

Abstract
During nuclear fuel reprocessing, radioiodine, can be released. The speciation of iodine drives its volatility, and partitioning processes are highly variable because they depend on facility operating conditions. Starting from iodine behavior in the fuel and progressing to its behavior in the environment, this review article describes the current understanding of iodine partitioning during aqueous fuel reprocessing. This review outlines knowledge gaps and describes the effects of state-of-the-art reprocessing techniques on iodine speciation and volatility. Whereas many review articles have described iodine behavior during specific reprocessing steps, this review provides a holistic overview of radioiodine, from the forms of iodine in different types of irradiated fuel to the forms of iodine released into the environment. The resultant behavior of radioiodine compared with stable iodine in the environment is also described.
During nuclear fuel reprocessing, radioiodine, can be released.
1. Introduction
Radioiodine is a highly regulated fission product that can be released during reprocessing of spent nuclear fuel (SNF). Iodine exhibits complex behavior driven by its many possible oxidation states and high reactivity. During fuel reprocessing, the volatility of the iodine determines the quantity of iodine released from the facility. This review article focuses on the current understanding of iodine partitioning during each reprocessing operation (as outlined in Fig. 1) and the conditions under which they are observed. The mass and activity of iodine in the fuel varies depending on irradiation conditions, which are not the focus of this review article; therefore, in this article, the irradiation of nuclear fuel is only discussed when it affects iodine speciation. Both oxide and metal fuels are discussed in this article. Oxides include ceramic UO2 fuels, which are commonly used in light-water reactors (LWRs) and pressurized heavy-water reactors (PHWR), as well as mixed uranium and plutonium oxide (MOX) fuels, which are used in fast reactors. Metal fuels have been used in both gas- and water-cooled graphite moderated reactors and fast breeder reactors. The fuel material and cladding impact processing conditions which in turn impact iodine speciation and volatility.
Fig. 1. Schematic of process steps that are covered in this review. Fuel bundle disassembly is covered in Section 2, dissolution and off gas treatment in Section 3, solvent extraction in Section 4, and environment in Section 5.

In reprocessing, one of the most problematic volatile radioisotopes is 129I. Other volatile radionuclides (e.g.85Kr, 14C, and 3H) are released during reprocessing but are not as hazardous as 129I. The long half-life of 129I (t1/2 = 1.57 × 107 years), its complex reactivity and the health concerns resulting from exposure, create the need for high decontamination factors to mitigate the release of 129I into the environment.1,2 However, understanding the complex chemistry is difficult and continues to be an active area of research.3–7
This paper discusses, factors that lead to the partitioning, speciation, and interdependence of physical, chemical, and radiological mechanisms. For additional information, readers are referred to previous literature cited throughout these sections. The primary differences between past reviews and the current review are that the current review aims to cover a more holistic overview of radioiodine behavior during reprocessing from iodine forms in irradiated fuel to environmental release as well as differences in environmental behaviors between stable iodine and radioiodine. Section 2 discusses contemporary nuclear fuel treatment and the processes that affect iodine chemistry. Section 3 investigates reactions and products observed during dissolution, as well as the complexities between scales in laboratory studies and facility environments. Section 4 discusses the distribution in iodine products in chemical phases through extractants, waste forms, and treatment steps. The movement of iodine throughout the nuclear fuels system ultimately results in potential cycling to the environment. Section 5 presents general transformation mechanisms characterizing reactions, and interactions with the environment. Section 6 summarizes these topics as the basis for a comprehensive model for the iodine system.
2. Iodine chemistry during fuel disassembly
2.1. Fuel types
A large portion of research focuses on UO2 fuel because it is the most common fuel type used worldwide. A variety of reactors, including (LWR's), boiling water reactors, pressurized water reactors, pressurized heavy-water reactors (PHWR's), fast-neutron reactors, advanced gas-cooled reactors, and high-temperature gas-cooled reactors can use UO2 fuel. This section focuses on iodine behavior in UO2 fuels in LWRs and PHWRs. Fuel cladding is an important consideration because it impacts the disassembly process chosen. Both reactor types use zirconium alloy (Zircaloy) cladding. Alternative cladding materials, such as those used in accident tolerant fuels are beyond the scope of this review.8,9 Metal fuels are often clad in magnesium alloy (magnox), as was done in gas cooled reactors, or with aluminum or stainless steel as was done in water cooled reactors.
2.2. Head-end processing for UO2 fuels
An advanced head-end process for uranium oxide fuel dissolution is shown in Fig. 2.10 Typical oxide fuel dissolution is similar but does not include voloxidation. The first head-end process is to remove the components of the bundle or assembly so individual fuel pins can be processed. Before fuel dissolution can occur, the cladding must be breached or removed from the fuel either mechanically or chemically. Mechanical decladding is typically performed for UO2 fuels and has the potential to release any volatile iodine that may have migrated to the gap between the fuel and the cladding. For Zircaloy or stainless-steel clad fuels, the fuel pins are sheared into small pieces to provide access to the uranium fuel for chemical dissolution. After shearing, voloxidation (O2-based) or advanced voloxidation (NO2-based) can be used to oxidize UO2 fuel to U3O8 powder and release volatile radionuclides.11,12 Both methods target the liberation of volatile radionuclides such as 129I, 85Kr, 14C, and 3H. These methods may be advantageous because they capture the vast majority of iodine in the head-end abatement systems and do not fractionate it throughout the reprocessing facility. Depending on how voloxidation is accomplished (including the temperature and gases used) a significant portion of iodine may be released from the fuel.
Fig. 2. Block flow diagram for aqueous head-end processing.10 This figure was reprinted with permission. Copyright 2008 CRESP.

Most reprocessing facilities currently move straight from shearing to dissolution. Many publications report the fraction of iodine that is volatilized to the dissolver off-gas (DOG) but exclude the shearing off-gas, which is often combined with the DOG. The exclusion of the shearing off-gas creates difficulty in determining what fraction of volatilized iodine, if any, is released during shearing. Two studies at the Wiederaufarbeitungsanlage Karlsruhe (WAK) reprocessing plant in Karlsruhe, Germany, specifically measured the amount of iodine released during shearing and reported that 0.15–0.3% of the total iodine in the fuel was released during shearing.13–15
The amount of iodine released during head end processes is driven by the species of iodine in the irradiated fuel. Many factors affect iodine speciation and concentration in fuel. Compared with metal fuel, which has a small heat gradient, UO2 fuel has a large heat gradient between the center of the fuel and the outer edge. This large heat gradient leads to differences in the oxygen-to-metal (O/M) ratio at different locations in the fuel. The O/M ratio is important because it changes the thermodynamically favorable species. Iodine is expected to segregate in fuels but an understanding of speciation and the form remains understudied.16 In general, the Cs/I molar ratio is ∼10, which leads to the prediction that CsI would be the predominate species.17 Even though CsI has been found on the inner cladding of irradiated LWR, micro-drilling of LWR pins has indicated that iodine migrates slightly faster than Cs. This contradicts the idea that higher amounts of CsI form at the surface of the fuel.18 At lower temperatures, <1000 K, thermal diffusion of iodine is directly proportional to the fission rates.19 At higher temperatures, fission products migrate primarily via thermally induced diffusion in the UO2.19 The vast literature surrounding reactor accidents provides a reliable source of data for understanding iodine speciation in irradiated fuel.20 For fuel failure, specifically stress corrosion cracking of Zircaloy clad fuels, I2 does not directly cause the cracking. Rather the formation of ZrI4 (and potentially other lower forms of iodide) can result in cracking.20
Studies have shown that in PHWR reactors the cesium and iodine are held firmly in the fuel and sheath surface.21 This arrangement may be due to higher fuel temperatures, which, compared with the temperatures used for LWR fuels, lead to more fission gas segregation.22
Fuel burnup plays an interesting role in the speciation of iodine. At burnups of less than 5 MWd per kgU, CsI does not form at the grain boundary. Generally, burnup increases the fuel's O/M ratio, which, changes the favored species.18 The volatile species, including I2(g), can accumulate in the annular region between the pellets and the cladding. A study at WAK showed that low- and high-burnup fuels have similar concentrations of undissolved iodine during fuel dissolution.23 Because more iodine is produced in high burnup fuels, this result indicates that in low-burn-up fuels, a higher portion of the iodine will be undissolved or released as gas than in higher-burn up fuels.23 Higher-burnup fuels cause iodine to be retained in a Ag–Pd halide phase, which is resistant to HNO3 dissolution.24 For high-burnup fuels, compounds of Ag, Pd and iodine have been measured, and the results indicate that these compounds were formed during irradiation and not during dissolution.24 Whether the same is true for low-burnup fuel still needs to be verified.
Stainless steel cladding is often used for MOX fuel. During reactor operation, iodine in MOX fuel is also expected to exist as CsI(g), which can migrate to the fuel cladding gap. The CsI will not attack the stainless-steel cladding, but if I2 is formed from either radiolytic decomposition of CsI or from high O/M ratios, then I2 can attack the stainless steel and form metal iodides with the metals in the steel.25–27 A passivation layer further inhibits the ability of I2 to interact with the stainless steel, and an O/M larger than 2.08 is needed.17 In defective fuel, volatile fission products such as iodine, xenon, and krypton migrate to the defect site and eventually into the coolant.28
2.3. Head-end processing for metal fuels
Depending on the cladding material, metal fuels can be chemically or mechanically declad. Metal uranium fuel clad in magnesium metal alloy was used in gas-cooled reactors. The fuel was decanned at the cooling pond location before being transported to the dissolver. A report detailing the iodine controls during processing at Windscale, states that no significant release of iodine occurred during the decanning process.29 Therefore, if volatile iodine species do form in metal fuel, then the assumption is that they do not migrate significantly to the surface of the fuel.
Metal uranium fuel clad in aluminum has also been used. The aluminum cladding is chemically separated from the fuel by dissolving the cladding in NaOH with NaNO3 added to control hydrogen evolution.30 As with mechanical decladding, very little iodine was released during dissolution of the cladding; however, measurements performed at facilities indicated that some iodine was evolved during this step.30 Limited information is available on iodine characterization in metal fuel. Instead, some information on the species can be inferred based on volatility during dissolution. Speciation during dissolution is discussed in Section 3.
3. Aqueous dissolution
3.1. Overview
The chemistry of iodine during the dissolution of UO2 and metal fuel is covered in this section, which focuses on HNO3 dissolution. As discussed in Section 2.2, UO2 fuel elements are sheared to the desired length and are dropped into a basket in the dissolver, resulting in the release of a small fraction of the iodine. Concentrated (∼11 M) HNO3 is added to dissolver and heated to near boiling. In a facility-scale dissolver, the sheared pieces of fuel cladding are typically added to rotating baskets submerged in HNO3. Before chemical separation, the uranium oxide fuel is dissolved away from the cladding hulls and the dissolver is routed to a tank for nuclear material accountability and chemical adjustment. Metal fuels can be mechanically or chemically declad but are then dissolved in HNO3 similar to UO2 fuels.
