

Abstract
In this study, a set of spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives 3a–p were synthesized starting from unsubstituted and N-methyl-substituted diendo- and diexo-2-aminonorbornene carboxamides, as well as various substituted isatins. The typical method involves a condensation reaction of alicyclic aminocarboxamide and isatin in the presence of a catalyst, using a solvent and an acceptable temperature. We developed a cost-effective and ecologically benign high-speed ball milling (HSBM), microwave irradiation (MW), and continuous flow (CF) technique to synthesize spiroquinazolinone molecule 3a. The structures of the synthesized compounds 3a–p were determined using 1D and 2D NMR spectroscopies. Furthermore, docking studies and absorption, distribution, metabolism, and toxicity (ADMET) predictions were used in this work. In agreement with the corresponding features found in the case of both the SARS-CoV-2 main protease (RCSB Protein Data Bank: 6LU7) and human mast cell tryptase (RCSB Protein Data Bank: 2ZA5) based on the estimated total energy and binding affinity, H bonds, and hydrophobicity in silico, compound 3d among our 3a–g, 3i–k, and 3m derivatives was found to be our top-rated compound.
Keywords: 2-aminonorbornene carboxamide, isatin, click reaction, HSBM, MW, docking studies
1. Introduction
Because of their biological properties, oxindole and spirooxindole derivatives are highly sought after in pharmaceutical research [1,2,3,4,5]. Oxindole derivatives were generated via the aldol reaction of isatin aldehydes [6] and ketones [7,8] by reacting isatin with aromatics in triflic acid [9,10] or isatins with barbituric acid [11]. Spirooxindole compounds were synthesized in the condensation reaction of 2-aminobenzamide and isatins [12,13,14,15,16,17,18]. Another technique to synthesize spirooxindoles is to react isatoic anhydride with ammonium acetate or amines, as well as aldehydes or ketones [19,20]. In addition, other procedures for synthesizing 2,3-dihydroquinazolin-4(1H)-one derivatives have been reported in the literature, including the spirocyclization of aldehydes or ketones with 2-aminobenzamide [12,16]. Several catalysts, including Amberlyst 15, CuCl2, ZrCl4, NH4Cl, KAl(SO4)2∙12H2O (alum), and phosphoric acid derivatives, were evaluated to improve yield in the reaction of isatin and 2-aminobenzamide. Overall, acidic catalysts such as sulfamic acid, phosphotungstic acid, or p-TSA were employed with loadings ranging from 10 to 30 mol% [12,15,16,17,18,21,22,23,24,25,26,27,28]. Several substituted isatins were studied as reaction partners in this condensation reaction, yielding the corresponding racemic spiro products [12,15,16,17,18,21,22,23,24,25,26,27,28]. In general, EtOH and ACN were used as solvents. In addition, there are examples of the use of toluene and glycerol, or applying solvent-free conditions at room temperature under reflux conditions or at −45 °C [12,15,16,17,18,21,22,23,24,25,26,27]. Several approaches, including chiral catalysts (chiral phosphoric acid derivatives), green solvents (acetonitrile, acetic acid, EtOH, deep eutectic solvent mixtures), and varied temperatures, were tested to produce racemic and enantiomeric spirooxindoles [12,15,18,21,22,23,24,25,27,28,29,30,31]. Furthermore, Bergman et al. obtained a racemate of 1′H-spiro[indoline-3,2′-quinazoline]-2,4′(3′H)-dione with a yield of 88%, and the enantiomer separation was noticeable using supercritical fluid chromatography (SFC). They separated the enantiomers and determined their optical rotation, but rapid racemization of the enantiomers were recognized [32].
Some greener approaches were tested in the case of the reaction of 2-amino-benzamide and isatins, such as using gentler conditions (room temperature), ultrasonic irradiation, microwave irradiation (MW), and the use of ionic liquids and deep eutectic solvents [17,24,27]. Shabaani et al. reported a straightforward technique for synthesizing the 2,3-dihydroquinazolin-4(1H)-one derivative. NH4Cl was used as a catalyst at room temperature in EtOH in a stirred solution of 2-aminobenzamide and isatin [17]. Pal et al. used ultrasonic irradiation and Amberlyst 15 as catalyst to produce spiro-2,3-dihydroquinazolin-4(1H)-ones at room temperature in a few minutes [24]. Nagarajan et al. described a green approach for synthesizing quinazolinone derivatives by cyclization with aldehydes or ketones mediated by deep eutectic solvent (DES) in good to excellent yields (62–95%). They investigated various deep eutectic solvent mixtures and discovered that the molten mixture of L-(+)-tartaric acid and N,N′-dimethylurea (DMU) (3:7) at 90 °C was the most effective in producing the highest yield of spirodihydroquinazolin product [27].
Continuous flow (CF) has become a key instrument for synthetic and medicinal chemistry. CF is a rapid and easy method for chemical synthesis, and the use of reagents, solvents, and waste can be reduced with these techniques. The most critical parameters, such as flow rate, residence time, pressure, temperature, and stoichiometry, can be simply adjusted or changed [33]. In the literature, few examples can be found for the synthesis of spirooxindole-containing compounds in CF [34,35].
Energy-efficient techniques such as MW irradiation, ultrasound, and ionic liquids are well-defined. High-speed ball milling (HSBM), in turn, has received little attention in organic synthesis [36]. Purifying and disposing of dangerous toxic solvents require a significant amount of energy and consequently, they are not cost-effective. HSBM requires no or minimal solvent during the milling process. Here, mechanical energy is conveyed inside the milling jar, where solid reactants are combined [37]. There are many factors to consider, including milling time, the number and size of balls, weight, vessel radius, and the heat capacity of compounds [38].
Ten years ago, co-workers of our department used environmentally benign ways to create 2,2-disubstituted- and 2-spiroquinazolinone derivatives in either aqueous or solventless media reacting anthranilamide and a number of ketones with poor water solubility. The ring closure reaction was carried out under HSBM circumstances, with molecular iodine as catalyst [39]. However, no studies have been conducted on the analogous reaction involving alicyclic β-amino amides and ketones under HSBM conditions.
While this area has received significant attention, discovering environmentally friendly synthesis pathways to generate oxindole derivatives, such as tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione derivatives or comparable compounds, remains a difficult task. Further research is needed to optimize the synthesis of these derivatives using spirocondensation processes, including purifying procedures as well, in order to increase yield and improve product selectivity. Furthermore, there is still a need for innovation in obtaining spiroquinazolinone derivatives using greener methodologies. Our primary goal was to synthesize novel spiroquinazolinone derivatives while simultaneously optimizing the synthesis method. We intended to investigate the reaction between racemic alicyclic diexo- or diendo-β-amino amides and isatins using more ecologically friendly approaches. Another aim was to perform an in silico study on the molecular docking of our novel compounds to two different macromolecules in order to help design presumably pharmaceutically useful molecules. Quinazolinone and spiroquinazoline derivatives possess a wide range of biological activities, such as anti-inflammatory, anticonvulsant, antiallergic, antibacterial, antihistamine, antitubercolosis, antimutagenic, and respiratory features [40,41,42]. Spiroquinazoline derivatives are ligands for several macromolecules, for example, serine/threonine kinase, tubulin, plant hormone, transferase, and hydrolase [43,44,45,46,47,48,49]. The binding of quinazolinone derivatives was also examined through docking studies to various targets [24,40,41,50,51,52]. In this study, complementing the synthesis of 3a–3p, we utilized molecular docking to identify the ligands with promising binding potential to two relevant biological targets, a main protease of SARS-CoV-2 and human mast cell tryptase.
2. Results and Discussion
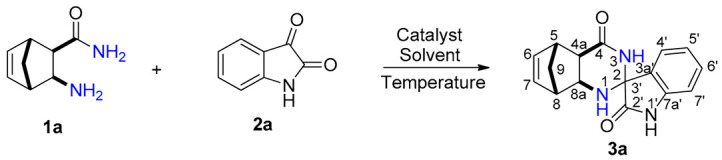
The syntheses of starting amides were performed according to the literature procedure. The diendo- and diexo-amino acids were esterified with ethanol and the ester bases, liberated from the hydrochlorides, were treated with methanolic ammonia or methyl amine to furnish amides 1a–d [53,54]. We initially studied the influence of the solvent, catalyst, and temperature in the spirocondensation reaction of unsubstituted diexo-amide 1a with unsubstituted isatin 2a as a standard and model to produce 4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3a). Different catalysts (30 mol% in each case), such as NH4Cl, KAl(SO4)2·12H2O (alum), LiOH, p-TsOH, Amberlyst 15, and I2, were tested at ambient temperature and under heating in different solvents. EtOH, glycerol, 2M2B (2-methyl-2-butanol), and water were tested as green solvents (Table 1).
Table 1.
Synthesis of 4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3a).
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Time (h) | Yield a (%) |
| 1 | NH4Cl | EtOH | Rt b | 24 | 29 c |
| 2 | NH4Cl | EtOH | 78 | 12 | 37 c |
| 3 | NH4Cl | 2M2B d | 100 | 9 | 35 |
| 4 | LiOH | EtOH | Rt b | 72 | 10 |
| 5 | LiOH | EtOH | 78 | 72 | 21 |
| 6 | p-TsOH | EtOH | Rt b | 168 | – |
| 7 | p-TsOH | EtOH | 78 | 168 | – |
| 8 | Amberlyst 15 | EtOH | Rt b | 120 | 20 c |
| 9 | Amberlyst 15 | EtOH | 78 | 10 | 25 c |
| 10 | I2 | EtOH | Rt b | 215 | – |
| 11 | I2 | EtOH | 78 | 14 | 35 |
| 12 | Alum | Glycerol | Rt b | 168 | – |
| 13 | Alum | Glycerol | 100 | 5 | – |
| 14 | Alum | 2M2B d | Rt b | 72 | – |
| 15 | Alum | 2M2B d | 100 | 168 | – |
| 16 | Alum | EtOH | 78 | 5 | 42 |
| 17 | Alum | Water | rt | 168 | – e |
| 18 | Alum | Water | 100 | 8 | – e |
a Isolated yield after purification by flash chromatography. b Room temperature. c The reaction was not selective, since the product contained the mixture of two diastereomers according to the 1H-NMR spectrum. d 2M2B: 2-methyl-2-butanol. e The starting materials did not dissolve in water and there was no transformation.
Screening several catalysts, solvents, and temperatures in the reaction between 1a and 2a showed that the most effective ones were NH4Cl in 2M2B at 100 °C (after 9 h), iodine (14 h), and alum in EtOH at 78 °C (within 5 h), which afforded the product selectively (Table 1 entries 3, 11, and 16). It was necessary to heat the reaction mixture, otherwise the reaction was not completed even after a long period of time.
The reaction time was significantly shorter with alum in comparison with iodine. In addition, because alum in EtOH gave the fastest reaction affording the highest yield, subsequent syntheses were carried out in this solvent. In order to obtain novel spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives 3a–p, the syntheses were performed under optimized reaction conditions through a conventional method, starting from unsubstituted and methyl-substituted diexo- and diendo-2-aminonorbornene carboxamides 1a–d and unsubstituted, 5-methyl, 5-iodo-, and 7-chloro-substituted isatins 2a–d (Scheme 1, Figure 1, Table 2).
Scheme 1.

