCN; Ep/2 = half-peak potential). The lack of stable (L4)Ni(I) carboxylate complexes is consistent with our stoichiometric experiments that showed no formation of (L4)Ni(OPiv) by either disproportionation of [(L4)2Ni] Cl (4a-Cl) with KOPiv or comproportionation between (L4)2Ni and (L4)Ni(OPiv)2 (3d).
Fig. 5 |. Electrochemical studies and stoichiometric reduction studies.
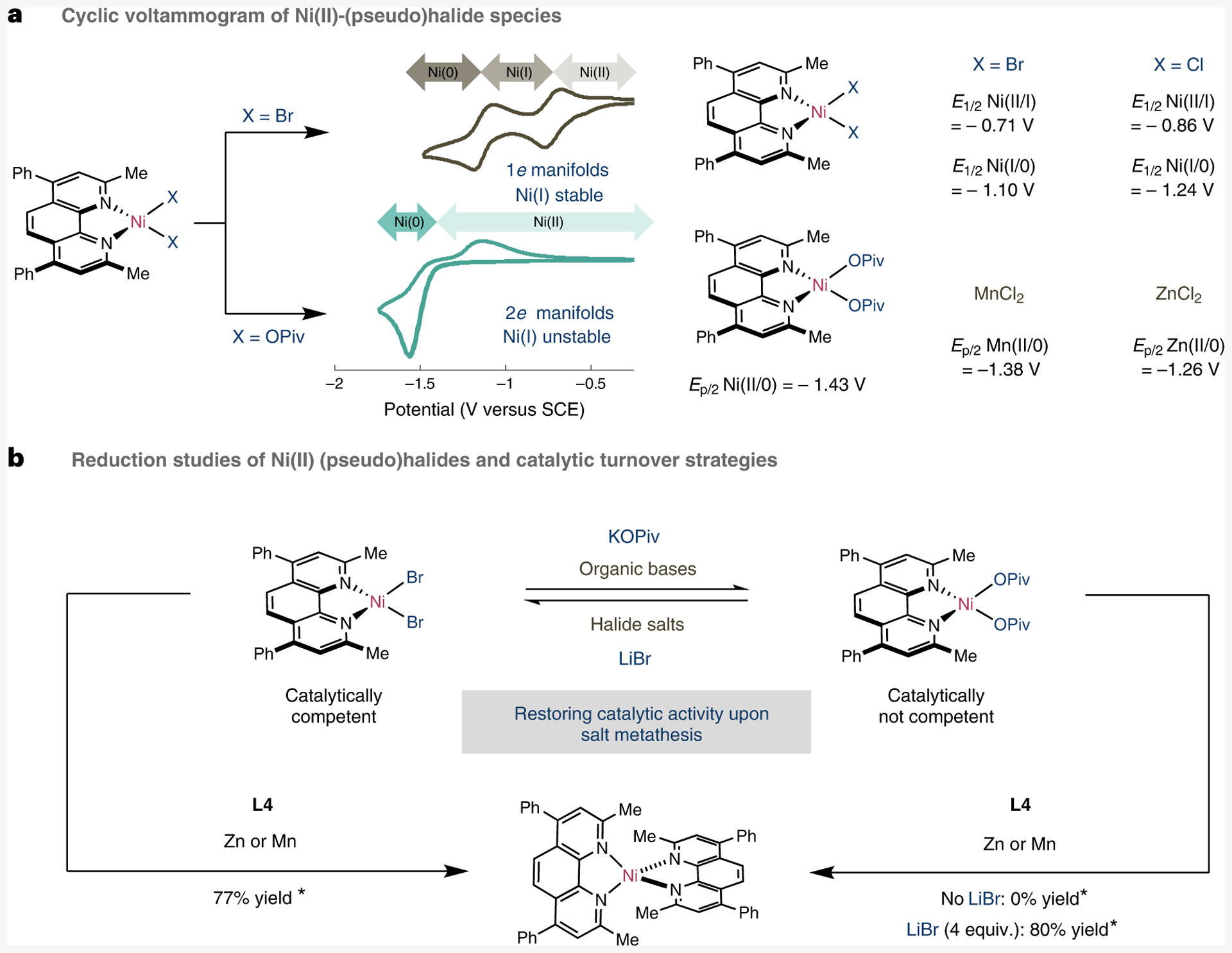
a, CVs of (L4)NiBr2 and (L4)Ni(OPiv)2 and comparison of redox potentials. b, Implications of halide exchange on reduction. *Additional L4 added to stabilize (L4)2Ni. SCE, saturated calomel electrode. E1/2 = half-wave potential; Ep/2 = half-peak potential; OPiv = Pivalate.