3.2. Chemistry in the dissolver
The majority of iodine in UNF is assumed to be in the form of CsI,31,32 which is highly soluble in aqueous media owing to its low crystal lattice energy: CsI solubility in water is 85.5 g/100 g H2O at 25 °C. This value increases with increasing temperatures to >205 g/100 g H2O at 100 °C.33 Like other alkali iodides, CsI is soluble in HNO3 at a range of concentrations and temperatures,11,34–37 where the following reaction occurs.
| 2CsI(aq) + 4HNO3(aq) + 2H2O(l) ↔ I2(aq) + 2NO2(aq) + 2CsNO3(aq) + 4H2O(l) | 1 |
Reaction (1) is a simplistic reaction and does not show that CsI must first disassociate to Cs+ and I−. The reaction also does not show the additional reactions that can occur, including the oxidation of I− to IO3− and the formation of compounds such as PdI2 and AgI which are discussed in Section 3.5. Thermodynamic data are available for the formation of these species; however, kinetics drive many of these reactions, and additional work is needed to fully understand this system. Although dissolution of I2 crystals in water is not favorable (3.4 g/100 g H2O at 25 °C),38 many reports support the solubility of I2 crystals in concentrated HNO3 solutions (3.12 mmol L−1 69.95% HNO3 at 25 °C).38–40 The volatility of iodine from aqueous solutions is a known route for the release of iodine from the dissolver solution.5 The primary volatile species is assumed to be I2 but HOI and some organic iodides are also volatile. Several studies that monitored the dissolution of alkali metal iodides (e.g., KI) in concentrated nitric acid solutions have confirmed that the reaction occurs rapidly,11 and is proportional to the rate of fuel dissolution when tested within a fuel matrix.37 The low concentrations of iodate ions (IO3−) are also expected, and can be further reduced to I2 by available NO2 species except at very low pH ([HNO3] > 16 M).34 However, studies from Cathers and Kibbey, which are documented in a report by Unger et al.,41 suggest that iodate ions are easily reduced to I2 as the concentration of HNO3 decreases to a concentration range (∼3 M) consistent with fuel dissolution. The primary reactive species of iodine (I− and I2) form organic iodides (e.g., CH3(CH2)xI where x = 0–12) or other volatile products, and their distributions in later processing steps depends on the total concentration of iodine.37,42,43
Experiments have shown that >90% of the molecular iodine is volatilized when UNF is dissolved in HNO3 with the addition of air sparging.44,45 Oxidation reactions of iodide in HNO3 with the resulting HNO2 content assist in the release of iodine from the dissolver solution. The oxidation reactions of iodide by HNO3 and HNO2 are shown in reactions (2) and (3). As shown in reaction (4), aqueous molecular iodine then transitions to gaseous iodine, which is swept out of the dissolver by the sparge air.46
| I− + 4H+ + NO2− + 2NO3− ⇌ 0.5I2 + 3NO2 + 2H2O | 2 |
| I− + 2H+ + NO3− ⇌ 0.5I2(aq) + NO2 + H2O | 3 |
| I2(aq) ⇌ I2(g) | 4 |
For metal fuels, Burger reported that elemental iodine is the most stable iodine species in the dissolver solution (3–10 M HNO3), and that all iodine should eventually evolve as I2. The only exception occurs in solutions with high HNO3 concentrations (>10 M). In these solutions, iodate is the main iodine species present.30 Later studies showed that elemental iodine can be further oxidized to iodate in HNO3 solutions. The equilibrium reaction between elemental iodine and iodate in HNO3 solutions is shown in reaction (5).47 Sakurai initially believed that iodate was the main species remaining in solution after the volatilization of elemental iodine; however, his research group found that in solutions of 4 M HNO3, the main species of iodine remaining in solution is colloidal iodine.47 Gaps in the understanding of the composition of iodine remaining in solution still exist, and the actual iodine composition is clearly more complex than Burger or Sakurai described.
| I2(aq) + 8H+ + 10NO3− ⇌ 2IO3− + 10NO2(g) + 4H2O | 5 |
3.3. Alternative acids
Some studies investigated replacing HNO3 with HCl. These studies cite several advantages, including better separation and dissolution of fission products, recyclability, and better control of oxidation states. Mailen and Bell48 point out that, although the iodine compounds (in the form of I2, CsI, and RbI) are soluble, iodine volatilization from HCl may be additionally troublesome because of the formation of a highly reactive interhalogen species, the I2Cl− ion.
3.4. Radiolysis in dissolution
The chemistry of iodine during UNF dissolution involves numerous chemical processes. Dissolution involves high concentrations of HNO3 which lowers the pH and introduces a powerful oxidizing agent (the nitrate anion). The ionizing radiation that results from the radioactive decay of the radioisotopes in the dissolver involves a complicated web of chemical reactions.49,50 The radioactive decay emits energy into the solution in the form of β particles, α particles, and γ-rays. The ionizing radiation from the radioactive decay causes radiolysis, the breaking of the chemical bonds of the molecules in solution and the production of several reactive species. The β particles and γ-rays emitted by decay of the fission products are expected to provide most of the dose rate within the solution, but the energy from α decay is deposited in a short distance and contributes significantly to radiolysis in the dissolver solution.
Species in solution may undergo direct radiolysis by interacting with a photon or β particle or may undergo secondary reactions with direct radiolysis products. The radiation energy deposited in the solution is generally distributed according to the electron fraction of each species.51 This quantity is derived from the mole fraction (or concentration) of each species along with the number of electrons in the species (e.g., 10 electrons for H2O, 32 electrons for HNO3). Because of the low concentration of iodine present in the dissolver solution, its electron fraction is essentially zero and direct radiolysis may be neglected; however, the direct effect of radiolysis on water and nitric acid is relevant to the chemistry of iodine in the dissolver solution.
The conversion of dose rate to the number of radicals formed is described using a quantity called the G value. For pure H2O, the radiolysis products and corresponding G values are provided in Table 1. The immediate products include solvated electrons (eaq−), hydrogen (H˙) and hydroxyl radicals (OH˙), protons (H+), hydroxide ions (OH−), molecular hydrogen (H2), and hydrogen peroxide (H2O2). These products further react with water and each other to form numerous other species (e.g., O−, O2, O2−, O3, O3−, HO2−, 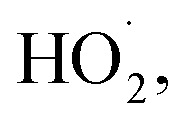 and HO3).52 Exposures on the order of an hour or longer lead to a quasi-steady state distribution of radiolysis products. In this state, the most stable species (H2, O2, and H2O2) accumulate to appreciable levels.
and HO3).52 Exposures on the order of an hour or longer lead to a quasi-steady state distribution of radiolysis products. In this state, the most stable species (H2, O2, and H2O2) accumulate to appreciable levels.
G values (in molecules per 100 eV) for γ-irradiation of water from Pastina and LaVerne52 as well as from HNO3 from Jiang et al.51 Part of this table was recreated with permission from Pastina and LaVerne.52 Copyright 2001 American Chemical Society.
| Species | Source | G (molecules per 100 eV) | G (μmol J−1) |
|---|---|---|---|
| eaq− | From H2O | 2.60 | 0.27 |
| H˙ | 0.66 | 0.068 | |
| H2 | 0.45 | 0.047 | |
| OH˙ | 2.70 | 0.28 | |
| H2O2 | 0.02 | 0.002 | |
| H+ | 3.10 | 0.32 | |
| OH− | 0.50 | 0.052 | |
| NO3˙ | From HNO3 | 4.8 | 0.50 |
| NO2− | 1.5 from NO3− | 0.16 from NO3− | |
| 2.0 from HNO3 | 0.21 from HNO3 |
The HNO3 undergoes direct radiolysis to form two reactive derivatives: 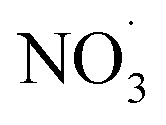 and NO2−. In acidic solution, the nitrite will rapidly protonate to form nitrous acid, which undergoes further decomposition to NO2 and NO.53 The nitrate radical,
and NO2−. In acidic solution, the nitrite will rapidly protonate to form nitrous acid, which undergoes further decomposition to NO2 and NO.53 The nitrate radical, 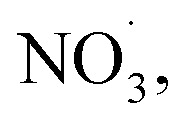 subsequently breaks down to form
subsequently breaks down to form 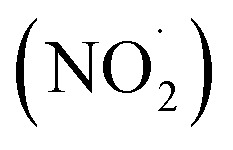 and (O˙). The highly reactive O˙ then extracts an oxygen atom from NO3− to produce O2 and NO2−.
and (O˙). The highly reactive O˙ then extracts an oxygen atom from NO3− to produce O2 and NO2−.
Direct radiolysis products along with the radicals produced by subsequent reactions have extensive chemical interactions with iodine in the dissolver solution.49,50 First, I− is oxidized into I2 by the presence of the OH˙ and 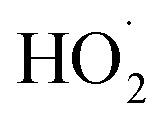 radicals. The I3− specie appears as an intermediate because dissolved I2 readily associates with I−.54 Once the iodine has been oxidized into I2, two phenomena may occur, the I2 may volatilize or it may undergo further oxidation to form HOI and HIO3. The interplay between vapor evolution and oxidation depends on the conditions within the dissolver, the time over which the dissolution occurs, and the time over which the diffusion through the solution into the headspace occurs.
radicals. The I3− specie appears as an intermediate because dissolved I2 readily associates with I−.54 Once the iodine has been oxidized into I2, two phenomena may occur, the I2 may volatilize or it may undergo further oxidation to form HOI and HIO3. The interplay between vapor evolution and oxidation depends on the conditions within the dissolver, the time over which the dissolution occurs, and the time over which the diffusion through the solution into the headspace occurs.
3.5. Insoluble products from dissolution
Small fractions of UNF may not completely dissolve under the acidic conditions described previously. UNF can form solid aggregates, colloids, microspheres, or sludge within the dissolver vessel. The total amount and composition of insoluble residue that remains from the dissolution process varies based on fuel type, burnup, and length of storage.11,55 Elements that remain in this undissolved material include ruthenium, molybdenum, technetium, rhodium, palladium, and iodine.56 In the 1990s, Sakurai pioneered isolated studies of iodine interactions with fission products to provide a better understanding of this reaction mechanism.47,57 By reacting solubilized iodides with Ag and Pd, Sakurai created colloidal species in the forms of AgI and PdI2.47,57 Similar studies using copper, aluminum, and zinc as reactive surfaces also lead to the formation of insoluble metal iodides.58
In an effort to further study the speciation in the dissolved fuel, Sakurai et al.45 dissolved 131I labelled KI and UO2(NO3)2 in 3.4 M or 6.1 M HNO3, with and without the presence of Ag and Pd among other fission products. In the 3.4 M HNO3–UO2(NO3)2 system, they found that the major iodine species remaining in solution was iodate, but only at NO2 pressures of less than 3 × 10−2 atm and the pressure in dissolved fuels is expected to be >7 × 10−2 atm.59 However, the simulated UNF system containing fission products produced unknown iodine colloids. Furthermore, the quantity of these colloids increased with increased concentration of Ag and Pd. Therefore, it seemed likely that in dissolved fuel the major iodine species would be colloidal and not iodate,45 these results were further confirmed in subsequent studies.47,59
Sakurai et al.47,60 further tested the colloids hypothesis by dissolving used fuel pellets and actual fuel specimens. From these studies, Sakurai concluded that until the amount of iodide decreases to less than 10−10 M, the amount of Ag, Pd, and I− present in dissolved fuel is sufficiently high that the formation of colloids is possible. The researchers confirmed that colloidal species are the most prominent form of iodine in dissolved fuel because iodide is initially on the order of 10−5 M in dissolved fuel.46,47 Non-volatile organic iodides are theorized to form as a results of organic impurities in HNO3.61 The primary organic impurity is CH3I, which is hypothesized to be able to undergo a similar decomposition reaction as colloidal iodine.7,61
In used nuclear fuel solutions, there is a retention of iodine species, which are suggested to be a mixture of I2(aq), IO3−, and colloidal iodine. Colloidal iodine is produced by reaction (6)–(10),46 however, multiple additional metals in solution could also form iodine species. Because these known reactions, researchers modified early studies to account for remaining iodine species, as shown in Fig. 3.46 The updated studies rely upon the iodine behavior that was present during a lab-scale demonstration dissolution at the Japan Atomic Energy Research Institute. This demonstration suggested that ≤10% of the total iodine from dissolution could be in the fuel solution, ≤3% being in insoluble residues, and the remaining being off-gasses as molecular iodine (i.e., I2(g)).
| AgI(s) + 2H+ + NO3− ↔ 0.5I2(aq) + Ag+ + NO2(g) + H2O | 6 |
| PdI2(s) + 4H+ + 2NO3− ↔ I2 + Pd2+ + 2NO2(g) + 2H2O | 7 |
| 5AgI(s) + HIO3 + 5HNO3 ↔ 3I2 + 5AgNO3 + 3H2O | 8 |
| 5PdI2(s) + 2HIO3 + 10HNO3 ↔ 6I2(aq) + 5Pd(NO3)2 + 6H2O | 9 |
| 3I− + Ag+ + Pd2+ ↔ AgI(s) + PdI2(s) | 10 |
Fig. 3. Distribution of iodine (129I) from dissolution of spent fuels in laboratory scale experiments. This figure was recreated from Sakurai et al.46 and was reprinted with permission.