Spirocondensation reaction of diexo- and diendo-2-aminonorbornene carboxamides 1a–d with unsubstituted, 5-methyl, 5-iodo- and 7-chloro-substituted isatins 2a–d.
Figure 1.

Structure of novel spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives 3a–p.
Table 2.
Synthesis of spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives 3a–p a.
| Entry | Amide | Ketone | Time (h) | Product | Yield b (%) | de c (Major/Minor) |
|---|---|---|---|---|---|---|
| 1 | 1a | 2a | 6 | 3a | 42 | 1:0 |
| 2 | 1a | 2b | 5 | 3b | 46 | 1:0 |
| 3 | 1a | 2c | 6 | 3c | 29 | 1:0 |
| 4 | 1a | 2d | 6 | 3d | 30 | 1:0 |
| 5 | 1b | 2a | 12 | 3e d | 35 | 1:0 |
| 6 | 1b | 2b | 10 | 3f | 37 | 1:0 |
| 7 | 1b | 2c | 24 | 3g d | 30 | 1:0 |
| 8 | 1b | 2d | 24 | 3h d | 28 | 1:0.3 |
| 9 | 1c | 2a | 5 | 3i | 45 | 1:0 |
| 10 | 1c | 2b | 5 | 3j | 42 | 1:0 |
| 11 | 1c | 2c | 6 | 3k | 24 | 1:0 |
| 12 | 1c | 2d | 6 | 3l | 38 | 1:0.4 |
| 13 | 1d | 2a | 14 | 3m | 31 | 1:0 |
| 14 | 1d | 2b | 20 | 3n | 24 | 1:0.4 |
| 15 | 1d | 2c | 24 | 3o e | 25 | 1:1 |
| 16 | 1d | 2d | 24 | 3p e | 34 | 1.3:1 |
a A mixture of the corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), alum (20 mg, 30 mol%), and EtOH (5 mL) was stirred under reflux for the specified time. b Isolated yield after purification by flash chromatography. c The diastereoselectivity major/minor ratio was calculated by NMR measurements. d A mixture of the corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), I2 (19 mg, 30 mol%), and EtOH (5 mL) was stirred under reflux for the specified time. e A mixture of corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), NH4Cl (5 mg, 30 mol%), and 2M2B (5 mL) was stirred under reflux for the specified time.
In most cases, the reaction of diexo-β-amino amide 1a and 1c with isatins 2a–d was selective and resulted in only a single diastereomer of spiroquinazolinone derivative 3a–d and 3i–k in low to moderate yields. The only exception is 7-chloroisatin 2d, where the minor diastereomer of 3l was also formed. (Figure 1). The reaction of diendo-β-amino amide 1b and 1d with isatin derivatives 2a–d was less diastereoselective, resulting in the formation of only one diastereomer of spiroquinazolinone derivative 3e–g, 3m. In contrast, a mixture of two diastereomers of spiro derivatives 3h* and 3n*–p* were isolated in varied ratios (Figure 1).
Starting from diexo-β-amino amide 1a, the reaction with isatin 2a and 5-methylisatin 2b afforded the best yields of 42% (3a) and 46% (3b) (Table 2 entries 1, 2). Starting from methyl-substituted diexo-β-amino amide 1c, we could obtain compounds 3i and 3j in a 42 and 45% yield under reflux (Table 2 entries 9, 10) similar to 3a and 3b. It was interesting to note that I2 was a more effective catalyst than alum in EtOH in the synthesis of diendo-spiro compounds 3e, 3g, and 3h (Table 2 entries 5, 7, and 8). Reactions were not completed after a long period of time using alum in EtOH under reflux. While we could obtain compounds 3o and 3p utilizing NH4Cl as catalyst, the reaction in 2M2B was not completed either.
The relative configuration of the newly built stereocenter of 3a and 3e was determined by 2D NMR spectroscopy. For 3a, medium-intensity NOE interactions were detected between the H4′ proton (7.29 ppm) and the H1 amine (3.54 ppm) and between H4′ and the H3 amide (8.35 ppm). In contrast, NOE signals could not be observed between the indoline protons (aromatic signals) and H4a or H8a. This indicates that the six-membered ring of indoline is located far away (>5 Å) from these hydrogens and pyrrolidone is positioned in the proximity of H4a and H8a (Figure 2). A weak NOESY cross-peak between H4′ and one of the methylene protons (1.82 ppm) provided further support that the aromatic hydrogens are closer to the methylene bridge. In line with the diexo configuration, NOEs were not detected between the methylene bridge and the H4a and H8a protons of norbornene. For 3e, strong NOEs between one of the methylene bridge protons (1.41 ppm) and H4a (2.81 ppm) and H8a (4.05 ppm), respectively, supported the diendo arrangement. The absence of NOE cross-peaks between the aromatic protons of the indoline ring and the H4a and H8a protons indicated that they are not in close proximity. Furthermore, NOESY cross-peaks between the methylene and the aromatic hydrogens were not observable, justifying the relative configuration shown in Figure 1. Medium-intensity H4′–H1 (7.14 ppm and 2.38 ppm) and H4′–H3 (7.14 ppm and 8.18 ppm) NOE contacts were also detected.
Figure 2.

NOE interactions proving the relative configuration of 3a and 3e. Red arrows show the detected NOESY cross-peaks; black crossed arrows represent missing NOE contacts.
We attempted our investigations under CF and HSBM and MW irradiation in order to make the spirocondensation reaction more sustainable and environmentally benign. First, the synthesis of 3a was carried out (Table 3). Furthermore, we studied the synthesis of the other products 3b–d and 3i–l under greener conditions (Table 3). Under MW irradiation, we could obtain diexo spiro products 3b–d and 3i–l in good to excellent yields of 60–85% (Table 3). Under HSBM conditions, 3k and 3l did not react (Table 3 entries 12, 13). Under the CF condition, diexo 3a, 3b, and 3d as well as diendo 3n* and 3p* were synthesized in good yields. The synthesis of 3c was unsuccessful because 5-iodoisatin was insoluble in EtOH. Since the pressure increased during the synthesis, the pump had to be stopped.
Table 3.
Synthesis of spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives 3 under microwave irradiation, HSBM, and CF conditions.
| Entry | Method | Product | Time (h) | Yield a (%) |
|---|---|---|---|---|
| 1 | MW b | 3a | 0.5 | 85 |
| 2 | MW b | 3b | 1.5 | 83 |
| 3 | MW b | 3c | 1 | 85 |
| 4 | MW b | 3d | 2 | 71 |
| 5 | MW b | 3i | 1 | 70 |
| 6 | MW b | 3j | 1.5 | 65 |
| 7 | MW b | 3k | 2.5 | 59 |
| 8 | MW b | 3l | 2.5 | 60 |
| 9 | HSBM c | 3a | 6 | 45 |
| 10 | HSBM c | 3i | 6 | 45 |
| 11 | HSBM c | 3j | 6 | 37 |
| 12 | HSBM c | 3k | 4 | - |
| 13 | HSBM c | 3l* | 4 | - |
| 14 | CF d | 3a | 0.08 d | 70 |
| 15 | CF d | 3b | 0.08 d | 62 |
| 16 | CF d | 3d | 0.08 d | 70 |
| 17 | CF d | 3n* | 0.08 d | 52 |
| 18 | CF d | 3p* | 0.08 d | 44 |
a Isolated yield after purification by flash chromatography. b Reaction conditions for microwave irradiation: a mixture of the corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), alum (20 mg, 30 mol%), and ethanol (4 mL) was stirred for the specified time at 100 °C. c Reaction conditions for high-speed ball milling: a mixture of the corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), and alum (20 mg, 30 mol%) was milled solvent-free for the specified time; use of 3 agate balls utilizing 15 Hz frequency. * mixtures of diastereomers. d Reaction conditions for CF reactions: 0.1 mL min−1 flow rate (residence time: 5 min), 100 °C, β-amino amide (c = 0.1 M, 1 equiv), and isatin derivative (1 equiv) dissolved in ethanol (4 mL).
In the spirocyclization reaction of 1a–d with isatin derivatives 2a–d, first, unstable intermediate A is produced [32]. Then, it undergoes a water elimination reaction to give imine B, which is formed by a ring-closure reaction yielding epimers of spiroquinazolinone derivative C (Scheme 2). In a few cases, intermediate B, which is the chain tautomer of spiroquinazoline C, could easily be obtained in good yield in the CF reaction.
Scheme 2.

The spirocondensation reaction pathway.
2.1. Docking Studies
A challenging global health problem is the new coronavirus, which is responsible for coronavirus disease 2019 (COVID-19) [55]. Another demanding task for drug design and development is the inhibition of tryptase, which has promising therapeutic features for treating allergic or inflammatory disorders, for example, asthma and inflammatory bowel disease [56]. Hence, considering the structural similarities of their ligands with the 3a–g, 3i–k, and 3m molecules, we have selected the following macromolecules for investigating the potential inhibitory activity of spiroquinazolinones:
(i) A main protease (Mpro) of SARS-CoV-2, which is a key enzyme of coronaviruses and a mediator of viral replication and transcription. Our first assumption was that the indole ring of our molecules might fit into the binding pocket of the protease where the 2-oxopyrrolidine moiety of the N3 inhibitor binds in the complex structure 6LU7 [55];
(ii) A protease tryptase, which has a significant role in mediating mast cell-dependent allergic and inflammatory responses and making it an attractive drug target [56]. Spirocyclic piperidine derivatives are potent tryptase inhibitors; a benzofurane analog was also crystallized with the protein. We assumed that spiro-quinazolinones 3a–p can mimic their binding mode.
The docking studies of spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives were carried out in silico using SARS-CoV-2 main protease (PDB: 6LU7) and human mast cell tryptase (PDB: 2ZA5) as targets. First, to validate our docking method, the protein’s co-crystallized ligands were redocked to the macromolecules. The docking accuracy requirement of 1.0–3.0 Å root mean square deviation (rmsd) between docked and X-ray poses is recommended for a well-docked structure [57]. We found an rmsd of less than 2 Å (Figures S1 and S2), which is regarded as satisfactory [57,58].
Then, we docked our 3a–g, 3i–k, and 3m molecules to the macromolecules. Where the spirocondensation reaction was not selective and resulted in mixtures of diastereomers (3h*, 3l*, and 3n*–p*), docking studies were not performed.
In the case of the SARS-CoV-2 main protease, most of our molecules preferred the same binding pocket (Supplementary Information, Figure S1). Based on the comparison of estimated total energy and binding affinity of ligands 3a–3p to the SARS-CoV-2 main protease (PDB: 6LU7), compounds 3b, 3c, 3d, and 3e could be promising according to in silico results (Table 4). By visualization of the docking results, we could see the H-bond and van der Waals interactions between molecules 3a–g, 3i–k, and 3m and the macromolecule (6LU7) on a 2D diagram. Molecular interactions and binding poses with the SARS-CoV-2 main protease for compounds are included in the (Supplementary Information, Figures S3–S10).
Table 4.