Given that a myriad of Ni-catalysed cross-electrophile couplings or reductive coupling reactions are commonly conducted with Mn or Zn as single-electron transfer reductants, we wondered whether the striking difference in redox potentials of Ni(II) halide and Ni(II) carboxylate complexes could be translated into a different reactivity profile in the presence of metallic reductants. According to the CVs of both ZnCl2 (Ep/2 = −1.26 V versus SCE in MeCN) and MnCl2 (Ep/2 = −1.38 V versus SCE in MeCN), we anticipated that Mn and Zn would be competent for triggering single-electron transfer reduction of (L4)Ni(II) halides en route to Ni(I) and subsequently to Ni(0); however, it was unclear whether (L4)Ni(OPiv)2 (3d) would be reduced due to its similar redox potential with either Mn or Zn (Fig. 5a). This hypothesis was corroborated by stoichiometric reduction of (L4)NiBr2 (3a-Br) with Zn or Mn in the presence of excess amounts of L4 to stabilize low-coordinate Ni species and bind Zn(II) or Mn(II) salts obtained after single-electron transfer. As anticipated (L4)2Ni was obtained equally well by using either Mn or Zn (Fig. 5b). In sharp contrast, no reaction was observed upon simple exposure of (L4)Ni(OPiv)2 (3d) to Mn or Zn; the starting Ni(II) precursor was recovered unaltered. These results indirectly suggested that formation of (L)Ni(OR)2 in Ni-catalysed reactions of C–O electrophiles or organic halides requiring pseudohalide bases operating via single-electron transfer might be particularly problematic for catalytic turnover. However, we speculated that addition of inorganic halide salts such as LiBr or ZnBr2 might be beneficial for turnover by generating (L4) NiX2 in situ (X = halide). This hypothesis was indirectly corroborated by stoichiometric studies that showed clean formation of (L4)NiBr2 (3a-Br) upon exposure of (L4)Ni(OPiv)2 (3d) to LiBr and quantitative formation of (L4)2Ni(0) (5a) by reduction of (L4)Ni(OPiv)2 (3d) with Zn in the presence of ZnBr2 or in 80% yield with LiBr. These results can hardly be underestimated, as they highlight the non-negligible impact that a priori innocent inorganic salts might have in aiding reduction events when Ni(II) pseudohalide complexes are formed during catalytic reactions. Although these findings allow the enigmatic, yet beneficial, role exerted by exogenous halide salts in a series of recent Ni-catalysed reductive coupling reactions of C–O electrophiles to be rationalized, it was unclear whether the inclusion of Zn(II) salts might populate other conceivable pathways. Indeed, recent findings from our group showed the formation of unorthodox Ni/Zn clusters via single-electron transfer and ligand-sequestering events when combining well-defined Ni(0) [PCy3] (Cy = cyclohexyl) complexes with ZnCl2 (ref.11). Interestingly, while no heterobimetallic Ni/Zn species were obtained upon exposure of (L4)2Ni to ZnCl2, we identified (L4)ZnCl2 in the crude mixtures, thus suggesting an inevitable ligand-sequestering event between hard Ni(0) and Zn(II) centres. Although tentative, we believe this divergent reactivity is likely to originate from L4 being a poor σ-donor ligand when compared to PCy3, thus making the corresponding (L4)2Ni complex a weaker reductant.
