

Abstract
The clinical relevance of the colorectal cancer serrated pathway is evident, but the screening of serrated lesions remains challenging. We aimed to characterize the serum methylome of the serrated pathway and to evaluate circulating cell‐free DNA (cfDNA) methylomes as a potential source of biomarkers for the non‐invasive detection of serrated lesions. We collected serum samples from individuals with serrated adenocarcinoma (SAC), traditional serrated adenomas, sessile serrated lesions, hyperplastic polyps and individuals with no colorectal findings. First, we quantified cfDNA methylation with the MethylationEPIC array. Then, we compared the methylation profiles with tissue and serum datasets. Finally, we evaluated the utility of serum cfDNA methylation biomarkers. We identified a differential methylation profile able to distinguish high‐risk serrated lesions from no serrated neoplasia, showing concordance with tissue methylation from SAC and sessile serrated lesions. Serum methylation profiles are pathway‐specific, clearly separating serrated lesions from conventional adenomas. The combination of ninjurin 2 (NINJ2) and glutamate‐rich 1 (ERICH1) methylation discriminated high‐risk serrated lesions and SAC with 91.4% sensitivity (64.4% specificity), while zinc finger protein 718 (ZNF718) methylation reported 100% sensitivity for the detection of SAC (96% specificity). This is the first study exploring the serum methylome of serrated lesions. Differential methylation of cfDNA can be used for the non‐invasive detection of colorectal serrated lesions.
Keywords: circulating cell‐free DNA, DNA methylation, non‐invasive biomarkers, screening, serrated colorectal cancer, serrated lesions
We analyzed the serum methylome of precursory lesions of the colorectal cancer serrated pathway and found a differential methylation profile between high‐risk serrated lesions and no serrated neoplasia. Since the methylation profiles in serum cfDNA are pathway‐specific at early stages, they may serve as a source of non‐invasive biomarkers for the detection of serrated lesions in screening programs.
Abbreviations
- AA
advanced adenoma
- AUC
area under the curve
- cfDNA
circulating cell‐free DNA
- CGI
CpG island
- CRC
colorectal cancer
- DMP
differentially methylated position
- DMR
differentially methylated region
- FDR
false discovery rate
- FIT
fecal immunochemical test
- FWER
family‐wise error rate
- HP
hyperplastic polyp
- HR‐HP
high‐risk hyperplastic polyp
- HR‐SL
high‐risk serrated lesion
- HR‐SP
high‐risk serrated polyp
- MS‐qPCR
methylation‐specific quantitative PCR
- NAA
non‐advanced adenomas
- NCF
no colorectal findings
- NMP
normalized methylation percentage
- NSN
no serrated neoplasia
- RMP
raw methylation percentage
- ROC
receiver‐operating characteristic
- SAC
serrated adenocarcinoma
- SP
serrated polyp
- SSL
sessile serrated lesion
- TSA
traditional serrated adenoma
1. Introduction
Colorectal cancer (CRC) is a very heterogeneous disease that develops via the stepwise accumulation of multiple genetic and epigenetic alterations [1]. While two‐thirds of CRC arise from the classical adenoma‐to‐carcinoma sequence, also known as the chromosome instability (CIN) pathway, the serrated pathway accounts for 15–30% of CRC. The clinical and biological relevance of this alternative pathway has been pointed out in the last years [2, 3, 4]. Serrated lesions are a heterogeneous group of saw‐toothed lesions, characterized histologically by a stellate pattern of crypt in‐folding [1, 5]. The most recent classification of the World Health Organization further defines serrated lesions into four main categories, namely, hyperplastic polyps (HP), sessile serrated lesions (SSL, with and without dysplasia), unclassified serrated adenomas/polyps (SP), and traditional serrated adenomas (TSA) [5, 6]. HP are the most‐frequently occurring serrated lesions (60–75%) and rarely undergo malignant transformation [7]. SSL account for 20–35% of all serrated lesions; dysplastic SSL are associated with advanced age, female sex, and proximal colon location [7, 8]. TSA are the rarest form of serrated lesion (1–7%), found predominantly in the distal colon. Overall, TSA have a protuberant growth pattern with villiform projections, challenging the distinction between TSA and classical tubulovillous adenomas [7, 9].
A two‐arm model has been proposed to describe the progression of the serrated pathway, characterized by genetic and epigenetic alterations that initiate and drive malignant transformation. The V600E activating BRAF mutation is a distinguishing trait of the serrated pathway and one of its first detected events, present in 70–80% of HP, > 90% of SSL, and 20–40% of TSA [4, 10]. After initiating BRAF mutations, serrated tumors may develop via two different routes: (a) one converging with the MSI pathway, characterized by mutations in the DNA mismatch machinery repair or by MLH1 hypermethylation, leading to SSL that may progress to tumors with MSI‐high phenotype; alternatively, (b) lesions with BRAF mutations can acquire TP53 mutations, activating oncogenic signaling such as Wnt and TGF‐β, and epithelial‐to‐mesenchymal transition, resulting in TSA and microsatellite stable (MSS) tumors [1, 11]. Tumors developing by either arm usually present high levels of CpG island methylation. The CpG island methylation phenotype (CIMP) is characterized by the hypermethylation of gene promoter regions causing loss of tumor suppressor gene function and can be already detected at early tumor stages [12, 13]. CIMP status correlates with poorly differentiated tumors containing BRAF mutations and MSI, commonly located in the proximal colon, and mostly in older female patients [12, 14].
Detection and removal of premalignant lesions is one of the principal objectives of CRC screening. Large or dysplastic SSL and TSA are considered the precursor lesions of the serrated pathway [9, 15]. While TSA are similar to conventional adenomas regarding the development of cancer, the subset of serrated lesions acquiring MSI, such as SSL, have an accelerated transition to carcinomas (1–3 years) [11]. This, together with the fact that SSL are flat, with a mucus cap and indistinct borders that make them likely to be missed and incompletely resected during colonoscopy, suggests that a large proportion of interval CRC, those developed within the recommended surveillance window of 3–5 years, arise from the serrated pathway [5, 8, 16]. The similarities in histological appearance between SSL and HP also result in misclassification and misdiagnosis [5].
Additionally, SSL are less prone to bleed due to their subtle morphology and thick but diffuse vascularity; thus, the fecal immunochemical test (FIT) commonly used for CRC screening has limited sensitivity for the detection of serrated lesions regardless of size or presence of dysplasia [17, 18, 19]. Hence, the successful screening of serrated lesions is still unmet.
In this study, we performed an epigenome‐wide analysis of serum circulating cell‐free DNA (cfDNA) pools to explore the methylation signatures in patients with serrated lesions, aiming to characterize the serum methylome as a potential source of non‐invasive biomarkers that could be implemented in screening programs to improve the rate of detection of serrated lesions and reduce the incidence of serrated CRC.
2. Materials and methods
2.1. Aim and study design
We aimed to characterize the serum methylome of the serrated pathway, and to evaluate cfDNA methylation as a source of biomarkers for the non‐invasive screening and diagnosis of serrated lesions. We collected a total of 186 serum samples from individuals with precursory lesions of the serrated pathway, serrated CRC and colonoscopically confirmed healthy controls. The study was conducted in two phases: (a) first, genome‐wide methylation was quantified in cfDNA pooled samples with the MethylationEPIC array. The differentially methylated profile between high‐ and low‐risk serrated lesions was characterized and were compared to external serum and tissue methylation datasets. Then, (b) the utility of serum cfDNA methylation for the non‐invasive detection of serrated lesions was evaluated in an independent cohort of individual serum samples.
2.2. Patient characteristics and samples
Individuals were recruited from January 2018 to October 2021 from the following Spanish Hospitals: Hospital Donostia (San Sebastián), Hospital Clínic de Barcelona (Barcelona), Hospital General Universitario de Alicante (Alicante), Complexo Hospitalario Universitario de Ourense (Ourense), Hospital Álvaro Cunqueiro (Vigo). Written informed consent was obtained from all patients with approval by the Ethics Committee for Clinical Research of Galicia (2018/008). The study was conducted according to the clinical and ethical principles of the Spanish Government and the Declaration of Helsinki. The study followed the “Strengthening the Reporting of Observational Studies in Epidemiology” (STROBE) reporting guideline. Individuals with incomplete colonoscopy or suboptimal bowel preparation, personal history of CRC, digestive cancer, inflammatory bowel disease, gastrointestinal disease, serrated polyposis syndrome, previous colectomy, or with a severe synchronic illness were excluded. The age range of the patients matches the USPSTF guideline recommendation for CRC screening (50–75 years) [20, 21].
All individuals underwent a colonoscopy, which was performed by experienced endoscopists following the Spanish clinical practice guidelines for colonoscopy procedures [22]. Blood samples were obtained before the colonoscopy from 186 individuals between 51 and 76 years old. Serum was collected after coagulation and centrifugation and was stored at −20 °C until cfDNA extraction.