Docking results of compounds 3a–g, 3i–k, and 3m with SARS-CoV-2 main protease (PDB: 6LU7) in silico.
| Molecules | Estimated Total Energy (kcal/mol) a | Binding Affinity (kcal/mol) b | Active Site Residue Chain A c |
|---|---|---|---|
| 3a | −62.559 | −7.8 | LEU141, SER144, CYS145 |
| 3b | −63.773 | −8.1 | LEU141, SER144, CYS145 |
| 3c | −59.252 | −8.2 | SER144, CYS145 |
| 3d | −64.985 | −8.1 | LEU141, GLY143, SER144, CYS145 |
| 3e | −55.856 | −8.6 | GLY143, SER144, CYS145 |
| 3f | −58.697 | −7.7 | GLY143, SER144, CYS145 |
| 3g | −57.707 | −7.7 | LEU141, GLY143, SER144, CYS145 |
| 3i | −64.442 | −7.3 | SER158 |
| 3j | −65.518 | −7.4 | GLY143, HIS164 |
| 3k | −57.736 | −7.1 | GLY143, HIS164 |
| 3m | −56.195 | −7.8 | HIS144 |
a Estimated total energy values were calculated by iGEMDOCK [59]. b Binding affinity of ligands to the macromolecule was calculated with the use of PyRx [60]. c The residues which took part in H bonding are represented from the residues of the active site: residues 141–143, 145, 164, long strand on one side of residues 155–168 and on the other side of residues 189–191 [55].
The residues LEU141, GLY143, CYS145, HIS163, and HIS164 at the chain A interface of the SARS-CoV-2 main protease are known to interact with most of its ligands and are critical for binding. Molecule 3d established H bonds with GLY143 in 2.5 Å, SER 144 in 2.1 Å, and CYS145 in 2.3 Å through its carbonyl group of the isatin ring, as well as with LEU141 in 2.7 Å with the NH group of the isatin ring (Figure 3). Moreover, compound 3d could also create van der Waals interactions with chain A residues THR25, THR26, HIS41, PHE140, ASN142, HIS164, and GLU166, and alkyl interaction with the chloro substituent of the isatin ring with HIS163 and HIS172. On the other hand, 3d has unfavorable interaction with the residues of chain C, e.g., PHE, LEU, and VAL. The highest binding affinity was revealed for the diendo compound 3e. By visualization, it turned out that it could fit into a special binding pocket (Figure 4). Compound 3e can form H bonds through its carbonyl group of the isatin ring with SER144 and GLY143 in 2.3 Å, 2.7 Å, and 2.1 Å. It can also establish a H bond with CYS145 through the NH group of the isatin ring.
Figure 3.

(a) Molecular interactions and binding pose of compound 3d at the interface of SARS-CoV-2 main protease (PDB: 6LU7); H bonds between the macromolecule and compound 3d are shown as yellow dashes and distances are in Å units; (b) Connelly surface of docking pose of 6LU7 with 3d shown as stick model; (c) receptor surface: H-bond donor vs. acceptor ability of surrounding amino acids; (d) 2D interaction map between 6LU7 and 3d.
Figure 4.

(a) Molecular interactions and binding pose of compound 3e at the interface of SARS-CoV-2 main protease (PDB: 6LU7); H bonds between the macromolecule and compound 3e are shown as yellow dashes and distances are in Å units; (b) Connelly surface of docking pose of 6LU7 with 3e shown as stick model; (c) receptor surface: H-bond donor vs. acceptor ability of surrounding amino acids; (d) 2D interaction map between 6LU7 and 3e.
For the in silico studies of human mast cell tryptase, we used the 2ZA5 PDB structure [56]. This X-ray structure of human mast cell tryptase is a tetramer containing hydrophobic binding pockets in the monomeric chains A and B. We used chain A of 2ZA5 for the studies. All of our spiroquinazolinones (3a–g, 3i–k, 3m) bound to the same binding site of human mast cell tryptase (Supplementary Information, Figure S2). Based on the comparison of the estimated total energy and binding affinity of ligands 3a–g, 3i–k, and 3m to the human mast cell tryptase (PDB: 2ZA5), compounds 3a, 3b, 3d, and 3f seem promising candidates for inhibiting the enzyme (Table 5). Compound 3d could interact with residues of the 2ZA5 macromolecule of chain A through H-bond interactions with ASP147 2.4 Å in 3 Å, excellent with HIS42 at 1.8 Å, and very well with GLN206 at a 1.9 Å distance. In addition, several van der Waals contacts were formed with the residues of 2ZA5, e.g., TRP145, GLY146, ARG153, and LEU154 (Figure 5). Molecular interactions and binding poses with mast cell tryptase for all compounds are included in the Supplementary Information (Supplementary Information, Figures S11–S18).
Table 5.
Docking results of compounds 3a–g, 3i–k, and 3m with human mast cell tryptase (PDB: 2ZA5) in silico.
| Molecules | Estimated Total Energy (kcal/mol) a | Binding Affinity (kcal/mol) b | Residue Chain A c |
|---|---|---|---|
| 3a | −100.957 | −8.7 | HIS42, ARG153 |
| 3b | −100.858 | −9.0 | ASP147, GLN206 |
| 3c | −97.333 | −8.6 | HIS42, ASP147 ARG153, GLN206 |
| 3d | −100.224 | −9.1 | HIS42, ASP146, GLN206 |
| 3e | −87.828 | −8.6 | TRP145, GLN206 |
| 3f | −98.296 | −8.7 | HIS42, TYR81, TRP145, ASP147, GLN206 |
| 3g | −95.208 | −8.7 | HIS42, TYR81, TRP145, ASP147, GLN206 |
| 3i | −100.404 | −8.0 | HIS42, ASP147 |
| 3j | −100.032 | −8.3 | HIS42, ARG153 |
| 3k | −97.9363 | −7.5 | TYR39, TYR81, ARG153, LEU154 |
| 3m | −86.146 | −8.6 | HIS42, TYR81, TRP145, ASP147 |
Figure 5.

(a) Molecular interactions and binding pose of compound 3d at the interface of human tryptase with potent non-peptide inhibitor (PDB: 2ZA5); H bonds between the macromolecule and compound 3d are shown as yellow dashes and distances are in Å units; (b) Connelly surface of docking pose of 2ZA5 with 3d shown as stick model; (c) receptor surface: H-bond donor vs. acceptor ability of surrounding amino acids; (d) 2D interaction map between 2ZA5 and 3d.
Summarizing the in silico results, molecules 3b, 3d, and 3f proved to be promising ligands for targeting the SARS-CoV-2 main protease and human mast cell tryptase. The main driving force of the binding between the SARS-CoV-2 main protease and 3b, 3d, 3e, and 3f was hydrophobic interaction (Supplementary Information, Figure S19). On the contrary, the driving force of binding between tryptase and compounds 3a, 3b, 3d, and 3f included both hydrophobic and hydrophilic interactions (Supplementary Information, Figure S20). We speculated that the other reason can be connected to the methyl substituent of 3b and 3f and to the chlorine atom of 3d. The methyl group established alkyl interactions, e.g., in the case of 6LU7, whereas the methyl group of 3b formed an alkyl interaction with VAL104. Our best-fitting ligands were 3b, 3d, and 3f, which could form interactions with residues in hydrophobic as well as hydrophilic environments. We suggested that compound 3d is the most compelling ligand from 3a–g, 3i–k, and 3m because the chlorine atom has high electronegativity and can establish H bonds and electrostatic interaction with amino acid residues. In addition, 3d had the highest binding affinity values and high estimated total energy values concerning both macromolecules. The results of the present work could give an understanding of the inhibitory mechanism of spiroquinazolinone derivatives on tryptase and the SARS-CoV-2 main protease, and they may assist in developing novel compounds with biological activities.
2.2. Prediction of Pharmacokinetic Properties by SwissADME
The pharmacokinetic properties were obtained by using the SwissADME web tool in order to forecast the absorption study (water solubility), distribution study (gastrointestinal absorption and BBB permeability), and metabolism study (CYP2C9 inhibitor, CYP3A4 inhibitor, CYP2D6 substrate, CYP1A2 inhibitor, and CYP2C19 inhibitor) (Supplementary Information, Table S1) similar to that in the literature [54]. The lipophilicity of compounds 3b, 3d, and 3f were in the range of 1.78 to 2.12 (recommended < 5) [61], and the logSw values were between −4.07 and −4.29, which indicated moderate water solubility and a rather lipophilic nature of the above-mentioned compounds. The data showed that the tested compounds are not able to cross the BBB. The predicted human intestinal absorption of compounds 3b, 3d, and 3f was high, indicating good absorption. Moreover, compounds 3b, 3d, and 3f presented good affinity toward the metabolizing enzyme CYP2D6 inhibitor. The absorption of the compounds is connected to skin permeation (logKp) and Caco-2 permeability. The skin permeation of 3b, 3d, and 3f was below −7.5, which is less than −2.5, suggesting that the compounds can penetrate well into the skin. Our most promising novel compounds 3b, 3d, and 3f passed the Lipinski and Veber parameters to be orally active (Supplementary Information, Table S2).
3. Materials and Methods
3.1. Experimental Procedures
General methods
All reactions were conducted using magnetic stirring in oven-dried glassware. To purify the products using column chromatography, Kieselgel 60 (Merck KGaA Darmstadt, Germany, 0.063–0.200 mm 70–230 mesh ASTM) was employed. Thin-layer chromatography was carried out on silica-coated aluminum backing plates 60A F254, using a UV lamp to see the spots (254 nm). Sulfuric acid and vanillin were utilized for detection.
Microwave-promoted reactions were carried out in sealed reaction vials (10 mL) in the cavity of a microwave equipment (CEM, Discover, SP, CEM Corporation, Matthwes, NC, USA).
All high-speed ball milling measurements were carried out with Retsch 400 Mixer Mill with 10 mL agate jars and 5 mm agate balls (Retsch Gmbh, Haan, Germany).
Ethyl acetate (EtOAc), n-hexane, chloroform, acetone, ethanol, and diethyl ether (Et2O) were among the solvents of the highest analytical grade purchased from Molar Chemicals Kft (Halásztelek, Hungary).
HPLC grade i-PrOH, n-hexane, and ethyl acetate were purchased from VWR Chemicals (Fontenary-sous-Bois, France). Isatin and alum (KAl(SO4)2·12H2O) were bought from Merck KGaA (Sigma Aldrich Chemie Gmbh, Steinheim, Germany). Compounds 3a–p were purified with a Teledyne ISCO Combi Flash Nextgen300+ flash chromatograph (Teledyne ISCO, Lincoln, NE, USA). Purifying methods on the flash chromatograph were optimized casewise.
1H NMR spectra were recorded at 500.20 MHz, while 13C NMR spectra were measured at 125.62 MHz in CDCl3 or in DMSO-d6 at ambient temperature with a Bruker Avance NEO Ascend 500 spectrometer (Bruker Biospin, Karlsruhe, Germany) with a 5 mm Prodigy BBO probe. Chemical shifts are given, relative to tetramethylsilane (Me4Si) as an internal standard, in δ (ppm). For signal assignment and for determining the relative configuration, 2D COSY, HSQC, HMBC, and NOESY measurements were carried out. Some 2D spectra were recorded on a Bruker Avance 600 MHz spectrometer (Bruker Biospin, Karlsruhe, Germany) equipped with a 5 mm z-gradient CP-TCI triple-resonance cryoprobe operating at 600.2 MHz and 150.92 MHz for 1H and 13C, respectively.
The HRMS flow injection analysis was performed with a Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ (Waters, Manchester, UK). MS measurements were performed with a Thermo Fisher LTQ XL instrument using UltraZoom scan rate.
