

Summary
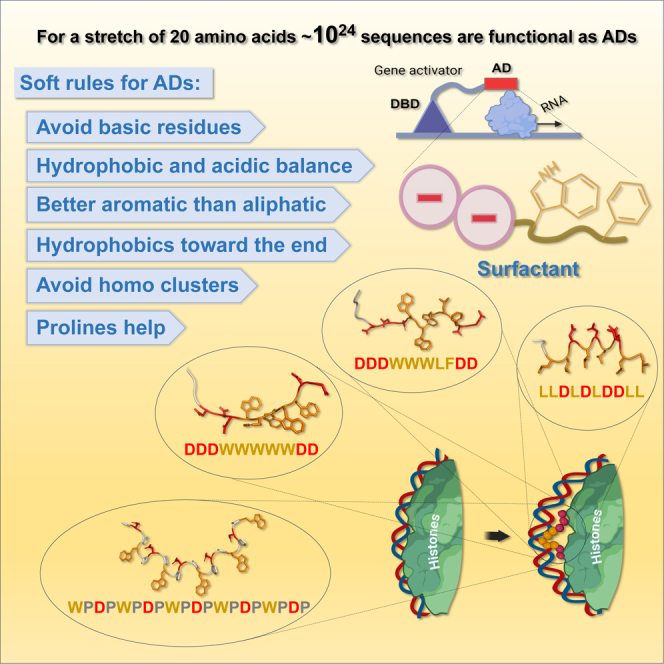
Transcriptional activation domains (ADs) of gene activators have remained enigmatic for decades as short, extremely variable, and structurally disordered sequences. Using a rational design and high throughput in vivo experimentation, we determine the grammar rules and exceptions for the language of ADs. According to identified rules, billions of highly active ADs can be composed of balanced amounts of acidic/aromatic amino acids, with either mixed composition of aromatic residues, or using only one aromatic residue mixed with acidic residues. However, equally active sequences can be composed of only aliphatic leucine and aspartic acid residues. The much rarer LD exceptions have a higher ratio of hydrophobic/acidic balance and display a specific LDL(L/D)DLL motif. For aromatic/acidic Ads, the intermixing of proline residues in context of amphipathic α-helix structures significantly increases the AD activity. The identified grammar rules and exceptions are interpreted in application to the biochemistry of AD function and eukaryotic gene expression.
Subject areas: Natural sciences, Biological sciences, Biochemistry, Genetics, Bioinformatics
Graphical abstract
Highlights
-
•
Sequences of only W and D, or F and D, or L and D work equally well as ADs in vivo
-
•
Only LD sequences have a consensus L(D/L)DLL
-
•
Increased number of Ps in acidic-hydrophobic ADs is favorable for function
-
•
Rules of AD language are soft with numerous exceptions
Natural sciences; Biological sciences; Biochemistry; Genetics; Bioinformatics
Introduction
Transcriptional activation domains (ADs) of gene activators are the key molecules determining activation and expression levels of eukaryotic genes. ADs have remained enigmatic for decades as extremely variable short sequences, which are usually intrinsically disordered, and interact with an uncertain number of potential targets during transcription activation. Along with other intrinsically disordered protein regions (IDRs), ADs are critical for the transcriptional dynamics and associated phase transitions during liquid-liquid phase separation.1 Solving the long standing riddle of AD sequences remains an important goal of molecular biology,2 and understanding the language of AD sequences is an important step in solving the enigma.
It was suggested previously3,4 and confirmed5 that AD sequences usually follow certain general rules: absence of basic residues, presence of acidic and hydrophobic (mostly aromatic) residues, balance between acidic and hydrophobic residues, minimal formation of homo-amino acid clusters by acidic or hydrophobic residues, and preferential terminal location for hydrophobic residues and internal location within AD for acidic residues. Machine learning analyses of AD sequences selected in vivo to function within the Gcn4 and HSF contexts in yeast showed that the preference for aromatic residues is W > F > Y and for acidic residues D > E. This preference was consistent with findings of several publications.6,7 It was shown further that for in vivo AD activity it is sufficient to have only aromatic and acidic residues,4 including hundreds of sequences composed entirely from Ws and Ds.8
To clarify features and properties of ADs, in addition to confirming the high functionality of the combinations containing only Ws and Ds, we explored if it is possible to have active in vivo AD sequences comprised entirely from F and D, Y and D, or L and D residues. We asked if aromatic residues are exclusively important for AD activity, or if the acidic-aliphatic amino acid combinations can also form active ADs. In addition, we investigated the impact of mixing aromatic residues in comparison to sequences having only one specific hydrophobic residue in combinations with acidic residues. We showed that hundreds of active ADs can be constructed from either: acidic residues intermixed with a variety of combinations of W, F, Y, and L residues, or acidic residues paired with only W, F, or L, but not Y residues. In addition, we found that while the acidic-aromatic requirement for functional ADs generally dominates, it is possible to have short highly active sequences comprised entirely from Ls and Ds. Importantly, the functional LD sequences, unlike sequences with aromatic residues, have a specific LDL(D/L)DLL mini-motif. Based on our results we formulate specific grammar rules the AD sequences follow and analyze exceptions to these rules.
Results
Experimental setup
The experimental setup was adopted from the our previous analysis of AD sequences containing all possible combinations of W and D for all positions for a stretch up to 12 positions8 and described in details in the STAR Methods section. Briefly, thousands of individual nucleotide sequences (Figure 1A) were parallel-synthesized and cloned into a previously constructed8 yeast centromeric parental shuttle vector in which the library sequences were fused individually to the Gal4 DNA-binding domain sequence. The resulting plasmid library was transformed into the yeast Y2HGold strain, where the individual Gal4 hybrid activator proteins were expressed and screened for functionality as ADs driving the expression of the Gal-Aureobasidin antibiotic resistance reporter gene (Figure 1A). The library DNA for different growth time points was isolated and sequenced to determine the number of reads for each individual sequence and its change over time. The growth slope for each sequence was calculated and served as a measure of the AD functionality. Since the results were obtained within the scope of the whole library pool screening, the results for individual sequences and for all different sequence sets can be considered to be obtained under identical experimental conditions and thus to be accurately comparable. In addition, each sequence was labeled by multiple individual barcodes; thus, the results for each individual sequence are the mean of multiple (typically five) independent experimental repeats (Figure 1B). Counts for each barcode were first normalized to 0 time point (Figure 1B), and then normalized to the slope of null (stop codon) sequences (see STAR Methods for details). As a part of the library, we used the sequences of known AD regions, such as Gal4(840–857), Gal4(860–872) and VP16 sequences,9,10 as internal positive controls (Figure 1C). As negative controls, we used null sequences that contained a stop codon after the DBD. To ensure the high stringency, the cutoff for a “functional AD” was defined as a growth slope two standard deviations above the mean of 97 individual stop codon-null sequences (see STAR Methods).
Figure 1.

Experimental setup and controls
(A) Experimental setup: oligo pool synthesis, followed by cloning in bacteria, then isolation of plasmid library and transformation in yeast, followed by screening for growth phenotype determined by expression of the reporter gene regulated by the activator with a specific AD, then isolation of DNA pool, NGS sequencing, and data analysis. For more details see STAR Methods section and.8.
(B) Raw (not normalized to null sequences) data for three inactive (top) and three active (bottom) sequences. X axis: time of cell growth on the medium containing Aureobasidin (days). Y axis: CPM(Log2) normalized to 0 time point. Each dot represents the value for an individual biorep barcode (see STAR Methods).
(C) Library internal controls. X axis: null sequences and previously characterized individual sequences; Y axis: growth slope for cells carrying the individual sequences. Error bars defined as mean +/− two standard deviations normalized to 0 time point and to null sequences.
(D) Internal controls functional reproducibility. X axis: slope value for individual sequences in HSF1 library4; Y axis: normalized slope value for individual sequences in current Gal4 library. Banded region around regression line indicates 95% confidence interval of slope.
(E) W and D residue containing control AD sequences functional reproducibility; X axis: slope value for individual sequences in previous WD design library8; Y axis: same as in C. Banded region around regression line indicates 95% confidence interval of slope.
To test the reproducibility of our assay, we compared the slopes for individual control sequences in our current study and previously published results4 and see good correlation (Figure 1C, Spearman’s rho = 0.718). In addition, we similarly looked at the correlation of other sequences which are shared between the library of the current study and the independent library analyzed previously.8 The results show an excellent correlation (Figures 1D and 1E Spearman’s rho = 0.944) indicating reproducibility of this assay across experiments.
Functional ADs can be composed of varying combinations of W, F, Y, or L residues balanced with D residues
Since it was shown previously that simple repetition of the WD dipeptide is sufficient to create highly active in vivo ADs,4,8 we asked if other dipeptide combinations could also be active, testing monotonous di-peptide repeats of 8 or 10 amino acids pairs composed from D and another one of 20 amino acid residues. Confirming previous observations,4,8 the di-amino acid peptides containing D and W, or D and F had the highest functionality scores (Figure 2A). Additional activity was observed for the L and a minor activity for T containing peptides. Since previous reports repeatedly highlighted the importance of aromatic residues and Ls,3,7,11 we focused on W, F, Y and L residues balancing them with D residues, asking if each individual hydrophobic residue alone or in combinations with other hydrophobic residues could create an in vivo functional AD.
Figure 2.

Functionality of balanced acidic/hydrophobic sequences in different arrangement contexts
(A) Functionality of dipeptide repeats with an aspartic acidic (D) residue in the first position of the dipeptide. Two dipeptide templates: 8 repeats (dotted borders) and 10 repeats (solid borders). X axis: individual amino acid used in X positions (blue – basic, red – acidic, green – hydrophilic neutral, yellow/orange hydrophobic, pink – others); Y axis: growth slope of cells carrying the individual sequences.