We grouped samples into five main categories: (a) serrated adenocarcinoma (SAC; including CRC with MSI, CIMP‐high, or BRAF mutation; mucinous adenocarcinoma and signet‐ring cell adenocarcinoma), (b) high‐risk serrated polyps (HR‐SP; comprising TSA, SSL, and SP with dysplasia or ≥ 10 mm), (c) high‐risk hyperplasic polyps (HR‐HP; HP ≥ 10 mm), (d) low‐risk serrated lesions (LR‐SL; SP without dysplasia < 10 mm and HP < 10 mm), and (e) individuals with no colorectal findings (NCF). Individuals with multiple lesions were classified according to the most advanced colorectal finding [23]. Serum samples were separated into two independent subsets: 106 serum samples (30 NCF, 30 LR‐SL, 16 HR‐HP, and 30 HR‐SP) for the genome‐wide methylation analysis and 80 samples (20 NCF, 25 LR‐SL, 10 HR‐HP, 20 HR‐SP, and 5 SAC) for the targeted evaluation of methylation biomarkers. Formalin‐fixed paraffin‐embedded (FFPE) tumor tissue from six SAC cases and matched normal mucosa from the same patient were also used to evaluate the methylation biomarkers (Table 1).
Table 1.
Clinical and epidemiological characteristics of the patients included in the study. The number of patients, age median and range, sex, and colorectal findings is provided. CRC was staged according to the American Joint Committee on Cancer (AJCC) system. CRC, colorectal cancer; FFPE, formalin‐fixed paraffin‐embedded; HGD, high‐grade dysplasia; HP, hyperplastic polyp; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; LGD, low‐grade dysplasia; LR‐SL, low‐risk serrated lesion; MS‐qPCR, methylation‐specific quantitative PCR; NCF, no colorectal findings; SAC, serrated adenocarcinoma; SP, serrated polyp; SSL, sessile serrated lesion; TSA, traditional serrated adenoma.
| Epigenome‐wide analysis (n = 106, serum) | Biomarker evaluation by MS‐qPCR (n = 80, serum) | Matched tumor‐mucosa (n = 6, FFPE tissue) | |||
|---|---|---|---|---|---|
| HR‐SL | NSN | HR‐SL | NSN | SAC a | |
| Total (n) | 46 | 60 | 35 | 45 | 6 |
| Age median (range) | 62.0 (51–73) | 61.5 (51–74) | 64.0 (52–73) | 60.0 (52–76) | 73.5 (63–90) |
| Sex | |||||
| Male | 23 | 30 | 16 | 25 | 2 |
| Female | 23 | 30 | 19 | 20 | 4 |
| NCF | – | 30 | – | 20 | – |
| LR‐SL | – | 30 | – | 25 | – |
| HP < 10 mm | – | 27 | – | 18 | – |
| SP < 10 mm | – | 0 | – | 3 | – |
| SSL < 10 mm | – | 3 | – | 4 | – |
| HR‐HP (HP > 10 mm) | 16 | – | 10 | – | – |
| Distal location | 8 | – | 5 | – | – |
| Proximal location | 8 | – | 5 | – | – |
| HR‐SP | 30 | – | 20 | – | – |
| TSA | 4 | – | 4 | – | – |
| Large/dysplastic SSL | 14 | – | 13 | – | – |
| SP > 10 mm | 26 | – | 12 | – | – |
| HGD | 13 | – | 9 | – | – |
| LGD | 2 | – | 8 | – | – |
| Distal location | 15 | – | 8 | – | – |
| Proximal location | 15 | – | 12 | – | – |
| SAC | – | – | 5 | – | 6 |
| Stage I | – | – | 2 | – | 1 |
| Stage II | – | – | 0 | – | 1 |
| Stage III | – | – | 2 | – | 4 |
| Stage IV | – | – | 1 | – | 0 |
| Distal location | – | – | 2 | – | 3 |
| Proximal location | – | – | 3 | – | 3 |
SAC tissue samples are paired with healthy mucosa from the same patient.
2.3. cfDNA isolation, sample pooling, and DNA extraction from FFPE samples
cfDNA was extracted from 0.5 to 2 mL serum, according to availability. For the epigenome‐wide analysis, we followed a sample pooling approach. First, cfDNA was isolated from serum samples with a phenol‐chloroform protocol [24] and was quantified with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Then, 11 independent pooled samples were prepared combining equal amounts of cfDNA from 10 individuals (half male, half female) as previously described [25]. The factors considered to match between pooled samples were sex, age, and recruitment hospital (Table S1). Pooled samples were stored at −20 °C.
For the targeted evaluation, cfDNA was extracted from serum samples with the QIAmp Circulating Nucleic Acids Kit (Qiagen, Hilden, Germany). DNA was extracted from FFPE tissue specimens with the NucleoSpin® DNA FFPE XS DNA Isolation kit (Macherey‐Nagel, Allentown, PA, USA) with xylene deparaffinization. DNA and cfDNA samples were bisulfite treated with the EZ DNA Methylation‐Direct Kit (Zymo Research, Irvine, CA, USA) and stored at −80 °C.
2.4. Genome‐wide methylation profiling of cfDNA
The cfDNA pooled samples were sent to the Josep Carreras Leukemia Research Institute (Badalona, Spain) for processing and methylation quantification. Pools were bisulfite‐treated in the same batch and were hybridized to Infinium MethylationEPIC BeadChip arrays (Illumina, San Diego, CA, USA) following the manufacturer's instructions. Samples from different pathological groups were carefully allocated to each slide to minimize confounder variability due to technical batch effects. Methylation levels were derived from a total of 866 091 CpG sites.
Illumina methylation data were preprocessed and analyzed using the r environment (versions 3.6.1 and 4.0.0; Vienna, Austria) with r and bioconductor packages. Raw methylated and unmethylated signal intensities were background and dye‐bias corrected applying a normal‐exponential convolution based on the non‐specific fluorescence of Infinium I probes (single‐sample out‐of‐band normalization method implemented in the minfi package) [26]. The technical BeadChip batch effect was adjusted by an empirical Bayes method implemented in the ComBat function from the sva package [27]. The DNA methylation level was summarized for each CpG probe as the fraction of the methylated signal intensity over the total signal intensity (beta‐value). Detection P‐values were computed with the minfi package. Probes with a P‐value > 0.01 and a bead count < 3 were discarded, and mean detection P‐values were examined across all samples to identify any failed sample. After excluding cross‐reactive probes [28], probes violating any assumption for linear regression model fitting (linearity, homoscedasticity, uncorrelatedness, and normality of the standardized residuals) [29], probes annotated with genetic variants, and those located on the X and Y chromosomes, a total of 734 739 CpG sites remained for analysis. No samples were removed due to quality issues.
Probes were mapped to the human genome assembly GRCh37/hg19, annotated to genomic regions, and RefSeq genes according to the MethylationEPIC Manifest. CpG sites were assigned to CpG island (CGI), CGI‐shores and shelves or opensea regions. CpG sites were also annotated as in promoter regions (5′UTR, TSS200, TSS1500, and first exons), intragenic regions (gene body and 3′UTR), and intergenic regions. Beta‐values (methylation levels) were reported for interpretability and M‐values (logit‐transformation of beta‐values) were used for statistical analyses, as they approximate a Gaussian distribution [30].
2.5. Differential methylation analyses
Differentially methylated CpG positions (DMPs) were identified by the standard workflow of the limma package [31]. Briefly, linear models were fitted for each CpG across all samples by generalized least squares, and an empirical Bayes moderated t‐test was used to compute the P‐values. A P‐value < 0.01 and at least 10% difference in the methylation levels were used as the threshold to select DMPs. We identified differentially methylated regions (DMRs) using the bumphunter method [32, 33], which searches for contiguous CpG sites consistently hypomethylated or hypermethylated between groups. Clusters were defined by neighboring probes within a window of 250 bp, then linear regressions were fitted to each probe, and 100 permutations were used to generate a null distribution of regions for establishing significance. The false positive rate was controlled by the family‐wise error rate (FWER), a more conservative approach compared with the BH and FDR methods [34]. Significant DMRs were selected as those with FWER < 10%, P‐value < 0.01, and with at least two adjacent CpG sites.
Serrated pathway differential methylation profiles were explored in cfDNA pooled samples from the conventional CRC pathway (GSE186381). This dataset includes 23 cfDNA pooled samples grouped in NCF, non‐advanced adenomas, advanced adenomas and CRC [35]. Serum differential methylation profiles were also tested in tissue samples from healthy colorectal mucosa (n = 16) and serrated tumors (n = 38) from the dataset GSE68060 [36], and from SSL (n = 13) from the dataset E‐MTAB‐7854 [37]. This analysis was limited to probes shared by the Methylation450k and MethyationEPIC BeadChip arrays.
2.6. Functional annotation of differential methylation
One‐sided Fisher's exact tests were used to assess the significance of the enrichment of the DMPs to functionally annotated elements such as CpG islands, promoter regions, and intergenic regions, using the annotation of the complete array as background. To determine the biological functions of the DMPs associated with serrated neoplasia, we conducted gene ontologies (GO) enrichment analyses using the gometh function from the missmethyl package [38], which accounts for the bias derived from the differing number of probes targeting each gene and from probes annotated to multiple genes, applying a Wallenius' non‐central hypergeometric test. Significantly enriched GO terms were obtained for promoter regions based on hypomethylated and hypermethylated DMPs separately. Overrepresentation results within a P‐value < 0.005 were considered statistically significant. Biological process (BP), cellular component (CC), and molecular function (MF) ontologies were included. GO term semantic similarity analyses were performed with the enrichplot package [39] using the Jaccard's similarity index [40].