Melting points were determined with a Hinotex-X4 (Hinotek, Ningbo, China) and Kofler melting point apparatus and are uncorrected.
The synthesis of diexo and diendo 3-aminobicyclo[2.2.1]hept-5-ene-2-carboxamide 1a,b were performed according to the literature [53].The syntheses of diexo and diendo 3-amino-N-methylbicyclo[2.2.1]hept-5-ene-2-carboxamide 1c,d were performed according to the literature [54].
3.2. General Procedure for the Synthesis of Spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione Derivatives 3a–p
Conventional method: A mixture of the corresponding β-amino amide 1a–d (0.13 mmol), isatin 2a–d (0.13 mmol), and the corresponding catalyst (NH4Cl, LiOH, p-TsOH, Amberlyst 15, I2, alum (30 mol%)) in an appropriate solvent (5 mL) was stirred under reflux or at room temperature for the specified time.
Continuous flow condition: For each reaction, the corresponding β-amino amide (c = 0.13 mmol, 1 equiv.) and isatin (0.13 mmol, 1 equiv.) were dissolved in ethanol, and the solution was pumped continuously under the following conditions: 0.1 mL min−1 flow rate (residence time: 5 min) and 100 °C. In each run, 4 mL of product solution was collected under steady-state conditions. Between two experiments, the system was washed for 20 min by pumping ethanol at a flow rate of 0.5 mL min−1. At the completion of the reaction, the product crystallized out from the solution after standing at room temperature. To carry out the synthesis of spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives under continuous flow conditions, a simple flow set-up was assembled. The system contains an HPLC pump (JASCO PU-2085), a stainless-steel HPLC column with internal dimensions of 4.6 × 100 mm as a catalyst bed, which encompassed approximately 1.5 g of alum and a 100 psi backpressure regulator (BPR) from IDEX to prevent solvent boil over.
Microwave irradiation: A mixture of the corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), alum (30 mol%), and ethanol (4 mL) was stirred for the specified time at 100 °C.
High-speed ball milling (HSBM) condition: A mixture of the corresponding isatin (0.13 mmol), β-amino amide (0.13 mmol), and alum (30 mol%) was milled solvent-free for the specified time, using 3 agate balls at a 15 Hz frequency. After completion of the reaction (monitored by TLC), the crude mixture was purified by normal phase flash chromatography on a silica gel column as the stationary phase in mixtures of n-hexane and ethyl acetate with different ratios (1:1, 1:2, 2:1) as the mobile phase utilizing a 8–13 mL/min flow rate. After solvent evaporation, the pure fractions were collected and crystallized from diethyl ether.
3.3. NMR Spectra
3.3.1. (2R*,4aR*,5R*,8S*,8aS*)-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3a)
Yield (15 mg, 42%), white solid, mp 262–268 °C (decomposition), 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.24 (1H, d, J = 8.3 Hz, 9-H), 1.82 (1H, d, J = 8.4 Hz, 9-H), 2.12 (1H, d, J = 6.9 Hz, 4a-H), 2.55 (1H, s, 8-H), 3.15 (1H, s, 5-H), 3.37 (1H, t, J = 6.2 Hz, 8a-H), 3.47 (1H, d, J = 5.3 Hz, 1-NH), 6.08 (1H, dd, J = 2.9, 5.3 Hz, 7-H), 6.29 (1H, dd, J = 2.9, 5.3 Hz, 6-H), 6.80 (1H, d, J = 7.7 Hz, Ar), 6.99 (1H, t, J = 7.5 Hz, Ar), 7.22–7.31 (2H, m, Ar), 8.29 (1H, s, 3-NH), 10.23 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 43.69, 44.07, 44.93, 48.31, 52.22, 71.01, 110.15, 122.24, 125.46, 129.35, 130.59, 135.39, 138.86, 142.53, 172.95, 177.05. HRMS (ESI) [M + H]+ m/z calcd. for C16H15N3O2: 282.11643, found: 282.12376, HPLC-MS: 282.17.
3.3.2. (2R*,4aR*,5R*,8S*,8aS*)-5′-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3b)
Yield (18 mg, 46%), beige solid, mp = 318–322 °C (decomposition). 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.27 (1H, d, J = 8.5 Hz, 9-H), 1.81 (1H, d, J = 8.4 Hz, 9-H), 2.10 (1H, d, J = 7.8 Hz, 4a-H), 2.26 (3H, s, CH3), 2.54 (1H, s, 8-H), 3.15 (1H, s, 5-H), 3.37 (1H, t, J = 6.2 Hz, 8a-H), 3.49 (1H, d, J = 5.4 Hz, 1-NH), 6.03–6.13 (1H, m, 7-H), 6.26–6.32 (1H, m, 6-H), 6.69 (1H, d, J = 7.8 Hz, Ar), 7.07 (1H, d, J = 7.9 Hz, Ar), 7.12 (1H, s, Ar), 8.32 (1H, s, 3-NH), 10.22 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 21.01, 43.62, 44.05, 44.89, 48.25, 52.23, 71.04, 109.95, 126.05, 129.29, 130.77, 131.20, 135.42, 138.83, 139.05, 173.04, 177.09. HRMS (ESI) [M + H]+ m/z calcd. for C17H17N3O2: 296.13935, found: 296.13802, HPLC-MS: 296.22.
3.3.3. (2R*,4aR*,5R*,8S*,8aS*)-5′-iodo-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3c)
Yield (21 mg, 40%), beige solid, mp 290–302 °C (decomposition). 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.28 (1H, d, J = 8.8 Hz, 9-H), 1.83 (1H, d, J = 8.6 Hz, 9-H), 2.11 (1H, d, J = 7.3 Hz, 4a-H), 2.57 (1H, brs, 8-H), 3.17 (1H, brs, 5-H), 3.36 (1H, t, J = 6.3 Hz, 8a-H), 3.60 (1H, d, J = 5.9 Hz, 1-NH), 6.10 (1H, dd, J = 2.9, 5.3 Hz, 7-H), 6.31 (1H, dd, J = 2.9, 5.3 Hz, 6-H), 6.68 (1H, d, J = 8.2 Hz, Ar), 7.58 (1H, s, Ar), 7.63 (1H, d, J = 8.3 Hz, Ar), 8.39 (1H, s, 3-NH), 10.43 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 43.60, 44.12, 44.91, 48.22, 52.29, 70.87, 84.81, 112.71, 131.72, 133.83, 135.45, 138.86, 139.0, 142.29, 172.79, 176.36. HRMS (ESI) [M + H]+ m/z calcd. for C16H14IN3O2: 408.02035, found: 408.01833, HPLC-MS: 408.13.
3.3.4. (2R*,4aR*,5R*,8S*,8aS*)-7′-chloro-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3d)
Yield (14 mg, 30%), beige solid, mp 159–163 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.27 (1H, d, J = 7.8 Hz, 9-H), 1.81 (1H, d, J = 8.4 Hz, 9-H), 2.12 (1H, d, J = 7.0 Hz, 4a-H), 2.54 (1H, s, 8-H), 3.15 (1H, s, 5-H), 3.30-3.36 (1H, dd, J = 4.2, 6.4 Hz, 8a-H), 3.75 (1H, d, J = 5.1 Hz, 1-H), 6.09 (1H, dd, J = 3.0, 5.6 Hz, 7-H), 6.30 (1H, dd, J = 3.0, 5.6 Hz, 6-H), 7.03 (1H, t, J = 7.9 Hz, Ar), 7.25 (1H, d, J = 7.0 Hz, Ar), 7.35 (1H, d, J = 8.3 Hz, Ar), 8.40 (1H, s, 3-NH), 10.75 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 43.69, 44.05, 44.89, 48.37, 52.02, 71.50, 114.33, 123.68, 124.24, 130.50, 131.17, 135.31, 138.91, 140.24, 172.78, 177.08. HRMS (ESI) [M + H]+ m/z calcd. for C16H14ClN3O2: 316.08528, found: 316.0976, HPLC-MS: 316.11.
3.3.5. (2S*,4aS*,5R*,8S*,8aR*)-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3e)
Yield (13 mg, 35%), white solid, mp 274–278 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.36–1.46 (2H, m, 9-H, 9-H), 2.39 (1H, d, J = 7.9 Hz, 1-NH), 2.81 (1H, dd, J = 8.6, 3.8 Hz, 4a-H), 2.85 (1H, s, 8-H), 3.19 (1H, s, 5-H), 4.04 (1H, ddd, J = 8.3, 8.3, 3.6 Hz, 8a-H), 6.22 (2H, s, 6-H, 7-H), 6.79 (1H, d, J = 7.7 Hz, Ar), 6.97 (1H, t, J = 7.5 Hz, Ar), 7.13 (1H, d, J = 7.3 Hz, Ar), 7.25 (1H, t, J = 7.7 Hz, Ar), 8.18 (1H, s, 3-NH), 10.34 (1H, s, 1′-NH).13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 43.58, 46.07, 46.73, 46.89, 53.94, 71.36, 110.15, 122.27, 124.69, 129.40, 130.55, 134.79, 137.05, 142.14, 173.03, 176.73. HRMS (ESI) [M + H]+ m/z calcd. for C16H15N3O2: 282.12370, found: 282.12319, HPLC-MS: 282.15.
3.3.6. (2S*,4aS*,5R*,8S*,8aR*)-5′-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3f)
Yield (14 mg, 37%), beige solid, mp 311–315 °C (decomposition). 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.38–1.45 (2H, m, 9-H, 9-H), 2.24 (3H, s, CH3), 2.30 (1H, d, J = 8 Hz, 1-NH), 2.80 (1H, dd, J = 8.6, 3.9 Hz, 4a-H), 2.86 (1H, s, 8-H), 3.20 (1H, s, 5-H), 4.04 (1H, ddd, J = 8.3, 8.3, 3.6 Hz, 8a-H), 6.19–6.26 (2H, m, 6-H, 7-H), 6.69 (1H, d, J = 7.8 Hz, Ar), 6.97 (1H, s, Ar), 7.06 (1H, d, J = 7.8 Hz, Ar), 8.17 (1H, s, 3-NH), 10.26 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 21.0, 43.53, 46.05, 46.77, 46.84, 53.99, 71.46, 109.96, 125.21, 129.33, 130.71, 131.26, 134.77, 137.14, 139.57, 173.15, 176.67. HRMS (ESI) [M + H]+ m/z calcd. for C17H17N3O2: 296.13990, found: 296.1643, HPLC-MS: 296.25.
3.3.7. (2S*,4aS*,5R*,8S*,8aR*)-5′-iodo-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3g)
Yield (16 mg, 30%), beige solid, mp 275–278 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.36–1.43 (2H, m, 9-H, 9-H), 2.64 (1H, d, J = 7.7 Hz, 1-NH), 2.80 (1H, dd, J = 8.6, 3.9 Hz, 4a-H), 2.84 (1H, s, 8-H), 3.18 (1H, s, 5-H), 4.02 (1H, ddd, J = 8.2, 8.2, 3.5 Hz, 8a-H), 6.17–6.20 (1H, m, 6-H), 6.22–6.26 (1H, m, 7-H), 6.64 (1H, d, J = 8.1 Hz, Ar), 7.39 (1H, d, J = 1.4 Hz, Ar), 7.60 (1H, dd, J = 8.2, 1.4 Hz, Ar), 8.20 (1H, s, 3-NH), 10.47 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 43.62, 45.96, 46.65, 46.91, 53.89, 71.20, 84.86, 112.67, 131.68, 133.22, 135.02, 136.70, 138.98, 142.00, 172.94, 176.21. HRMS (ESI) [M + H]+ m/z calcd. for C16H14IN3O2: 408.02035, found: 408.01997, HPLC-MS: 408.14.