(B–G) Functionality of sequences with only one type of hydrophobic residue (W, F, L, or Y) within three sequence templates: flanked template (DDDXXXXXDD, panel B), intermixed template (DXDXDXDXDX, panel C), and amphipathic helix template (XGDGXGDGXGDGXGDGXGDG, panel D). Axes same as in A. (E–G) Distributions of AD activities of mixed amino acids (W, F, L, and Y) within five X positions in each sequence template: flanked template (panel E), intermixed template (panel F), and amphipathic helix template (panel G) (1024 individual sequences for each template). X axis: template used; Y axis: same as in A. Values above violin plots indicate percentage of functional sequences within each template dataset. Blue bar indicates the average growth slope for sequences above the 0 threshold. Inset sequence logos depict proportion of each amino acid at each position, for the top 5% (top) and bottom 5% (bottom) of sequences for each template dataset.
We decided to test three different sequence templates each with balanced amounts of acidic and hydrophobic amino acids: flanked (DDDXXXXXDD), intermixed (DXDXDXDXDX), or an amphipathic helix (XGDGXGDGXGDGXGDGXGDG) indicated below as (XGDG)5, where Xs were W, F, Y, or L amino acids. The rationale behind template choices was that (i) the flanked template with all Xs as Ws, as previously shown8 and Figure 1E, produces the AD with one of the highest activity among thousands identified, (ii) the intermixed template Figure 1E is a simple balanced hydrophobic-acidic template which also was previously reported producing in vivo active ADs,3,4,8 while (iii) the amphipathic helix template sequence with hydrophobic Xs, if folded into a canonical α-helix, always creates a short amphipathic helix segment that was repeatedly reported to be important for functionality of ADs.7,12,13,14
When we used individual W, F, Y, or L amino acids for all X positions within the flanked template context (Figure 2B), only tryptophan produced an in vivo active AD, which is consistent with the previous report.8 For the intermixed template (Figure 2C) both Ws and Fs were able to create in vivo active AD, with slightly higher activity level for Fs over Ws, while Ls and Ys did not produce activity in this context. The amphipathic α-helix template (Figure 2D) did not have ADs with a significant activity level for any of four individual W, F, Y, or L amino acids.
To determine the effect of mixing different hydrophobic amino acids within an AD, which is more typical for natural ADs, we created a library with all combinations of W, F, Y, and L across five X positions, which amounts to 1024 sequence variants for each template: flanked (Figure 2E), intermixed (Figure 2F), and amphipathic helix (Figure 2G). For the flanked template 47.2% (483/1024) of sequence variants were above the threshold for functionality, for the intermixed template 57.4% (587/1023) of sequences are functional, while for the amphipathic helix template only 16.2% (166/1024) of sequences were above the threshold. The average slope of functional sequences is 2.2 for the flanked template, 1.6 for the intermixed template, and only 0.5 for the amphipathic helix template. For all three templates the highest functionality was recorded for sequences containing a mixture of different hydrophobic residues especially in the context of the flanked and intermixed templates (Figures 2E and 2F) in comparison to sequences containing only one hydrophobic residue for all X positions (Figures 2B–2D). If templates are compared to each other (Figures 2E–2G), the flanked template produced sequences with highest activity, while the intermixed template produced the highest number of active ADs.
The number of each specific hydrophobic residue in the sequence affects AD activity differently. Generally, an increase in the number of Ws is beneficial regardless of template (Figure S1A), whereas for Fs this trend is only observed for the intermixed template (Figure S1B). However, while Ls and Ys can be present in highly active ADs, an increased number of L residues is not necessarily beneficial (Figure S1C), and for Y it is detrimental (Figure S1D). These trends are consistent with the previous ML analysis of the unbiased random sequence AD libraries3,7 showing the W > F > Y preference among aromatics and a mild near-neutral effect of L residues within tens of thousands of analyzed active ADs. Analyzing the effects of positions for a specific hydrophobic residue in different templates, we observe that functional sequences with the flanked template often have L or F residues present at the most C-terminal position (Figure S1E).
The compositional analysis of mixed sequences revealed that instead of one specific composition, a diverse set of compositions of W, F, Y, and L residues produce functional ADs across these three sequence templates (Table S1). These functional compositions, with 5–60 possible sequence arrangements each, often have high functionality. In general, W and F residues contribute most to function across all three templates, L residues are less common than W or F across functional sequences, and Y residues are rarely present in functional sequences. The flanked template is most functional when enriched in W residues. The intermixed template is most functional when enriched in F residues. The amphipathic template is most functional when depleted of L and Y residues.
To uncover any residue position preferences among functional sequences, we calculated the proportion of each residue at each position among the most functional (top 5%) for each set of sequences. The outcomes summarized in Figure 2E (sequence logos), indicate that for the flanked template there is some preference for Ws toward the N-terminus of the active sequences and some preference for Ls more internally. For the intermixed template (Figure 2F) there is some preference for Fs over Ws and even more so over Ls. For the amphipathic helix template (Figure 2G) there is a preference for Ws over Fs, which both dominate over Ls. The only position where L is preferred is toward the N-terminus of AD. The investigation of non-functional sequences (bottom 5%) suggests that Y is significantly enriched and generally is detrimental for AD functionality. Overall, the minimotif analysis performed using the above approaches, as well as SlimFinder,15,16 and MEME suite,17,18 indicates that a wide variety of short sequences containing balanced acidic/hydrophobic composition can function in vivo as ADs with no specific mini-motif dominating any of tested libraries. The non-functional sequences also do not contain any specific minimotif.
In summary
we analyzed a total of 3071 sequences with W, F, Y, L amino acid sequences balanced with an equal number of Ds using three templates and found 40% (1236/3071) of sequences above the functionality threshold in the context of our assay. Within functional sequences for each template, we found that a wide variety of different sequences can function as an active in vivo AD with varying levels of activity. The active AD sequences similar to previous observations4,7,11 do not have any specific dominant minimotifs, but instead have a wide variety of functional sequence compositions.
The presence of proline significantly increases the functionality of sequences as ADs in the context of an amphipathic helix template
The outcomes for the amphipathic helix template analyses (Figure 2G) indicating extremely low functionality of this template are in slight disagreement with a previous report8 demonstrating that similar amphipathic helix-forming template generated 19.6% (21,163/107,975) of functional sequences. The difference between templates, however, is that in the previous study the analyzed template was WXDXWXDXWXDXWXDXWXDX designed to determine what X amino acids could facilitate or hinder the activity, while in the current study the analyzed amphipathic template (XGDG)5 was XGDGXGDGXGDGXGDGXGDG designed to determine what combinations of hydrophobic amino acids are conducive for the AD activity within the amphipathic helix context. Using the glycine in current study is justified by the fact that it was shown previously as being neutral for AD activity.3 However, glycine residues are also known as helix destabilizers, thus could contribute to the low yield of AD functional sequences in the (XGDG)5 template. To test the effect of the helix formation for this template we tested all individual amino acids instead of glycine. The outcomes (Figure 3) indicate that although some amino acids are predicted to stabilize the hydrophobic/acidic amphipathic helix, especially L, M, and W (Figure 3B) the only amino acid that created significant AD activity was P - the known α-helix breaker (Figure 3A). The level of activity in this context evidently correlates with the number of Ps used in this template (Figure 3C, Spearman’s rho = 0.466), with a slight trend for Ps being more advantageous for the N-terminus of the sequence (Figure 3D). As in previous study,8 these results suggest that breaking the amphipathic hydrophobic/acidic helix and thus preventing aromatic residues residing on one side of the helix from interacting with each other is beneficial for AD activity in vivo.
Figure 3.

The presence of proline significantly increases the functionality of sequences as ADs in the context of amphipathic helix template
(A) Functionality of sequences with the amphipathic helix template (WXDXWXDXWXDXWXDXWXDX) where X in all positions represents one of the 20 amino acids. X axis: individual amino acids; Y axis: growth slope of cells carrying the individual sequences.
(B) α-helix predictions for each individual sequence. X axis: helicity probability predicted using Agadir19; Y axis: same as in A.
(C) Effect of number of proline residues in the G-amphipathic helix template (WGDGWGDGWGDGWDGDWGDG). X axis: number of proline residues within the sequence replacing glycine residues. Y axis: same as in A.
(D) Effect of position of proline residues in the G-amphipathic helix template. X axis: average position of proline residues, the actual position for sequences with a single proline residue or the average for sequences with two or three proline residues. Y axis: same as in A. Banded region around regression line indicates 95% confidence interval of slope.
Robust in vivo AD activity displayed by sequences with only a single type of hydrophobic residue balanced with aspartic acid
Our results and previous investigations8 showed that sequences containing only Ws (Figures 2B and 2C) or Fs (Figure 2C) balanced with acidic residues can create ADs functional in vivo. We wanted to investigate if different combinations of Ws, Fs, Ys, or Ls alone balanced with Ds can create functional ADs in vivo and how numerous those combinations can be. Evidently (Figure 4A) and consistent with previously published results,8 34.3% of sequences in the WD10 set containing combinations of Ws and Ds for 10 positions (350/1021 sequences) are above the functionality threshold, 35.5% (363/1022 sequences, FD10 set) are functional if all Ws are replaced with Fs (Figure 4B), and 22% (225/1023 sequences, LD10 set) are functional if Ls are the only hydrophobics (Figure 4C), while only 0.8% (8/997 sequences, YD10 set) are barely above the threshold for D and Y combinations (Figure 4D).
Figure 4.

Robust in vivo AD activity displayed by sequences with only a single type of hydrophobic residue balanced with aspartic acid
(A–D) Distributions of AD activities for sequences containing all combinations across ten positions of D with W (WD10, panel A), F (FD10, panel B), L (LD10, panel C), or Y (YD10, panel D) (1024 individual sequences for each dataset). X axis: dataset used; Y axis: growth slope of cells carrying the corresponding sequences. Values above violin plots indicate percentage of functional sequences within each dataset. Blue bar indicates the average growth slope for sequences above the 0 threshold. Inset sequence logos depict proportion of each amino acid at each position, for the top 5% (top) and bottom 5% (bottom) of sequences for each dataset.
(E–G) Effect of number of hydrophobic residues for WD10 (panel E), FD10 (panel F), and LD10 (panel G) AD sequences. X axis: number of W, F, or L residues. Y axis same as in A. Values above boxplots indicate percentage of functional sequences within each data subset.