Relevance in nickel-catalysed cross-coupling reactions
Altogether, the results of Figs. 3–5 stand as a testament to the unique reactivity exerted by polypyridine nickel complexes in electron-transfer events and the relevance that these findings might have in Ni-catalysed cross-coupling reactions. Most apparent is the non-negligible influence that commonly employed pseudohalide bases and/or additives might have in catalyst speciation, inhibiting reduction from LNi(II) (pseudohalides) while preventing formation of Ni(I) species via disproportionation reactions. Driven by this perception, we turned our attention to unravel the implications that these findings might have in Ni-catalysed reductive coupling reactions operating via the intermediacy of Ni(I) complexes. The deleterious effect that pseudohalide bases have on Ni-catalysed reductive couplings was apparent by the inclusion of KOPiv after 60 minutes in the cross-electrophile coupling of alkyl bromides and aryl bromides reported by Yin based on the Ni/L4 couple (Fig. 6a–teal trace)46. As shown in Fig. 6a, a significant reduction in both reaction rate and yield was observed when compared to the standard reaction, suggesting the formation of inactive (L4)Ni(OPiv)2 species (3d). The latter observation was corroborated by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of the crude mixture, resulting in a rather distinctive m/z pattern for 3d (Supplementary Fig. 36). As expected, no reaction was observed when utilizing solely 3d as the precatalyst whereas catalytic activity was significantly restored upon addition of nBu4NBr after 90 minutes (Fig. 6a–grey trace). However, note that a lower rate was found when compared to that of the standard reaction using the NiI2/L4 couple due to the presence of pivalate anions. Similar findings were observed when studying the site-selective Ni-catalysed carboxylation of allylic alcohols with CO2 (1 atm) based on a Ni/L4 regime and Zn as metallic reductant, where addition of MgCl2 was found to be critical for success (Fig. 6b)40. In line with our expectations, removal of MgCl2 resulted in a significant loss in efficiency (15% yield). It is likely that the minor quantities of 8 formed under these conditions can be attributed to the formation of ZnBr2 upon reduction of NiBr2, allowing the formation of (L4)NiBr2 (3a-Br) upon reaction of (L4)Ni pseudohalides with ZnBr2· This observation was indirectly corroborated by the lack of reaction found when utilizing Ni(COD)2 as the precatalyst in the absence of either ZnBr2 or MgCl2 whereas reactivity was restored upon addition of the latter, resulting in 61% yield of 8. Similarly, a catalytic system based on 3a without MgCl2 resulted in no formation of 8. Extending these findings to ligand systems beyond L4 that form Ni carboxylate species on-cycle, we studied the reductive carboxylation of dienes employing 2-methyl-4,7-diphenyl-1,10-phenanthroline (L5) and bathrophenanthroline (L6). We found similar reactivity to that observed in the allylic carboxylation in which Ni(COD)2 without a halide source resulted in no carboxylated product, but reactivity could be restored upon adding inorganic halide source nBu4NBr (Supplementary Fig. 39)47. Interested in the possibility that these results may extend to other ligand systems we first compared the CVs of bipyridine-ligated nickel complexes (L2) NiBr2 and (L2)Ni(OPiv)2 (Supplementary Figs. 52 and 53)48. Supporting the notion that the investigations with L4 could be extended to L2 were the similar CVs between the analogous complexes, with (L2) NiBr2 displaying distinct Ni(II/I) and Ni(I/0) redox waves while (L2) Ni(OPiv)2 displayed a single Ni(II/0) redox wave. Turning to study the generality of these findings and the impact of forming other on-cycle pseudohalide species, we considered the reductive coupling of alkyl tosylates with aryl bromides35. Consistent with the previous findings, removal of KI from the standard conditions resulted in a significant drop in yield (Fig. 6c entries 1 and 2). Likewise, employing halide-free precatalyst Ni(COD)2 was less effective than the standard conditions but performing the reaction with Ni(COD)2 and removing KI resulted in trace product (5%). Putting all these findings into perspective, we believe these experiments do not only provide a rational explanation of the enigmatic role exerted by additives in existing Ni-catalysed reductive couplings but also offer a gateway for designing future Ni-catalysed reactions.
Fig. 6 |. Catalytic relevance of electron transfer.