Contaminated aqueous streams may also inhibit the complete dissolution of iodine in fuel. Although a study by Castleman and coworkers55 ruled out water impurities, Lieser and coworkers36 allude to potential causation in HNO3 purity. It is also difficult to dissolve colloidal iodine species even after NO2 sparging with concentrations remaining as high as 20 μg I/100 mL.59,62 A clarifying filtration step to remove these particulates may be necessary.
3.6. Partitioning
The complexity of iodine chemistry allows it to partition into gas, solid, and liquid phases during the dissolution of UNF in HNO3. Iodine partitions into the gas phase as volatile inorganic and organic species; iodine partitions into the solid phase as undissolved colloidal species, which are typically associated with Ag or Pd.15 Therefore, if radioiodine is not completely removed from the dissolver, it will spread into other off-gas and liquid-based waste streams throughout the facility. Dissolved residual iodate moves into the solvent extraction vessel, where radioiodine may partition into the off-gas or build in concentration in the organic solvents. This radioiodine may interfere with the intended extraction processes as the solvent is recycled. Undissolved solids move into solid waste processing where iodine may volatilize during evaporation processes and may need to be abated. Because the environmental and health concerns surrounding the release of radioiodine into the atmosphere, significant research has been devoted to ensuring that nearly all iodine is released during the dissolution step so that all iodine is abated from the dissolver off-gas.
3.6.1. Experimental iodine partitioning
Over the past three decades, experimental studies and observations from operating reprocessing plants have attempted to quantify the percentage and speciation of iodine that is volatilized into the dissolver off-gas. The work of Sakurai et al.45,62 suggest that during dissolution in 100 °C HNO3 (∼4 M), between 90 and 99% of the iodine inventory is volatilized into the dissolver off-gas as I2. In general, the dissolution in HNO3 and the evolution of gaseous iodine can be summarized by reaction (11).
| 2I− + 4H+ + 2NO3− ↔ I2 + 2NO2 + 2H2O | 11 |
Because only 90% of the UNF iodine inventory may be volatilized, up to 10% of the iodine may remain in the dissolver as dissolved iodate (IO3−) or colloidal iodide (AgI and PdI2).62 The reaction of iodine with the fission products Ag and Pd to form colloidal AgI and PdI2 is routinely observed in experiments, as shown in reaction (10).45,60,62 The quantity of colloidal species is likely determined by the amount of HNO2 in the solution. This HNO2 forms during the dissolution of UO2.57 As the dissolution rate increases, the concentration of HNO2 increases, thereby decreasing the mass of residual colloidal iodine species.62 Thus, a higher dissolution rate will likely yield more volatile I2.
To increase the amount of iodine volatilized from the solution, experimental studies have shown the need to sparge the dissolver solution with NOx gas and add excess iodate. Sakurai et al.45 found that after the dissolution of fuel is complete, the addition of NO2 gas to the dissolution solution converts any remaining aqueous IO3− to gaseous I2, as shown in reaction (12). Similarly, the addition of excess IO3− as a strip gas may convert colloidal AgI and PdI2 to gaseous I2, as shown in eqn (8) and (9), respectively. This process allows for a near quantitative release of the UNF iodine inventory.
| 2H+ + 2IO3− + 10NO2 + 4H2O ↔ 10HNO3 + I2 | 12 |
These experiments were repeated by Boukis and Henrich61 who found that the dissolution step may take >15 hours to remove >99% of the iodine inventory, and that both NO2 sparging and KIO3 stripping are necessary to achieve this removal efficiency. In that study, NO2 was generated by sparging the solution with NO (1.2 L h−1) and O2 (0.2 L h−1) diluted in N2. Excess NOx and HNO2 were removed from the dissolver with a subsequent air sparge.
Mineo et al.63 performed bench-scale dissolution tests of UNF to determine the percentage of radionuclides that were volatilized. The experiments found that, during dissolution at 90 °C in 4–5.5 M HNO3, 62–72% of the iodine inventory was released. After dissolution, the dissolver solution was stripped of iodine using a 2 hour KIO3 dissolution and 2 hour NO2 purge. The iodine stripping step likely released the residual dissolved iodine because no 129I was detected in the dissolver after this step, although the amount of iodine sequestered in solids could not be accurately quantified.
3.6.2. Observed iodine partitioning
Observations of iodine partitioning into the dissolver off-gas during UNF dissolution in reprocessing facilities broadly confirm experimental findings. In WAK Karlsruhe in Germany, approximately 94% of iodine was released to the dissolver off-gas during reprocessing activities in the 1970s and 1980s, and the remaining ∼5% moved to the solvent extraction vessel.13 This dissolver release efficiency was achieved after >8 hours of dissolution at 100 °C and NO2 sparging. However, Henrich et al.64 and Herrmann et al.65 suggested that by adding a NOx sparge and iodate carrier gas, WAK Karlsruhe could expel >99% of the iodine from the dissolver off-gas. Thus, a range of possible iodine release fractions have been observed in operating plants. These release fractions depend on dissolver conditions and analytical uncertainty.
3.7. Iodine speciation in the DOG
The primary volatile iodine species in the dissolver off-gas is elementary I2.45 However, organic iodoalkanes from methyl iodide to dodecyl iodide (i.e., CH3(CH2)xI, x = 0–11) have been observed or theoretically calculated to form in the dissolver off-gas.66 These organic iodides may form via reactions with solvents in recycled HNO3, or potentially even reaction with impurities in clean HNO3.15,67 Additionally, inorganic species such as HOI and HI have been theorized to form, but these iodine species are difficult to measure directly in the off-gas.67
Volatile iodine in the off gas has the potential to adsorb to the ducting surfaces. Most surfaces are expected to be either 304 or 316 austenitic stainless steel. Under a variety of conditions, I2 has been shown to form metal iodides with these materials.27,68,69 Due to the high humidity in these systems, highly deliquescent metal iodides (e.g., FeIx) are expected to readily dissolve. The HNO2 and HNO3 in the gas stream can then oxidize iodides back to I2 as shown in reactions (2) and (3), thereby enabling reversible reactions. However, not all metal iodides formed from the interaction between I2 and stainless steel are deliquescent and previous research has shown that the adsorption is not completely reversible.25–27 Ducting with lower humidity and NOx could result in the adsorbed metal iodides being favored, which would act as a sink for volatile iodine in the system.
Volatile iodine that is not trapped via adsorption with metal surfaces must be captured and retained by the facility's abatement system so that environmental regulations for safe release can be followed. A variety of abatement systems have been used ranging from liquid scrubbing (e.g., aqueous caustic scrubbing, molten hydroxide scrubbing, Iodox, Mercurex) to solid sorbent capture beds (e.g., silver mordenite, Clariant AC-6120).6,66,70–78 Wet scrubbing methods reduce the NOx and some forms of iodine; dry solid sorbent capture methods can be used separately or in tandem with wet methods.
4. Solvent extraction
Throughout history, several extraction techniques have been utilized for the separation of lanthanides and actinides from systems. These techniques include the Plutonium Uranium Extraction Process (PUREX), Bismuth Uranium Extraction (BUTEX), Reduction Oxidation (REDOX), and Mercury Extraction. The most popular of these systems is PUREX, which is shown in Fig. 4. Possible modifications to the process are shown in Fig. 5.79 The remaining I species in solution from the fuel dissolution can reach these extraction phase processes and lead to several issues.80 For PUREX, the remaining I species are theorized to be a small amount of iodate, colloidal species, and organoiodides. The small fraction of iodate is formed through reaction (5).15 The distribution of I species leads to contamination in both the aqueous and organic phases. Roughly 40–50% of the iodine will transfer to the organic phase. This leads to contamination in both feedstocks. Additionally, owing to the recycling of solutions, an accumulation of I species can lead to buildup in wash solutions leading to higher activities, breakdown or interaction of the organic phase, and uncontrolled discharge to low activity waste lines.7,15
Fig. 4. Traditional PUREX process.

Fig. 5. Basis of comparison flowsheet for UNF actinide recovery. This figure was recreated with permission from Arm.79.

Because I− oxidizes to I2 in the acidic fuel dissolution, the extraction of I2 with tributyl phosphate (TBP) has also been explored.81–83 Tsubomura and Kliegman81 reported the formation of the TBP·I2 in n-heptane with an absorption band at 460 nm. The band was attributed to the I2 bound to TBP, because free I2 absorbs at 525 nm.81 Tsubomura and Kliegman81 also stated that the I2 was likely bound to the oxygen in the P O group in TBP rather than the R–O group. The oxygen in the P O group is more negative than R–O, thereby allowing P O to act as a better electron donor than R–O. The presence of a charge transfer band from I2 to the oxygen donor in TBP was also speculated to be below 250 nm.
Zagorets et al.82 also reported the formation of a TBP·I2 complex. They confirmed both the appearance of the bound I2 to TBP at 460 nm and the charge transfer band below 250 nm. They also suggested the decomposition of TBP·I2 through radiolysis and subsequent formation of I3− through reactions (13)–(15).84 These low activity waste lines can lead to environmental contamination, as observed during fuel reprocessing at the Sellafield plan in the United Kingdom and the La Hague plant in France. At these sites, an estimated 1400 kg (Sellafield) and 3800 kg (La Hague), of radioactive iodine was released into the environment.7,85 Additionally, iodine insoluble residues build up in sediment and has an average of 2.34 × 109 atoms per L of sediment.86
| TBP + I2 ↔ TBPI2 | 13 |
| TBPI2 ↔ TBPI+ + I− | 14 |
| TBPI2 + I− ↔ I3− + TBP | 15 |
Historical observations of the exact fraction of iodine that remains in solution from fuel dissolution and eventually reaches the solvent extraction step have varied; however, all papers have reported some percentage of iodine being transported (even with extreme caution). For example, as reported by Henrich et al.,36,87 extensive treatment to remove the iodine from the dissolver solution by lowering the boiling temperature, and the addition of potassium iodate, still led to less than 1% of the iodine remaining with the recycled HNO3.87 In 1988, these results were further verified by Herrmann et al.65
5. Iodine transformation in the environment
5.1. General reviews
Many references provide introductory material regarding the characteristics of iodine and its environmental cycling. Perhaps the most consistently cross-referenced is a paper by Whitehead,88 that provides a synopsis of iodine sources, concentrations, and general transformations throughout the atmosphere, lithosphere, hydrosphere, and biosphere. Fig. 6 provides a schematic of these processes and reservoirs.
Fig. 6. Outline of the movement of iodine in the environment. Figure was recreated from Whitehead and reprinted with permission.88 Copyright 1984 Elsevier.

The 2004 Toxicological Profile for Iodine published by the Agency for Toxic Substances and Disease Registry (ATSDR) is a large volume (580 pages) that describes the effects to human health that result from exposure to various isotopic forms of iodine.89 A section of this publication provides a more quantitative description than Whitehead's review of release pathways and cycling processes. For example, Fig. 7 shows global annual fluxes and steady-state concentrations of iodine throughout various environmental reservoirs.
Fig. 7. Diagram of the global iodine cycle at steady state that shows environmental compartment inventories in grams (g), transport pathways, and fluxes in grams per year (g per year). This figure was taken from ATSDR (2004).89.