(H–J) Effect of position of hydrophobic (W, F, or L) and D residues for WD10 (panel H), FD10 (panel I), and LD10 (panel J) AD sequences. X axis: position of residues within the 10 positions of the sequences. Y axis: percent of cells carrying the corresponding sequences that have a growth slope above the functionality threshold. Horizontal lines correspond to the overall percent functionality of each template dataset.
(K–M) Effect of hydrophobic clusters for WD10 (panel K), FD10 (panel L), and LD10 (panel M) AD sequences. X axis: Patterning parameter20 where small values correspond to fewer clusters of the same residue (see STAR Methods). Y axis: same as in A.
The existence of a significant number of functional L and D combinations is especially interesting because the machine learning (ML) analysis of large unbiased random sequence pools (106-105 sequences) screened for functional ADs in vivo3 showed only minor importance of Ls for AD functionality, and L-rich regions in transcriptional activator IDRs are often not considered as ADs7,12,21 or more specific to human ADs.11 Even though LD combinations created fewer functional ADs than the WD and FD combinations (Figures 4A–4C), the LD combinations produced sequences with the highest overall functionality (Figure 4C). The compositional (Figures 4E–4G), positional (Figures 4I–4J), and amino acid clustering analyses (Figures 4K–4M) of functional WD10, FD10, and LD10 combinations revealed that all three sequence sets follow three previously reported3 sequence rules: maintaining the general balance between hydrophobic and acidic residues within each individual functional AD sequence (Figures 4E–4G), preferred C-terminal location of hydrophobic and internal location of acidic residues (Figures 4H–4J), and avoiding homo-amino acid clusters (Figures 4K–4M). For functional sequences, the average balance between Ds and Ws is 5.42 within the 10 amino acid stretch (slightly higher W presence) (Figure 4E), and 5.23 for Fs and Ds (Figure 4F), while for Ls and Ds it is 5.75 which is shifted toward excess of Ls (Figure 4G) making sequences with six Ls and four Ds the dominant functionality cohort among the LD10 combinations.
We next tested the frequency of a specific amino acid for each of ten positions for the most functional sequences (top 5%) and non-functional sequences (bottom 5%) (Figures 4A–4C, sequence logos). It is evident that for the WD10 and FD10 sets, there is a similar probability between hydrophobic and acidic residues for each position in the sequence with some preference of hydrophobic residues for the C-terminus, which is consistent with the positional analysis (Figures 4H–4J). Tested for sensitivity to the mono amino acid cluster formation (Figures 4K–4M) it is evident that in all three datasets the ADs had greater functionality if hydrophobic or acidic residues were not clustered (high patterning parameter value)20 but instead evenly intermixed (low patterning parameter value).
To identify mini-motifs enriched in the functional sequences in each dataset, we determined the average slope of sequences that contain all possible progressively longer combinations of amino acids (Figure S2). The mini-motif search within each of the WD10, FD10 and LD10 sequence sets revealed that there were no clear minimotifs enriched in functionality in WD10 and FD10 sets; however, the LD10 set has a small number of functionally enriched minimotifs, especially for the 7 amino acid long window, with LDLDDLL noticeably enriched in functional sequences (Figure S2). The FD10 set has the greatest diversity of functional sequences with 46.9% of 7 amino acid long sequences having a functional average growth slope while the LD10 set has the least diversity of functional sequences with only 28.1% of 7 amino acid long sequences having a functional average growth slope.
Highest activity LD10 AD sequences have an LDL(D/L)DLL motif with an important LDDLL core sequence
Because the top functional LD10 sequences have especially high functionality value, we extended the motif search analysis for these sequences. To test if active AD sequences have any Short Linear Motifs (SLiMs), we used the MEME motif discovery tool17,18. The motif search confirmed the findings for the LD10 set, identifying the SLiM LDL(D/L)DLL with very low E-value (4.2x10−8) (Figure 5A). In fact, among the top functional 5% of sequences 20 out of 52 sequences contained the LDLDDLL sequence, and 12 out of 52 sequences contained the LDLLDLL sequence. When we applied the same approach for the WD10 and FD10 sets, no motifs had significant E-values.
Figure 5.

Highest activity LD1AD 0 sequences have an LDL(D/L)DLL motif with an important LDDLL core sequence
(A) LDL(D/L)DLL motif (inset sequence logo) identified using the MEME motif discovery tool17 in 40 of the top 50 functional LD10 sequences. Activities of sequences containing a single amino acid variation within the LDL(D/L)DLL motif. X axis: sequence variants (substituted residue underlined). Y axis: growth slope of cells carrying the corresponding sequences.
(B) Activity discrimination of the LDL(D/L)DLL motif. X axis: presence or absence of the motif in the sequence. Y axis: same as in A.
(C) Activity discrimination of the inversed LLD(L/D)LDL motif. Axes: same as in A.
(D) Effect of position of the L(D/L)DLL core motif within the sequence. X axis: distance of motif from the N-terminus of the AD (color coded) within sequences containing LDDLL or LLDLL. Y axis: same as in A.
(E) Effect of number of L residues for sequences with the L(D/L)DLL core motif within the sequence. X axis: number of L residues (color coded) within sequences containing LDDLL or LLDLL motif. Y axis: same as in A.
(F) Effect of position of L and D residues for sequences with the L(D/L)DLL core motif (either LDDLL or LLDLL present) within the sequence. X axis: position of residues within the 10 positions of the sequences. Y axis: percent of cells carrying the corresponding sequences with a growth slope above the functionality threshold. Horizontal line corresponds to overall percent functionality of the L(D/L)DLL core motif containing sequences.
(G) Effect of hydrophobic clusters for sequences with the L(D/L)DLL core motif (either LDDLL or LLDLL present) within the sequence X axis: Patterning parameter20 where small values correspond to fewer clusters of same residue (see STAR Methods). Y axis: same as in A.
We then checked the importance of each specific position within the LDL(D/L)DLL sequence. By selecting within the LD10 set the sequences containing the LDL(D/L)DLL stretch with only one amino acid in a specific position deviating from the consensus and calculating the average functionality score for each subset, we see that the most important core of the sequence is L(D/L)DLL with LDDLL being the more common sequence (28 out of 50 functional sequences containing the LDL(D/L)DLL motif) (Figure 5A). An important quality of the LDL(D/L)DLL is that if it is inversed to LL(D/L)LDL it loses much of its functionality (Figures 5B and 5C). Similarly, sequences are inactive when the positions of only two letters within the LDDLL core are reversed, i.e., for the top functional sequence: LLDLDLDDLL (slope = 6.70) vs. LLDLDLDLDL (slope = −0.984) or second top functional sequence: DLLDLDDLLL (slope = 6.40) vs. DLLDLDLDLL (slope = 0.067). The effect of sequence inversion was tested for all functional WD10 FD10 and LD10 sequences in each set. Generally, the disruptive effect is clearly seen for the LD10 and WD10 sequences, but the FD10 sequences were significantly more tolerant to the inversions, which correlates with the functional sequences in the FD10 set being the most variable for specific sequence motifs.
Sequences containing the L(D/L)DLL core still varied in activity. To test what factors are affecting the activity level of LD10 set sequences, we tested if in addition to the presence of the motif, they also follow the rules we formulated before3 and confirmed in Figure 4. It is evident that sequences containing the core L(D/L)DLL motif generally are more functional if the amount of the acidic and hydrophobic residues is balanced and quickly lose activity with excess of either acidic or hydrophobic residues (Figure 5E). The activity level is also correlated with the proximity of hydrophobics toward the C-terminus of the molecule (Figure 5F). Notice, that the bump for the middle positions for Ds is likely the result of Ds being in the middle of the LDL(D/L)DLL motif. The presence of L-clusters in addition to the L(D/L)DLL core also negatively affected the level of activity (Figure 5G). Notice, the functional exceptions containing a relatively high clustering score all have balanced L/D content and the presence of the LDL(D/L)DLL motif.
Presence of positively charged residues negatively affects AD functionality
Another rule that was obvious from the previous ML analysis of the random pool sequences3 was the requirement for the absence of positively charged residues: R, K, and to a lesser degree H. We checked how sequences containing an otherwise active AD module (e.g., GGGGGDDDWWWWWDDGGGGG) are affected by presence of R residues, the most positively charged amino acid. Evidently (Figure 6A, Spearman’s rho = −0.755), presence of only one R replacing a G within varying positions significantly reduced the AD activity, two Rs in many cases reduced the AD activity closer to the threshold, while most of the sequences containing 3Rs in different positions eliminated the AD activity entirely. The position of the positively charged residues within the sequence correlated with the greatest negative effect at the N-terminus of the sequence (Figure 6B, R2 for 2 R residues = 0.58), and the AD activity was sensitive to clustering of Rs (Figure 6C).
Figure 6.

Presence of positively charged residues negatively affects AD functionality
(A) Effect of number of arginine residues in the flanked template (GGGGGDDDWWWWWDDGGGGG) with R residues replacing G residues. X axis: number of arginine residues within the sequence. Y axis: growth slope of cells carrying the corresponding sequences.
(B) Effect of position of arginine residues in the flanked template. X axis: average position of arginine residues. Y axis: same as in A. Banded region around regression line indicates 95% confidence interval of slope.
(C) Effect of clustered arginine residues for sequences containing two arginine residues. X axis: sequence groups based on position of residues: both residues upstream of the AD (N-cluster), both residues downstream of the AD (C-cluster), and residues on either side of the AD (Spaced). Y axis: same as in A.
Algorithms trained/designed on natural AD sequences are not accurate and often overpredict the functionality of “synthetic” ADs.
Since we used designed sequences in our libraries, which might not necessarily be present in natural gene activators, and because recent ML models were developed by training on natural AD sequences, we wanted to see how well these ML models predict the functionality of our “synthetic” ADs. We used the most comprehensive to date PADDLE model trained on AD sequences from all 162 yeast (S. cerevisiae S288C) transcription factors, broken into over 10,000 fragments.12 This model was demonstrated to have an accuracy of 92%12 for prediction of ADs in human activators. In addition, we compared the results of PADDLE prediction with the predictions made by the Attention AD model22 trained on an unbiased large (>10ˆ6) set of random sequences screened for AD activity in vivo in the context of Gcn4 DBD.7 The Attention AD model is an improved modification of the ADpred model7 which has high AUC 0.98 when tested on the fraction of the dataset not used for training.