2.7. Targeted evaluation of differentially methylated regions
Nested custom qPCR assays were used for targeted methylation analyses in serum (n = 81) and tissue samples (n = 12). First, bisulfite‐treated cfDNA or DNA was subjected to a pre‐amplification (pre‐PCR) with primers flanking the region of interest, followed by a MS‐qPCR (methylation‐specific qPCR) with a probe targeting the methylated sequence, using diluted pre‐amplification products as template (Table S2). Pre‐PCRs were performed in a 25 μL reaction mix containing 2 μL of bisulfite‐modified DNA, 0.72 μm forward and reverse primers, a 75 μm dNTPs mixture, a 1× Ex Taq Buffer, and 1 unit of Takara Ex Taq HotStart (Takara Bio Inc., Kusatsu, Japan), with the following cycling conditions: 95 °C for 5 min, 25 cycles of 95 °C for 30 s, 30 s at 60 °C for all amplicons, 72 °C for 30 s, and finally 72 °C for 7 min. MS‐qPCR was performed using the following dilutions of the previous PCR product: 1/20 for DMR2, DMR9 and ACTB, and 1/200 for DMR7. Real‐time PCR was carried out in triplicate in a 20 μL volume containing 2 μL of the diluted pre‐PCR, 600 nm of each primer, 200 nm of probe, and 1× TaqMan Gene Expression Master Mix (Thermo Fisher Scientific), with an annealing temperature of 60 °C for DMR7, DMR9 and ACTB, and of 58 °C for DMR2, during 40 cycles. Amplifications were carried out in 96‐well plates and run on a StepOne Plus instrument (Thermo Fisher Scientific).
A fully methylated control (methyltransferase‐treated DNA with M.SssI; New England Biolabs, Ipswich, MA, USA) and a fully unmethylated control (whole‐genome amplification of DNA with GenomiPhi V2 DNA Amplification kit; GE Healthcare, Chicago, IL, USA) were included in each run for normalization and to verify plate‐to‐plate consistency. DNA extracted from peripheral blood from a donor was used to prepare controls. Standard curves were elaborated for each amplicon using dilutions of the fully methylated control (100–0.01% methylation; amplification efficiency > 90%, R 2 > 0.99). Raw methylation percentages (RMP) were estimated based on the corresponding standard curve. Then, RMP were normalized for DNA input, obtained by targeting a methylation independent region of the β‐actin gene (ACTB). Normalized methylation percentages (NMP) were calculated as follows:
2.8. Statistical analyses
All statistical analyses were performed with the r environment (version 4.1.0). Differentially methylated positions P‐values were adjusted to control de false discovery rate (FDR) with the Benjamini–Hochberg procedure. For the differentially methylated regions, the false positive rate was controlled by the family‐wise error rate (FWER) [34]. In the Gene Ontology enrichment analysis, a P‐value < 0.005 was considered statistically significant. For qPCR validation, normalized methylation values were transformed to log10(NMP + 1) for analysis. Methylation levels were compared with the Wilcoxon rank‐sum test for individual serum samples (P‐value < 0.05), and the non‐parametric Wilcoxon signed‐rank test for matched tumor and healthy mucosa tissue samples (P‐value < 0.05). Multivariate logistic regressions were fitted to the NMP to derive classification models for the detection of HR‐HP, HR‐SP, and SAC. The classification performance was assessed by leave‐one‐out cross‐validation, ROC curves were elaborated, and AUC, sensitivity, and specificity values were estimated with their corresponding 95% confidence intervals. The Youden Index method was used to determine the best cut‐off values [41]. Fisher's exact tests were employed to compare the proportion of distal and proximal lesions detected.
3. Results
3.1. Study overview
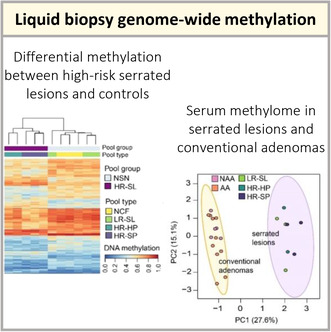
This study was conducted to explore the serum methylome of precancerous lesions belonging to the serrated pathway of colorectal carcinogenesis in a prospective multicenter cohort. Individuals were grouped into five main categories regarding the most advanced colorectal finding: SAC, HR‐SP, HR‐HP, LR‐SP, and NCF (Table 1). First, epigenome‐wide methylation levels were quantified in pooled cfDNA samples to characterize the differential methylation profile between no serrated neoplasia (NSN: NCF and LR‐SL) and high‐risk serrated lesions (HR‐SL: HR‐HP and HR‐SP); concordance with tissue methylation levels was evaluated using external datasets. Then, the pathway‐specific cfDNA methylation signature was assessed in comparison with cfDNA pools from the conventional CRC pathway. Finally, targeted assays were performed to evaluate the potential biomarker utility of serum cfDNA methylation to detect serrated lesions in an independent cohort.
3.2. cfDNA sample pooling
For the serum methylome profiling, 106 serum cfDNA samples were grouped into 11 cfDNA pools as described [25]. Briefly, five men and five women with the same colorectal pathology were included in each pool, matched by recruitment hospital and age (median 62, range 51–74) (Table S1). No pools were assembled for SAC cases due to limited sample availability. The final quantity of cfDNA in the pools ranged from 124 to 336 ng. There was no statistically significant difference in the mean age between pools (ANOVA, P‐value < 0.05).
3.3. Serum methylome profiling of the serrated pathway
Differential methylation analyses were performed across the total of 734 739 CpG‐targeting probes left after quality filtering and normalization. The global cfDNA methylation level of each pooled sample is shown in Fig. S1. Pairwise comparisons were carried out between the four pathological groups (NCF, LR‐SL, HR‐HP, and HR‐SP) according to the expected progression of the serrated pathway: either NCF – LR‐SL – HR‐HP, or NCF – LR‐SL – HR‐SP. Following this scheme, six independent differential methylation analyses were performed: NCF vs LR‐SL, NCF vs HR‐HP, NCF vs HR‐SP, LR‐SL vs HR‐HP, LR‐SL vs HR‐SP, and HR‐HP vs HR‐SP. Differential methylation results are detailed in Figs S2–S4.
To explore the differential methylation profile of precursor serrated lesions we compared NSN vs HR‐SL, obtaining only one CpG site with a significant false discovery rate (FDR, Bonferroni‐Hochberg correction) lower than 10%, likely due to the small group size available. This CpG site was hypomethylated in HR‐SL with a methylation difference of 14.66%. Thus, to explore serum methylome profiles, we used a less stringent statistical criterion: CpG sites with a nominal P‐value < 0.01 and at least 10% difference in methylation levels were considered as differentially methylated positions (DMPs) (Fig. S5). Applying these criteria, we identified 330 DMPs between HR‐SL and NSN (Table S3). Among them, 30.3% (100/330) were hypermethylated, while 69.7% (230/330) were hypomethylated in HR‐SL (Fig. 1A). One of the hypermethylated DMPs, cg24917382, is located within a CpG island of IGF2 promoter, a gene that is part of one of the proposed panels to classify CIMP tumors [42]. The methylation levels for each DMP and the sample clustering profile are shown in the heatmap in Fig. 1B. Regarding functional genomic elements, 39.1% of DMPs were related to CpG islands (CGI, CGI‐shores, and CGI‐shelves) and 26.7% were associated to gene promoter regions including TSS200, TSS1500 (200 bp and 200–1500 bp upstream of the transcription starting site, respectively), 5′UTR, and first exons (Fig. 1C). Hypermethylated DMPs were enriched in intergenic regions, while hypomethylated DMPs were enriched in intergenic regions and CGI‐shelves (one‐sided Fisher's exact test odds‐ratio > 1 and P‐value < 0.05) (Fig. 1D).
Fig. 1.