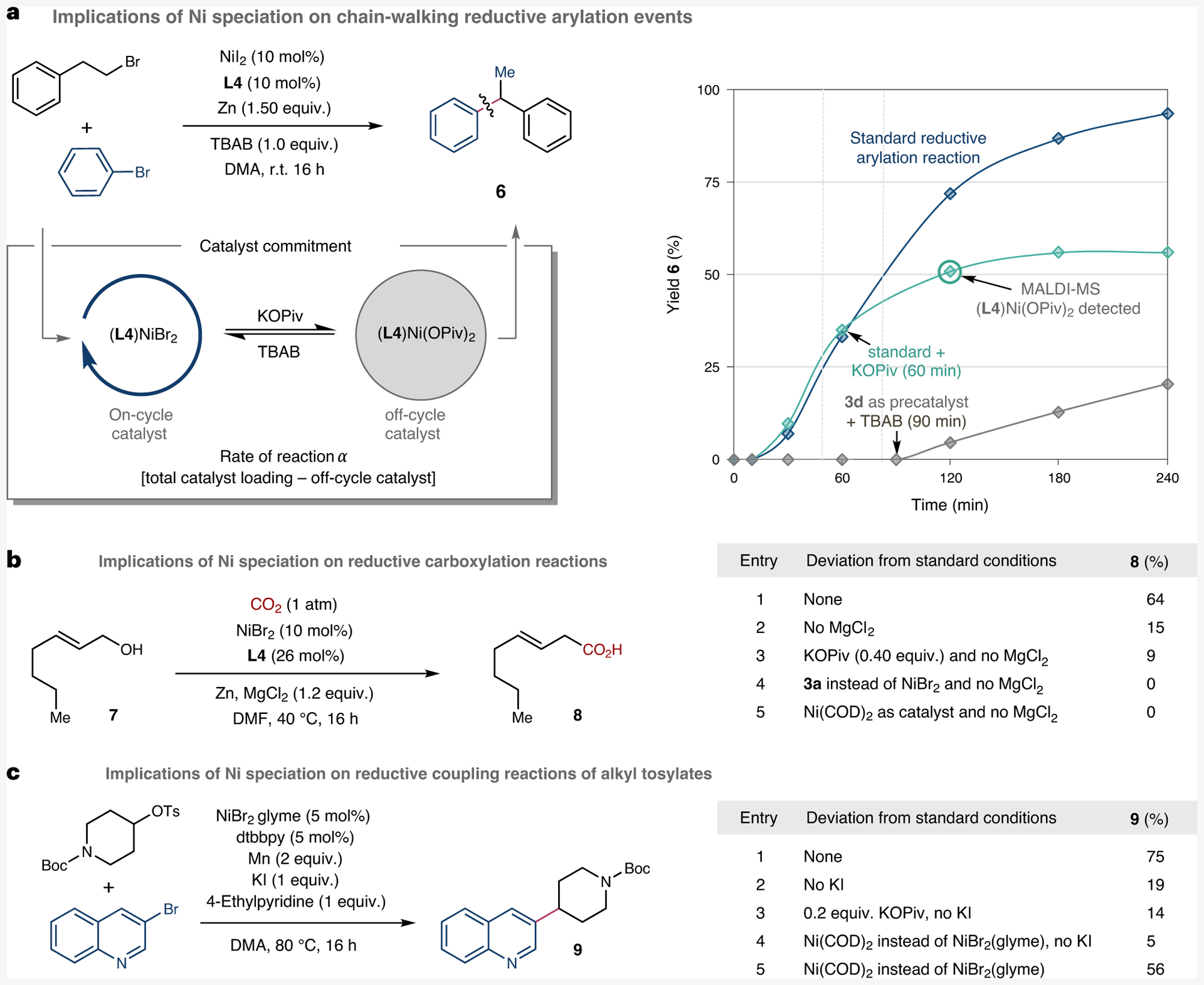
a, Monitoring chain-walking reductive coupling reactions, with added potassium pivalate (0.4 equiv.) or using (L4)Ni(OPiv)2 (3a) as a precatalyst. Blue trace, standard catalytic conditions; teal trace, standard catalytic conditions with KOPiv added after 60 minutes; grey trace, modified reaction conditions using 3d as a precatalyst and without TBAB until it is added after 90 minutes. b, Implications of Ni speciation on Ni-catalysed reductive carboxylation reactions in the presence of exogenous organic bases or in the absence of halide salts. The tables describe deviations from the standard reductive carboxylation reaction conditions with the yield of the corresponding carboxylic acid. c, Implications of Ni speciation of Ni-catalysed reductive coupling reactions of alkyl tosylates. The table describes deviations from the standard reductive coupling reaction conditions with the yield of the corresponding cross-coupled product. TBAB, tetrabutyl ammonium bromide; r.t., room temperature; COD, cyclooctadiene; dtbbpy, 4,4’-di-tert-butyl-2,2’-dipyridyl; Ts = tosyl; Boc = tert-butyloxycarbonyl; DMA = dimethylacetamide; DMF = dimethylformamide; TBAB = tetrabutylammonium bromide.