Applying PADDLE model to our current datasets (Figure 7 panels A, B, C and G, H, I, J) we see that the AUC value varies between 0.58 and 0.92 depending on the dataset. The Attention AD model (Figures 7, panels D, E, F, and K, L, M, N) has AUC values of 0.57–0.87, which is slightly lower consistently for all datasets. However, the Attention AD model has greater overall accuracy (Acc) in all datasets except for LD10. When we look at the correlation between the predictions made by models (X axes) and the in vivo functionality scores (Y axes) we see that PADDLE tends to overpredict (most of values are in the right two quadrants), with stark example of the flanked template (panel A) where all sequences are predicted to be functional, while in vivo ∼50% are not functional. A similar trend is observed for the intermixed template (panel B), while amphipathic template sequences (panel C) with the highest accuracy were predicted to be mostly inactive ADs. A similar trend of overprediction is observed (panels G, H, I, J) for the WD10, FD10, LD10 (with lesser degree), and for YD10 (mostly inactive sequences) datasets. As a comparison to the PADDLE model the Attention AD model, in addition to having lower AUC values, appears to make random mistakes in some cases, with wider distribution of values for all four quadrants (panels D, E, F, and K, L, M, N).
Figure 7.

Performance and comparison of ML models PADDLE and Attention AD, and Mechanistic Predictor
(A–N) PADDLE ML model7 (panels A, B, C, G, H, I, & J) and Attention AD model22 (panels D, E, F, K, L, M, & N) used to predict functionality across seven sets of sequences: Flanked WYFL (panels A-B), Intermixed WYFL (panels B-E), Amphipathic Helix WYFL (panels C–F), WD10 (panels G–K), FD10 (panels H–L), LD10 (panels I–M), and YD10 (panel J–N). X axis: Z-scores represent predicted probability of functionality using PADDLE (see STAR Methods) with scores greater than a threshold of 4 predicted to be functional.7 Probability values represent predicted probability of functionality using Attention_AD with scores greater than a threshold of 0.5 predicted to be functional. Y axis: growth slope of cells carrying the corresponding sequences. Values in the upper left corner of each panel are the correlation between the growth slope and prediction (r), area under the ROC curve (AUC) and the fraction accurately predicted (Acc).
(O) A modified Mechanistic Predictor6 used to predict functionality across seven sets of sequences. The modified predictor applied to sequences with a total length of 20 residues: Functional ADs = [-6.5 ≤ Net Charge ≤ −4 & Number of W, F, and L Residues ≥3]. Distributions of sequences predicted to be functional (solid borders) and sequences predicted to be non-functional (dotted borders). X axis: dataset used; Y axis: same as in A. Values on the graph represent the percent of experimentally determined functional sequences out of the sequences predicted to be functional using the modified mechanistic predictor.
(P–R) Average scores for three rules: balance (panel P), position (panel Q), and clustering (panel R) within sequences correctly predicted (true positives (TP) and true negatives (TN)) and incorrectly predicted (false positives (FP) and false negatives (FN)) to be activation domains by PADDLE (solid borders) and Attention AD (dotted borders). Deviation from balance is the absolute value of the difference in number of acidic and hydrophobic residues. C-terminal preference is the slope calculated from a best fit line where the X axis was 1–10 for the ten positions of each sequence and the Y axis was the number of sequences that had a hydrophobic residue at each position, with a greater preference indicating more hydrophobic residues toward the C-terminus (see Figures 4 panels H, I, J).
Another recently developed tool for prediction of AD functionality for a sequence is a mechanistic predictor based essentially on the composition of the sequence.6 It accurately predicted nonfunctional sequences (Figure 7, panel O, dotted lighter color violins), but has poor prediction with 53%–18% accuracy depending on the dataset. The likely reason for the poor performance of the mechanistic predictor is that it is based primarily on composition, counting charged (D, E) and hydrophobic residues (W, F, Y, L) and does not consider other rules.
PADDLE and the Attention AD model are neural network ML models, and thus do not clearly reveal the ML features used for the model building and predictions. However, understanding ML features and connecting them to AD biochemical features is important for deducing the mechanism of AD function. For this reason, we tested how each quadrant: true positive (TP), false positive (FP), true negative (TN), and false negative) in Figure 7 panels J, H, I, and K, L, M follow the basic rules we emphasized above (balance of acidic and hydrophobics, C-terminal preference of hydrophobic residues, and no-cluster formation). We used WD10, FD10, and LD10 datasets because only these sets are truly randomized using all possible combinations of specific hydrophobic residue and D. Evidently (panel P) for both ML models the lowest deviation from the hydrophobic/acidic balance is for TP and the highest is for TN, with intermediate level for FP and insufficient data for FN. Similar situation is for C-terminal preference of the hydrophobic residue (panel Q) and for clustering score (panel R). Thus, at least these three basic rules appear to be recognized by both ML models. Although we see this correlation, the rules are likely formulated mathematically differently by NN models and by us. In addition, to test the importance of each rule we performed logistic regression (see STAR Methods) testing the importance of each rule. All three rules contributed significantly (Table S2) showing importance of deviation from the acidic/hydrophobic balance, position of hydrophobics, and homo-amino acid clustering.
The computational feature development is not a trivial task because features might overlap and contain exceptions. For instance, balance and clustering rules are connected, as the more is the deviation from balance between hydrophobic and acidic residues, the higher is the probability of clustering. Although homo amino acid clustering has generally a negative impact on AD functionality (Figures 4K–4M; Figure 5G), it might depend on a specific context. For instance, we see that while clusters of 4–6 Ws flanked by Ds are functional, this type of clustering is not tolerable for function if instead of Ws, Fs or Ls are used (Figure S3). This observation might be a reflection of the hydrophobicity level. According to many hydrophobicity scales and the normalized best hydrophobicity scale,23 among W, F, and L, the amino acid W has the lowest hydrophobicity and thus while flanking Ws with Ds prevents intramolecular hydrophobic aggregation, for F and L the repulsion of flanking Ds is maybe not sufficient. Further ML feature development connecting them to biochemical features of ADs will be an important task for revealing more nuanced rules, allowing us to gain more insights in the mechanism of AD function.
Discussion
ADs’ functionality has remained enigmatic for decades as ADs do not follow the foundational biochemistry/molecular biology specificity triad: specific sequence determines specific structure, which in turn determines specific interactions. Although it was repeatedly demonstrated by screening of sequence libraries in vivo that ∼1% of all random sequences are functional as ADs,4,7,24,25 the extremely high variability of AD sequences is largely either ignored or underestimated. Considering the 20 amino acid optimal minimum length for ADs,7 the whole combinatorial space constitutes 20ˆ20 sequences and 1% of it is a staggering ∼10ˆ24 sequence possibilities for a single gene activator molecule. Absence of structural specificity is widely accepted, and it is by now a convention to characterize ADs as a typical example of intrinsically disordered protein regions. The spectrum of AD targets range from basal transcription factors such as TBP,26,27,28,29,30 TFIIB,31,32 TFIIH,33,34 TFIIA,35,36 RNA polymerase II,37 variable TAFs,38 Mediator subunits Med17 (Srb4), Srb10, Med15 (Gal11), Med2, and Med 25,39,40,41,42,43,44,45,46 as well as subunits of chromatin remodeling and histone-modifying complexes such as Ada2, Taf17, Tra1 (SAGA and NuA4 complexes),47,48,49,50,51,52,53 Swi1 and Snf2 (SWI/SNF complex),53,54,55 and CBP.56,57,58 Some ‘fuzzy’ interactions between Gal4 and Gcn4 ADs and Med15 were analyzed in detail,13 however it was stated that other equally fuzzy ‘free for all’ interactions are possible with other coactivators and are not investigated. Understanding ADs remains a difficult, long-standing fundamental scientific problem.2 Solving the AD enigma has foundational practical importance as multiple diseases including cancer are associated with changes in gene expression. The correction of the inappropriate expression level by modification of specific activator AD sequences by genome editing such as CRISPR-Cas9 technology seems to be increasingly feasible and beneficial; however, it requires first understanding AD sequence features and creation of ML models able to accurately predict the modified AD activity level. Decoding the language of ADs is one of the starting points in solving the AD enigma.
To be able to study the grammar of ADs, we intentionally used sequences which are enriched in hydrophobic amino acid residues (W, F, Y, L), previously identified to be important for AD function,3,5,11 so that we limit the number of nonfunctional AD sequences and to significantly increase the number of functional ADs in comparison to random sequence libraries.4,5,6,7 The main, immediate, and general finding of our study, consistent with previous reports,3,7,8,11 is that a significant number of active ADs can be as short as 10 amino acids and contain balanced acidic/hydrophobic sequences with mixtures of W, F, Y, L amino acids (1236 out of 3071 tested sequences using three different templates Figure 2). More surprisingly, for a 10 amino acid stretch a high AD activity is achievable by using Ds in combination with only Ws, or Fs, or Ls, but not Ys (350/1021 in WD10, 363/1022 in FD10 and 225/1023 in LD10 sets, Figure 4). In analyzing this large number of functional sequences, even when the composition is limited to a mix of W, F, Y, L as hydrophobics, or even for only a unique hydrophobic within a 10 amino acid stretch, no specific minimotif was identified among functional sequences, except for LD10 sequences.
Despite not having even short consensus sequences except for the LD10 set, ADs follow certain grammar rules that we3 and others5,6 started to formulate and refine based on high throughput experimental data and informatics analyses. The foundational rules that are clear from our current and previous3 study are formulated below, but not necessarily in a strict order of importance: (1) the absence of basic amino acid residues or at least a net negative charge, see Figure 6;3 (2) the balance between acidic and hydrophobic, preferably aromatic residues, see Figures 2, 3, 4, and 5;3,5 (3) with aromatic residues dominating in AD composition over aliphatic residues, see Figures 2 and 3; 3 (4) with acidic and hydrophobic residues preferably not forming significant acidic or hydrophobic clusters, see Figures 4, 5, and 6;3 (5) with hydrophobic residues preferably situated closer to the C-terminus and acidic residues situated more internally, see Figures 4, 5, and 6;3 and (6) additionally, for sequences following the above rules the functionality increases with an increased number of proline residues, see Figures 3 and 3.