3.3.8. (4aS*,5R*,8S*,8aR*)-7′-chloro-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3h*)
Mixtures of diastereomers of 2R* and 2S*, yield (12 mg, 28%), yellow/cream solid, mp 212-217 (decomposition) °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.38–1.45 (2H, m, 9-H, 9-H), 2.64 (1H, d, J = 7.2 Hz, 1-NH), 2.82–2.86 (2H, m, 4a-H, 8-H), 3.19 (1H, brs, 5-H), 4.01-4.06 (1H, m, 8a-H), 6.18-6.21 (2H, m, 6-H, 7-H), 7.00 (1H, t, J = 7.6 Hz, Ar), 7.10 (1H, d, J = 7.1 Hz, Ar), 7.32 (1H, d, J = 8.2 Hz, Ar), 8.16 (1H, s, 3-NH), 10.74 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 43.51, 45.97, 46.76, 46.84, 53.78, 71.88, 114.47, 123.34, 124.00, 130.60, 136.89, 138.07, 139.66, 173.84, 176.61. HRMS (ESI) [M + H+]+ m/z calcd. for C16H14ClN3O2: 316.08528, found: 316.0717.
3.3.9. (2R*,4aR*,5R*,8S*,8aS*)-3-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3i)
Yield (15 mg, 46%), white solid, mp 271–274 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.26 (1H, d, J = 8.5 Hz, 9-H), 1.87 (1H, d, J = 8.5 Hz, 9-H), 2.17 (1H, d, J = 7.0 Hz, 4a-H), 2.42 (3H, s, CH3), 2.57 (1H, s, 5-H), 3.18 (1H, s, 1-H), 3.30–3.35 (1H, m, 8a-H), 3.81 (1H, d, J = 4.8 Hz, 1-NH), 6.06–6.09 (1H, m, 7-H), 6.31–6.34 (1H, m, 6-H), 6.88 (1H, d, J = 7.7 Hz, Ar), 7.05 (1H, t, J = 7.5 Hz, Ar), 7.28–7.34 (2H, m, Ar), 10.54 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 29.9, 43.99, 44.57, 45.44, 48.46, 51.43, 75.84, 110.83, 122.60, 125.61, 127.78, 130.95, 135.20, 139.16, 142.41, 172.19, 176.11. HRMS (ESI) [M + H]+ m/z calcd. for C17H17N3O2: 296.13935, found: 296.13891, HPLC-MS: 296.20.
3.3.10. (2R*,4aR*,5R*,8S*,8aS*)-3,5′-dimethyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3j)
Yield (15 mg, 42%), beige solid, mp 221–226 °C (decomposition). 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.27 (1H, d, J = 8.4 Hz, 9-H), 1.87 (1H, d, J = 8.5 Hz, 9-H), 2.16 (1H, d, J = 7.0 Hz, 4a-H), 2.28 (3H, s, CH3), 2.42 (3H, s, HN-CH3), 2.57 (1H, s, 5-H), 3.18 (1H, s, 8-H), 3.30–3.38 (1H, m, 8a-H), 3.77 (1H, d, J = 4.8 Hz, 1-NH), 6.05–6.09 (1H, m, 6-H), 6.30–6.34 (1H, m, 7-H), 6.77 (1H, d, J = 8.1 Hz, Ar), 7.10–7.15 (2H, m, Ar), 10.44 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 21.03, 29.93, 43.99, 44.56, 45.43, 48.46, 51.48, 75.92, 110.57, 126.07, 127.80, 131.13, 131.66, 135.20, 139.14, 139.88, 172.21, 176.12. HRMS (ESI) [M + H]+ m/z calcd. for C18H19N3O2: 310.15500, found: 310.15458, HPLC-MS: 310.20.
3.3.11. (2R*,4aR*,5R*,8S*,8aS*)-5′-iodo-3-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3k)
Yield (13 mg, 24%), ecru solid, mp 237–241 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.27 (1H, d, J = 8.0 Hz, 9-H), 1.88 (1H, d, J = 8.6 Hz, 9-H), 2.15 (1H, d, J = 6.9 Hz, 4a-H), 2.43 (3H, s, CH3), 2.58 (1H, s, 5-H), 3.18 (1H, s, 8-H), 3.30–3.32 (1H, m, 8a-H), 3.86 (1H, s, 1-NH), 6.07–6.10 (1H, m, 7-H), 6.31–6.34 (1H, m, 6-H), 6.74 (1H, d, J = 8.2 Hz, Ar), 7.60–7.62 (1H, m, Ar), 7.66 (1H, dd, J = 8.2, 1.8 Hz, Ar), 10.68 (1H, s, NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 30.0, 44.05, 44.53, 45.44, 48.41, 51.52, 75.77, 85.41, 113.33, 130.12, 133.79, 135.23, 139.18, 139.44, 142.15, 172.07, 175.41. HRMS (ESI) [M + H]+ m/z calcd. for C17H16IN3O2:422.02872, found: 422.0232, HPLC-MS: 422.22.
3.3.12. (4aR*,5R*,8S*,8aS*)-7′-chloro-3-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3l*)
Mixture of diastereomers of 2R* and 2S*, where the major compound (2R*) can be identified at NMR spectra, yield (16 mg, 38%), ecru solid, mp 346–350 °C (decomposition). 1H-NMR δH (500.20 MHz, CDCl3, 30 °C, Me4Si) 1.48 (1H, d, J = 8.9 Hz, 9-H), 1.90 (1H, d, J = 9.0 Hz, 9-H), 2.53 (1H, d, J = 7.1 Hz), 2.64 (1H, s, 5-H), 2.66 (3H, s, CH3), 3.46 (1H, s, 8-H), 3.56 (1H, d, J = 7.2 Hz), 6.04–6.07 (1H, m, 7-H), 6.34–6.38 (1H, m, 6-H), 7.09 (1H, d, J = 7.9 Hz, Ar), 7.22 (1H, d, J = 7.4 Hz, Ar), 7.31–7.36 (1H, m, Ar), 8.45 (1H, s, NH), 8.77 (1H, s, NH) for the major compound. 13C-NMR δC (125.62 MHz, CDCl3, 30 °C, Me4Si) 30.46, 43.84, 44.81, 45.71, 48.70, 51.76, 76.57, 116.20, 122.99, 123.86, 124.60, 128.56, 130.86, 134.23, 138.26, 139.37, 139.53, 172.98, 174.67 for the major compound. HRMS (ESI) [M + H]+ m/z calcd. for C17H16ClN3O2: 330.10038, found: 330.09827, HPLC-MS: 341.38.
3.3.13. (2S*,4aS*,5R*,8S*,8aR*)-3-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3m)
Yield (12 mg, 31%), white solid, mp 255–257 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.37–1.44 (2H, m, 9-H, 9-H), 2.34 (3H, s, CH3), 2.78 (1H, d, J = 7.2 Hz, 1-NH), 2.86 (1H, s, 8-H), 2.88 (1H, dd, J = 8.6, 3.9 Hz, 4a-H), 3.22 (1H, s, 8-H), 4.00 (1H, ddd, J = 8.0, 8.0, 3.6 Hz, 8a-H), 6.22 (2H, brs, 6-H, 7-H), 6.87 (1H, d, J = 7.7 Hz, Ar), 7.02 (1H, t, J = 7.5 Hz, Ar), 7.14 (1H, d, J = 7.3 Hz, Ar), 7.30 (1H, t, J = 7.7 Hz, Ar), 10.58 (1H, s, 1′-NH). 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 29.74, 44.23, 46.49, 46.83, 47.17, 53.23, 76.21, 110.83, 122.61, 124.79, 127.83, 130.82, 135.14, 136.61, 142.04, 172.45, 175.85. HRMS (ESI) [M + H]+ m/z calcd. for C17H17N3O2: 296.13990, found: 296.1610, HPLC-MS: 296.21.
3.3.14. (4aS*,5R*,8S*,8aR*)-3,5′-dimethyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3n*)
Mixture of diastereomers of 2R* and 2S*, where the major compound can be identified with NMR spectra, yield (10 mg, 24%), yellow solid, mp 251–254 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.37–1.41 (2H, m, 9-H, 9-H), 2.26 (3H, s, CH3), 2.34 (3H, s, CH3), 2.72 (1H, d, J = 7.2 Hz, 1-NH), 2.85–2.89 (2H, m, 4a-H, 8-H), 3.23 (1H, s, 5-H), 4.01 (1H, ddd, J = 8.0, 8.0, 3.6 Hz, 8a-H), 6.19–6.25 (2H, m, 6-H, 7-H), 6.75 (1H, d, J = 7.8 Hz, Ar), 6.97 (1H, s, Ar), 7.09 (1H, d, J = 8.0 Hz, Ar), 10.47 (1H, s, NH) for the major compound. 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 21.01, 29.76, 44.21, 46.48, 46.69, 47.16, 53.27, 76.30, 110.57, 125.27, 127.85, 131.00, 131.66, 135.16, 136.66, 139.54, 172.41, 175.83 for the major compound. HRMS (ESI) [M + H]+ m/z calcd. for C18H19N3O2: 310.15555, found: 310.1568, HPLC-MS: 310.19.
3.3.15. (4aS*,5R*,8S*,8aR*)-5′-iodo-3-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3o*)
Mixture of diastereomers of 2R* and 2S*, where the major compound can be identified with NMR spectra, yield (11 mg, 25%), white solid, mp 179–183 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.35–1.45 (2H, m, 9-H, 9-H), 2.34 (3H, s, CH3), 2.84 (1H, brs, 8H), 2.88 (1H, dd, J = 8.7, 4.0 Hz, 4a-H), 3.07 (1H, d, J = 7.2 Hz, 1-NH), 3.20 (1H, s, 5-H), 3.98 (1H, ddd, J = 8.0, 8.0, 3.6 Hz, 8a-H), 6.16–6.20 (1H, m, 7H), 6.23–6.27 (1H, m, 6H), 6.72 (1H, d, J = 8.0 Hz, Ar), 7.41 (1H, s, Ar), 7.64 (1H, d, J = 8.2 Hz, Ar), 10.71 (1H, s, NH) for the major compound. 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 29.78, 44.27, 46.41, 46.76, 47.19, 53.21, 76.08, 85.35, 113.30, 130.11, 133.15, 135.35, 136.33, 139.37, 141.89, 172.35, 175.32 for the major compound. HRMS (ESI) [M + H]+ m/z calcd. for C17H16IN3O2: 422.02872, found: 422.0321, HPLC-MS: 422.16.