While the Whitehead88 and ATSDR89 references provide excellent overviews of iodine in all environmental compartments, a separate reference by Vogt90 provides a more in-depth account of iodine in the atmosphere. This reference describes the partitioning of iodine between organic, inorganic, and particulate (iodine solvated in airborne water droplets) forms and tabulates observations of each throughout the world. The text also shows that the photo-dissociation of iodine atoms from organic forms of iodine is highly dependent on speciation (Table 2). The diurnal lifetimes were calculated by scaling the photolysis rate constant by ZA at mid-latitudes throughout a 24 hour cycle during summer and winter. The rate constant is assumed to be zero at night (when ZA = 90). Straight chained n-alkyl-iodides are quite long lived whereas branched and multi-halide species have significantly shorter lifetimes. Vogt90 also introduces a schematic (without rate constants) mechanism that describes the reactive, photolytic, and heterogeneous exchange of iodine in the atmosphere. This mechanism is a prelude to the separate, extensively cross-referenced paper by Vogt et al.91
Properties of select organoiodide compounds including the photolysis rate constants, instantaneous atmospheric lifetimes at a solar zenith angle (ZA) of 40°, diurnal lifetime, and the corresponding ref. 90 and 92–94.
| Compound | Photolysis rate (s−1) | Instantaneous lifetime | Diurnal lifetime (summer–winter) | Ref. |
|---|---|---|---|---|
| CF3I | 2 × 10−5a | 14 ha | 1–3 daya | Solomon et al.92 |
| CH2ClI | 1 × 10−4 | 3 h | <1 day | Roehl et al.93 |
| CH2I2 | 5 × 10−3 | 3 min | <1 day | Roehl et al.93 |
| CH3(CH2)2I | 7 × 10−6 | 40 h | 4–8 day | Roehl et al.93 |
| CH3CHICH3 | 2 × 10−5 | 14 h | 1–3 day | Roehl et al.93 |
| CH3I | 5 × 10−6 | 55 h | 6–12 day | Jenkin94 |
The data for CF3I is calculated for midlatitudes in winter.
5.2. Iodine reaction mechanisms
Unlike other halogens (F, Cl, Br), iodine does not play a major role in hydrogen abstraction from volatile organic compounds (VOCs) in the atmosphere. Instead, iodine primarily reacts with ozone (O3) to produce iodine monoxide IO˙ radicals. Whereas some subsequent IO reactions lead to the restoration of O3 for a “null” cycle, other reaction cycles lead to the catalytic removal of O3 from the atmosphere.90 The significance of this pathway has led to extensive studies of iodine cycling in the marine boundary layer (MBL) where the biogenic emission of organic iodine from surface algae generates some of the highest concentrations of atmospheric iodine in the world. In 1999, Vogt et al. proposed mechanisms to describe these processes.91 In 2000, McFiggans et al. proposed a variation of the mechanism by adding an IO self-reaction channel in the gas phase and additional aqueous phase pathways to liberate IX (where X represents Cl, Br, or I) back to the gas phase.95 Variations of the mechanism (reversible and irreversible uptake into aqueous aerosol) were compared with MBL measurements of IO and a few other iodine species in two locations (Mace Head, Ireland at 53° N and Tenerife Island at 28° N).95 Both Vogt et al.91 and McFiggans et al.95 are widely cross-referenced in literature about iodine reaction schemes and their effects on O3 depletion in the MBL.
5.3. Source characterization
Numerous source characterization studies have been conducted to quantify the release of radioiodine from nuclear reprocessing centers in the United States. The purpose of many of these studies was to characterize the impact of emissions to the local environment and more specifically to assess any potential long-term health implications faced by human populations near these facilities. A reference by Kantelo et al.96 in 1993 is one such study. Because these reports are health related, most have focused on long-term (usually annual) accumulated total emissions and have generally described 131I monitoring. These reports can provide some insight into the chemical distribution of iodine emissions. For example, Kantelo et al.96 reported that 80–90% of iodine released in air effluent from Savannah River Site's H Canyon was characterized as organic with methyl, ethyl, and butyl forms being the most concentrated species observed.
5.4. Field measurement studies
Field measurements, ex situ of facilities, provide a direct account of the exchange of iodine between different chemical phases and different environmental compartments. These measurements can provide data that are useful for model development and validation. Several studies were identified that targeted characterization of iodine in meteoric water (i.e., rain).86,97,98 Moran et al.98 describe analysis of meteoric water and epiphyte (Spanish moss) samples collected at various locations throughout the United States. The mechanism for iodine content in rainwater is a combination of the heterogeneous nucleation of rain droplets formed around particles that may contain iodine, the chemical adsorption of gaseous iodine material into a droplet, and the physical washout of aqueous and/or nonaqueous aerosols as a droplet falls to the ground. In all cases, the iodine content is a direct result of the atmospheric iodine at the location and specific time of the rain event. Likewise, iodine content in epiphytes is a direct result of atmospheric iodine in the local area. Because these plants do not have a root system, they acquire their nutrients from direct cycling of the air. In contrast to rain measurements, epiphytes provide a long-term integrated collection mechanism that represents average atmospheric conditions for extended periods of time. Although, the absolute iodine concentrations in rain samples were much lower than those reported for epiphyte samples, the 129I/127I values were consistently in the 1 × 10−9 range for both media. These ratios were generally an order of magnitude greater than collections from fresh water and riverine systems in the vicinity suggesting an enhanced atmospheric signal. Moran et al.98 performed basic transport calculations to attribute these elevations to activity from the Sellafield, England and Cap de La Hague, France reprocessing facilities.
Hou et al.86 reported on another meteoric water study, that showed evidence of a preferential speciation for 129I (as iodide) vs.127I (as iodate) in rain water suggesting a native difference in the primary atmospheric species containing each isotope that was scavenged by the rain (Fig. 8). The authors86 suggested that the IO3−/I− ratio in collected rain could provide an attributable indicator of the origins of air masses that were present during the collection event.
Fig. 8. Variations of iodide (I−), iodate (IO3−), non-ionic iodine, and total iodine concentrations in precipitation from Roskilde, Denmark from 2001–2006 for (a) 127I and (b) 129I. The error bars show a one standard deviation analytical uncertainty. Reprinted with permission from Hou et al.86 Copyright 2009 American Chemical Society.

The work of Moran et al.98 and Hou et al.86 clearly indicate that rain is a major environmental scavenger of atmospheric iodine, regardless of source, and its effects must be included in any atmospheric transport modeling scheme. The 1988–1991 works of Robens, Wershofen, and Aumann99–106 reported on environmental iodine at a much closer range to a nuclear reprocessing operation. This reporting is an 8-volume compendium that documents nearly a decade of environmental data from the areas surrounding the Karlsruhe Nuclear Fuel Reprocessing Plant (WAK) in Germany. Chemical distributions (organic, inorganic, and particulate) were characterized at various downwind locations and contrasted with observations in Bonn, Germany, which is upwind of the facility. The distribution of 129I clearly favored organic forms whereas the 127I was much more distributed across all three chemical forms with only a moderately higher organic content.105Fig. 9 shows example data from summer collections between June and September 1987. Similar values were reported for off-gases at the Sellafield facility where 60% of the iodine was organic, 40% was inorganic, and <1% was particulate.98
Fig. 9. Air concentrations of (a) 129I and (b) 127I in various chemical forms at four sampling locations NNE from WAK in the dominant downwind direction, as measured between June and September 1987. This figure was modified with permission from Wershofen and Aumann.105 Copyright 1989 Elsevier.

5.4.1. Reactions
As previously discussed, the Vogt et al.91 and McFiggans et al.95 environmental fate descriptions for iodine in the MBL are widely cross-referenced sources and virtually no information was found that described characterization over land-based regions remote from the MBL.91,95 As a result, these two references were the primary basis for the mechanisms considered in this review. Reaction rates were supplemented with updates reported in the National Institute of Standards and Technology (NIST) and International Union of Pure and Applied Chemistry kinetics databases as well as with data from a few more recent experimental studies.107–116
5.4.2. Deposition (wet and dry)
Wet deposition is the loss of gas-phase and/or particulate material (PM) from the atmosphere due to adsorption in rain. The magnitude of the loss is calculated using Henry's law constants for each iodine species. Vogt et al.91 reported Henry law constants for many iodine species and these values were used directly. Where values were not reported, Henry's law constants were estimated. Model developers reviewed the literature for a wide range of atmospheric species to generate a relationship between Henry law constants and solubility in water. A power function was fit to the data and this relationship was used to fill in data gaps for iodine species.
Dry deposition is the loss of gas-phase material and/or PM from the atmosphere to ground level surfaces (e.g., soil, plants, and buildings). The magnitude of the loss depends on the gravitational settling velocity (for PM only), the flow of the air around or near the surface (calculated from meteorological conditions), the ability of the material to diffuse through air to a surface binding site (calculated from a diffusion coefficient), and the chemical attraction between the surface and the material (calculated from a canopy resistance).117
Diffusion coefficients were estimated using the US Environmental Protection Agency Estimation Program Interface Suite 4.10 program.118 Dry surface canopy resistance (dry Rc) values were estimated for organic and inorganic species respectively. Organic species are reported to have very slow deposition rates.88 As a result, dry Rc values for these species were set to 5000 to limit the deposition of these materials. For inorganic species, the dry Rc values were empirically fit to data summarized in a review by Sehmel,119 Sehmel119 reported deposition velocities of I2 ranging from 0.02–26 cm s−1 and the majority of these values fell between 0.05 and 7 cm s−1.119 The reactive atmospheric transport model, which is known as Chemical Calculations using Ordinary Differential Equations (ChemCODE),120 was modified to output deposition rates and plume height in order to manually calculate a deposition velocity. A dry Rc value of 1 injected into ChemCODE produced deposition velocities for a variety of land uses (e.g., forest, grass, water) that were the most consistent with those reported by Sehmel.119 This value was then applied to all inorganic iodine species. Wet surface canopy resistance (wet Rc) values were estimated to range from 1 to 5000 and were scaled between species based on their respective solubility values. Table 3 lists the physical property results that were generated by the authors for each iodine species.
Chemical properties of iodine-containing species.
| Species | Diffusion coefficient | MW (g mol−1) | Canopy resistance (dry deposition) | Henry's law constant (M atm−1) | Solubility (g L−1) | |
|---|---|---|---|---|---|---|
| Dry Rc | Wet Rc | |||||
| DMSa | 0.1220 | 62.14 | 5000 | 50 | 0.48 | 22 |
| CH3I | 0.1070 | 141.937 | 5000 | 100 | 0.14 | 13.8 |
| I2 | 0.0972 | 253.81 | 1 | 5000 | 3 | 0.33 |
| HI | 0.1270 | 127.913 | 1 | 1 | 5 × 103 | 425 |
| HOI | 0.1185 | 143.911 | 1 | 10 | 4.5 × 102 | 53.5 |
| C4H9I | 0.0789 | 184.012 | 5000 | 5000 | 6.30 × 10−2 | 0.2 |
| I | 0.1315 | 126.905 | 1 | 5000 | 4.32 × 10−2 | 1.00 |
| IO | 0.1220 | 142.904 | 1 | 1 | 4.50 × 102 | 450 |
| INO2 | 0.1031 | 172.91 | 1 | 400 | 0.3 | 5 |
| OIO | 0.0932 | 158.903 | 1 | 150 | 1 | 10 |
| IONO2 | 0.0983 | 188.909 | 1 | 1 | 5000 | 450 |
| IX | 0.1027 | 207.659 | 1 | 700 | 44.6667 | 3.51 |
| CH2I2 | 0.0873 | 267.835 | 5000 | 5000 | 2.55 | 0.833 |
| CH2IBr | 0.0921 | 220.834 | 5000 | 5000 | 3 | 0.759 |
| I2O2 | 0.0898 | 285.808 | 1 | 1 | 5000 | 450 |
DMS (dimethyl sulfide) is a common tracer for testing computational models of atmospheric transport.