The analysis of these rules and their relation to the existing models is important for gaining insights into the biochemical processes involved. Rule (1) suggests that the interacting targets or target binding sites contain positively charged amino acids; thus, if an AD also has an excess of positively charged amino acids it creates an electrostatic repulsion which negatively affects AD function. Rule (2) suggests that interactions with AD targets likely involve both electrostatic and hydrophobic interactions, because sequences not containing either hydrophobic or acidic amino acids are generally not functional.4,7,25 Additionally, the balance between acidic and hydrophobic amino acids is also consistent with the “stickers and spacers” idea for IDRs59 and the related “acidic exposure” model5 for ADs, whereby the balanced composition of hydrophobic and hydrophilic residues keeps hydrophobic residues from forming hydrophobic condensates, thus readying and exposing them for interactions with functional targets. The significant preference for aromatic residues in rule (3) suggests that the AD target interactions do not simply involve hydrophobic interactions but likely also encompass pi-pi interactions specific for aromatic residues. Rule (4) is related in interpretation to rule (2), as it also ensures that intramolecular hydrophobic aggregates are not formed. Rule (5) suggests that positioning the hydrophobics at the spatially free C-terminus ensures the best exposure of hydrophobics.
The results indicating the exceptional benefits of having proline residues within the AD sequence (Figure 3) suggest that contrary to the expectations of hydrophobic/acidic amphipathic helix being beneficial for AD function,7,12,13,14 the presence of proline as a “helix breaker” is highly beneficial (rule (6)). The likely explanation for this phenomenon is that by breaking the helix at least in the context of the amphipathic helix (Figure 3), Ps prevent intramolecular interactions between hydrophobic residues. In this respect, the proline phenomenon is in line with rules (2) and (4). Additionally, the proline phenomenon explains the existence of the entire proline rich class of ADs, which was observed long ago60 but never was fully explained.
There are, however, interesting exceptions from the above rules which in some cases can help us to understand the foundational rules more deeply. For instance, for rule (1) the occasional presence of positively charged amino acids, as long as it does not greatly reduce the net negative charge of AD (Figure 6), modulates the level of AD activity which might be important during the evolutionary adaptation of the biological object. Similarly, for rule (2) a deviation from the acidic/hydrophobic balance also modulates the AD level of activity (Figures 2, 4, and 5). For rule (3) all functional ADs within the frame of the flanked template DDDXXXXXDD (Figure 2E) constitute a deviation from the no-clusters rule. However, within this template the flanking Ds likely ensure the solvent exposure of hydrophobics within the cluster by repulsion, which is similar to the effect of an even and balanced distribution of acidic and hydrophobic residues in the intermixed template (Figure 2F). Deviations from rule (4) also modulate the AD level of activity, which might be important in evolutionary adaptations.
The example of ADs containing only Ls and Ds as a deviation from rule (3) deserves special consideration. Although LD sequences were reported several decades ago61 as possible candidates for ADs, they are rarely considered as a functional alternative to a typical aromatic/acidic ADs. It was shown that LXXLL motif is present in multiple coactivators such as CBP/p300, Rip140, TAFH and others62 playing a facilitating role for the recruitment of these and other coactivators for the proper transcription activation. In some cases, the L-enriched sequences are considered important for the function of the gene activator molecule, however not as ADs but rather as sequence modules influencing the sequence selection/discrimination by the DNA binding domain of the gene activator.21 According to modern high throughput experimentation studies in yeast and in human, although ADs are dominated by aromatic and acidic residues, and largely are consensus-less, the LXXLL motif was indicated to be associated with AD activities for some sequences.11,12 Previous work based on the ML analysis of large unbiased datasets of random AD sequence libraries3,7 showed that typical ADs overwhelmingly contain aromatic residues as hydrophobic representatives with Ls only occasionally facilitating aromatic residues. However, our current study shows that LD sequence combinations, although much rarer than aromatic/acidic sequences (Figure 4), can create ADs which surpass aromatic/acidic ADs in level of functionality. The rarity of LD ADs is based on the unique requirement in this case to have a specific LDL(L/D)DLL consensus (Figure 5) which is unusual for ADs as largely consensus-less sequences (Figures 2E–2G; Figures 4A–4C). In addition, the usual balance between hydrophobic and acidic residues is shifted in this case toward more hydrophobics (in this case Ls) due to the LDL(D/L)DLL motif. These facts suggest that the interacting target of LD AD has very specific structural characteristics. Consistent with this specificity and suggesting that chirality might be involved is also an observation that inversing of the LDLDDLL sequence to LLDDLDL (Figure 5C) or changing position of just two amino acids to LDLDLDL eliminates the functionality.
The key question regarding the mechanism of ADs’ function is the determination of actual AD target(s) and thus clarifying the mechanism of action. In this respect functional LD sequences seem as an exception from the general aromatic/acidic compositional rule and can be considered as a special case with a special type of target or special mode of interaction, e.g., the leucine zipper type of interactions with coactivators. However, the similarity in levels of activity of ADs with LD composition and ADs with a multitude of all other hydrophobic/acidic compositions (Figures 2, 3, 4, and 5) characterized in identical conditions for all sequences within our high-throughput experimental setup, suggests that the mechanism is possibly universal, because otherwise for thousands of very compositionally diverse sequence variations there are thousands of different targets and mechanisms, which is unlikely. The ADs with the LD composition, because they require more Ls for functionality, a specific motif, and a very special arrangement of amino acids, likely even in 3D, may then inform us about very special structural characteristics of the universal target. One such target suggested earlier3,63 might be the interface between the DNA and histones in the nucleosome (Figure 8). The hypothesis of AD interaction with the DNA/histone interface is consistent with the biochemical analysis of all rules discussed above. Importantly, the sequences identified in our screen are reminiscent by chemical nature (aromatic-hydrophilic) to the DNA groove binders such as netropsin and chromomycin, which recently were shown to initiate the histone octamer translocation in vitro.64 The universality of the AD mechanism between lower and higher eukaryotes is suggested by similar composition of ADs between yeast and human3,7,11,12 and the retention of AD functionality when transferred between biological phyla.65,66,67.
Figure 8.

Proposed mechanism for AD peptide action
On the nucleosome surface the AD peptide (e.g., DDDWWWWWDD or LLDLDLDDLL) interacts with DNA bases via hydrophobic residues (in case of aromatic residues – via intercalation) while interacting with histone tails by electrostatic contacts between acidic residues of the AD peptide and basic residues of histone tails. Created bulge of DNA later is propagated by a chromatin remodeler, similar to the proposed action of DNA groove binders,64 so that the histone octamer is translocated away from the gene promoter opening it for the transcription initiation complex assembly. Note: the structures of depicted peptides are predicted by AlphaFold2 ML model,68 which for the LD peptide suggests that the structure is amphipathic with all Ls situated on one side possibly aligning with the DNA groove. Elements of figure created with BioRender.com.
The ML approach and the ML feature development is a new way to get insights to the mechanistic aspects of AD function. The fact that PADDLE trained on natural AD sequences12 and the Attention AD model22 trained on the unbiased random sequence set, both perform suboptimal on our “synthetic” sequence sets (Figures 7A–7N) is likely because the total combinatorial space is enormous amounting to ∼10ˆ24, and both ML models were trained on comparably tiny fractions of sequences, with the designed sequences being outside of either training set. Thus, our sets can be used for additional training of future ML models likely improving their accuracy. The similarity of the overprediction outcomes for both PADDLE and Mechanistic Predictor which is based on the composition suggests that hidden ML features of PADDLE, are likely also dominated by compositional features. The noticeable overpredictions of functional ADs by PADDLE over more random mistakes by the Attention AD model might suggest that our design of AD sequences generally reflects the natural selection. The combination of designed AD approach and further development of ML models, and connection ML and biochemical AD features is potentially a prospective research direction.
Limitations of the study
Although we provide and analyze a massive library of variable sequences tested for functionality as ADs, our study is limited to in vivo characterizations. The actual mechanism of AD function—either via direct physical recruitment of coactivators and transcriptional machinery components, or by the AD action as a surfactant (Figure 8), or both—remains unclear, which highlights the fundamental long-standing enigma of eukaryotic gene regulation. The reasons for the AD problem to be so hard and potential ways to solve it are reviewed recently.69 To test potential AD target(s) additional in vitro modeling with identified AD sequences will be necessary in the future.
Resource availability
Lead contact
Requests for further information and resources should be directed to the lead contact, A.E. (aerkine@butler.edu).
Data and code availability
-
•
Data: The datasets generated for this study are available in the Gene Expression Omnibus (GEO) repository, in series GSE277056. Analyzed data are provided at https://doi.org/10.5281/zenodo.13351327
-
•
Code: Code used for data analysis and figure generation is provided at https://doi.org/10.5281/zenodo.13351327
-
•
Other items: This study did not generate new reagents. Items listed in the key resources table may be ordered from their supplier.
Materials availability
This study did not generate new reagents.
Acknowledgments
We thank Marcos Oliveira for discussions and suggestions. The work was supported by NSF grant MCB 1925646 (to A.M.E.).