3.3.16. (4aS*,5R*,8S*,8aR*)-7′-chloro-3-methyl-4a,5,8,8a-tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4(3H)-dione (3p*)
Mixture of diastereomers of 2R* and 2S*, yield (12 mg, 34%), white solid, mp 178–182 °C. 1H-NMR δH (500.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.36–1.54 (2H, m, 9-H, 9-H), 2.36 (3H, s, CH3), 2.39 (3H, ms, CH3), 2.84 (1H, s, 8-H), 2.88–2.93 (1H, m, 4a-H), 2.99 (1H, s, 8-H), 3.08 (1H, d, J = 6.7 Hz, 1-NH), 3.12–3.15 (1H, m, 4a-H), 3.21 (1H, m, 5-H), 3.32 (1H, m, 5-H), 6.19 (2H, brs, 6-H, 7-H), 6.27–6.30 (1H, m, 7-H), 6.39–6.42 (1H, m, 6-H), 7.02–7.07 (1H, m, Ar), 7.08–7.13 (3H, m, Ar), 7.37–7.40 (1H, m, Ar), 7.42–7.46 (1H, m, Ar), 7.47–7.52 (1H, m, Ar), 11.05 (1H, s, NH), 11.36 (1H, s, NH) for the major and minor compounds. 13C-NMR δC (125.62 MHz, DMSO-d6, 30 °C, Me4Si) 28.92, 29.73, 43.94, 44.29, 46.42, 46.68, 46.72, 46.87, 47.26, 47.50, 53.15, 54.29, 76.72, 77.59, 115.05, 115.46, 123.57, 123.71, 123.97, 124.28, 129.55, 129.66, 130.53, 130.78, 133.82, 135.32, 136.24, 136.26, 137.87, 139.55, 139.66, 139.88, 170.56, 172.33, 173.03, 176.01 for the major and minor compounds. HRMS (ESI) [M + H]+ m/z calcd. for C17H16ClN3O2: 330.10093, found: 330.1410, HPLC-MS: 330.23.
3.3.17. (1R*,2R*,3S*,4S*)-3-((Z)-(5-methyl-2-oxoindolin-3-ylidene)amino)bicyclo[2.2.1]hept-5-ene-2-carboxamide (Bb)
Yield (22 mg, 58%), white solid, mp 245–248 °C 1H-NMR δH (600.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.41 (1H, d, J = 8.2 Hz, 7-H), 2.26 (3H, s, CH3), 2.42 (1H, dd, J = 1.8, 7.9 Hz, 2-H), 2.44 (1H, d, J = 8.1 Hz, 7-H), 2.60 (1H, s, 4-H), 2.94 (1H, s, 1-H), 5.35 (1H, d, J = 8.0 Hz, 3-H), 6.23 (1H, dd, J = 3.0, 5.6 Hz, 5-H), 6.35 (1H, dd, J = 3.0, 5.6 Hz, 6-H), 6.57 (1H, s, NH), 6.63 (1H, s, NH), 6.73 (1H, d, J = 7.9 Hz, Ar), 7.16–7.19 (2H, m, Ar), 10.75 (1H, s, NH). 13C-NMR δC (150.92 MHz, DMSO-d6, 30 °C, Me4Si) 20.87, 44.92, 45.46, 50.53, 51.02, 61.54, 110.62, 122.36, 122.44, 131.48, 133.48, 136.76, 140.56, 142.35, 153.56, 160.43, 173.72. HRMS (ESI) [M + H]+ m/z calcd. for C17H17N3O2: 296.13935, found: 296.1607.
3.3.18. (1R*,2R*,3S*,4S*)-3-((Z)-(7-chloro-2-oxoindolin-3-ylidene)amino)bicyclo[2.2.1]hept-5-ene-2-carboxamide (Bd)
Yield (21 mg, 47%), white solid, mp 242–245 °C 1H-NMR δH (600.20 MHz, DMSO-d6, 30 °C, Me4Si) 1.42 (1H, d, J = 8.2 Hz, 7-H), 2.42–2.45 (2H, m, 2-H, 7-H), 2.65 (1H, s, 4-H), 2.95 (1H, s, 1-H), 5.35 (1H, d, J = 8.0 Hz, 3-H), 6.24 (1H, dd, J = 3.0, 5.6 Hz, 5-H), 6.37 (1H, dd, J = 3.0, 5.6 Hz, 6-H), 6.56 (1H, s, NH), 6.67 (1H, s, NH), 7.03 (1H, t, J = 7.7 Hz, Ar), 7.34 (1H, d, J = 7.6 Hz, Ar), 7.46 (1H, d, J = 8.1 Hz, Ar), 11.32 (1H, s, NH). 13C-NMR δC (150.92 MHz, DMSO-d6, 30 °C, Me4Si) 44.93, 45.47, 50.74, 50.92, 61.88, 115.10, 120.65, 123.78, 124.11, 132.79, 136.68, 140.69, 142.06, 152.87, 160.15, 173.58. HRMS (ESI) [M + H]+ m/z calcd. for C16H14ClN3O2: 316.08528, found: 316.0976.
3.4. Docking Studies
For docking studies, the macromolecules SARS-CoV-2 main protease (PDB: 6LU7) and human mast cell tryptase (PDB: 2ZA5) were downloaded in pdb format from RCSB Protein Data Bank (https://www.rcsb.org) (accessed on 1 August 2024). Preparing the macromolecules for docking was performed with the AutoDock4 software. Ligand structures were optimized and minimized through Avogadro software (version 1.95.0). Docking protocols were accomplished by the PyRx-Virtual [60] screening tool and iGEMDOCK version 2.1 software [59]. Blind docking was accomplished. The docking calculation was partially flexible (rigid protein, flexible ligand). Validation of the molecular docking method was accomplished by redocking the crystallographic ligand of the target enzymes. The ligand interactions were visualized and analyzed through PyMOL 2.5 (The PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC) and Discovery Studio Visualizer (BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio, 4.5, San Diego: Dassault Systèmes, 2021).
All figures representing the binding ligand position of binding pockets in the case of both macromolecules were visualized in PyMOL 2.5 software. H-bond interactions were signed with distances in Å. The 2D interaction diagrams of the ligand interaction with the residues of the macromolecule were created utilizing BIOVIA Discovery Studiodraw software.
3.5. SwissADME Predictions
Information with respect to pharmacokinetic properties was collected by using the SwissADME web tool in order to forecast the absorption study (water solubility), distribution study (gastrointestinal absorption and blood–brain barrier (BBB) permeability), metabolism study, etc. [62]. The SwissADME is a free and user-friendly web application, allowing a simple assessment of pharmacokinetics [62]. BBB permeability and HIA (human intestinal absorption) represent the absorption and the distribution of compounds. The drug likeness property envisions the molecules, which could improve an orally bioavailable drugs by contrasting their physicochemical or structural characteristics [63]. The five features of the Lipinski rule state that oral drugs should not have more than one violation, which include the following: molecular weight ≤500 g/mol; partition coefficient ≤ 5; number of H-bond accepters ≤ 10 and H-bond donors ≤ 5; and the number of rotatable bonds ≤ 10 [63]. The Veber rule requires compounds to have ≤10 rotatable bonds and a polar surface area of less than 140 Å [64].
4. Conclusions
With the aim of helping drug design and improvement, we have synthesized novel tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives. Furthermore, the synthesis of these spiro compounds was optimized by developing new methods and testing both the catalysts and solvents. The best methods were using alum as a catalyst in EtOH, I2 in EtOH at 78 °C, and NH4Cl in 2M2B at 100 °C in all cases. In order to make the synthesis route greener, different kinds of environmentally benign methods such as MW irradiation, HSBM, and CF were utilized. Of these, MW irradiation was found to be the most effective to achieve excellent yields up to 85%. Reaction times in each case were significantly reduced with the usage of a microwave reactor. In the case of HSBM conditions, we could develop a solvent-free method with moderate yields. Docking studies were performed in silico comparing two different macromolecules, namely the SARS-CoV-2 main protease (PDB: 6LU7) and human mast cell tryptase (PDB: 2ZA5). According to the estimated total energy and binding affinity, compounds 3b, 3d, and 3f of our novel derivatives were found to be good fitting ligands to both macromolecules (6LU7 and 2ZA5). We also suggest 3e as an outstanding ligand to the SARS-CoV-2 main protease (PDB: 6LU7). Thus, the present results are essential for the design, synthesis, and improvement in novel spiroquinazolinone derivatives with promising biological effects and additional modification of the structure in the future. We suggested that compound 3d is an outstanding ligand because of the high electronegativity of the chlorine atom. It can form H bonds and electrostatic interactions with amino acid residues in hydrophobic environments. The ADMET data predictions by the SwissADME web tool theoretically predicted that the present novel tetrahydro-1H-spiro[5,8-methanoquinazoline-2,3′-indoline]-2′,4-dione derivatives could be promising biologically active molecules. Moreover, they could be orally active, and they could penetrate the skin according to theoretical predictions by SwissADME.