Conclusions
In summary, this work uncovers the enigmatic role exerted by anionic ligands in electron-transfer events occurring in comproportionation, disproportionation and reduction reactions of polypyridine nickel complexes, elementary steps commonly encountered in the Ni-catalysed cross-coupling arena. Specifically, stoichiometric and quantum mechanical studies tacitly reveal that Ni(II) halides easily react by comproportionation with Ni(0) complexes to form Ni(I) species. In sharp contrast, Ni(II) pseudohalides are unreactive whereas their corresponding Ni(I) analogues react irreversibly by disproportionation. The implications of these findings are strongly related to the formation of off-cycle species that compromise catalytic turnover, thus leading to catalytic reactions with poor efficiency. Taking into consideration the popularity exerted by polypyridine nickel complexes in one- or two-electron pathways in a myriad of catalytic reactions and their rapid adoption in industrial work, our study provides long-awaited insights into mechanistic aspects of utmost relevance for designing future Ni-catalysed reactions.
Methods
General
Reactions were carried out under N2 in a glovebox or on a Schlenk line unless otherwise noted. Reagents and solvents were purified by standard means. See Supplementary Methods for detailed conditions and the characterization data.
Synthesis of (L4)Ni(OPiv)2
In the glovebox (L4)NiCl2 (224 mg, 0.46 mmol) was added to a 12 ml vial, together with potassium pivalate (206 mg, 1.66 mmol). A stir bar was added and the vial was charged with 6 ml toluene. This turned the pink powder to a pink suspension and this was stirred overnight. After 16 h the resulting green solution was filtered through a Celite plug with the salt being filtered off and a green solution was collected. The solvent was then removed to afford a green solid and this was washed 3 times with pentane (1 ml) to give (L4)Ni(OPiv)2 (199 mg, 70% yield) as a green powder.
Synthesis of (L4)Ni(OCOPh)2
In the glovebox (L4)NiCl2 (122 mg, 0.25 mmol) and potassium benzoate (83 mg, 0.52 mmol, 2.08 equiv.) were added to a 10 ml vial. A stir bar was added and the vial was then charged with 3 ml THF. The solution was stirred overnight, during which a colour change from pink to orange to green was observed. The solvent was then removed and the solid was dissolved in DCM. This mixture was filtered through a Celite plug where a white solid was filtered off. The green solution was concentrated to dryness. The solid was washed 3 times with pentane (3 ml) to afford (L4)Ni(OCOPh)2 as a green powder (128 mg, 78% yield).
Synthesis of [(L4)Ni(OPh)2]2
In the glovebox (L4)NiCl2 (158 mg, 0.32 mmol) and sodium phenoxide (116 mg, 1.00 mmol, 3.1 equiv.) were added to a 10 ml vial. A stir bar was added and the vial was then charged with 5 ml toluene and the solution was stirred overnight. The solvent was then removed and the residue was redissolved in DCM (sparingly soluble). This mixture was filtered through a Celite plug. The solvent was then removed and the solid was washed with 3 times with pentane (3 ml) to afford [(L4)Ni(OPh)2]2 as a brown powder (88 mg, 45 % yield).