Author contributions
A.M.E. conceived the project. T.Y.E. performed in vivo part of the work. D.C., C.A.C., and A.M.E. performed data analysis. D.C. and C.A.C. oversaw methods and visualizations. A.M.E. wrote the manuscript. All authors edited and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Raw sequencing data | This paper | GEO: GSE277056 |
| Normalized data | This paper | Zenodo: https://doi.org/10.5281/zenodo.6461744 |
| Experimental models: Organisms/strains | ||
| Yeast strain Y2HGold | Takara | Cat# 630498 |
| Recombinant DNA | ||
| Plasmid pGBKT7 | Takara | Cat# 630489 |
| Plasmid pRS314 | Addgene | Addgene Plasmid # 11004 |
| Software and algorithms | ||
| Analysis code | This paper | Zenodo: https://doi.org/10.5281/zenodo.6461744 |
| cutadapt | Martin, M.70 | https://pypi.org/project/cutadapt/ |
| STAR | Dobin, A. et al.71 | https://github.com/alexdobin/STAR |
| PADDLE | Sanborn, A.L. et al.12 | https://github.com/asanborn/PADDLE |
| LSTM Attention-AD | Wang, X. & Kihara, D.22 | $https://github.com/kiharalab/Attention_AD |
Experimental model and study participant details
East strain Y2HGold (Takara) was maintained on a standard YPD growth medium and after transformation with the sequence library, screened on the –trp synthetic yeast growth medium containing 200 μg/mL of aureobasidin. See details in the method details section below.
Method details
Library construction, cloning, and screening
The parental library plasmid was constructed by cloning the fragment containing the ADH1 promoter and the Gal4(1–147) DBD cassette, PCR amplified from the commercially available pGBKT7 vector. The PCR fragment was cloned into the SacI and KpnI sites of the centromeric yeast shuttle vector pRS314.
The design library containing 12,400 individual sequences each 60 nucleotide long. Individual sequences are described for each figure. If sequences are less than 20 amino acid residues the remaining space was filled by glycine codons, as it was shown previously that glycine is neutral for the AD activity.3,8 Each library sequence had an individual 20-nucleotide barcode directly following the stop codon to improve alignment performance. The library was synthesized at the GenScript commercial facility, amplified by PCR five times (each time appending a unique Biological Replicate (BioRep) barcode), quantitated for the DNA content, and mixed in equal proportions into a single pool. For description of more detailed steps, see Figure S4. The pool was cloned into the NcoI and SalI restriction sites remaining from the pGBKT7 fragment of the parental library plasmid. The library complexity was estimated by individual colony counts after transformation for a fraction of the total transformation mix, then multiplying by the fraction factor. Total complexity was estimated to be ∼10ˆ6. The total content of individual sequences within the library was determined by NGS at GenScript. The NGS also confirmed the in-frame fusion of AD sequences to the Gal4 DBD region. After the bacterial cloning and verification, the plasmid library was isolated for the following yeast transformation.
The isolated plasmid library was transformed into the yeast strain Y2HGold, available commercially from Clontech/Takara. The maintenance of the library complexity was determined by the individual colony count for a fraction of a transformation mix as described above for the bacterial transformation. The number of individual yeast transformants for the entire library was estimated to be ∼10ˆ6. After transformation, the whole-library cell culture was transferred into the –trp synthetic yeast growth medium containing 200 μg/mL of aureobasidin and grown for four days with daily 1/100 dilution to maintain the culture in the mid-log phase. Cell culture samples were taken at 0, 1, 2, 3, and 4 days. DNA was isolated using a Thermo Scientific Pierce Yeast DNA Extraction Reagent Kit. The library component was isolated by PCR using the Invitrogen AccuPrime SuperMix I kit with primers containing Illumina adapters and barcodes unique for each DNA sample. DNA samples were controlled for purity, repeatedly quantitated for DNA content, and sequenced at the NovoGene commercial facility.
Quantification and statistical analysis
Sequence processing
For each sample, cutadapt70 was used to demultiplex by BioReps, remove adapter barcodes, and remove sequence reads shorter than 73 residues. Reads from the design library were mapped using STAR71 to a pseudo-genome of 12,400 sequences. The pseudo-genome was built using the 12,400 designed sequences, with their unique barcodes, each represented as its own “chromosome.” The pseudo-genome is a general feature format file with one “gene” assigned to each “chromosome.” Successful mapping in STAR required a perfect match (allowing no mismatches). For each sample, the output read (gene) count file was used as the input for further data processing.
Estimation of sequence growth rates
Data processing and analyses were conducted in R version 4.2.3.72 Correlations among the read counts of biological replicates (BioReps) were calculated to ensure reasonable consistency. For each sample, counts for each sequence were converted to counts per million and log-transformed. The counts were then normalized to the average count for each sequence at the initial timepoint. Sequences detected in fewer than two of the five BioReps at the initial timepoint were removed (366 sequences). Sequences where counts drop to 0 at the first growth timepoint were removed (78 additional sequences), as no accurate growth slope could be calculated. Robust linear regression (implemented in the MASS package in R) was used to estimate the growth slope of each sequence over time; this was our final estimate for the functionality of each sequence. Growth slopes were calculated for the remaining 11,956 sequences using all BioReps at the initial timepoint and first growth timepoint. The intercept was not defined, though expected to be ∼0 based on normalization. A functionality threshold was determined as the mean growth slope +2 standard deviations for the non-functional stop-codon controls (n = 97). The functionality threshold was subtracted from all slopes to set the threshold to zero. Sequences with adjusted growth slopes greater than zero are considered functional. Percent functionality corresponds to the percentage of sequences in a group with a slope greater than zero.
Sequence sets
The library contained several control sequence sets: sequences comprised solely of a stop-codon (n = 97), 8 replicates of 24 natural AD sequences (n = 192), and 8 replicates of 3 previously tested sequences containing W and D residues (n = 24). In addition to the controls, the library was comprised of various sequence sets that cover the combinatorial space for a given sequence template. All dipeptide combinations repeated 4, 6, 8, or 10 times (n = 1421). Aromatic/hydrophobic (W, Y, F, or L) and aspartic acid (D) residues in a ten-residue sequence: WD10 (n = 1022), YD10 (n = 997), FD10 (n = 1022), and LD10 (n = 1023). Flanked template (GGGGGGGGGGDDDXXXXXDD) with all combinations of W, Y, F, and L residues in place of X (n = 1020). Flanked template (GGGGDDDWWWWWDDGGGGG) with 1, 2, or 3 glycine residues replaced with arginine residues (n = 176). Intermixed template (GGGGGGGGGGDXDXDXDXDX) with all combinations of W, Y, F, and L residues in place of X (n = 1019). Amphipathic template (XGDGXGDGXGDGXGDGXGDG) with all combinations of W, Y, F, and L residues in place of X (n = 1024). Amphipathic template (WGDGWGDGWGDGWGDGWGDG) with all glycine residues replaced with other amino acids (n = 18) or 1, 2, or 3 glycine residues replaced with proline residues (n = 175).
Sequence feature analyses
Among each set of sequences, the effects of the following sequence features on function were determined for aromatic/hydrophobic and basic residues: number of residues, balance of different residues, position of residues, clustering of residues, and multiresidue motifs. Balance refers to an equal number of aromatic/hydrophobic and basic residues. The effect of the position of residues along each AD was visualized as the percent of all sequences with the specified residue at the specified position that were functional or through sequence logos for groups of sequences. Sequence logos (prepared using ggseqlogo package in R) depict proportion of each residue at each position within a set of sequences.
Clustering refers to grouping of the same consecutive residue. Clustering was calculated using the “patterning parameter” metric defined by Martin et al.20 In brief, the “patterning parameter” was calculated as the average deviation of local sequence aromatic asymmetry from total sequence aromatic asymmetry normalized to the maximally clustered score. “Aromatic” (W, F, or L residues in this study) asymmetry is the difference between the fraction of the AD sequence that is W, F, or L residues and the fraction of the AD sequence that is D residues. The total sequence is 10 residues while the local sequences are 5 residue wide windows scanning through each sequence. A maximally clustered sequence in this context would have 5 consecutive W, F, or L residues and then 5 consecutive D residues (or the inverse). This sequence has the lowest possible total sequence aromatic asymmetry (0.5–0.5, perfectly symmetrical) and the highest possible local sequence aromatic asymmetry (1–0 or 0–1) with two of six windows with only one type of residue. Normalizing to the maximally clustered sequence sets this sequence to a “patterning parameter” value of 1 while the least clustered sequences will have scores approaching 0.
For sequences with 1–3 proline or arginine residues inserted across AD sequences, the average position of these residues was calculated for each sequence by first assigning the 20 positions of each AD with values from 1 to 20 and then averaging the values that correspond to the position of the proline or arginine residues.
Logistic regression
For the WD10, FD10, and LD10 sets, the number of hydrophobic residues, deviation from balance, average position, and clustering were converted into features scaled to scores between 0 and 1. The number of hydrophobic residues is a count of the number of residues within each 10-position long sequence. The deviation from balance was calculated by taking the absolute value of the difference between the number of hydrophobic and acidic residues in each sequence. The average position of hydrophobic residues was calculated the same way as the average position of proline or arginine residues described above. For these three features the maximum value was 10 (10 hydrophobic residues, a completely imbalanced sequence with a deviation of 10, or a sequence with a single hydrophobic residue at position 10), so these features were all scaled by dividing each score by 10. The “patterning parameter” was directly used as the clustering feature. The effect of each of these features on predicting functionality were determined using logistic regression (stats package in R). Coefficient estimates for the model for each set represent the average change in the log odds of a sequence being functional per increase in each rule value from a value of 0–1.
Mini-motif analysis
Minimotifs enriched in functional sequences were determined systematically by calculating the average slope for all sequences containing each combination of residues for 3–8 residue wide windows. Minimotifs were also identified among the top 50 functional LD10 sequences using MEME.17,18 To ensure identification of all significant minimotifs using MEME, default settings were used expect for minimum motif width that was decreased to 3 and number of motifs to search for increased to 20. Subsequent analyses were conducted on sequence subsets that were prepared using regular expressions to select sequences that contain specified motifs.
Secondary structure prediction and analysis
For the SPOT-1D secondary structure,73 the prediction for each unique 20-amino-acid-long AD sequence was performed using the SPOT-1D-Single program. The software was installed and run locally, as obtained from https://github.com/jas-preet/SPOT-1D-Single (accessed 07/20/23). The SS3 prediction component of SPOT-1D-Single was utilized to estimate the percentage of alpha-helical content for each AD sequence.