Acknowledgments
The high-resolution mass spectrometric (HRMS) analysis was performed by Robert Berkecz. Thanks to Vivien Resch (Department of Medicinal Chemistry, University of Szeged, H-6720 Szeged, Hungary) for making useful suggestions for the docking studies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29215112/s1, Figure S1: Docking accuracy validation and binding poses of co-ligand of 6LU7 and the redocked one of SARS-CoV-2 main protease (PDB: 6LU7) and binding poses of molecules 3a–g, 3i–k and 3m with 6LU7; Figure S2: Docking accuracy validation and binding poses of co-ligand of 2ZA5 and the redocked one human tryptase with potent non-peptide inhibitor (PDB: 2ZA5) and binding poses of molecules 3a–g, 3i–k and 3m with 2ZA5; Figures S3–S10: Molecular interactions and binding poses and H bonds and connelly surface of docked pose of compound 3a, 3c, 3e, 3g, 3i, 3j, 3k and 3m at the interface of SARS-CoV-2 main protease (PDB: 6LU7); Figures S11–S18: Molecular interactions and binding poses and H bonds and connelly surface of docked pose of compound 3a, 3c, 3e, 3g, 3i, 3j, 3k and 3m at the interface of SARS-CoV-2 main protease (PDB: 6LU7); Figure S19: Receptor surface: Hydrophobicity of the surrounding amino acids around the binding pose of compound 3b, 3d, 3e and 3f at the upper binding pocket of SARS-CoV-2 main protease (PDB: 6LU7); Figure S20: Receptor surface: Hydrophobicity of the surrounding amino acids around the binding pose of compound 3a, 3b, 3d and 3f at the binding pocket of human mast cell tryptase (PDB: 2ZA5). Table S1: Prediction of in-silico lipophilicity, water solubility and pharmacokinetic parameters of novel spiro[5,8-methanoquinazoline-2,3’-indoline]-2’,4-dione derivatives 3a–g, 3i–k, 3m by SwissADME webtool; Table S2: Prediction of in silico druglikeness properties of novel spiro[5,8-methanoquinazoline-2,3’-indoline]-2’,4-dione derivatives 3a–g, 3i–k, 3m by SwissADME web tool. References [59,60,65,66,67] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, M.P. and T.F.; methodology, M.P.; software T.F.; formal analysis, T.F. and E.W.; investigation, T.F. and R.M.; writing—original draft preparation, T.F. and M.P.; writing—review and editing, E.W. and M.P.; visualization, T.F.; docking studies, T.F.; supervision, M.P. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or Supplementary Material.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
The authors’ thank the Hungarian Research Foundation (OTKA No. K-138871) and the Ministry of Human Capacities, Hungary grant TKP-2021-EGA-32. Tünde Faragó was supported by the ÚNKP-22-3 -SZTE-188 New National Excellence Program of the Ministry for Innovation and Technology from the National Research, Development and Innovation Fund.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Dhokne P., Sakla A.P., Shankaraiah N. Structural Insights of Oxindole Based Kinase Inhibitors as Anticancer Agents: Recent Advances. Eur. J. Med. Chem. 2021;216:113334. doi: 10.1016/j.ejmech.2021.113334. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J., Zhao J., Wang L., Liu J., Ren D., Ma Y. Design, Synthesis and Docking Studies of Some Spiro-Oxindole Dihydroquinazolinones as Antibacterial Agents. Tetrahedron. 2016;72:936–943. doi: 10.1016/j.tet.2015.12.055. [DOI] [Google Scholar]
- 3.Mane R., Yaraguppi D.A., Ashok A.K., Gangadharappa B., Chandrakala K.B., Kamanna K. Glutamic Acid-Catalyzed Synthesis of Dihydroquinazolinone: Anticancer Activity, Electrochemical Behavior, Molecular Docking, Dynamics, Simulations and Drug-Likeness Studies. Res. Chem. Intermed. 2024;50:3271–3303. doi: 10.1007/s11164-024-05305-6. [DOI] [Google Scholar]
- 4.Saraswat P., Jeyabalan G., Hassan M.Z., Rahman M.U., Nyola N.K. Review of Synthesis and Various Biological Activities of Spiro Heterocyclic Compounds Comprising Oxindole and Pyrrolidine Moities. Synth. Commun. 2016;46:1643–1664. doi: 10.1080/00397911.2016.1211704. [DOI] [Google Scholar]
- 5.Kaur M., Singh M., Chadha N., Silakari O. Oxindole: A Chemical Prism Carrying Plethora of Therapeutic Benefits. Eur. J. Med. Chem. 2016;123:858–894. doi: 10.1016/j.ejmech.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Huang X., Wang H., Cao Q., Li Y., Zhang J. Access to 3,3-Disubstituted Oxindoles via Microwave-Assisted Cannizzaro and Aldol Reactions of Formaldehyde with Isatins and Their Imines. RSC Adv. 2021;11:17320–17323. doi: 10.1039/D1RA02150H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y., Gao P., Wang J., Sun Q., Ge Z., Li R. Primary 1,2-Diamine Catalysis (V): Efficient Asymmetric Aldol Reactions of Isatins with Cyclohexanone. Synlett. 2012;23:1031–1034. doi: 10.1002/chin.201233125. [DOI] [Google Scholar]
- 8.Lv X.-X., Liu N., Chen F., Zhang H., Du Z.-H., Wang P., Yuan M., Da C.S. Highly Asymmetric Aldol Reaction of Isatins and Ketones Catalyzed by Chiral Bifunctional Primary-Amine Organocatalyst on Water. Org. Biomol. Chem. 2023;21:8695–8701. doi: 10.1039/D3OB01227A. [DOI] [PubMed] [Google Scholar]
- 9.Klumpp D.A., Yeung K.Y., Prakash G.K.S., Olah G.A. Preparation of 3,3-Diaryloxindoles by Superacid-Induced Condensations of Isatins and Aromatics with a Combinatorial Approach. J. Org. Chem. 1998;63:4481–4484. doi: 10.1021/jo980588g. [DOI] [Google Scholar]
- 10.Rokade B.V., Guiry P.J. Synthesis of α-Aryl Oxindoles by Friedel–Crafts Alkylation of Arenes. J. Org. Chem. 2020;85:6172–6180. doi: 10.1021/acs.joc.0c00370. [DOI] [PubMed] [Google Scholar]
- 11.Vinoth N., Lalitha A. Catalyst-Free Three-Component Synthesis, Antibacterial, Antifungal, and Docking Studies of Spiroindoline Derivatives. Polycyclic Aromat. Compd. 2022;42:517–533. doi: 10.1080/10406638.2020.1744025. [DOI] [Google Scholar]
- 12.Cheng X., Vellalath S., Goddard R., List B. Direct Catalytic Asymmetric Synthesis of Cyclic Aminals from Aldehydes. J. Am. Chem. Soc. 2008;130:15786–15787. doi: 10.1021/ja8071034. [DOI] [PubMed] [Google Scholar]
- 13.Kinsella M., Duggan P.G., Lennon C.M. Screening of Simple N-Aryl and N-Heteroaryl Pyrrolidine Amide Organocatalysts for the Enantioselective Aldol Reaction of Acetone with Isatin. Tetrahedron: Asymmetry. 2011;22:1423–1433. doi: 10.1016/j.tetasy.2011.07.016. [DOI] [Google Scholar]
- 14.Gavendova M., Lennon C.M., Coffey L., Manesiotis P., Kinsella M. Novel β -amino Amide Organocatalysts for the Synthesis of Pharmaceutically Relevant Oxindoles. ChemistrySelect. 2019;4:8246–8254. doi: 10.1002/slct.201901360. [DOI] [Google Scholar]
- 15.Dabiri M., Mohammadi A.A., Qaraat H. An Efficient and Convenient Protocol for the Synthesis of Novel 1′H-Spiro[Isoindoline-1,2′-Quinazoline]-3,4′(3′H)-Dione Derivatives. Monatsh Chem. 2009;140:401–404. doi: 10.1007/s00706-008-0040-3. [DOI] [Google Scholar]
- 16.Mustaque K.M., Subramani A., Shabeer T.K., Thajudeen H., Ahamed V.S.J. Amino Acid Catalyzed Synthesis of 2,3-Dihydroquinazolin-4(1H)-One Derivatives. LOC. 2018;15:246–250. doi: 10.2174/1570178614666171130154814. [DOI] [Google Scholar]
- 17.Shaabani A., Maleki A., Mofakham H. Click Reaction: Highly Efficient Synthesis of 2,3-Dihydroquinazolin-4(1 H)-Ones. Synth. Commun. 2008;38:3751–3759. doi: 10.1080/00397910802213802. [DOI] [Google Scholar]
- 18.Revathy K., Lalitha A. P-TSA-Catalyzed Synthesis of Spiroquinazolinones. J. Iran. Chem. Soc. 2015;12:2045–2049. doi: 10.1007/s13738-015-0680-2. [DOI] [Google Scholar]
- 19.Safaei-Ghomi J., Teymuri R. A Three-component Process for the Synthesis of 2,3-dihydroquinazolin-4(1H)-one Derivatives Using Nanosized Nickel Aluminate Spinel Crystals as Highly Efficient Catalysts. J. Chin. Chem. Soc. 2019;66:1490–1498. doi: 10.1002/jccs.201800450. [DOI] [Google Scholar]
- 20.Narasimhulu M., Lee Y.R. Ethylenediamine Diacetate-Catalyzed Three-Component Reaction for the Synthesis of 2,3-Dihydroquinazolin-4(1H)-Ones and Their Spirooxindole Derivatives. Tetrahedron. 2011;67:9627–9634. doi: 10.1016/j.tet.2011.08.018. [DOI] [Google Scholar]
- 21.Mane M.M., Pore D.M. Sulfamic Acid as Energy Efficient Catalyst for Synthesis of Flurophores, 1-H-Spiro [Isoindoline-1,2′-Quinazoline]-3,4′(3′H)-Diones. J. Chem. Sci. 2016;128:657–662. doi: 10.1007/s12039-016-1047-7. [DOI] [Google Scholar]
- 22.Fallah-Mehrjardi M., Kalantari S. A Brønsted Acid Ionic Liquid Immobilized on Fe3O4@SiO2 Nanoparticles as an Efficient and Reusable Solid Acid Catalyst for the Synthesis of 2,3-Dihydroquinazolin-4(1H)-Ones. Russ. J. Org. Chem. 2020;56:298–306. doi: 10.1134/S1070428020020207. [DOI] [Google Scholar]
- 23.Subba Reddy B.V., Venkateswarlu A., Madan C., Vinu A. Cellulose-SO3H: An Efficient and Biodegradable Solid Acid for the Synthesis of Quinazolin-4(1H)-Ones. Tetrahedron Lett. 2011;52:1891–1894. doi: 10.1016/j.tetlet.2011.02.030. [DOI] [Google Scholar]
- 24.Rambabu D., Raja G., Yogi Sreenivas B., Seerapu G.P.K., Lalith Kumar K., Deora G.S., Haldar D., Rao M.V.B., Pal M. Spiro Heterocycles as Potential Inhibitors of SIRT1: Pd/C-Mediated Synthesis of Novel N-Indolylmethyl Spiroindoline-3,2′-Quinazolines. Bioorg. Med. Chem. Lett. 2013;23:1351–1357. doi: 10.1016/j.bmcl.2012.12.089. [DOI] [PubMed] [Google Scholar]
- 25.Hemalatha K., Madhumitha G., Vasavi C.S., Munusami P. 2,3-Dihydroquinazolin-4(1H)-Ones: Visible Light Mediated Synthesis, Solvatochromism and Biological Activity. J. Photochem. Photobiol. B. 2015;143:139–147. doi: 10.1016/j.jphotobiol.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 26.Novanna M., Kannadasan S., Shanmugam P. Phosphotungstic Acid Mediated, Microwave Assisted Solvent-Free Green Synthesis of Highly Functionalized 2′-Spiro and 2,3-Dihydro Quinazolinone and 2-Methylamino Benzamide Derivatives from Aryl and Heteroaryl 2-Amino Amides. Tetrahedron Lett. 2019;60:201–206. doi: 10.1016/j.tetlet.2018.12.011. [DOI] [Google Scholar]
- 27.Ghosh S.K., Nagarajan R. Deep Eutectic Solvent Mediated Synthesis of Quinazolinones and Dihydroquinazolinones: Synthesis of Natural Products and Drugs. RSC Adv. 2016;6:27378–27387. doi: 10.1039/C6RA00855K. [DOI] [Google Scholar]