Synthesis of [NiLi3Cl2(OtBu)3]2·4THF
In the glovebox (L4)NiCl2 (113 mg, 0.23 mmol) and lithium tert-butoxide (41 mg, 0.51 mmol) were added to a 10 ml vial. A stir bar was added and the vial was charged with 3 ml THF and the solution was stirred overnight. The solvent was then removed and the residue was dissolved in minimal THF and this mixture was then filtered through a Celite plug. The solvent was then removed and the residue was washed 3 times with pentane (3 ml) to afford [NiLi3Cl2(OtBu)3]2·4THF as a blue powder (88 mg, quantitative).
Supplementary Material
Acknowledgements
We thank ICIQ and FEDER/MCI−AEI/PGC2018-096839-B-I00 for financial support. C.S.D. thanks the European Union’s Horizon 2020 under the Marie Curie PREBIST grant agreement 754558. S.J.T. thanks Marie Sklodowska-Curie grant agreement No. 859910. O.G. thanks the NIGMS NIH (R35GM137797), the Camille and Henry Dreyfus Foundation and the Welch Foundation (A-2102-20220331) for funding and Texas A&M University HPRC resources (https://hprc.tamu.edu) for computational resources. We sincerely thank T. Skrydstrup for allowing revisions to be completed using his laboratory space and equipment, J. Benet for X-ray crystallographic data and G. Stoica for assistance with EPR experiments.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41929-023-00925-4.
Data availability
Experimental procedures and characterization data for the stochiometric experiments, catalysts and the synthesized compounds along with computational information are included in the Supplementary Information. Crystallographic data are available from the Cambridge Crystallographic Data Centre with the following codes: 3d (CCDC-2175355), 3c (CCDC-2175356), 3b (CCDC-2175354), 4a-Br (CCDC-2175353), 3a-Br (CCDC-2175357), [NiLi3Cl2(OtBu)3·2THF]2 (CCDC-2175358) and 3e (CCDC-2175352). Other data are available from the corresponding authors upon reasonable request.
References
- 1.Diccianni JB & Diao T Mechanisms of nickel-catalyzed cross-coupling reactions. Trends Chem. 1, 830–844 (2019). [Google Scholar]
- 2.Diccianni J, Lin Q & Diao T Mechanisms of nickel-catalyzed coupling reactions and applications in alkene functionalization. Acc. Chem. Res 53, 906–919 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasker SZ, Standley EA & Jamison TF Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazari N, Melvin PR & Beromi MM Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem 1, 0025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Everson DA & Weix DJ Cross-electrophile coupling: principles of reactivity and selectivity. J. Org. Chem 79, 4793–4798 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohadjer Beromi M et al. Mechanistic study of an improved Ni precatalyst for Suzuki–Miyaura reactions of aryl sulfamates: understanding the role of Ni(I) species. J. Am. Chem. Soc 139, 922–936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barth EL et al. Bis(dialkylphosphino)ferrocene-ligated nickel(II) precatalysts for Suzuki–Miyaura reactions of aryl carbonates. Organometallics 38, 3377–3387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohadjer Beromi M, Banerjee G, Brudvig GW, Hazari N & Mercado BQ Nickel(I) aryl species: synthesis, properties, and catalytic activity. ACS Catal. 8, 2526–2533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohadjer Beromi M et al. Modifications to the aryl group of dppf-ligated Ni σ-aryl precatalysts: impact on speciation and catalytic activity in Suzuki–Miyaura coupling reactions. Organometallics 37, 3943–3955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yanagi T, Somerville RJ, Nogi K, Martin R & Yorimitsu H Ni-catalyzed carboxylation of C(sp2)–S bonds with CO2: evidence for the multifaceted role of Zn. ACS Catal. 10, 2117–2123 (2020). [Google Scholar]
- 11.Day CS, Somerville RJ & Martin R Deciphering the dichotomy exerted by Zn(II) in the catalytic sp2 C–O bond functionalization of aryl esters at the molecular level. Nat. Catal 4, 124–133 (2021). [Google Scholar]