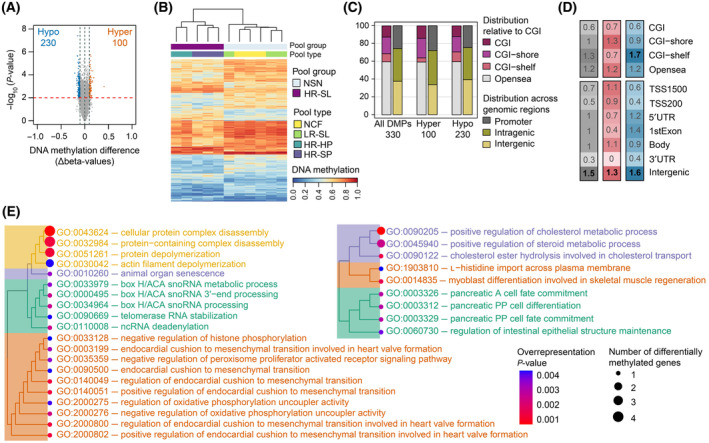
Characterization of the 330 differentially methylated positions (DMPs) between no serrated neoplasia and high‐risk serrated lesions. (A) Volcano plot showing the differential methylation −log10(P‐value) for the 741 739 probes analyzed versus differences in methylation levels (Δbeta: obtained by subtracting the beta‐values of NSN from HR‐SL). Significant hypermethylated (Δbeta > 0.1) and hypomethylated (Δbeta < −0.1) positions appear highlighted in orange or blue, respectively, above the red dashed line (P‐value < 0.01). (B) Hierarchical clustering and heatmap showing the methylation levels across the 13 cfDNA pooled samples for the 330 DMPs. Dendrograms were computed and reordered using Euclidean distance and a complete clustering agglomeration. Methylation levels (beta‐values) range from 0 (blue, unmethylated) to 1 (red, fully methylated). (C) Distribution of the 330 DMPs relative to CGI and functional genomic locations. (D) Enrichment of 330 DMPs in relation to CGI annotation and functional genomic regions. The color scale indicates the fold enrichment of all DMPs (gray), hypermethylated (red), and hypomethylated (blue) positions. The bolded numbers indicate annotations that are enriched with respect to the distribution of probes on the MethylationEPIC array (odds‐ratio > 1 and one‐sided Fisher's exact test P‐value < 0.05). (E) Hierarchical clustering of BP ontology terms based on semantic similarity. Treeplots of the 20 BP terms enriched in the hypermethylated promoter‐associated DMPs (left) and 9 BP terms enriched in the hypomethylated promoter‐associated DMPs. Nodes are colored with respect to the overrepresentation P‐value and sized relatively to the number of differentially methylated genes annotated to each term. CGI (CpG island): region of at least 200 bp with a CG content > 50% and an observed‐to‐expected CpG ratio ≥ 0.6; CGI‐shore: sequences 2 kb flanking the CGI, CGI‐shelf: sequences 2 kb flanking shore regions, opensea: sequences located outside these regions, promoter regions (5′UTR, TSS200, TSS1500, and first exons), intragenic regions (gene body and 3′UTR), and intergenic regions. TSS200, TSS1500: 200 and 200–1500 bp upstream of the transcription start site, respectively. BP, biological process; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; LR‐SL, low‐risk serrated lesion; NCF, no colorectal findings; ncRNA, non‐coding RNA; NSN, no serrated neoplasia; snoRNA, small nucleolar RNA.
To identify biological functions annotated to the DMPs, we computed gene ontology (GO) enrichment analysis on the promoter‐associated hypermethylated and hypomethylated DMPs separately. We identified 25 enriched GO terms associated with the DMPs that were hypermethylated in HR‐SL compared to NSN. Differentially methylated genes overlapping with those GO terms were PARN, TWIST1, COMP, ADD2, MICAL3, MRPL28, NCKAP5, CPLX2, RPEL1, CHST10, and LRRC26. The hypomethylated DMPs reported the enrichment of 12 GO terms, including the genes NEUROD1, WNT10B, CES1, FGF1, SLC7A1, NCEH1, GHR, and MRI1 (Table S4). Within the BP (biological process) ontology, 20 GO terms were enriched in the hypermethylated DMPs, which were related to protein complex depolymerization and disassembly, animal organ senescence, box H/ACA snoRNA processing, and regulation of endocardial cushion to mesenchymal transition. On the other hand, 9 BP terms were enriched in the hypomethylated DMPs, related to the positive regulation of cholesterol metabolic process, l‐histidine import across the membrane, and pancreatic cell fate commitment. Hierarchical clustering of BP terms according to semantic similarity is shown in Fig. 1E.
To test whether there were regional‐specific differences in cfDNA associated with the different lesions belonging to the serrated pathway, significant differentially methylated regions (DMRs) were defined as regions with at least two adjacent CpG sites yielding methylation differences in the same direction, with a family‐wise error rate (FWER) < 10%, and P‐value < 0.01. Altogether, the results from all the comparisons performed rendered a total of 9 DMRs (Fig. 2), some of them resulting significant in more than one comparison (Table 2). Two of these hypomethylated DMRs were identified comparing NSN and HR‐SL: DMR1 annotated to the promoter (TSS200) of the PRRT3 gene, and DMR2 located on the gene body of NINJ2. The largest methylation differences were found in DMR2 and DMR9 for the comparisons NCF vs HR‐HP and HR‐HP vs HR‐SP, respectively.
Fig. 2.
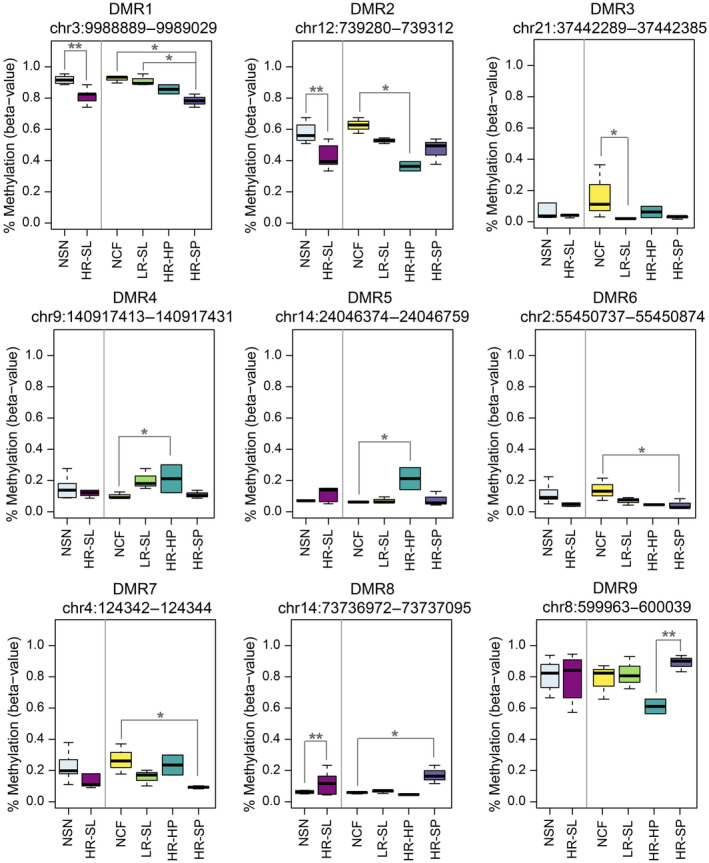
Methylation levels of the nine differentially methylated regions (DMRs) in the cfDNA pooled samples. Methylation levels are shown as beta‐values ranging from 0‐unmethylated to 1‐fully methylated (**Wilcoxon rank‐sum test P‐value < 0.0001; *Wilcoxon rank‐sum test P‐value < 0.001). The boxplot represents the median (bold line across the box), inter‐quartile ranges, and maximum and minimum values (whiskers). DMR, differentially methylated region; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; LR‐SL, low‐risk serrated lesion; NCF, no colorectal findings; NSN, no serrated neoplasia.
Table 2.
Differentially methylated regions. Annotation of the regions to regulatory features and CGI according to the Methylation EPIC Manifest. CGI: CpG island, region of at least 200 bp with a CG content > 50% and an observed‐to‐expected CpG ratio ≥ 0.6; Opensea: sequences located outside CGI regions; Body: gene body (intragenic region); TSS200, TSS1500: 200 and 200–1500 bp upstream the transcription start site, respectively. FWER, family‐wise error rate; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; LR‐SL, low‐risk serrated lesion; NCF, no colorectal findings; NSN, no serrated neoplasia.
| DMR | Location (GRCh37/hg19) | Length (bp) | Gene symbol | Regulatory feature | Relation to CGI | Methylation differences (%) | Methylation status (most severe lesion) | P‐value | FWER |
|---|---|---|---|---|---|---|---|---|---|
| DMR1 | chr3:9988889–9989029 | 141 | PRRT3 | TSS200 | CGI | −10.5% (NSN vs HR‐SL) | Hypomethylated | < 0.0001 | 0.02 |
| −13.9% (NCF vs HR‐SP) | Hypomethylated | 0.00043 | 0.08 | ||||||
| −12.9% (LR‐SL vs HR‐SP) | Hypomethylated | 0.00019 | 0.07 | ||||||
| DMR2 | chr12:739280–739312 | 33 | NINJ2 | Body | Opensea | −14.9% (NSN vs HR‐SL) | Hypomethylated | < 0.0001 | 0.04 |
| −26.2% (NCF vs HR‐HP) | Hypomethylated | 0.00014 | 0.08 | ||||||
| DMR3 | chr21:37442289–37442385 | 97 | CBR1 | 5′UTR | CGI | −14.8% (NCF vs LR‐SL) | Hypomethylated | 0.0001 | 0.05 |
| DMR4 | chr9:140917413–140917431 | 19 | CACNA1B | TSS1500 | CGI | 10.9% (NCF vs HR‐HP) | Hypermethylated | 0.00014 | 0.08 |
| DMR5 | chr14:24046374–24046759 | 386 | JPH4 | 5′UTR | CGI | 15.0% (NCF vs HR‐HP) | Hypermethylated | 0.00014 | 0.08 |
| DMR6 | chr2:55450737–55450874 | 138 | C2orf63 | 5′UTR | CGI | −9.4% (NCF vs HR‐SP) | Hypomethylated | 0.00021 | 0.08 |
| DMR7 | chr4:124342–124344 | 3 | ZNF718 | TSS200 | CGI | −17.7% (NCF vs HR‐SP) | Hypomethylated | 0.00043 | 0.08 |
| DMR8 | chr14:73736972–73737095 | 124 | PAPLN | Body | Opensea | 11.5% (NCF vs HR‐SP) | Hypermethylated | 0.00043 | 0.08 |
| 10.5% (LR‐SL vs HR‐SP) | Hypermethylated | < 0.0001 | 0.07 | ||||||
| DMR9 | chr8:599963–600039 | 77 | ERICH1 | Intergenic | CGI | 27.9% (HR‐HP vs HR‐SP) | Hypermethylated | < 0.0001 | 0.09 |
3.4. Pathway‐specific cfDNA methylome
The concordance between the differential methylation profile identified in serum cfDNA and tissue methylation was assessed. Two external datasets (GSE68060 and E‐MTAB‐7854) [36, 37] were combined to obtain microarray methylation data from serrated CRC (n = 38), SSL (n = 13), and healthy mucosa (n = 16) tissue samples. This exploration was restricted to 188 out of the 330 DMPs targeted by probes shared by the Methylation450k and MethylationEPIC arrays. The principal component analysis (PCA) and heatmap performed on tissue samples revealed that the differential methylation profile found between NSN and HR‐SL cfDNA can clearly discriminate serrated pathological groups in tissue samples (Fig. 3A,B).