- 28.Ramesh R., Nagasundaram N., Meignanasundar D., Vadivel P., Lalitha A. Glycerol Assisted Eco-Friendly Strategy for the Facile Synthesis of 4,4′-(Arylmethylene)Bis(3-Methyl-1H-Pyrazol-5-Ols) and 2-Aryl-2,3-Dihydroquinazolin-4(1H)-Ones under Catalyst-Free Conditions. Res. Chem. Intermed. 2017;43:1767–1782. doi: 10.1007/s11164-016-2728-z. [DOI] [Google Scholar]
- 29.Jiang Y., Liu Y., Tu S.-J., Shi F. Enantioselective Synthesis of Biologically Important Spiro[Indoline-3,2′-Quinazolines] via Catalytic Asymmetric Isatin-Involved Tandem Reactions. Tetrahedron: Asymmetry. 2013;24:1286–1296. doi: 10.1016/j.tetasy.2013.08.019. [DOI] [Google Scholar]
- 30.Wang L., Jiang T., Li P., Sun R., Zuo Z. Asymmetric Syntheses of Spirooxindole-dihydroquinazolinones by Cyclization Reactions between N-substituted Anthranilamides and Isatins. Adv. Synth. Catal. 2018;360:4832–4836. doi: 10.1002/adsc.201801329. [DOI] [Google Scholar]
- 31.Nakamura S., Wada T., Takehara T., Suzuki T. Catalytic Enantioselective Synthesis of N, N -Acetals from α-Dicarbonyl Compounds Using Chiral Imidazoline-Phosphoric Acid Catalysts. Adv. Synth. Catal. 2020;362:5374–5379. doi: 10.1002/adsc.202001128. [DOI] [Google Scholar]
- 32.Bergman J., Arewång C.-J., Svensson P.H. Oxidative Ring Expansion of Spirocyclic Oxindole Derivatives. J. Org. Chem. 2014;79:9065–9073. doi: 10.1021/jo501269f. [DOI] [PubMed] [Google Scholar]
- 33.Ötvös S.B., Georgiádes Á., Mészáros R., Kis K., Pálinkó I., Fülöp F. Continuous-Flow Oxidative Homocouplings without Auxiliary Substances: Exploiting a Solid Base Catalyst. J. Catal. 2017;348:90–99. doi: 10.1016/j.jcat.2017.02.012. [DOI] [Google Scholar]
- 34.Bisht G.S., Dunchu T.D., Gnanaprakasam B. Synthesis of Quaternary Spirooxindole 2H -Azirines under Batch and Continuous Flow Condition and Metal Assisted Umpolung Reactivity for the Ring-Opening Reaction. Chem. Asian J. 2021;16:656–665. doi: 10.1002/asia.202001418. [DOI] [PubMed] [Google Scholar]
- 35.Liu D., Xu H. Electrochemical Rearrangement of Indoles to Spirooxindoles in Continuous Flow. Eur. J. Org. Chem. 2023;26:e202200987. doi: 10.1002/ejoc.202200987. [DOI] [Google Scholar]
- 36.Stolle A., Szuppa T., Leonhardt S.E.S., Ondruschka B. Ball Milling in Organic Synthesis: Solutions and Challenges. Chem. Soc. Rev. 2011;40:2317–2329. doi: 10.1039/c0cs00195c. [DOI] [PubMed] [Google Scholar]
- 37.James S.L., Adams C.J., Bolm C., Braga D., Collier P., Friščić T., Grepioni F., Harris K.D.M., Hyett G., Jones W., et al. Mechanochemistry: Opportunities for New and Cleaner Synthesis. Chem. Soc. Rev. 2012;41:413–447. doi: 10.1039/C1CS15171A. [DOI] [PubMed] [Google Scholar]
- 38.Rodríguez B., Bruckmann A., Rantanen T., Bolm C. Solvent-Free Carbon-Carbon Bond Formations in Ball Mills. Adv. Synth. Catal. 2007;349:2213–2233. doi: 10.1002/adsc.200700252. [DOI] [Google Scholar]
- 39.Miklós F., Hum V., Fülöp F. Eco-Friendly Syntheses of 2,2-Disubstituted- and 2-Spiroquinazolinones. Arkivoc. 2014;2014:25–37. doi: 10.3998/ark.5550190.p008.717. [DOI] [Google Scholar]
- 40.Zhang J., Liu J., Ma Y., Ren D., Cheng P., Zhao J., Zhang F., Yao Y. One-Pot Synthesis and Antifungal Activity against Plant Pathogens of Quinazolinone Derivatives Containing an Amide Moiety. Bioorg. Med. Chem. Lett. 2016;26:2273–2277. doi: 10.1016/j.bmcl.2016.03.052. [DOI] [PubMed] [Google Scholar]
- 41.Gamal Al-kaf A. Introductory Chapter: The Newest Research in Quinazolinone and Quinazoline Derivatives. In: Gamal Al-kaf A., editor. Quinazolinone and Quinazoline Derivatives. IntechOpen; London, UK: 2020. [Google Scholar]
- 42.Dutta A., Sarma D. Recent Advances in the Synthesis of Quinazoline Analogues as Anti-TB Agents. Tuberculosis. 2020;124:101986. doi: 10.1016/j.tube.2020.101986. [DOI] [PubMed] [Google Scholar]
- 43.Pettus L.H., Andrews K.L., Booker S.K., Chen J., Cee V.J., Chavez F., Chen Y., Eastwood H., Guerrero N., Herberich B., et al. Discovery and Optimization of Quinazolinone-Pyrrolopyrrolones as Potent and Orally Bioavailable Pan-Pim Kinase Inhibitors. J. Med. Chem. 2016;59:6407–6430. doi: 10.1021/acs.jmedchem.6b00610. [DOI] [PubMed] [Google Scholar]
- 44.Dohle W., Jourdan F.L., Menchon G., Prota A.E., Foster P.A., Mannion P., Hamel E., Thomas M.P., Kasprzyk P.G., Ferrandis E., et al. Quinazolinone-Based Anticancer Agents: Synthesis, Antiproliferative SAR, Antitubulin Activity, and Tubulin Co-Crystal Structure. J. Med. Chem. 2018;61:1031–1044. doi: 10.1021/acs.jmedchem.7b01474. [DOI] [PubMed] [Google Scholar]
- 45.Hamiaux C., Larsen L., Lee H.W., Luo Z., Sharma P., Hawkins B.C., Perry N.B., Snowden K.C. Chemical Synthesis and Characterization of a New Quinazolinedione Competitive Antagonist for Strigolactone Receptors with an Unexpected Binding Mode. Biochem. J. 2019;476:1843–1856. doi: 10.1042/BCJ20190288. [DOI] [PubMed] [Google Scholar]
- 46.Gero T.W., Heppner D.E., Beyett T.S., To C., Azevedo S.C., Jang J., Bunnell T., Feru F., Li Z., Shin B.H., et al. Quinazolinones as Allosteric Fourth-Generation EGFR Inhibitors for the Treatment of NSCLC. Bioorg. Med. Chem. Lett. 2022;68:128718. doi: 10.1016/j.bmcl.2022.128718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwashita A., Hattori K., Yamamoto H., Ishida J., Kido Y., Kamijo K., Murano K., Miyake H., Kinoshita T., Warizaya M., et al. Discovery of Quinazolinone and Quinoxaline Derivatives as Potent and Selective Poly(ADP-ribose) Polymerase-1/2 Inhibitors. FEBS Lett. 2005;579:1389–1393. doi: 10.1016/j.febslet.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 48.Bouley R., Kumarasiri M., Peng Z., Otero L.H., Song W., Suckow M.A., Schroeder V.A., Wolter W.R., Lastochkin E., Antunes N.T., et al. Discovery of Antibiotic (E)-3-(3-Carboxyphenyl)-2-(4-Cyanostyryl)Quinazolin-4(3H)-One. J. Am. Chem. Soc. 2015;137:1738–1741. doi: 10.1021/jacs.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janardhanan J., Bouley R., Martínez-Caballero S., Peng Z., Batuecas-Mordillo M., Meisel J.E., Ding D., Schroeder V.A., Wolter W.R., Mahasenan K.V., et al. The Quinazolinone Allosteric Inhibitor of PBP 2a Synergizes with Piperacillin and Tazobactam against Methicillin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 2019;63:e02637-18. doi: 10.1128/AAC.02637-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei M., Chai W.-M., Wang R., Yang Q., Deng Z., Peng Y. Quinazolinone Derivatives: Synthesis and Comparison of Inhibitory Mechanisms on α-Glucosidase. Bioorg. Med. Chem. 2017;25:1303–1308. doi: 10.1016/j.bmc.2016.09.042. [DOI] [PubMed] [Google Scholar]
- 51.Taayoshi F., Iraji A., Moazzam A., Soleimani M., Asadi M., Pedrood K., Akbari M., Salehabadi H., Larijani B., Adibpour N., et al. Synthesis, Molecular Docking, and Cytotoxicity of Quinazolinone and Dihydroquinazolinone Derivatives as Cytotoxic Agents. BMC Chem. 2022;16:35. doi: 10.1186/s13065-022-00825-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhananjaya G., Venkateshwarlu R., Madhubabu M.V., Raghunadh A., Murthy V.N., Reddy S.P., Anna V.R., Kapavarapu R., Pal M. Synthesis of 2,3-Dihydroquinazolin-4(1H)-One Derivatives as Potential Inhibitors of TNF-α. J. Mol. Struct. 2023;1287:135668. doi: 10.1016/j.molstruc.2023.135668. [DOI] [Google Scholar]
- 53.Stájer G., Szabó A.E., Fülöp F., Bernáth G., Sohár P. Stereochemical Studies, 106.–Saturated Heterocycles, 110 Synthesis of Methylene-bridged Partially Saturated Quinazolones. Chem. Ber. 1987;120:259–264. doi: 10.1002/cber.19871200302. [DOI] [Google Scholar]
- 54.Stájer G., Szabó A.E., Bernáth G., Sohár P. Stereochemical Studies. Part 103. Saturated Heterocycles Part 107. Preparation of 3-Mono- and 2,3-Di-Substituted Pyrimidin-4(3H)-Ones in Retro–Diels–Alder Reactions. The Correct 1,2-Disubstituted Structure of the Compounds Previously Described as 2,3-Disubstituted Derivatives. J. Chem. Soc. Perkin Trans. 1. 1987:237–240. doi: 10.1039/P19870000237. [DOI] [Google Scholar]
- 55.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 56.Costanzo M.J., Yabut S.C., Zhang H.-C., White K.B., De Garavilla L., Wang Y., Minor L.K., Tounge B.A., Barnakov A.N., Lewandowski F., et al. Potent, Nonpeptide Inhibitors of Human Mast Cell Tryptase. Synthesis and Biological Evaluation of Novel Spirocyclic Piperidine Amide Derivatives. Bioorg. Med. Chem. Lett. 2008;18:2114–2121. doi: 10.1016/j.bmcl.2008.01.093. [DOI] [PubMed] [Google Scholar]
- 57.Lohning A.E., Levonis S.M., Williams-Noonan B., Schweiker S.S. A Practical Guide to Molecular Docking and Homology Modelling for Medicinal Chemists. Curr. Top. Med. Chem. 2017;17:2023–2040. doi: 10.2174/1568026617666170130110827. [DOI] [PubMed] [Google Scholar]
- 58.Erickson J.A., Jalaie M., Robertson D.H., Lewis R.A., Vieth M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem. 2004;47:45–55. doi: 10.1021/jm030209y. [DOI] [PubMed] [Google Scholar]
- 59.Hsu K.-C., Chen Y.-F., Lin S.-R., Yang J.-M. iGEMDOCK: A Graphical Environment of Enhancing GEMDOCK Using Pharmacological Interactions and Post-Screening Analysis. BMC Bioinf. 2011;12:S33. doi: 10.1186/1471-2105-12-S1-S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dallakyan S., Olson A.J. Small-Molecule Library Screening by Docking with PyRx. Chem. Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 61.Alamri M.A., Afzal O., Akhtar M.J., Karim S., Husain M., Alossaimi M.A., Riadi Y. Synthesis, in Silico and in Vitro Studies of Novel Quinazolinone Derivatives as Potential SARS-CoV-2 3CLpro Inhibitors. Arabian J. Chem. 2024;17:105384. doi: 10.1016/j.arabjc.2023.105384. [DOI] [Google Scholar]
- 62.Daina A., Michielin O., Zoete V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings 1PII of Original Article: S0169-409X(96)00423-1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997) 3–25. 1. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 64.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 65.Huey R., Morris G.M., Olson A.J., Goodsell D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 66.Schrödinger, LLC . The PyMOL Molecular Graphics System, Version 2.5. Schrödinger, LLC.; New York, NY, USA: 2021. [Google Scholar]
- 67.BIOVIA, Dassault Systèmes . BIOVIA Discovery Studio, 4.5. Dassault Systèmes; San Diego, CA, USA: 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article or Supplementary Material.