- 12.Somerville RJ, Hale LVA, Gómez-Bengoa E, Burés J & Martin R Intermediacy of Ni–Ni species in sp2 C–O bond cleavage of aryl esters: relevance in catalytic C–Si bond formation. J. Am. Chem. Soc 140, 8771–8780 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Somerville RJ et al. Ni(I)–alkyl complexes bearing phenanthroline ligands: experimental evidence for CO2 insertion at Ni(I) centers. J. Am. Chem. Soc 142, 10936–10941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heimbach P Changes in the coordination number of Ni(0) and Ni(I) compounds. Angew. Chem. Int. Ed 3, 648 (1964). [Google Scholar]
- 15.Tsou TT & Kochi JK Mechanism of oxidative addition. Reaction of nickel(0) complexes with aromatic halides. J. Am. Chem. Soc 101, 6319–6332 (1979). [Google Scholar]
- 16.Cundy CS & Nöth H Metal-boron compounds: XI. Complexes derived from reactions of bis(triphenylphosphine)(π-ethylene) nickel with alkyl and boron halides. J. Organomet. Chem 30, 135–143 (1971). [Google Scholar]
- 17.Porri L, Gallazzi MC & Vitulli G Complexes of nickel(I) with triphenylphosphine. Chem. Commun, 228–228 (1967). [Google Scholar]
- 18.Beattie DD, Lascoumettes G, Kennepohl P, Love JA & Schafer LL Disproportionation reactions of an organometallic Ni(I) amidate complex: scope and mechanistic Investigations. Organometallics 37, 1392–1399 (2018). [Google Scholar]
- 19.Dible BR, Sigman MS & Arif AM Oxygen-induced ligand dehydrogenation of a planar bis-μ-chloronickel(I) dimer featuring an NHC ligand. Inorg. Chem 44, 3774–3776 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Schunn RA Preparation and reactions of triethylphosphine complexes of zerovalent nickel, palladium, and platinum. Inorg. Chem 15, 208–212 (1976). [Google Scholar]
- 21.Dürr AB, Fisher HC, Kalvet I, Truong K-N & Schoenebeck F Divergent reactivity of a dinuclear (NHC)nickel(I) catalyst versus nickel(0) enables chemoselective trifluoromethylselenolation. Angew. Chem. Int. Ed 56, 13431–13435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalvet I, Guo Q, Tizzard GJ & Schoenebeck F When weaker can be tougher: the role of oxidation state (I) in P- vs N-ligand-derived Ni-catalyzed trifluoromethylthiolation of aryl halides. ACS Catal. 7, 2126–2132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapat A, Sperger T, Guven S & Schoenebeck F E-olefins through intramolecular radical relocation. Science 363, 391–396 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Yuan M, Song Z, Badir SO, Molander GA & Gutierrez O On the nature of C(sp3)–C(sp2) bond formation in nickel-catalyzed tertiary radical cross-couplings: a case study of Ni/photoredox catalytic cross-coupling of alkyl radicals and aryl halides. J. Am. Chem. Soc 142, 7225–7234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phapale VB, Guisán-Ceinos M, Buñuel E & Cárdenas DJ Nickel-catalyzed cross-coupling of alkyl zinc halides for the formation of C(sp2)—C(sp3) bonds: scope and mechanism. Chem. Eur. J 15, 12681–12688 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Lin Q & Diao T Mechanism of Ni-catalyzed reductive 1,2-dicarbofunctionalization of alkenes. J. Am. Chem. Soc 141, 17937–17948 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohadjer Beromi M, Brudvig GW, Hazari N, Lant HMC & Mercado BQ Synthesis and reactivity of paramagnetic nickel polypyridyl complexes relevant to C(sp2)–C(sp3)coupling reactions. Angew. Chem. Int. Ed 58, 6094–6098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ting SI, Williams WL & Doyle AG Oxidative addition of aryl halides to a Ni(I)-bipyridine complex. J. Am. Chem. Soc 144, 5575–5582 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Till NA, Oh S, MacMillan DWC & Bird MJ The application of pulse radiolysis to the study of Ni(I) intermediates in Ni-catalyzed cross-coupling reactions. J. Am. Chem. Soc 143, 9332–9337 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Ting SI et al. 3d-d Excited states of Ni(II) complexes relevant to photoredox catalysis: spectroscopic identification and mechanistic implications. J. Am. Chem. Soc 142, 5800–5810 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Huang