Fig. 3.
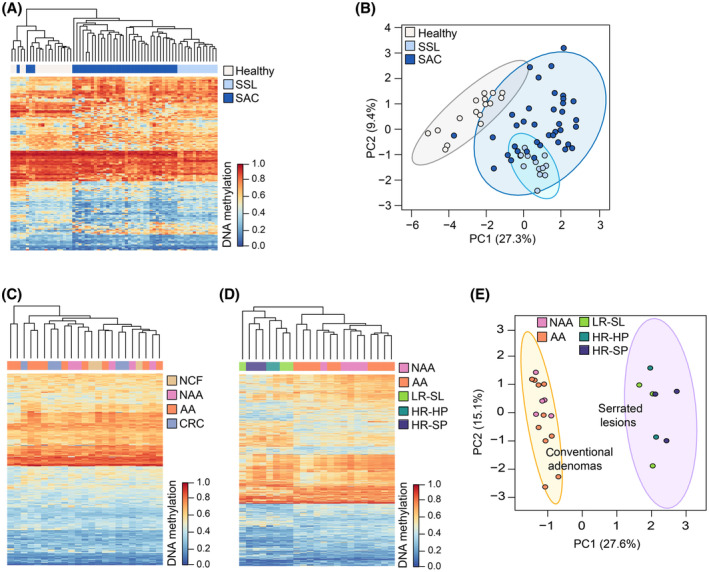
Differences in CRC pathways cfDNA methylation profiles. (A) Heatmap and hierarchical clustering performed on tissue samples from the serrated pathway, based on the 188 DMPs (differentially methylated positions) shared by the 450k and EPIC arrays. (B) PCA performed on tissue samples from the serrated pathway, based on the 188 DMPs shared by the 450k and EPIC arrays. (C) Heatmap and hierarchical clustering showing the methylation levels of the 330 DMPs of the serrated pathway in 23 cfDNA pooled samples from the CRC conventional pathway. (D) Heatmap and hierarchical clustering based on the 1000 most‐variable CpG positions in cfDNA pools. (E) PCA based on the 1000 most‐variable CpG positions in cfDNA pools. For heatmaps, dendrograms were computed and reordered using Euclidean distance and a complete clustering agglomeration and methylation levels are expressed as beta‐values ranging from 0 (blue, unmethylated) to 1 (red, fully methylated). PCA plots show the 95% confidence ellipses. AA, advanced adenomas; CRC, colorectal cancer; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; LR‐SL, low‐risk serrated lesion; NAA, non‐advanced adenoma; NCF, no colorectal findings; PCA, principal component analysis; SSL, sessile serrated lesions.
To check whether the cfDNA methylation profiles are specific to the different CRC carcinogenic pathways, cfDNA methylation data from the serrated pathway were compared with cfDNA methylation data from the conventional CRC pathway (GSE186381). This dataset includes MethylationEPIC data of cfDNA pooled samples of NCF (n = 3), non‐advanced adenomas (n = 5), advanced adenomas (n = 10), and CRC (n = 5) [43]. None of the 330 DMPs from the serrated pathway was differentially methylated in samples from the conventional pathway (10% FDR). Unsupervised clustering and heatmap from Fig. 3C shows no difference in the methylation levels of the 330 DMPs from the serrated pathway and no ability to group advanced neoplasia samples from the conventional pathway. Only the CpG cg08779649 (chr13:50194554) met the criteria of P‐value < 0.01 and at least 10% difference in the methylation levels. This CpG site is annotated to an opensea region located downstream of the CGI chr13:49092410–49092680. cg08779649 is hypermethylated (14.4%) in HR‐SL, while hypomethylation (−13.8%) was observed in advanced neoplasia from the conventional pathway. Altogether these results suggest that the cfDNA differential methylation profile identified is specific to the serrated pathway. The pathway‐specific differential methylation profile in serum can also be observed from early stages of carcinogenesis, as the 1000 most‐variables CpG sites display a different methylation profile between precursor lesions from both pathways (Fig. 3D). PCA analysis based on the most‐variable positions shows a clear separation between conventional adenomas and serrated lesions (Fig. 3E).
3.5. Evaluation of DMRs as potential biomarkers
MS‐qPCR assays were successfully developed for DMR2, DMR7, and DMR9 (Table S2). To explore the utility of the DMRs as biomarkers, their methylation status was evaluated in an independent cohort of 80 individual serum cfDNA samples (Table 1). The methylation levels of the DMRs analyzed did not follow a normal distribution. Methylation levels of DMR7 were skewed toward 0%, while those of DMR2 and DMR9 were skewed toward 100%. DMR2, DMR7, and DMR9 were hypermethylated in HR‐SL compared to NSN, with methylation differences of 2.4%, 5.8%, and 1.6%, respectively, although none of them was statistically significant. Statistically significant differences for DMR2 were found between NCF, LR‐SL, and HR‐HP vs SAC (methylation differences of 18.7%, 18.9%, and 37.9%, respectively) and between HR‐HP and HR‐SP (28.1%); between LR‐SL and HR‐HP for DMR7 (13.49%); and between HR‐SP and SAC for DMR9 (−4.2%) (Fig. 4A). No statistically significant differences were found for any of the DMRs between tumor tissue and matched healthy mucosa.
Fig. 4.
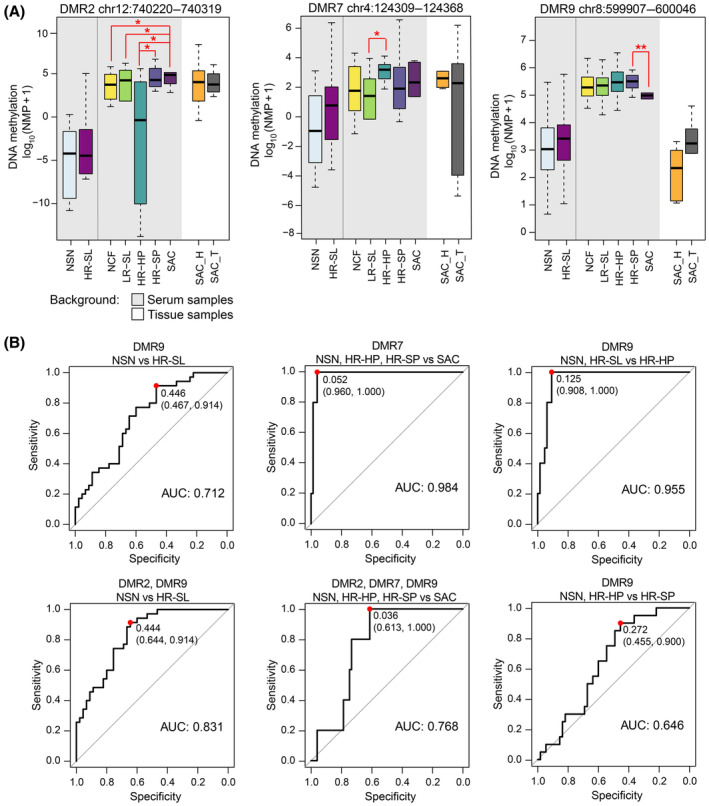
Targeted evaluation of the differentially methylated regions (DMRs) in serum. (A) Methylation levels of the DMRs in individual serum cfDNA samples and matched SAC tumor‐mucosa tissue samples, expressed as log10(NMP + 1) (*Wilcoxon rank‐sum test P‐value < 0.05, **Wilcoxon rank‐sum test P‐value < 0.01). The boxplot represents the median (bold line across the box), inter‐quartile ranges, and maximum and minimum values (whiskers). (B) ROC curve analysis and AUC for the best models obtained with single or combinations of DMRs for the detection of HR‐SL, SAC, HR‐HP, or HR‐SP, derived by leave‐one‐out cross‐validation in the individual serum samples (n = 80). The red dots indicate the sensitivity and specificity values for the best cut‐offs based on the Youden Index method. DMR, differentially methylated region; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; LR‐SL, low‐risk serrated lesion; NCF, no colorectal findings; NSN, no serrated neoplasia; SAC, serrated adenocarcinoma; SAC_H, healthy mucosa from SAC patients; SAC_T, tumor tissue from SAC patients.