5.4.3. Heterogeneous chemistry
Vogt et al.91 and McFiggans et al.95 describe particulate iodine in the MBL as water droplets (predominately sea spray) that have dissolved iodine content. Both references use a series of reactions to account for the adsorption, transformation, and release of iodine species from this heterogeneous particulate phase. Because the phase transfer and aqueous reactions have rate constants with different units than those in the gas phase, they cannot be solved simultaneously by a single ordinary differential equation solver like the construct in ChemCODE. As a result, these portions of the reference mechanisms were not used. ChemCODE does have a heterogeneous module that can account for gas phase interactions with particulate matter. The physics-based module allows for a first order treatment of the uptake, reactive loss, and desorption of species onto and from a fixed size distribution of particles with generic composition (i.e., the model does not resolve a differentiation between water, dry aerosols, soot, and other components). The main advantage of using this approach to heterogeneous chemistry was that it did not require a significant modification to the existing ChemCODE architecture during this prototype development to account for what is generally considered to be a minor fraction (<25%) of the total atmospheric iodine content.88,90,96,98 This approach is also more flexible if future studies suggest that gaseous iodine has a strong interaction with non-aqueous aerosols that are more typical over land-based transport regimes compared with the MBL. The main disadvantage was that this treatment could not easily leverage the aqueous phase chemistry presented by Vogt et al.91 and McFiggans et al.95 Although the particulate fraction of iodine is considered to be small relative to the gas fraction, both sources have shown that this aqueous chemistry can affect speciation within the gas phase.
5.5. Meteorology
Iodine is transported globally by atmospheric circulations. For example, increased concentrations of 131I released from the damaged Fukushima Daiichi nuclear power plant were detected across the northern hemisphere.121 Regardless, both observations and models indicate that the tropics have the highest concentration of iodine as a result of global distribution.122,123 Temporally, in the northern hemisphere, iodine concentrations are generally found to be elevated during the summer months.123–125
The reactive capability of various inorganic species aids in the formation and growth of particles. In aerosol chamber reactions investigated at the Cosmics Leaving Outdoor Droplets (CLOUD) chamber in the European Organization for Nuclear Research (Conseil européen pour la Recherche nucléaire, CERN), iodic acid (HIO3) was shown to have higher particle nucleation and growth rates at low temperature (−10 °C) than sulfuric acid. HIO3 particle nucleation rates continued to increase while growth rates were similar to those of sulfuric acid at 10 °C.126 Photolysis rates of iodine are likely affected by the cloud field influence on actinic flux.127 Additionally, aqueous phase reactions within cloud droplets could play a role in gas to particle conversions.128 Models that included HIO3 particle formation compared better with particle size distribution measurements than those that only used sulfuric acid.129 Additionally, most organic species are short lived and are processed through reactions with OH and photolysis.130,131 Droste et al.132 found that coarse (>1 μm) sea spray and mineral dust particles are more likely to contain IO3− because they tend to be more alkaline. However polluted fine mode (<1 μm) particles tend to have more I- and soluble organic iodine due to higher acidity. Increased concentrations of iodine monoxide (IO) were also found in layers of dust plumes off the coast of South America.133 The tendency of iodine to adhere to particles can result in long distance transport through the atmosphere134 as well as vertically through convection.135,136 However, chemical processes within the particles could lead to iodine cycling, which would release the iodine from the particles.132,137
Iodine can be deposited to the surface via the particle settling (dry deposition) and via cloud and precipitation scavenging (washout). Estimated global wet and dry deposition of iodine species is approximately 1.65 Tg per year.122 Deposition of iodine has been increasing across the globe since the mid-20th century. These increases have been found in the Greenland ice cap, ice cores from a glacier in France, and in tree rings from spruce trees on the Qinghai-Tibet Plateau.124,138,139 Cuevas et al. attributed the increase in iodine deposition to increases in anthropogenic ozone pollution which induces increased iodine emissions.138 However, Zhao et al. attributed increases to human nuclear weapons testing.139 Because the deposition of airborne radioiodine onto pasture grass significantly affects the ingestion doses in produce, meat, and dairy, the magnitude of its deposition velocity is extremely important.140 Elemental iodine has a significantly higher deposition velocity than other inorganic forms due to its considerable reactivity. Organic forms of iodine are the least reactive and have the lowest relative deposition velocity.
Iodine particles in the atmosphere can act as cloud condensation nuclei that condense cloud water or as ice nuclei that form cloud ice, subsequently leading to precipitation and wet deposition. Falling rain and drizzle drops scavenge additional iodine particles and gases from the atmosphere. On average, precipitation contains 2.0 μg L−1 of iodine.88 Rainwater iodine concentrations in France varied from 0.8 to 2.7 μg L−1. The samples with the highest concentrations were collected in oceanic marine environments and in areas of transition from marine to continental.125 However, the lowest lying coastal areas in the United Kingdom were found to have less iodine content in rainwater than was observed 12–35 km inland in an elevated area.141 Baker et al.134 observed wet deposition fluxes to be 2.7 μmol per m2 per year at a coastal site near the marine environment.
The removal of iodine through dry deposition occurs when iodine particles settle or when iodine gases adhere to the surface. Estimates of dry deposition of elemental iodine, other inorganic iodine, and iodine particles have been reported to be 1 cm s−1, <0.1 cm s−1, and 0.1–0.2 cm s−1, respectively.140,142–144 However, organic forms of iodine, such as CH3I, have a significantly lower reported approximate deposition velocities, (0.0001–0.005 cm s−1).140,145 Dry deposition of elemental iodine depends on the type of vegetation. Tschiersch et al.146 reported deposition velocities of 131I for a variety of leafy vegetables to be 0.16–1.6 mm s−1. At a coastal United Kingdom site, Baker et al.134 reported deposition from aerosol sedimentation as 3.6 to 6.5 μmol per m2 per year and direct deposition of CH3I as 0.003–0.17 μmol per m2 per year.
5.6. Geographic considerations
The atmospheric processing and deposition of iodine vary depending on the environment. Iodine in the atmosphere is primarily released from the oceans. A significant portion of the iodine in the oceans resides in ionic forms (I− and IO3−). Iodine can be oxidized by ozone at the ocean's surface and can be released into the atmosphere as I2 and HOI.147 Organic forms of iodine (i.e., CH3I, CH2I2, and other halogenated organics) from biological sources are also released from the ocean's surface.141 Soluble organic iodine fractions of marine aerosol are largest near the equator and smallest in less biologically productive regions.148 Higher humidity near water bodies likely increases reactions between iodine and OH. Marine boundary layer aerosols participate in iodine recycling, in which soluble organic iodine and HOI react and collect onto particles that then release gaseous inorganic iodine.132,149 Environments near the coast can be influenced by the transport of iodine from the marine boundary layer.
Because iodine is primarily released from the ocean, terrestrial sources of iodine in the atmosphere are fewer, likely because the low iodine content in crustal rocks limits iodine production from the weathering of rocks.141 Iodocarbons can be emitted from terrestrial biogenic processes.150 Additionally, iodine is more commonly found in sedimentary rocks and dust.133,141 Koenig et al.133 identified larger concentrations of iodic acid in atmospheric layers containing elevated concentrations of windblown dust off the coast South America. These layers coincided with reduced tropospheric ozone. Iodine could also be released by the burning of vegetation that had previously taken up the iodine.151 Thus, local iodine speciation depends on geographic location.
6. Summary and conclusions
Before dissolution, the predominate species of iodine in both metal and oxide fuels is expected to be CsI. Depending on burnup, some fraction may form insoluble Pd–Ag iodide compounds in oxide fuels. Less data is available on the forms of iodine in metal fuels. The species of iodine generated during dissolution has been the focus of many research efforts, but owing to the multiple variables, including acid concentration, radiolysis, and sparging, continued research is needed to fully understand this complex system. A large fraction of the iodine partitions to the gas phase during dissolution and, depending on conditions, has the potential to interact with various surfaces in the off-gas system. Some fraction of the iodine is expected to adsorb or absorb to surfaces, although the specific amount remains a multi-variable problem. Abatement is also a well-studied step, but its effectiveness hinges on understanding the quantities and species present in the off-gas system. The iodine that stays in solution after dissolution can move to the extraction step. During extraction, I2, I−, and IO3− have all been shown to extract at varying degrees to the organic phase. The specific extraction behavior depends on the species present in the aqueous phase, which is variable. Many studies have attempted to elucidate the species and quantities of iodine at various steps in a reprocessing facility; however, the interdependence of the steps necessitates an overall process model to understand the complete system.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgments
Pacific Northwest National Laboratory (PNNL), Oak Ridge National Laboratory, Savannah River National Laboratory, and ENSCO's contributions were funded through the Office of Defense Nuclear Nonproliferation within the US Department of Energy's (DOE's) National Nuclear Security Administration. PNNL is operated by Battelle Memorial Institute for the DOE under contract DE-AC05-76RL01830. This manuscript has been authored in part by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with DOE. Savannah River National Laboratory is operated by Battelle Savannah River Alliance under Contract No. 89303321CEM000080 with DOE's Office of Environmental Management. Publisher acknowledges the U.S. Government license to provide public access under the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
References
- 40 CFR 61.92, Environmental Protection Agency, Washington, D.C [Google Scholar]
- Standards for Protection Against Radiation, Nuclear Regulatory Commission, Washington D.C: [PubMed] [Google Scholar]
- Marković S. Influence of the acidity of the iodous acid solution system on the kinetics of the disproportionation reaction. Bulletin of Natural Sciences Research. 2021;11:20–24. [Google Scholar]
- Luther G. W. Review on the physical chemistry of iodine transformations in the oceans. Front. Mar. Sci. 2023;10:1085618. [Google Scholar]
- Greaney A. T. Ngelale R. O. Bruffey S. H. Martin L. R. Abatement of radioiodine in aqueous reprocessing off-gas. Front. Chem. 2023;10:1078668. doi: 10.3389/fchem.2022.1078668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley B. J. Vienna J. D. Strachan D. M. McCloy J. S. Jerden Jr J. L. Materials and processes for the effective capture and immobilization of radioiodine: a review. J. Nucl. Mater. 2016;470:307–326. [Google Scholar]
- Kulyukhin S. A. Fundamental and applied aspects of the chemistry of radioactive iodine in gas and aqueous media. Russ. Chem. Rev. 2012;81:960–982. [Google Scholar]
- Naceur A. Marleau G. Neutronic analysis for accident tolerant cladding candidates in CANDU-6 reactors. Ann. Nucl. Energy. 2018;113:147–161. [Google Scholar]
- NRC, Accident Tolerant Fuel Technologies, https://www.nrc.gov/reactors/power/atf/technologies.html, accessed 6/18/2024
- Jubin R. T., Spent Fuel Reprocessing, Consortium for Risk Evaluation with Stakeholder Participation (CRESP), Vanderbilt University, Nashville, TN, 2008 [Google Scholar]
- Unger W. E., Blanco R. E., Crouse D. J., Irvine A. R. and Watson C. D., Aqueous Fuel Reprocessing Quarterly Report for Period Ending December 31, 1972, Report ORNL-TM-4141, Oak Ridge National Laboratory, Oak Ridge, TN, 1973 [Google Scholar]
- Jubin R. T., Greaney A. T., Spencer B. B. and Bruffey S. H., Testing of an Iodine and Tritium Capture System for an NO2-Based Tritium Pretreatment Process, Report ORNL/TM-2019/1220, Oak Ridge National Laboratory, Oak Ridge, TN, 2019 [Google Scholar]
- Herrmann F. J., Motoi V., Fang D., Finsterwalder L., Kuhn K. D., van Shoor A., Beyer C., Furrer J. and Knoch W., 22nd DOE/NRC Nucl. Air Clean. Treat. Conf., 1993, pp. 75–90
- Herrmann F., Herrmann B., Kuhn K., Van Schoor A., Weishaupt M., Furrer J. and Knoch W., 24th DOE/NRC Nuclear Air Cleaning and Treatment Conference (NUREG/CP-0153, CONF-960715), 1997, pp. 618–627
- Jubin R. T., Strachan D. M. and Soelberg N. R., Iodine Pathways and Off-Gas Stream Characteristics for Aqueous Reprocessing Plants – A Literature Survey and Assessment, Report INL/EXT-13-30119, FCRD-SWF-2013-000308, ORNL/LTR-2013/383, Idaho National Laboratory, Idaho Falls, ID, 2013 [Google Scholar]
- Lewis B. J., Thompson W. T. and Iglesias F. C., in Comprehensive Nuclear Materials, ed. R. J. M. Konings, Elsevier, Amsterdam, 2012, vol. 2, pp. 515–546 [Google Scholar]
- Viswanathan R. Fuel clad chemical interactions in fast reactor MOX fuels. J. Nucl. Mater. 2014;444:101–111. [Google Scholar]
- Kleykamp H. The chemical state of the fission products in oxide fuels. J. Nucl. Mater. 1985;131:221–246. [Google Scholar]
- Saidy M. Hocking W. H. Mouris J. F. Garcia P. Carlot G. Pasquet B. Thermal diffusion of iodine in UO2 and UO2+x. J. Nucl. Mater. 2008;372:405–415. [Google Scholar]
- Cox B. Pellet-clad interaction (PCI) failures of zirconium alloy fuel cladding—a review. J. Nucl. Mater. 1990;172:249–292. [Google Scholar]
- Johnson L. H. Burns K. I. Joling H. H. Moore C. J. Leaching of 137Cs, 134Cs, and 129I from Irradiated UO2 Fuel. Nucl. Technol. 1983;63:470–475. [Google Scholar]
- Oversby V. M., in Actinides and the Environment, ed. P. A. Sterne, A. Gonis and A. A. Borovoi, Kluwer Academic Publishers, Netherlands, 1998, pp. 245–265 [Google Scholar]
- Umadevi K. Mandal D. Performance of radio-iodine discharge control methods of nuclear reprocessing plants. J. Environ. Radioact. 2021;234:106623. doi: 10.1016/j.jenvrad.2021.106623. [DOI] [PubMed] [Google Scholar]
- Buck E. C. Mausolf E. J. McNamara B. K. Soderquist C. Z. Schwantes J. M. Sequestration of radioactive iodine in silver-palladium phases in commercial spent nuclear fuel. J. Nucl. Mater. 2016;482:229–235. [Google Scholar]
- Beck C. L. Riley B. J. Chong S. Karkamkar A. Seiner D. R. Clark S. B. Molecular iodine interactions with metal substrates: towards the understanding of iodine interactions in the environment following a nuclear accident. J. Nucl. Mater. 2021;546:152771. [Google Scholar]
- Beck C. L. Riley B. J. Chong S. Smith N. Seiner D. R. Seiner B. N. Engelhard M. H. Clark S. B. Molecular Iodine Interactions with Fe, Ni, Cr, and Stainless Steel Alloys. Ind. Eng. Chem. Res. 2021;60:2447–2454. [Google Scholar]
- Beck C. L. Smith N. P. Riley B. J. Clark S. B. Adsorption of iodine on metal coupons in humid and dry environments. J. Nucl. Mater. 2021;556:153204. [Google Scholar]
- Lewis B. J. Fundamental aspects of defective nuclear fuel behaviour and fission product release. J. Nucl. Mater. 1988;160:201–217. [Google Scholar]
- Bryant P. M. and Warner B. F., Control of Radioiodine Release from Reprocessing Plants, International Atomic Energy Agency, Vienna (Austria), 1973 [Google Scholar]
- Burger L. L., Fission product iodine during early Hanford-site operations: its production and behavior during fuel processing, off-gas treatment and release to the atmosphere, Report PNL-7210-HEDR, UC-707, Pacific Northwest National Laboratory, Richland, WA, 1991 [Google Scholar]
- Guenther R. J., Blahnik D. E., Campbell T. K., Jenquin U. P., Mendel J. E., Thomas L. E. and Thornhill C. K., Characterization of Spent Fuel Approved Testing Material – ATM-105, Report PNL-5109-105, UC-802, Pacific Northwest Laboratory, Richland, WA, 1991 [Google Scholar]
- Campbell T. K. Guenther R. J. Jenson E. D. Ceram. Trans. 1990:409–422. [Google Scholar]
- Lide D. R., CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 88th edn, 2008 [Google Scholar]
- Mailen J. C. Tiffany T. O. The reaction of iodine with concentrated nitric acid. J. Inorg. Nucl. Chem. 1975;37:127–132. [Google Scholar]
- LMFBR Fuel Cycle Studies Progress Report for January 1970, No. 11, Report ORNL-TM-2871, Oak Ridge National Laboratory, Oak Ridge, TN, 1970 [Google Scholar]
- Lieser K. H. Georgoulas P. Hoffmann P. Radioiodine in Nitric Acid. Radiochim. Acta. 1989;48:193–200. [Google Scholar]
- Sakurai T. Izumo M. Takahashi A. Komaki Y. Behavior of Iodine-131 in Dissolution of Irradiated Uranium Dioxide. J. Nucl. Sci. Technol. 1987;24:931–936. [Google Scholar]
- Smith M. B., in Encyclopedia of Reagents for Organic Synthesis (EROS), ed. A. Charette, J. Bode, T. Rovis and R. Shenvi, John Wiley & Sons, Ltd., 2005, 10.1002/047084289X.ri005.pub3 [DOI] [Google Scholar]
- Carter J. S. CCCXCVI—the salting-out effect. The influence of electrolytes on the solubility of iodine in water. J. Chem. Soc., Trans. 1925;127:2861–2866. [Google Scholar]
- Lutsyk A. I. Suikov S. Y. Chuprina V. S. Portnyanskii V. Y. Solubility of Iodine in the System Water-Nitric Acid. Russ. J. Appl. Chem. 2004;77:1195–1197. [Google Scholar]
- Unger W. E., Blanco R. E., Crouse D. J., Irvine A. R. and Watson C. D., LMFBR fuel cycle studies progress report for July 1972, no. 41. Aqueous fuel reprocessing, Report ORNL-TM-3952, Oak Ridge National Laboratory, Oak Ridge, TN, 1972 [Google Scholar]
- Richard L. Gaona X. Thermodynamic properties of organic iodine compounds. Geochim. Cosmochim. Acta. 2011;75:7304–7350. [Google Scholar]
- Schmitz G. Inorganic reactions of iodine(+1) in acidic solutions. Int. J. Chem. Kinet. 2004;36:480–493. [Google Scholar]
- Berry F. J. Collins R. D. Parish R. V. Moore L. S. Studies of iodine in nitric acid and of iodine adsorbed onto silver-impregnated silica. Inorg. Chim. Acta. 1987;126:119–124. [Google Scholar]
- Sakurai T. Takahashi A. Ishikawa N. Komaki Y. The Behavior of Iodine in a Simulated Spent-Fuel Solution. Nucl. Technol. 1989;85:206–212. [Google Scholar]
- Sakurai T., Komatsu K. and Takahashi A., 24th DOE/NRC Nucl. Air Clean. Treat. Conf., 1996, pp. 550–562
- Sakurai T. Takahashi A. Ishikawa N. Komaki Y. Ohnuki M. Adachi T. The Iodine Species and Their Behavior in the Dissolution of Spent-Fuel Specimens. Nucl. Technol. 1992;99:70–79. [Google Scholar]
- Mailen J. C. Bell J. T. Potential for the Use of Hydrochloric Acid in Fission Reactor Fuel Recycle. Sep. Sci. Technol. 1987;22:347–360. [Google Scholar]
- Cripps R. C. Güntay S. Jäckel B. The PSIodine code: a computer program to model experimental data on iodine and other species in irradiated CsI solutions sparged with argon, air, or nitrous oxide. Nucl. Eng. Des. 2011;241:4306–4325. [Google Scholar]
- Wren J. C. Ball J. M. LIRIC 3.2 an updated model for iodine behaviour in the presence of organic impurities. Radiat. Phys. Chem. 2001;60:577–596. [Google Scholar]
- Jiang P.-Y. Nagaishi R. Yotsuyanagi T. Katsumura Y. Ishigure K. γ-Radiolysis study of concentrated nitric acid solutions. J. Chem. Soc., Faraday Trans. 1994;90:93–95. [Google Scholar]
- Pastina B. LaVerne J. A. Effect of Molecular Hydrogen on Hydrogen Peroxide in Water Radiolysis. J. Phys. Chem. A. 2001;105:9316–9322. [Google Scholar]
- Park J. Y. Lee Y. N. Solubility and decomposition kinetics of nitrous acid in aqueous solution. J. Phys. Chem. 1988;92:6294–6302. [Google Scholar]
- Palmer D. A. Ramette R. W. Mesmer R. E. The hydrolysis of iodine: equilibria at high temperatures. J. Nucl. Mater. 1985;130:280–286. [Google Scholar]
- Castleman A. W. Tang I. N. Munkelwitz H. R. The chemical states of fission-product iodine emanating into a high temperature aqueous environment. J. Inorg. Nucl. Chem. 1968;30:5–13. [Google Scholar]
- Adachi T. Ohnuki M. Yoshida N. Sonobe T. Kawamura W. Takeishi H. Gunji K. Kimura T. Suzuki T. Nakahara Y. Muromura T. Kobayashi Y. Okashita H. Yamamoto T. Dissolution study of spent PWR fuel: dissolution behavior and chemical properties of insoluble residues. J. Nucl. Mater. 1990;174:60–71. [Google Scholar]
- Marc P. Magnaldo A. Vaudano A. Delahaye T. Schaer É. Dissolution of uranium dioxide in nitric acid media: what do we know? EPJ Nucl. Sci. Technol. 2017;3:13. [Google Scholar]