For the Agadir prediction of helical content,19 each unique 20-amino-acid-long AD sequence was submitted to the Agadir prediction server (http://agadir.crg.es/). Agadir was executed with the default parameters: pH = 7, temperature = 278 K, ionic strength = 0.01, N-terminus = free, and C-terminus = free. The output provided the percentage of each peptide predicted to adopt an alpha-helical conformation.
Functional AD prediction models
Three separate models trained and designed based on natural AD sequences were applied to the design library to predict functionality: PADDLE ML model,12 Long Short-Term Memory model with Attention AD mechanism,22 and the Mechanistic Predictor.6 To apply the PADDLE and Attention AD models to the library, each sequence was supplied alongside a set of 20 background sequences so that each sequence could be embedded in the background sequences to produce sequences of the required residue lengths (53 for PADDLE, 30 for Attention). The background sequences were generated randomly to contain equal amounts of the residues A, G, S, T, N, Q, and V. The no secondary structure PADDLE model was then applied to those inputs which generates Z-scores based on the probability of being an active transcription activation domain. The Attention AD model provides probabilities of functionality, for which we selected 0.5 as a cutoff.
The Mechanistic Predictor (MP) is an experimentally informed and empirically modified set of conditions to predict functional ADs. The MP, based on a library of sequences with 39 residues: Functional ADs = [-13 ≤ Net Charge ≤ −8 & Number of W, F, and L Residues ≥6]. As our library consists of sequences with a total length of 20 residues, the MP had to be modified to be applied to our library by halving all the numeric cutoffs. The modified predictor applied to our library with sequences with a total length of 20 residues: Functional ADs = [-6.5 ≤ Net Charge ≤ −4 & Number of W, F, and L Residues ≥3]. Net Charge is determined for each sequence by adding 1 for each basic residue (R, K, and H) and subtracting 1 for each acidic residue (D and E). The modified MP was then applied to each sequence in the library resulting in either a prediction of a functional or nonfunctional AD.
Published: September 26, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111057.
Supplemental information
References
- 1.Boija A., Klein I.A., Sabari B.R., Dall'Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M., et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell. 2018;175:1842–1855.e16. doi: 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strader L.C., Staller M.V., Willis A.E., Faulkner G.J., Beggs J.D., Cech T.R. The complexity of transferring genetic information. Mol. Cell. 2023;83:320–323. doi: 10.1016/j.molcel.2023.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Broyles B.K., Gutierrez A.T., Maris T.P., Coil D.A., Wagner T.M., Wang X., Kihara D., Class C.A., Erkine A.M. Activation of gene expression by detergent-like protein domains. iScience. 2021;24:103017. doi: 10.1016/j.isci.2021.103017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravarani C.N., Erkina T.Y., De Baets G., Dudman D.C., Erkine A.M., Babu M.M. High-throughput discovery of functional disordered regions: investigation of transactivation domains. Mol. Syst. Biol. 2018;14:e8190. doi: 10.15252/msb.20188190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staller M.V., Ramirez E., Kotha S.R., Holehouse A.S., Pappu R.V., Cohen B.A. Directed mutational scanning reveals a balance between acidic and hydrophobic residues in strong human activation domains. Cell Syst. 2022;13:334–345.e5. doi: 10.1016/j.cels.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotha S.R., Staller M.V. Clusters of acidic and hydrophobic residues can predict acidic transcriptional activation domains from protein sequence. Genetics. 2023;225:iyad131. doi: 10.1093/genetics/iyad131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erijman A., Kozlowski L., Sohrabi-Jahromi S., Fishburn J., Warfield L., Schreiber J., Noble W.S., Söding J., Hahn S. A High-Throughput Screen for Transcription Activation Domains Reveals Their Sequence Features and Permits Prediction by Deep Learning. Mol. Cell. 2020;78:890–902.e6. doi: 10.1016/j.molcel.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broyles B.K., Erkina T.Y., Maris T.P., Gutierrez A.T., Coil D.A., Wagner T.M., Class C.A., Erkine A.M. Sequence features of transcriptional activation domains are consistent with the surfactant mechanism of gene activation. bioRxiv. 2023 doi: 10.1101/2023.06.18.545482. Preprint at. [DOI] [Google Scholar]
- 9.Wu Y., Reece R.J., Ptashne M. Quantitation of putative activator-target affinities predicts transcriptional activating potentials. EMBO J. 1996;15:3951–3963. [PMC free article] [PubMed] [Google Scholar]
- 10.Piskacek S., Gregor M., Nemethova M., Grabner M., Kovarik P., Piskacek M. Nine-amino-acid transactivation domain: establishment and prediction utilities. Genomics. 2007;89:756–768. doi: 10.1016/j.ygeno.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 11.DelRosso N., Tycko J., Suzuki P., Andrews C., Fordyce P., et al. Mukund A., Mukund A., Liongson I., Ludwig C., Spees K. Large-scale mapping and mutagenesis of human transcriptional effector domains. Nature. 2023;616:365–372. doi: 10.1038/s41586-023-05906-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanborn A.L., Yeh B.T., Feigerle J.T., Hao C.V., Townshend R.J., Lieberman Aiden E., Dror R.O., Kornberg R.D. Simple biochemical features underlie transcriptional activation domain diversity and dynamic, fuzzy binding to Mediator. Elife. 2021;10:e68068. doi: 10.7554/eLife.68068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuttle L.M., Pacheco D., Warfield L., Wilburn D.B., Hahn S., Klevit R.E. Mediator subunit Med15 dictates the conserved "fuzzy" binding mechanism of yeast transcription activators Gal4 and Gcn4. Nat. Commun. 2021;12:2220. doi: 10.1038/s41467-021-22441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staller M.V., Holehouse A.S., Swain-Lenz D., Das R.K., Pappu R.V., Cohen B.A. A high-throughput mutational scan of an intrinsically disordered acidic transcriptional activation domain. Cell Syst. 2018;6:444–455.e6. doi: 10.1016/j.cels.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards R.J., Davey N.E., Shields D.C. SLiMFinder: a probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins. PLoS One. 2007;2:e967. doi: 10.1371/journal.pone.0000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davey N.E., Haslam N.J., Shields D.C., Edwards R.J. SLiMFinder: a web server to find novel, significantly over-represented, short protein motifs. Nucleic Acids Res. 2010;38:W534–W539. doi: 10.1093/nar/gkq440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey T.L., Johnson J., Grant C.E., Noble W.S. The MEME Suite. Nucleic Acids Res. 2015;43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nystrom S.L., McKay D.J. Memes: A motif analysis environment in R using tools from the MEME Suite. PLoS Comput. Biol. 2021;17:e1008991. doi: 10.1371/journal.pcbi.1008991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz V., Serrano L. Development of the multiple sequence approximation within the AGADIR model of alpha-helix formation: comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 20.Martin E.W., Holehouse A.S., Peran I., Farag M., Incicco J.J., Bremer A., Grace C.R., Soranno A., Pappu R.V., Mittag T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. 2020;367:694–699. doi: 10.1126/science.aaw8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonas F., Carmi M., Krupkin B., Steinberger J., Brodsky S., Jana T., Barkai N. The molecular grammar of protein disorder guiding genome-binding locations. Nucleic Acids Res. 2023;51:4831–4844. doi: 10.1093/nar/gkad184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X., Kihara D. Activation of gene expression by nucleosome detergents. Purdue University; 2021. Attention_AD is a computational tool using deep learning that can identify gene activation, which is for the dataset.https://github.com/kiharalab/Attention_AD [Google Scholar]
- 23.Simm S., Einloft J., Mirus O., Schleiff E. 50 years of amino acid hydrophobicity scales: revisiting the capacity for peptide classification. Biol. Res. 2016;49:31. doi: 10.1186/s40659-016-0092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma J., Ptashne M. A new class of yeast transcriptional activators. Cell. 1987;51:113–119. doi: 10.1016/0092-8674(87)90015-8. [DOI] [PubMed] [Google Scholar]
- 25.Abedi M., Caponigro G., Shen J., Hansen S., Sandrock T., Kamb A. Transcriptional transactivation by selected short random peptides attached to lexA-GFP fusion proteins. BMC Mol. Biol. 2001;2:10. doi: 10.1186/1471-2199-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horikoshi M., Hai T., Lin Y.S., Green M.R., Roeder R.G. Transcription factor ATF interacts with the TATA factor to facilitate establishment of a preinitiation complex. Cell. 1988;54:1033–1042. doi: 10.1016/0092-8674(88)90118-3. [DOI] [PubMed] [Google Scholar]
- 27.Stringer K.F., Ingles C.J., Greenblatt J. Direct and selective binding of an acidic transcriptional activation domain to the TATA-box factor TFIID. Nature. 1990;345:783–786. doi: 10.1038/345783a0. [DOI] [PubMed] [Google Scholar]
- 28.Hermann S., Berndt K.D., Wright A.P. How transcriptional activators bind target proteins. J. Biol. Chem. 2001;276:40127–40132. doi: 10.1074/jbc.M103793200. [DOI] [PubMed] [Google Scholar]
- 29.Capella M., Ré D.A., Arce A.L., Chan R.L. Plant homeodomain-leucine zipper I transcription factors exhibit different functional AHA motifs that selectively interact with TBP or/and TFIIB. Plant Cell Rep. 2014;33:955–967. doi: 10.1007/s00299-014-1576-9. [DOI] [PubMed] [Google Scholar]
- 30.Khan S.H., Ling J., Kumar R. TBP binding-induced folding of the glucocorticoid receptor AF1 domain facilitates its interaction with steroid receptor coactivator-1. PLoS One. 2011;6:e21939. doi: 10.1371/journal.pone.0021939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Y.S., Ha I., Maldonado E., Reinberg D., Green M.R. Binding of general transcription factor TFIIB to an acidic activating region. Nature. 1991;353:569–571. doi: 10.1038/353569a0. [DOI] [PubMed] [Google Scholar]