Logistic regression models were elaborated for the detection of HR‐SL (joint detection of HR‐HP, HR‐SP, and SAC) and HR‐HP, HR‐SP, and SAC separately. The discriminatory capacity of individual or combined DMRs was assessed by ROC curve analysis and leave‐one‐out cross‐validation (Fig. 4B and Fig. S6). Results from the best‐performing models are summarized in Table 3. The combination of DMR2 and DMR9 discriminated HR‐SL from NSN with 64.4% specificity and 91.4% sensitivity (AUC 0.831, 95% CI: 0.745–0.917), and detected all the SAC cases. DMR7 also discriminated all the patients with SAC (100% sensitivity) from patients with NSN, HR‐HP and HR‐SL (96% specificity) (AUC 0.984, 95% CI: 0.959–1). Regarding models for the discrimination of precancerous lesions, DMR9 showed the best performance for the detection of HR‐HP and HR‐SP, yielding 90.7% specificity and 100% sensitivity (AUC 0.955) for HR‐HP, while HR‐SP were identified with 45.5% specificity and 90% sensitivity (AUC 0.646).
Table 3.
Performance of the DMRs as biomarkers for serrated lesions. Data based on 80 individual serum cfDNA from an independent cohort. The detection rates of HR‐SL and SAC are also shown. No significant differences were found between the detection of distal versus proximal lesions for all models (Fisher's exact test P‐value > 0.05). ROC curves and performance parameters were derived by the leave‐one‐out cross‐validation approach. AUC, area under the curve; CI, confidence interval; HR‐HP, high‐risk hyperplastic polyp; HR‐SL, high‐risk serrated lesion; HR‐SP, high‐risk serrated polyp; NSN, no serrated neoplasia; SAC, serrated adenocarcinoma.
| Model performance | Detection rates (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUC (95% CI) | Specificity % (95% CI) | Sensitivity % (95% CI) | HR‐HP distal | HR‐HP proximal | All | HR‐SP distal | HR‐SP proximal | All | SAC | |
| NSN vs HR‐SL | ||||||||||
| DMR9 | 0.712 (0.601–0.825) | 46.7 (32–62) | 91.43 (77–98) | 100 | 60 | 80 | 100 | 91.7 | 95 | 100 |
| DMR2, DMR9 | 0.831 (0.745–0.917) | 64.4 (49–78) | 91.4 (77–98) | 100 | 80 | 90 | 100 | 83.3 | 90 | 100 |
| NSN, HR‐HP, HR‐SL vs SAC | ||||||||||
| DMR7 | 0.984 (0.959–1) | 96 (89–99) | 100 (48–100) | – | – | – | – | – | – | 100 |
| DMR2, DMR7, DMR9 | 0.768 (0.633–0.903) | 61.3 (49–72) | 100 (48–100) | – | – | – | – | – | – | 100 |
| NSN, HR‐SP vs HR‐HP | ||||||||||
| DMR9 | 0.955 (0.912–0.999) | 90.7 (81–97) | 100 (69–100) | 100 | 100 | 100 | – | – | – | – |
| NSN, HR‐HP vs HR‐SP | ||||||||||
| DMR9 | 0.646 (0.520–0.773) | 45.5 (32–59) | 90 (68–99) | – | – | – | 100 | 83.3 | 90 | – |
4. Discussion
Although CRC represents the classical model of epithelial neoplasm development through the so‐called adenoma‐to‐carcinoma sequence [44], it is now well established that there are many molecular mechanisms driving CRC development [45]. The serrated neoplasia pathway accounts for 15–30% of CRC cases [2, 3, 4], with SAC having a worse prognosis compared to conventional CRC due to weak immune response, high tumor budding and an infiltrative tumor pattern [46, 47].
The early detection of CRC and precancerous lesions is determinant for the success of screening programs. Several biological characteristics of the serrated lesions have clinical relevance for the screening and colonoscopic surveillance of SAC [6]. Thus, an accurate early detection of serrated lesions could reduce both the incidence of SAC and the serrated‐lesion related interval CRC. To the best of our knowledge, this study explored for the first time the serum methylome in patients with precursor lesions from the serrated pathway, aiming its characterization and evaluation of cfDNA methylation as a potential source of biomarkers for the non‐invasive screening and diagnosis of the serrated pathway.
First, we conducted an epigenome‐wide methylation analysis of serum cfDNA, combining the MethylationEPIC array with a sample pooling approach. We identified a cfDNA differential methylation profile between HR‐SL (large HP, large SSL, SSL with dysplasia, and TSA) and NSN (individuals with no colorectal findings, small HP, and small non‐dysplastic SSL and SP), reporting 330 DMPs of which 39.1% are associated to CpG islands. All the different serrated lesions included were pairwise compared to obtain 9 significant DMRs, of which 7 were located within the CpG islands of the genes PRRT3, CBR1, CACNA1B, JPH4, C2orf63, ZNF718, and ERICH1.
Several high‐throughput methylation analyses of tissue samples have identified the gradual accumulation of methylation changes at all steps of the progression of the serrated pathway [10, 13, 37, 48], reporting significant hypomethylation even at higher frequency than hypermethylation. This is consistent with the differential methylation profile we report in serum cfDNA, where 69.7% of the DMPs were hypomethylated in HR‐SL and DMRs were mostly hypomethylated in the most‐severe lesion. The serum cfDNA methylation profile was explored in tissue samples from healthy mucosa, SSL and SAC, combining external methylation microarray data [36, 37]. The differential methylation pattern found between HR‐SL and NSN cfDNA can also discriminate serrated pathological groups in tissue samples. Though this verification is limited, some degree of concordance between serum and tissue methylation can be observed. It is worth mentioning that discrepancy in the frequencies of methylation alterations in tumor tissue and cfDNA has been reported, showing the latter considerably lower frequencies [49].
Different oncogene mutation and expression profiles, MSI status, methylome signatures, and epigenetic regulation have been observed between SAC and conventional and sporadic CRC tissue samples [36, 50, 51]. Here we evidenced that the serum differential methylation profile is also pathway‐specific, as the DMPs identified for HR‐SL are unable to distinguish conventional colorectal neoplasia in cfDNA pooled samples. Moreover, when pooled cfDNA samples from both pathways are merged, the most‐variable CpG sites exhibit different methylation levels in conventional compared to serrated precursor lesions, suggesting that pathway‐specific serum methylation profiles can already be detected from early stages of carcinogenesis.
We also made a preliminary evaluation of three DMRs as non‐invasive biomarkers for the detection of HR‐SL and SAC in an independent cohort of individual serum samples. The methylation levels were quantified in serum and tissue samples, targeting DMR2 (NINJ2 gene body, chr12:740220–740319), DMR7 (ZNF718 CpG island, chr4:124309–124368), and DMR9 (ERICH1 CpG island, chr8:599907–600046). Logistic regression models were cross‐validated to derive classification rules for the detection of serrated lesions in serum. The combination of DMR2 and DMR9 detected HR‐SL (HR‐HP, HR‐SP, and SAC) with 91.4% sensitivity and 64.4% specificity, and reported detection rates of 90%, 90%, and 100%, for HR‐HP, HR‐SP, and SAC, respectively. For the discrimination of SAC from all the other lesions, DMR7 showed 100% sensitivity and 96% specificity. We also explored the ability of the DMRs to specifically detect precursor lesions. DMR9 showed the best performance for the independent detection of HR‐HP and HR‐SL, reporting sensitivities of 100% and 90% for HR‐HP and HR‐SP, respectively, at 90.7% and 45.5% specificity. No statistically significant differences were found for any of the DMRs between tumor tissue and matched healthy mucosa, probably due to the small sample size (n = 6).
From the DMRs studied, only NINJ2 gene has been previously related to CRC development. NINJ2 (encode ninjurin2) can activate NF‐κB and c‐jun pathways through interacting with TLR4, therefore promoting cell proliferation and survival [52], and its overexpression, both in vivo and in vitro, promotes CRC cells proliferation [53]. Although methylation levels of NINJ2 gene body are not directly related to gene expression regulation, the aforementioned studies suggest a possible oncogenic role of NINJ2 in CRC.
The morphology of serrated lesions makes them less prone to bleed, which limits the sensitivity of FIT for detecting SSL regardless of their size and grade of dysplasia [17, 18, 19, 54]. The sensitivities showed by our DMRs are superior to that of FIT for the detection of serrated lesions, which ranges from 6.2% to 20.4% for SSL at 87.4% to 96.8% specificity [17, 18], and shows 7.4% sensitivity for HR‐SL when the specificity is fixed to 95% [19]. Although most screening programs are based on FIT, the aforementioned studies suggest its limited value to detect serrated lesions, resulting ineffective to prevent interval cancers arising from these lesions.