L, Ackerman LKG, Kang K, Parsons AM & Weix DJ LiCl-accelerated multimetallic cross-coupling of aryl chlorides with aryl triflates. J. Am. Chem. Soc 141, 10978–10983 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powers DC, Anderson BL & Nocera DG Two-electron HCl to H2 photocycle promoted by Ni(II) polypyridyl halide complexes. J. Am. Chem. Soc 135, 18876–18883 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Juliá-Hernández F, Moragas T, Cornella J & Martin R Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature 545, 84–88 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Li Z et al. Electrochemically enabled, nickel-catalyzed dehydroxylative cross-coupling of alcohols with aryl halides. J. Am. Chem. Soc 143, 3536–3543 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Molander GA, Traister KM & O’Neill BT Engaging nonaromatic, heterocyclic tosylates in reductive cross-coupling with aryl and heteroaryl bromides. J. Org. Chem 80, 2907–2911 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Dong Z & Macmillan DWC Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 598, 451–456 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Xu X & Zhu S Nickel-catalysed selective migratory hydrothiolation of alkenes and alkynes with thiols. Nat. Commun 10, 1752 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith RT et al. Metallaphotoredox-catalyzed cross-electrophile Csp3-Csp3 coupling of aliphatic bromides. J. Am. Chem. Soc 140, 17433–17438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng L, Li Z & Yin G Photochemical nickel-catalyzed reductive migratory cross-coupling of alkyl bromides with aryl bromides. Org. Lett 20, 1880–1883 (2018). [DOI] [PubMed] [Google Scholar]
- 40.van Gemmeren M et al. Switchable site-selective catalytic carboxylation of allylic alcohols with CO2. Angew. Chem. Int. Ed 56, 6558–6562 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Tortajada A, Börjesson M & Martin R Nickel-catalyzed reductive carboxylation and amidation reactions. Acc. Chem. Res 54, 3941–3952 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Tortajada A, Juliá‐Hernández F, Börjesson M, Moragas T & Martin R Transition‐metal‐catalyzed carboxylation reactions with carbon dioxide. Angew. Chem. Int. Ed 57, 15948–15982 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Harris CM & McKenzie ED Nitrogenous chelate complexes of transition metals–III: bis-chelate complexes of nickel (II) with 1,10-phenanthroline, 2,2′-bipyridyl and analogous ligands. J. Inorg. Nucl. Chem 29, 1047–1068 (1967). [Google Scholar]
- 44.Kinnunen T-JJ, Haukka M, Pakkanen TT & Pakkanen TA Four-coordinated bipyridine complexes of nickel for ethene polymerization–the role of ligand structure. J. Organomet. Chem 613, 257–262 (2000). [Google Scholar]
- 45.Fagnou K & Lautens M Halide effects in transition metal catalysis. Angew. Chem. Int. Ed 41, 26–47 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Peng L et al. Ligand-controlled nickel-catalyzed reductive relay cross-coupling of alkyl bromides and aryl bromides. ACS Catal. 8, 310–313 (2018). [Google Scholar]
- 47.Tortajada A, Ninokata R & Martin R Ni-catalyzed site-selective dicarboxylation of 1,3-dienes with CO2. J. Am. Chem. Soc 140, 2050–2053 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Eremenko IL et al. Bi- and mononuclear nickel(II) trimethylacetate complexes with pyridine bases as ligands. Inorg. Chem 38, 3764–3773 (1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental procedures and characterization data for the stochiometric experiments, catalysts and the synthesized compounds along with computational information are included in the Supplementary Information. Crystallographic data are available from the Cambridge Crystallographic Data Centre with the following codes: 3d (CCDC-2175355), 3c (CCDC-2175356), 3b (CCDC-2175354), 4a-Br (CCDC-2175353), 3a-Br (CCDC-2175357), [NiLi3Cl2(OtBu)3·2THF]2 (CCDC-2175358) and 3e (CCDC-2175352). Other data are available from the corresponding authors upon reasonable request.