- 32.Choy B., Green M.R. Eukaryotic activators function during multiple steps of preinitiation complex assembly. Nature. 1993;366:531–536. doi: 10.1038/366531a0. [DOI] [PubMed] [Google Scholar]
- 33.Xiao H., Pearson A., Coulombe B., Truant R., Zhang S., Regier J.L., Triezenberg S.J., Reinberg D., Flores O., Ingles C.J., et al. Binding of basal transcription factor TFIIH to the acidic activation domains of VP16 and p53. Mol. Cell Biol. 1994;14:7013–7024. doi: 10.1128/mcb.14.10.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chabot P.R., Raiola L., Lussier-Price M., Morse T., Arseneault G., Archambault J., Omichinski J.G. Structural and functional characterization of a complex between the acidic transactivation domain of EBNA2 and the Tfb1/p62 subunit of TFIIH. PLoS Pathog. 2014;10:e1004042. doi: 10.1371/journal.ppat.1004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stargell L.A., Struhl K. The TBP-TFIIA interaction in the response to acidic activators in vivo. Science. 1995;269:75–78. doi: 10.1126/science.7604282. [DOI] [PubMed] [Google Scholar]
- 36.Ozer J., Bolden A.H., Lieberman P.M. Transcription factor IIA mutations show activator-specific defects and reveal a IIA function distinct from stimulation of TBP-DNA binding. J. Biol. Chem. 1996;271:11182–11190. doi: 10.1074/jbc.271.19.11182. [DOI] [PubMed] [Google Scholar]
- 37.Tan Q., Linask K.L., Ebright R.H., Woychik N.A. Activation mutants in yeast RNA polymerase II subunit RPB3 provide evidence for a structurally conserved surface required for activation in eukaryotes and bacteria. Genes Dev. 2000;14:339–348. [PMC free article] [PubMed] [Google Scholar]
- 38.Goodrich J.A., Tjian R. TBP-TAF complexes: selectivity factors for eukaryotic transcription. Curr. Opin. Cell Biol. 1994;6:403–409. doi: 10.1016/0955-0674(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 39.Koh S.S., Ansari A.Z., Ptashne M., Young R.A. An activator target in the RNA polymerase II holoenzyme. Mol. Cell. 1998;1:895–904. doi: 10.1016/s1097-2765(00)80088-x. [DOI] [PubMed] [Google Scholar]
- 40.Myers L.C., Gustafsson C.M., Hayashibara K.C., Brown P.O., Kornberg R.D. Mediator protein mutations that selectively abolish activated transcription. Proc. Natl. Acad. Sci. USA. 1999;96:67–72. doi: 10.1073/pnas.96.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeong C.J., Yang S.H., Xie Y., Zhang L., Johnston S.A., Kodadek T. Evidence that Gal11 protein is a target of the Gal4 activation domain in the mediator. Biochemistry. 2001;40:9421–9427. doi: 10.1021/bi010011k. [DOI] [PubMed] [Google Scholar]
- 42.Ansari A.Z., Koh S.S., Zaman Z., Bongards C., Lehming N., Young R.A., Ptashne M. Transcriptional activating regions target a cyclin-dependent kinase. Proc. Natl. Acad. Sci. USA. 2002;99:14706–14709. doi: 10.1073/pnas.232573899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu H., Hu C., Yoon S., Natarajan K., Swanson M.J., Hinnebusch A.G. An array of coactivators is required for optimal recruitment of TATA binding protein and RNA polymerase II by promoter-bound Gcn4p. Mol. Cell Biol. 2004;24:4104–4117. doi: 10.1128/MCB.24.10.4104-4117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warfield L., Tuttle L.M., Pacheco D., Klevit R.E., Hahn S. A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. USA. 2014;111:E3506–E3513. doi: 10.1073/pnas.1412088111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguilar X., Blomberg J., Brännström K., Olofsson A., Schleucher J., Björklund S. Interaction studies of the human and Arabidopsis thaliana Med25-ACID proteins with the herpes simplex virus VP16- and plant-specific Dreb2a transcription factors. PLoS One. 2014;9:e98575. doi: 10.1371/journal.pone.0098575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuttle L.M., Pacheco D., Warfield L., Luo J., Ranish J., Hahn S., Klevit R.E. Gcn4-Mediator specificity is mediated by a large and dynamic fuzzy protein-protein complex. Cell Rep. 2018;22:3251–3264. doi: 10.1016/j.celrep.2018.02.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knutson B.A., Hahn S. Domains of Tra1 important for activator recruitment and transcription coactivator functions of SAGA and NuA4 complexes. Mol. Cell Biol. 2011;31:818–831. doi: 10.1128/MCB.00687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown C.E., Howe L., Sousa K., Alley S.C., Carrozza M.J., Tan S., Workman J.L. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science. 2001;292:2333–2337. doi: 10.1126/science.1060214. [DOI] [PubMed] [Google Scholar]
- 49.Bhaumik S.R., Raha T., Aiello D.P., Green M.R. In vivo target of a transcriptional activator revealed by fluorescence resonance energy transfer. Genes Dev. 2004;18:333–343. doi: 10.1101/gad.1148404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barlev N.A., Candau R., Wang L., Darpino P., Silverman N., Berger S.L. Characterization of physical interactions of the putative transcriptional adaptor, ADA2, with acidic activation domains and TATA-binding protein. J. Biol. Chem. 1995;270:19337–19344. doi: 10.1074/jbc.270.33.19337. [DOI] [PubMed] [Google Scholar]
- 51.Thut C.J., Chen J.L., Klemm R., Tjian R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science. 1995;267:100–104. doi: 10.1126/science.7809597. [DOI] [PubMed] [Google Scholar]
- 52.Henriksson A., Almlöf T., Ford J., McEwan I.J., Gustafsson J.A., Wright A.P. Role of the Ada adaptor complex in gene activation by the glucocorticoid receptor. Mol. Cell Biol. 1997;17:3065–3073. doi: 10.1128/mcb.17.6.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neely K.E., Hassan A.H., Wallberg A.E., Steger D.J., Cairns B.R., Wright A.P., Workman J.L. Activation domain-mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Mol. Cell. 1999;4:649–655. doi: 10.1016/s1097-2765(00)80216-6. [DOI] [PubMed] [Google Scholar]
- 54.Neely K.E., Hassan A.H., Brown C.E., Howe L., Workman J.L. Transcription activator interactions with multiple SWI/SNF subunits. Mol. Cell Biol. 2002;22:1615–1625. doi: 10.1128/MCB.22.6.1615-1625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prochasson P., Neely K.E., Hassan A.H., Li B., Workman J.L. Targeting activity is required for SWI/SNF function in vivo and is accomplished through two partially redundant activator-interaction domains. Mol. Cell. 2003;12:983–990. doi: 10.1016/s1097-2765(03)00366-6. [DOI] [PubMed] [Google Scholar]
- 56.Kundu T.K., Palhan V.B., Wang Z., An W., Cole P.A., Roeder R.G. Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol. Cell. 2000;6:551–561. doi: 10.1016/s1097-2765(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 57.Lau O.D., Kundu T.K., Soccio R.E., Ait-Si-Ali S., Khalil E.M., Vassilev A., Wolffe A.P., Nakatani Y., Roeder R.G., Cole P.A. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 58.Mukherjee S.P., Behar M., Birnbaum H.A., Hoffmann A., Wright P.E., Ghosh G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 2013;11:e1001647. doi: 10.1371/journal.pbio.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi J.M., Holehouse A.S., Pappu R.V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 2020;49:107–133. doi: 10.1146/annurev-biophys-121219-081629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitchell P.J., Tjian R. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science. 1989;245:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 61.Feng B., Marzluf G.A. The regulatory protein NIT4 that mediates nitrate induction in Neurospora crassa contains a complex tripartite activation domain with a novel leucine-rich, acidic motif. Curr. Genet. 1996;29:537–548. doi: 10.1007/BF02426958. [DOI] [PubMed] [Google Scholar]
- 62.Plevin M.J., Mills M.M., Ikura M. The LxxLL motif: a multifunctional binding sequence in transcriptional regulation. Trends Biochem. Sci. 2005;30:66–69. doi: 10.1016/j.tibs.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 63.Erkine A.M. Nonlinear' Biochemistry of Nucleosome Detergents. Trends Biochem. Sci. 2018;43:951–959. doi: 10.1016/j.tibs.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 64.Lorch Y., Kornberg R.D., Maier-Davis B. Disruption of nucleosomes by DNA groove binders of clinical significance and implications for chromatin remodeling. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2216611120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Escher D., Bodmer-Glavas M., Barberis A., Schaffner W. Conservation of glutamine-rich transactivation function between yeast and humans. Mol. Cell Biol. 2000;20:2774–2782. doi: 10.1128/mcb.20.8.2774-2782.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yanagisawa S. The transcriptional activation domain of the plant-specific Dof1 factor functions in plant, animal, and yeast cells. Plant Cell Physiol. 2001;42:813–822. doi: 10.1093/pcp/pce105. [DOI] [PubMed] [Google Scholar]
- 67.Ma J., Przibilla E., Hu J., Bogorad L., Ptashne M. Yeast activators stimulate plant gene expression. Nature. 1988;334:631–633. doi: 10.1038/334631a0. [DOI] [PubMed] [Google Scholar]
- 68.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erkine A.M., Oliveira M.A., Class C.A. The enigma of transcriptional activation domains. J. Mol. Biol. 2024;436 doi: 10.1016/j.jmb.2024.168766. [DOI] [PubMed] [Google Scholar]
- 70.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011;17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 71.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.R Development Team . R Foundation for Statistical Computing; 2023. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 73.Hanson J., Paliwal K., Litfin T., Yang Y., Zhou Y. Improving prediction of protein secondary structure, backbone angles, solvent accessibility and contact numbers by using predicted contact maps and an ensemble of recurrent and residual convolutional neural networks. Bioinformatics. 2019;35:2403–2410. doi: 10.1093/bioinformatics/bty1006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: The datasets generated for this study are available in the Gene Expression Omnibus (GEO) repository, in series GSE277056. Analyzed data are provided at https://doi.org/10.5281/zenodo.13351327
-
•
Code: Code used for data analysis and figure generation is provided at https://doi.org/10.5281/zenodo.13351327
-
•
Other items: This study did not generate new reagents. Items listed in the key resources table may be ordered from their supplier.