- Glänneskog H. Liljenzin J.-O. Sihver L. Reactions between reactive metals and iodine in aqueous solutions. J. Nucl. Mater. 2006;348:87–93. [Google Scholar]
- Sakurai T. Takahashi A. Ishikawa N. Komaki Y. Ohnuki M. Adachi T. Thermochemical and Experimental Considerations of NOx Composition and Iodine Species in the Dissolution of Spent PWR-Fuel Specimens. J. Nucl. Sci. Technol. 1993;30:533–541. [Google Scholar]
- Sakurai T. Takahashi A. Ishikawa N. Komaki Y. The Interaction of Iodine with Insoluble Residue in the Dissolution of Simulated Spent-Fuel Pellets. Nucl. Technol. 1991;94:99–107. [Google Scholar]
- Boukis N. Henrich E. Two-Step Procedure for the Iodine Removal from Nuclear Fuel Solutions. Radiochim. Acta. 1991;55:37–42. [Google Scholar]
- Sakurai T. Takahashi A. Ishikawa N. Komaki Y. Ohnuki M. Influence of NOx and HNO2 on Iodine Quantity in Spent-Fuel Solutions. Nucl. Technol. 1996;116:319–326. [Google Scholar]
- Mineo H., Iizuka M., Fukisaki S., Kihara T. and Uchiyama G., 26th Int. Nucl. Air Clean. Conf., 2000
- Henrich E., Hufner R. and Sahm A., Management of Gaseous Wastes from Nuclear Facilities, 1980, pp. 139–156
- Herrmann F. J., Motoi V., Fies H., Stojanik B., Furrer J. and Kaempffer R., 20th DOE/NRC Nucl. Air Clean. Conf., 1988, pp. 234–245
- Pan T. Yang K. Dong X. Han Y. Adsorption-based capture of iodine and organic iodides: status and challenges. J. Mater. Chem. A. 2023;11:5460–5475. [Google Scholar]
- Bruffey S. H., Spencer B. B., Jubin R. T., Strachan R. M., Soelberg N. R. and Riley B. J., A Literature Survey to Identify Potentially Problematic Volatile Iodine-Bearing Species Present in Off-gas Streams, Report ORNL-SPR-2015/290, Oak Ridge National Laboratory, Oak Ridge, TN, 2015 [Google Scholar]
- Evans G. J. and Panyan E. J., Iodine volatilization from irradiated CsI solutions, CSNI Workshop on the Chemistry of Iodine in Reactor Safety, PSI-197-102; CONF-9606320, 1996, pp. 111–122 [Google Scholar]
- Tyler J. W. Surface analysis using X-ray photoelectron spectroscopy of iodine deposits on 17% Cr/12% Ni and mild steel surfaces oxidised in CO2CH3I gas mixtures. J. Nucl. Mater. 1989;161:72–88. [Google Scholar]
- Holladay D. W., A literature survey: methods for the removal of iodine species from off-gases and liquid waste streams of nuclear power and nuclear fuel reprocessing plants, with emphasis on solid sorbents, Report ORNL/TM-6350, Oak Ridge National Laboratory, Oak Ridge, TN, 1979 [Google Scholar]
- Haefner D. R. and Tranter T. J., Methods of Gas Phase Capture of Iodine from Fuel Reprocessing Off-Gas: A Literature Survey, Report INL/EXT-07-12299, Idaho National Laboratory, Idaho Falls, ID, 2007 [Google Scholar]
- Huve J. Ryzhikov A. Nouali H. Lalia V. Augé G. Daou T. J. Porous sorbents for the capture of radioactive iodine compounds: a review. RSC Adv. 2018;8:29248–29273. doi: 10.1039/c8ra04775h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pénélope R. Campayo L. Fournier M. Gossard A. Grandjean A. Solid sorbents for gaseous iodine capture and their conversion into stable waste forms. J. Nucl. Mater. 2022;563:153635. [Google Scholar]
- Kurisingal J. F. Yun H. Hong C. S. Porous organic materials for iodine adsorption. J. Hazard. Mater. 2023;458:131835. doi: 10.1016/j.jhazmat.2023.131835. [DOI] [PubMed] [Google Scholar]
- Moore R. C. Pearce C. I. Morad J. W. Chatterjee S. Levitskaia T. G. Asmussen R. M. Lawter A. R. Neeway J. J. Qafoku N. P. Rigali M. J. Saslow S. A. Szecsody J. E. Thallapally P. K. Wang G. Freedman V. L. Iodine immobilization by materials through sorption and redox-driven processes: a literature review. Sci. Total Environ. 2020;716:132820. doi: 10.1016/j.scitotenv.2019.06.166. [DOI] [PubMed] [Google Scholar]
- Riley B. J. McFarlane J. DelCul G. D. Vienna J. D. Contescu C. I. Forsberg C. W. Molten salt reactor waste and effluent management strategies: a review. Nucl. Eng. Des. 2019;345:94–109. [Google Scholar]
- Andrews H. B. McFarlane J. Chapel A. S. Ezell N. D. B. Holcomb D. E. de Wet D. Greenwood M. S. Myhre K. G. Bryan S. A. Lines A. Riley B. J. Felmy H. M. Humrickhouse P. W. Review of molten salt reactor off-gas management considerations. Nucl. Eng. Des. 2021;385:111529. [Google Scholar]
- Jie K. Zhou Y. Li E. Li Z. Zhao R. Huang F. Reversible Iodine Capture by Nonporous Pillar[6]arene Crystals. J. Am. Chem. Soc. 2017;139:15320–15323. doi: 10.1021/jacs.7b09850. [DOI] [PubMed] [Google Scholar]
- Arm S. T., Flowsheet Evaluation of Dissolving Used Nuclear Fuel in PUREX Solvent, Report PNNL-31863, Pacific Northwest National Laboratory, Richland, WA, 2021 [Google Scholar]
- Kulyukhin S. A. Kamenskaya A. N. Konovalova N. A. Chemistry of radioactive iodine in aqueous media: basic and applied aspects. Radiochemistry. 2011;53:123–141. [Google Scholar]
- Tsubomura H. Kliegman J. M. Molecular Complexes and their Spectra. XI. The Interaction of Iodide with Tri-n-Butyl Phosphate1. J. Am. Chem. Soc. 1960;82:1314–1317. [Google Scholar]
- Zagorets P. A. Raskina Z. I. Bulgakova G. P. Makarov V. M. Radiolysis of solutions of iodine in tributyl phosphate. Sov. At. Energy. 1972;32:494–496. [Google Scholar]
- Sakurai T. Takahashi A. Ishikawa N. Komaki Y. Interaction of Iodine with an Extractant of 30% TBP/70% n-Dodecane. J. Nucl. Sci. Technol. 1995;32:664–670. [Google Scholar]
- Nowak Z. The influence of iodine on the radiolysis of PUREX solvent. J. Radioanal. Nucl. Chem. Lett. 1988;127:309–317. [Google Scholar]
- Zhou W. J., Hou X. L., Chen N., Zhang L. Y., Liu Q., He C. H., Luo M. Y., Liang W. G., Fan Y. K., Wang Z. W., Fu Y. C. and Li H. B., Preliminary Study of radioisotope 129I application in China using Xi'an Accelerator Mass Spectrometer, INCS News, 2010, vol. 7, pp. 9–23
- Hou X. Aldahan A. Nielsen S. P. Possnert G. Time Series of 129I and 127I Speciation in Precipitation from Denmark. Environ. Sci. Technol. 2009;43:6522–6528. doi: 10.1021/es9012678. [DOI] [PubMed] [Google Scholar]
- Henrich E. and Hufner R., 16th DOE Nuclear Air Cleaning Conference, 1980, pp. 612–625
- Whitehead D. C. The distribution and transformations of iodine in the environment. Environ. Int. 1984;10:321–339. [Google Scholar]
- Toxicological Profile for Iodine, U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances & Disease Registry (ATSDR), Atlanta, Georgia [Google Scholar]
- Vogt R., in Reactive Halogen Compounds in the Atmosphere, ed. P. Fabian and O. N. Singh, Springer Berlin Heidelberg, Berlin, Heidelberg, ch. 4, 1999, pp. 113–128, 10.1007/10628761_4 [DOI] [Google Scholar]
- Vogt R. Sander R. von Glasow R. Crutzen P. J. Iodine Chemistry and its Role in Halogen Activation and Ozone Loss in the Marine Boundary Layer: A Model Study. J. Atmos. Chem. 1999;32:375–395. [Google Scholar]
- Solomon S. Burkholder J. B. Ravishankara A. R. Garcia R. R. Ozone depletion and global warming potentials of CF3I. J. Geophys. Res. 1994;99:20929–20935. [Google Scholar]
- Roehl C. M. Burkholder J. B. Moortgat G. K. Ravishankara A. R. Crutzen P. J. The temperature dependence of the UV absorption cross sections and the atmospheric implications of several alkyl iodides. J. Geophys. Res. 1997;102:12819–12829. [Google Scholar]
- Jenkin M. E., The photochemistry of iodine-containing compounds in the marine boundary layer, Report AEA-EE-0405, United Kingdom Atomic Energy Authority, Harwell Laboratory, Oxon, OX11 0RA, U.K., 1992 [Google Scholar]
- McFiggans G. Plane J. M. C. Allan B. J. Carpenter L. J. Coe H. O'Dowd C. A Modeling Study of Iodine Chemistry in the Marine Boundary Layer. J. Geophys. Res.: Atmos. 2000;105:14371–14385. [Google Scholar]
- Kantelo M. V., Bauer L. R., Marter W. L., Murphy Jr C. E. and Zeigler C. C., Radioiodine in the Savannah River Site Environment, Report WSRC-RP-90-424-2, DE93-006329, Westinghouse Savannah River Company, Aiken, SC, 1993 [Google Scholar]
- Michel R., Ernst T., Jakob D., Klipsch K., Szidat S., Synal H.-A. and Schnabel C., Long-lived Radionuclides in the Environment: the Case of Iodine-129, EUROSAFE, 2002, pp. 1-9 [Google Scholar]
- Moran J. E. Oktay S. Santschi P. H. Schink D. R. Atmospheric Dispersal of 129Iodine from Nuclear Fuel Reprocessing Facilities. Environ. Sci. Technol. 1999;33:2536–2542. [Google Scholar]
- Robens E. Aumann D. C. Iodine-129 in the environment of a nuclear fuel reprocessing plant: I. 129I and 127I contents of soils, food crops and animal products. J. Environ. Radioact. 1988;7:159–175. [Google Scholar]
- Robens E. Hauschild J. Aumann D. C. Iodine-129 in the environment of a nuclear fuel reprocessing plant: II. Iodine-129 and iodine-127 contents of soils, forage plants and deer thyroids. J. Environ. Radioact. 1988;7:265–274. [Google Scholar]
- Robens E. Hauschild J. Aumann D. C. Iodine-129 in the environment of a nuclear fuel reprocessing plant: III. Soil-to-plant concentration factors for iodine-129 and iodine-127 and their transfer factors to milk, eggs and pork. J. Environ. Radioact. 1988;8:37–52. [Google Scholar]
- Robens E. Hauschild J. Aumann D. C. Iodine-129 in the environment of a nuclear fuel reprocessing plant: IV. 129I and 127I in undisturbed surface soils. J. Environ. Radioact. 1989;9:17–29. [Google Scholar]
- Hauschild J. Aumann D. C. Iodine-129 in the environment of a nuclear fuel reprocessing plant: V. The transfer of 129I and 127I in the soil-pasture-cow-milk/meat pathway, as obtained by field measurements. J. Environ. Radioact. 1989;9:145–162. [Google Scholar]
- Robens-Palavinskas E. Hauschild J. Aumann D. C. Iodine-129 in the Environment of a nuclear fuel reprocessing plant: VI. Comparison of measurements of 129I concentrations in soil and vegetation with predictions from a radiological assessment model. J. Environ. Radioact. 1989;10:67–78. [Google Scholar]
- Wershofen H. Aumann D. C. Iodine-129 in the environment of a nuclear fuel reprocessing plant: VII. Concentrations and chemical forms of 129I and 127I in the atmosphere. J. Environ. Radioact. 1989;10:141–156. [Google Scholar]
- Wershofen H. Aumann D. C. Hübschmann W. G. Iodine-129 in the environment of a nuclear fuel reprocessing plant: VIII. Comparison of measured 129I concentrations in the atmosphere with those predicted by a radiological assessment code. J. Environ. Radioact. 1991;13:93–101. [Google Scholar]
- NIST Chemical Kinetics Database, Standard Reference Database 17, Version 7.1 (Web Version), Release 1.6.8, Data Version 2024, https://kinetics.nist.gov/kinetics/
- Task Group on Atmospheric Chemical Kinetic Data Evaluation, https://iupac.aeris-data.fr/en/home-english/
- Carter W. P. L. Development of the SAPRC-07 chemical mechanism. Atmos. Environ. 2010;44:5324–5335. [Google Scholar]
- Mössinger J. C. Rowley D. M. Cox R. A. The UV-visible absorption cross-sections of IONO2. Atmos. Chem. Phys. 2002;2:227–234. [Google Scholar]