A few studies have evaluated the performance of DNA methylation non‐invasive tests for the detection of precursory serrated lesions, such as the FDA‐approved methylated‐SEPT9 plasma test or the multitarget fecal test. In an opportunistic screening study, the plasma SEPT9 assay showed a sensitivity of 27.8% for the detection of serrated polyps, 21.3% for HP, and 40.9% for conventional adenomas, at 78.4% specificity [55]. The FDA‐approved multitarget fecal test detected serrated lesions with 40.7–42.4% sensitivity and 86.6–89.1% specificity [19, 54]. Another study evaluating methylation of BCAT1/IKZF1 in plasma reported a sensitivity of 8.8% for the detection of SSL at 93% specificity [18].
It is worth mentioning that our multicenter cohort includes the whole pathological range of the serrated pathway, from small serrated and hyperplastic polyps, to TSA and large and dysplastic SSL, as well as SAC and colonoscopically confirmed healthy controls. The sample size is justified by the low prevalence of serrated lesions, with colonoscopy detection rates of 1.8% for large HP, 0.8–4.6% for SSL, 0.8–1.6% for large or dysplastic SSL, and 0.2–4.4% for TSA [56, 57]. In relation to CRC, its prevalence in colonoscopic studies ranges from 0.28% to 0.42% for all types of CRC [58, 59]. As the serrated pathway accounts for up to 30% of CRC, the small number of SAC serum samples available for our study was expected. This precluded the construction of cfDNA pooled samples from SAC cases, that therefore were retained for the biomarker evaluation phase.
5. Conclusions
As far as we are concerned, this is the only study available to date exploring the serum methylome of precursory lesions of the serrated pathway. We have reported a differential methylation profile that can distinguish HR‐SL from NSN, showing concordance with tissue methylation from different external datasets. The methylation profiles in serum cfDNA are pathway‐specific, and may serve as a source of non‐invasive biomarkers for the detection of HR‐SL and SAC in screening programs.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
LDC conceived and designed the study. LDC, JC, and ME supervised the study. MG‐G, LC‐R, LDC, LA‐R, and CAG‐P contributed to the sample preparation and data acquisition. MG‐G and LDC performed the analysis and interpretation of data. MG‐G, LC‐R and LDC contributed to the experimental design. JC, LB, MB, AC, FB, RJ, ATB, and JG‐CF provided clinical advice for the study design, collection, and management of clinical data. MG‐G and LDC prepared the manuscript. All authors critically reviewed and approved the final manuscript.
Peer review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1002/1878‐0261.13573.
Supporting information
Fig. S1. Global methylation levels of the cfDNA pooled samples.
Fig. S2. Results of the differential methylation analysis at probe level.
Fig. S3. Distribution of the DMPs obtained from all the pairwise comparisons, relative to CGI and functional genomic locations.
Fig. S4. Enrichment of DMPs obtained from all the pairwise comparisons, in relation to CGI annotation and functional genomic regions.
Fig. S5. Manhattan plots of differential methylation.
Fig. S6. ROC curve analysis and AUC.
Table S1. Description of cfDNA pooled samples.
Table S2. Primers, probes, and amplicon details for the evaluation of the DMRs in individual serum samples.
Table S3. List of the 330 differentially methylated positions (DMPs) between high‐risk serrated lesions (HR‐SL) and no serrated neoplasia (NSN) cfDNA pooled samples.
Table S4. List of significantly enriched gene ontology (GO) terms in the hypermethylated and hypomethylated DMPs between high‐risk serrated lesions (HRSL) and no serrated neoplasia (NSN).
Acknowledgements
This work received funding from Plan Nacional I + D + I 2015‐2018 (Acción Estratégica en Salud) Instituto de Salud Carlos III (Spain)‐FEDER (PI15/02007), “Fundación Científica de la Asociación Española contra el Cáncer” (GCB13131592CAST), and support from Centro Singular de Investigación de Galicia (Consellería de Cultura, Educación e Ordenación Universitaria) (ED431G/02, Xunta de Galicia and FEDER‐European Union). María Gallardo‐Gómez was supported by a predoctoral fellowship from Ministerio de Educación, Cultura y Deporte (Spanish Government) (FPU15/02350). We would like to thank Dr Martínez Zorzano, and Professors Rodríguez Berrocal and Páez de la Cadena for their support and scientific advice. The samples and clinical data of patients included in this study were provided by: the HGUA Biobank (PT13/0010/0044), integrated into the Red Nacional de Biobancos and in the Red Valenciana de Biobancos; Basque Biobank/Biodonostia Node; Biobank at the Galicia Sur Health Research Institute; and Biobank of Hospital Clínic, Barcelona – IDIBAPS, with the approval of the respective Ethical and Scientific Committees, and have been processed following standard procedures. We acknowledge CESGA (Fundación Pública Galega Centro Tecnolóxico de Supercomputación de Galicia) for providing access to computing facilities to analyze methylation microarray data. Figure color palettes were adapted from Blake Robert Mills (2022): MetBrewer: Color Palettes Inspired by Works at the Metropolitan Museum of Art. R package version 0.1.0.
Data accessibility
The Infinium MethylationEPIC data from all the pooled samples generated and analyzed during this study have been deposited in the NCBI Gene Expression Omnibus (GEO) (www.ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE199173.
References
- 1. Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology. 2020;158(2):291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh R, Tao Pu LZC, Koay D, Burt A. Sessile serrated adenoma/polyps: where are we at in 2016? World J Gastroenterol. 2016;22(34):7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. East JE, Atkin WS, Bateman AC, Clark SK, Dolwani S, Ket SN, et al. British Society of Gastroenterology position statement on serrated polyps in the colon and rectum. Gut. 2017;66(7):1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pai RK, Bettington M, Srivastava A, Rosty C. An update on the morphology and molecular pathology of serrated colorectal polyps and associated carcinomas. Mod Pathol. 2019;32(10):1390–1415. [DOI] [PubMed] [Google Scholar]
- 5. Trovato A, Turshudzhyan A, Tadros M. Serrated lesions: a challenging enemy. World J Gastroenterol. 2021;27(34):5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology. 2020;76(2):182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim SY, Kim TI. Serrated neoplasia pathway as an alternative route of colorectal cancer carcinogenesis. Intest Res. 2018;16(3):358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murakami T, Sakamoto N, Nagahara A. Clinicopathological features, diagnosis, and treatment of sessile serrated adenoma/polyp with dysplasia/carcinoma. J Gastroenterol Hepatol. 2019;34(10):1685–1695. [DOI] [PubMed] [Google Scholar]
- 9. McCarthy AJ, Serra S, Chetty R. Traditional serrated adenoma: an overview of pathology and emphasis on molecular pathogenesis. BMJ Open Gastroenterol. 2019;6(1):e000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dehghanizadeh S, Khoddami V, Mosbruger TL, Hammoud SS, Edes K, Berry TS, et al. Active BRAF‐V600E is the key player in generation of a sessile serrated polyp‐specific DNA methylation profile. PLoS One. 2018;13(3):e0192499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tornillo L, Lehmann FS, Garofoli A, Paradiso V, Ng CKY, Piscuoglio S. The genomic landscape of serrated lesion of the colorectum: similarities and differences with tubular and tubulovillous adenomas. Front Oncol. 2021;11:668466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Advani SM, Advani P, DeSantis SM, Brown D, VonVille HM, Lam M, et al. Clinical, pathological, and molecular characteristics of CpG island methylator phenotype in colorectal cancer: a systematic review and meta‐analysis. Transl Oncol. 2018;11(5):1188–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parker HR, Orjuela S, Martinho Oliveira A, Cereatti F, Sauter M, Heinrich H, et al. The proto CpG island methylator phenotype of sessile serrated adenomas/polyps. Epigenetics. 2018;13(10–11):1088–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang M, Ou J, Hutchinson L, Green MR. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG island methylator phenotype. Mol Cell. 2014;55(6):904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li D, Doherty AR, Raju M, Liu L, Lei NY, Amsden LB, et al. Risk stratification for colorectal cancer in individuals with subtypes of serrated polyps. Gut. 2022;71(10):2022–2029. [DOI] [PubMed] [Google Scholar]
- 16. Burgess NG, Tutticci NJ, Pellise M, Bourke MJ. Sessile serrated adenomas/polyps with cytologic dysplasia: a triple threat for interval cancer. Gastrointest Endosc. 2014;80(2):307–310. [DOI] [PubMed] [Google Scholar]
- 17. Chang L‐C, Shun C‐T, Hsu W‐F, Tu C‐H, Tsai P‐Y, Lin B‐R, et al. Fecal immunochemical test detects sessile serrated adenomas and polyps with a low level of sensitivity. Clin Gastroenterol Hepatol. 2017;15(6):872–879.e1. [DOI] [PubMed] [Google Scholar]