- Saiz-Lopez A. Saunders R. W. Joseph D. M. Ashworth S. H. Plane J. M. C. Absolute absorption cross-section and photolysis rate of I2. Atmos. Chem. Phys. 2004;4:1443–1450. [Google Scholar]
- Atkinson R. Baulch D. L. Cox R. A. Crowley J. N. Hampson R. F. Hynes R. G. Jenkin M. E. Rossi M. J. Troe J. Evaluated kinetic and photochemical data for atmospheric chemistry: volume III – gas phase reactions of inorganic halogens. Atmos. Chem. Phys. 2007;7:981–1191. [Google Scholar]
- Baulch D. L. Campbell I. M. Chappel J. M. Formation of organic nitro-compounds in flowing H2O2 + NO2 + N2 + organic vapour systems. Part 3—effects of O2 addition on H2O2 + NO2 + N2 + alkane systems. J. Chem. Soc., Faraday Trans. 1984;80:617–628. [Google Scholar]
- Knyazev V. D. Slagle I. R. Kinetics of the Reactions of n-Alkyl (C2H5, n-C3H7, and n-C4H9) Radicals with CH3. J. Phys. Chem. A. 2001;105:6490–6498. [Google Scholar]
- Darwin D. C. Moore C. B. Reaction Rate Constants (295 K) for 3CH2 with H2S, SO2, and NO2: Upper Bounds for Rate Constants with Less Reactive Partners. J. Phys. Chem. 1995;99:13467–13470. [Google Scholar]
- Atakan B. Kocis D. Wolfrum J. Nelson P. Direct investigations of the kinetics of the reactions of CN radicals with N atoms and 3CH2 radicals with NO. Symp. (Int.) Combust., [Proc.] 1992;24:691–699. [Google Scholar]
- Finlayson-Pitts B. J. and Pitts Jr J. N., Chemistry of the Upper and Lower Atmosphere, Academic Press, San Diego, 2000 [Google Scholar]
- EPA, EPI Suite™-Estimation Program Interface, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface
- Sehmel G. A. Particle and gas dry deposition: a review. Atmos. Environ. 1980;14:983–1011. [Google Scholar]
- Evans R. J., Chynwat V., Dreher J. and Kienzle M., Use of virtual scenarios for meteorological, disperson, and atmospheric chemistry modeling, 15th Joint Conference on the Applications of Air Pollution Meteorology with the A&WMA, 8.5-1-8, 2008 [Google Scholar]
- Thakur P. Ballard S. Nelson R. An overview of Fukushima radionuclides measured in the northern hemisphere. Sci. Total Environ. 2013;458–460:577–613. doi: 10.1016/j.scitotenv.2013.03.105. [DOI] [PubMed] [Google Scholar]
- Carpenter L. J. Chance R. J. Sherwen T. Adams T. J. Ball S. M. Evans M. J. Hepach H. Hollis L. D. J. Hughes C. Jickells T. D. Mahajan A. Stevens D. P. Tinel L. Wadley M. R. Marine iodine emissions in a changing world. Proc. R. Soc. A. 2021;477:20200824. doi: 10.1098/rspa.2020.0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez Martín J. C. Saiz-Lopez A. Cuevas C. A. Fernandez R. P. Gilfedder B. Weller R. Baker A. R. Droste E. Lai S. Spatial and Temporal Variability of Iodine in Aerosol. J. Geophys. Res.: Atmos. 2021;126:e2020JD034410. [Google Scholar]
- Legrand M. McConnell J. R. Preunkert S. Arienzo M. Chellman N. Gleason K. Sherwen T. Evans M. J. Carpenter L. J. Proc. Natl. Acad. Sci. U. S. A. 2018:12136–12141. doi: 10.1073/pnas.1809867115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulier M. Bueno M. Coppin F. Nicolas M. Thiry Y. Rigal F. Le Hécho I. Pannier F. Atmospheric iodine, selenium and caesium depositions in France: I. Spatial and seasonal variations. Chemosphere. 2021;273:128971. doi: 10.1016/j.chemosphere.2020.128971. [DOI] [PubMed] [Google Scholar]
- He X.-C. Tham Y. J. Dada L. Wang M. Finkenzeller H. Stolzenburg D. Iyer S. Simon M. Kürten A. Shen J. Rörup B. Rissanen M. Schobesberger S. Baalbaki R. Wang D. S. Koenig T. K. Jokinen T. Sarnela N. Beck L. J. Almeida J. Amanatidis S. Amorim A. Ataei F. Baccarini A. Bertozzi B. Bianchi F. Brilke S. Caudillo L. Chen D. Chiu R. Chu B. Dias A. Ding A. Dommen J. Duplissy J. El Haddad I. Gonzalez Carracedo L. Granzin M. Hansel A. Heinritzi M. Hofbauer V. Junninen H. Kangasluoma J. Kemppainen D. Kim C. Kong W. Krechmer J. E. Kvashin A. Laitinen T. Lamkaddam H. Lee C. P. Lehtipalo K. Leiminger M. Li Z. Makhmutov V. Manninen H. E. Marie G. Marten R. Mathot S. Mauldin R. L. Mentler B. Möhler O. Müller T. Nie W. Onnela A. Petäjä T. Pfeifer J. Philippov M. Ranjithkumar A. Saiz-Lopez A. Salma I. Scholz W. Schuchmann S. Schulze B. Steiner G. Stozhkov Y. Tauber C. Tomé A. Thakur R. C. Väisänen O. Vazquez-Pufleau M. Wagner A. C. Wang Y. Weber S. K. Winkler P. M. Wu Y. Xiao M. Yan C. Ye Q. Ylisirniö A. Zauner-Wieczorek M. Zha Q. Zhou P. Flagan R. C. Curtius J. Baltensperger U. Kulmala M. Kerminen V.-M. Kurtén T. Donahue N. M. Volkamer R. Kirkby J. Worsnop D. R. Sipilä M. Role of iodine oxoacids in atmospheric aerosol nucleation. Science. 2021;371:589–595. doi: 10.1126/science.abe0298. [DOI] [PubMed] [Google Scholar]
- Bouet C. Szczap F. Leriche M. Benassi A. What is the effect of cloud inhomogeneities on actinic fluxes and chemical species concentrations? Geophys. Res. Lett. 2006;33:L01818. [Google Scholar]
- Saiz-Lopez A. Plane J. M. C. Baker A. R. Carpenter L. J. von Glasow R. Gómez Martín J. C. McFiggans G. Saunders R. W. Atmospheric Chemistry of Iodine. Chem. Rev. 2012;112:1773–1804. doi: 10.1021/cr200029u. [DOI] [PubMed] [Google Scholar]
- Xavier C. de jonge R. W. Jokinen T. Beck L. Sipilä M. Olenius T. Roldin P. Role of Iodine-Assisted Aerosol Particle Formation in Antarctica. Environ. Sci. Technol. 2024;58:7314–7324. doi: 10.1021/acs.est.3c09103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordóñez C. Lamarque J. F. Tilmes S. Kinnison D. E. Atlas E. L. Blake D. R. Sousa Santos G. Brasseur G. Saiz-Lopez A. Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources. Atmos. Chem. Phys. 2012;12:1423–1447. [Google Scholar]
- Sherwen T. Evans M. J. Carpenter L. J. Andrews S. J. Lidster R. T. Dix B. Koenig T. K. Sinreich R. Ortega I. Volkamer R. Saiz-Lopez A. Prados-Roman C. Mahajan A. S. Ordóñez C. Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem. Atmos. Chem. Phys. 2016;16:1161–1186. [Google Scholar]
- Droste E. S. Baker A. R. Yodle C. Smith A. Ganzeveld L. Soluble Iodine Speciation in Marine Aerosols Across the Indian and Pacific Ocean Basins. Front. Mar. Sci. 2021;8:788105. [Google Scholar]
- Koenig T. K. Volkamer R. Apel E. C. Bresch J. F. Cuevas C. A. Dix B. Eloranta E. W. Fernandez R. P. Hall S. R. Hornbrook R. S. Pierce R. B. Reeves J. M. Saiz-Lopez A. Ullmann K. Ozone depletion due to dust release of iodine in the free troposphere. Sci. Adv. 2021;7:eabj6544. doi: 10.1126/sciadv.abj6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A. R. Tunnicliffe C. Jickells T. D. Iodine speciation and deposition fluxes from the marine atmosphere. J. Geophys. Res.: Atmos. 2001;106:28743–28749. [Google Scholar]
- Dix B. Baidar S. Bresch J. F. Hall S. R. Schmidt K. S. Wang S. Volkamer R. Proc. Natl. Acad. Sci. U. S. A. 2012:2035–2040. doi: 10.1073/pnas.1212386110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz-Lopez A. Baidar S. Cuevas C. A. Koenig T. K. Fernandez R. P. Dix B. Kinnison D. E. Lamarque J.-F. Rodriguez-Lloveras X. Campos T. L. Volkamer R. Injection of iodine to the stratosphere. Geophys. Res. Lett. 2015;42:6852–6859. [Google Scholar]
- Baker A. R. Thompson D. Campos M. L. A. M. Parry S. J. Jickells T. D. Iodine concentration and availability in atmospheric aerosol. Atmos. Environ. 2000;34:4331–4336. [Google Scholar]
- Cuevas C. A. Maffezzoli N. Corella J. P. Spolaor A. Vallelonga P. Kjær H. A. Simonsen M. Winstrup M. Vinther B. Horvat C. Fernandez R. P. Kinnison D. Lamarque J.-F. Barbante C. Saiz-Lopez A. Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century. Nat. Commun. 2018;9:1452. doi: 10.1038/s41467-018-03756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Hou X. Zhou W. Atmospheric Iodine (127I and 129I) Record in Spruce Tree Rings in the Northeast Qinghai-Tibet Plateau. Environ. Sci. Technol. 2019;53:8706–8714. doi: 10.1021/acs.est.9b01160. [DOI] [PubMed] [Google Scholar]
- Till J. E., Aanenson J. W., Boelter P. J., Case M. J., Dreicer M., Grogan H. A., Langan M. O., McGavran P. D., Meyer R. M., Mohler H. J., Rood A. S., Rope R. C., Rope S. K., Stetar L. A., Voilleque P. G., Windsor T. F. and Yang W., Savannah River Site Environmental Dose Reconstruction Project Phase II: Source Term Calculation and Ingestion Pathway Data Retrieval Evaluation of Materials Released from the Savannah River Site, Report 1-CDC-SRS-1999-Final, Risk Assessment Corporation, Neeses, SC, 2001 [Google Scholar]
- Fuge R. Johnson C. C. Iodine and human health, the role of environmental geochemistry and diet, a review. Appl. Geochem. 2015;63:282–302. [Google Scholar]
- Chamberlain A. C. Chadwick R. C. Transport of iodine from atmosphere to ground. Tellus. 1966;18:226–237. [Google Scholar]
- Parache V. Pourcelot L. Roussel-Debet S. Orjollet D. Leblanc F. Soria C. Gurriaran R. Renaud P. Masson O. Transfer of 131I from Fukushima to the Vegetation and Milk in France. Environ. Sci. Technol. 2011;45:9998–10003. doi: 10.1021/es202242g. [DOI] [PubMed] [Google Scholar]
- Telly Bah O. Hebert D. Connan O. Solier L. Laguionie P. Bourlès D. Maro D. Measurement and modelling of gaseous elemental iodine (I2) dry deposition velocity on grass in the environment. J. Environ. Radioact. 2020;219:106253. doi: 10.1016/j.jenvrad.2020.106253. [DOI] [PubMed] [Google Scholar]
- Muramatsu Y. Uchida S. Sumiya M. Ohmomo Y. Deposition Velocity of Gaseous Organic Iodine from the Atmosphere to Rice Plants. Health Phys. 1996;71:757–762. doi: 10.1097/00004032-199611000-00018. [DOI] [PubMed] [Google Scholar]
- Tschiersch J. Shinonaga T. Heuberger H. Dry deposition of gaseous radioiodine and particulate radiocaesium onto leafy vegetables. Sci. Total Environ. 2009;407:5685–5693. doi: 10.1016/j.scitotenv.2009.06.025. [DOI] [PubMed] [Google Scholar]
- Carpenter L. J. MacDonald S. M. Shaw M. D. Kumar R. Saunders R. W. Parthipan R. Wilson J. Plane J. M. C. Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 2013;6:108–111. [Google Scholar]
- Gómez Martín J. C. Saiz-Lopez A. Cuevas C. A. Baker A. R. Fernández R. P. On the Speciation of Iodine in Marine Aerosol. J. Geophys. Res.: Atmos. 2022;127:e2021JD036081. doi: 10.1029/2021JD036081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. Tham Y. J. Fernandez R. P. He X.-C. Cuevas C. A. Saiz-Lopez A. Role of Iodine Recycling on Sea-Salt Aerosols in the Global Marine Boundary Layer. Geophys. Res. Lett. 2022;49:e2021GL097567. doi: 10.1029/2021GL097567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sive B. C. Varner R. K. Mao H. Blake D. R. Wingenter O. W. Talbot R. A large terrestrial source of methyl iodide. Geophys. Res. Lett. 2007;34:L17808. [Google Scholar]
- Negri A. E. Fernández Niello J. O. Wallner A. Arazi A. Fifield L. K. Tims S. G. 129I Dispersion in Argentina: Concentrations in Fresh and Marine Water and Deposition Fluences in Patagonia. Environ. Sci. Technol. 2013;47:9693–9698. doi: 10.1021/es400610h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.