- 18. Cock C, Anwar S, Byrne SE, Meng R, Pedersen S, Fraser RJL, et al. Low sensitivity of fecal immunochemical tests and blood‐based markers of DNA hypermethylation for detection of sessile serrated adenomas/polyps. Dig Dis Sci. 2019;64(9):2555–2562. [DOI] [PubMed] [Google Scholar]
- 19. Bosch LJW, Melotte V, Mongera S, Daenen KLJ, Coupé VMH, van Turenhout ST, et al. Multitarget stool DNA test performance in an average‐risk colorectal cancer screening population. Am J Gastroenterol. 2019;114(12):1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bibbins‐Domingo K, Grossman DC, Curry SJ, Davidson KW, Epling JW, García FAR, et al. Screening for colorectal cancer. JAMA. 2016;315(23):2564–2575. [DOI] [PubMed] [Google Scholar]
- 21. Davidson KW, Barry MJ, Mangione CM, Cabana M, Caughey AB, Davis EM, et al. Screening for colorectal cancer. JAMA. 2021;325(19):1965–1977. [DOI] [PubMed] [Google Scholar]
- 22. Jover R, Herráiz M, Alarcón O, Brullet E, Bujanda L, Bustamante M, et al. Clinical practice guidelines: quality of colonoscopy in colorectal cancer screening. Endoscopy. 2012;44(4):444–451. [DOI] [PubMed] [Google Scholar]
- 23. Mangas‐Sanjuan C, Jover R, Cubiella J, Marzo‐Castillejo M, Balaguer F, Bessa X, et al. Endoscopic surveillance after colonic polyps and colorrectal cancer resection. 2018 Update. Gastroenterol Hepatol. 2019;42(3):188–201. [DOI] [PubMed] [Google Scholar]
- 24. Hufnagl C, Stöcher M, Molk M, Geisberger R, Greil R. A modified phenol‐chloroform extraction method for isolating circulating cell free DNA of tumor patients. J Nucleic Acids Investig. 2013;4:e1. [Google Scholar]
- 25. Gallardo‐Gómez M, Moran S, Páez de la Cadena M, Martínez‐Zorzano VS, Rodríguez‐Berrocal FJ, Rodríguez‐Girondo M, et al. A new approach to epigenome‐wide discovery of non‐invasive methylation biomarkers for colorectal cancer screening in circulating cell‐free DNA using pooled samples. Clin Epigenetics. 2018;10(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fortin J‐P, Triche T, Hansen K. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33(4):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina methylationEPIC BeadChip microarray for whole‐genome DNA methylation profiling. Genome Biol. 2016;17:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mansell G, Gorrie‐Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 2019;20(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Du P, Zhang X, Huang C‐C, Jafari N, Kibbe WA, Hou L, et al. Comparison of beta‐value and M‐value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11(1):587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41(1):200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aryee MJ, Jaffe AE, Corrada‐Bravo H, Ladd‐Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shaffer JP. Multiple hypothesis testing. Annu Rev Psychol. 1995;46:561–584. [Google Scholar]
- 35. Gallardo‐Gómez M, Rodríguez‐Girondo M, Planell N, Moran S, Bujanda L, Etxart A, et al. Serum methylation of GALNT9, UPF3A, WARS, and LDB2 as noninvasive biomarkers for the early detection of colorectal cancer and advanced adenomas. Clin Epigenetics. 2023;15(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conesa‐Zamora P, García‐Solano J, Turpin MC, Sebastián‐León P, Torres‐Moreno D, Estrada E, et al. Methylome profiling reveals functions and genes which are differentially methylated in serrated compared to conventional colorectal carcinoma. Clin Epigenetics. 2015;7(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu C, Fennell LJ, Bettington ML, Walker NI, Dwine J, Leggett BA, et al. DNA methylation changes that precede onset of dysplasia in advanced sessile serrated adenomas. Clin Epigenetics. 2019;11(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics. 2016;32(2):286–288. [DOI] [PubMed] [Google Scholar]
- 39. Yu G. enrichplot: Visualization of Functional Enrichment Result. 2022.
- 40. Pesquita C, Faria D, Falcão AO, Lord P, Couto FM. Semantic similarity in biomedical ontologies. PLoS Comput Biol. 2009;5(7):e1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3(1):32–35. [DOI] [PubMed] [Google Scholar]
- 42. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. [DOI] [PubMed] [Google Scholar]
- 43. Gallardo‐Gómez M, Rodríguez‐Girondo M, Planell N, Moran S, Bujanda L, Etxart A, et al. Serum methylation of GALNT9, UPF3A, WARS, and LDB2 as non‐invasive biomarkers for the early detection of colorectal cancer and premalignant adenomas. medRxiv. 2022. 10.1101/2021.11.11.21266182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. [DOI] [PubMed] [Google Scholar]
- 45. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. García‐Solano J, Conesa‐Zamora P, Trujillo‐Santos J, Mäkinen MJ, Pérez‐Guillermo M. Tumour budding and other prognostic pathological features at invasive margins in serrated colorectal adenocarcinoma: a comparative study with conventional carcinoma. Histopathology. 2011;59(6):1046–1056. [DOI] [PubMed] [Google Scholar]
- 47. Phipps AI, Limburg PJ, Baron JA, Burnett‐Hartman AN, Weisenberger DJ, Laird PW, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148(1):77–87.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andrew AS, Baron JA, Butterly LF, Suriawinata AA, Tsongalis GJ, Robinson CM, et al. Hyper‐methylated loci persisting from sessile serrated polyps to serrated cancers. Int J Mol Sci. 2017;18(3):535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jung K, Fleischhacker M, Rabien A. Cell‐free DNA in the blood as a solid tumor biomarker – a critical appraisal of the literature. Clin Chim Acta. 2010;411(21–22):1611–1624. [DOI] [PubMed] [Google Scholar]
- 50. García‐Solano J, Conesa‐Zamora P, Carbonell P, Trujillo‐Santos J, Torres‐Moreno DD, Pagán‐Gómez I, et al. Colorectal serrated adenocarcinoma shows a different profile of oncogene mutations, MSI status and DNA repair protein expression compared to conventional and sporadic MSI‐H carcinomas. Int J Cancer. 2012;131(8):1790–1799. [DOI] [PubMed] [Google Scholar]
- 51. Conesa‐Zamora P, García‐Solano J, García‐García F, Turpin MC, Trujillo‐Santos J, Torres‐Moreno D, et al. Expression profiling shows differential molecular pathways and provides potential new diagnostic biomarkers for colorectal serrated adenocarcinoma. Int J Cancer. 2013;132(2):297–307. [DOI] [PubMed] [Google Scholar]
- 52. Wang J, Fa J, Wang P, Jia X, Peng H, Chen J, et al. NINJ2 – a novel regulator of endothelial inflammation and activation. Cell Signal. 2017;35:231–241. [DOI] [PubMed] [Google Scholar]
- 53. Li G, Zhou L‐N, Yang H, He X, Duan Y, Wu F. Ninjurin 2 overexpression promotes human colorectal cancer cell growth in vitro and in vivo. Aging (Albany NY). 2019;11(19):8526–8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget stool DNA testing for colorectal‐cancer screening. N Engl J Med. 2014;370(14):1287–1297. [DOI] [PubMed] [Google Scholar]
- 55. Song L, Peng X, Li Y, Xiao W, Jia J, Dong C, et al. The SEPT9 gene methylation assay is capable of detecting colorectal adenoma in opportunistic screening. Epigenomics. 2017;9(5):599–610. [DOI] [PubMed] [Google Scholar]
- 56. Meester RGS, van Herk MMAGC, Lansdorp‐Vogelaar I, Ladabaum U. Prevalence and clinical features of sessile serrated polyps: a systematic review. Gastroenterology. 2020;159(1):105–118.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Desai M, Anderson JC, Kaminski M, Thoguluva Chandrasekar V, Fathallah J, Hassan C, et al. Sessile serrated lesion detection rates during average risk screening colonoscopy: a systematic review and meta‐analysis of the published literature. Endosc Int Open. 2021;9(4):E610–E620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wernly S, Wernly B, Semmler G, Bachmayer S, Niederseer D, Stickel F, et al. A sex‐specific propensity‐adjusted analysis of colonic adenoma detection rates in a screening cohort. Sci Rep. 2021;11(1):17785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ding H, Lin J, Xu Z, Chen X, Wang HHX, Huang L, et al. A global evaluation of the performance indicators of colorectal cancer screening with fecal immunochemical tests and colonoscopy: a systematic review and meta‐analysis. Cancers (Basel). 2022;14(4):1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Global methylation levels of the cfDNA pooled samples.
Fig. S2. Results of the differential methylation analysis at probe level.
Fig. S3. Distribution of the DMPs obtained from all the pairwise comparisons, relative to CGI and functional genomic locations.
Fig. S4. Enrichment of DMPs obtained from all the pairwise comparisons, in relation to CGI annotation and functional genomic regions.
Fig. S5. Manhattan plots of differential methylation.
Fig. S6. ROC curve analysis and AUC.
Table S1. Description of cfDNA pooled samples.
Table S2. Primers, probes, and amplicon details for the evaluation of the DMRs in individual serum samples.
Table S3. List of the 330 differentially methylated positions (DMPs) between high‐risk serrated lesions (HR‐SL) and no serrated neoplasia (NSN) cfDNA pooled samples.
Table S4. List of significantly enriched gene ontology (GO) terms in the hypermethylated and hypomethylated DMPs between high‐risk serrated lesions (HRSL) and no serrated neoplasia (NSN).
Data Availability Statement
The Infinium MethylationEPIC data from all the pooled samples generated and analyzed during this study have been deposited in the NCBI Gene Expression Omnibus (GEO) (www.ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE199173.