

In this issue of the Journal of Thrombosis and Haemostasis, Choi et al. [1] report a novel role of von Willebrand factor (VWF) in the pathogenesis of venous thrombosis associated with endotoxemia. In an inferior vena cava (IVC)-stenosis model, venous thrombi were significantly increased in the presence of lipopolysaccharides (LPSs). Importantly, however, this increased risk was mitigated by polyclonal anti-VWF antibodies. On the basis of their findings, the authors proposed that VWF may be more important in venous thrombus initiation than clot propagation. Collectively, these data move us closer to understanding the central role of VWF in linking inflammation and hemostasis in vivo.
VWF plays a crucial role in normal hemostasis by enabling platelet plug formation at the sites of vascular injury [2]. VWF also contributes to thrombin generation by acting as a carrier for procoagulant factor VIII (FVIII). Interaction with VWF protects FVIII against proteolytic degradation and premature clearance [3]. Beyond these established hemostatic functions, recent studies have described novel additional biological functions of VWF. Specific roles of VWF in regulating angiogenesis [4], wound healing [5], and tumor cell biology [6] have been proposed. In addition, previous studies have shown that plasma VWF antigen (VWF:Ag) levels are significantly increased in patients with severe sepsis and that VWF levels correlate inversely with clinical outcomes [7,8]. Furthermore, accumulating data have suggested that VWF does not just serve as a marker of endothelial cell activation and Weibel Palade body exocytosis but instead that VWF plays a direct role in modulating inflammation [6]. Critically, however, the molecular mechanisms through which VWF exerts immunomodulatory effects remain poorly understood.
Under static and flow conditions, VWF has been shown to bind to both polymorphonuclear leucocytes and activated monocytes [9]. Polymorphonuclear interaction with VWF was mediated, at least in part, through P-selectin glycoprotein ligand-1 and β2-integrins expressed on the leucocyte surface [9]. Similarly, several macrophage receptors bind to VWF [10]. These include the low-density lipoprotein receptor-related protein-1 [11], scavenger receptor class A member 1 [12], and macrophage galactose-type lectin [13]. Recently, Drakeford et al. [14] reported that VWF binding directly triggers proinflammatory intracellular signaling in macrophages. Specifically, VWF induced activation of the MAPinase signaling pathway to induce the phosphorylation of p38 and Janus kinase. In addition, VWF also promoted the activation of nuclear factor kappa B by the phosphorylation of its regulatory subunit IKBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha). VWF-dependent signaling resulted in macrophage polarization that resembled the proinflammatory “M1” phenotype, characterized by enhanced glycolysis and increased expression of proinflammatory cytokines and chemokines [14]. Collectively, these data define a biological role of VWF in linking primary hemostasis to innate immunity at the sites of vascular injury. A potent proinflammatory role of VWF is supported by previous in vivo studies that demonstrated that VWF also contributes to the regulation of endothelial cell barrier permeability and neutrophil extravasation.
Clinical studies have shown that elevated plasma levels of the VWF-FVIII complex are associated with a dose-dependent increased risk of venous thromboembolism (VTE) [15].
Similarly, ABO blood group affects plasma VWF levels, with significantly reduced levels in individuals with blood group O, who are consequently at a lower risk of VTE [16]. Because VWF can impact thromboinflammation through multiple mechanisms, previous studies have investigated the role of VWF in the pathogenesis of deep vein thrombosis (DVT). Brill et al. [17] reported that VWF-deficient mice were significantly protected against thrombosis in a murine IVC thrombosis model. Interestingly, this protective effect was shown to be independent of reduced plasma FVIII levels in these mice because repeated infusions of FVIII did not reverse the protective effect in VWF-deficient mice. Conversely, inhibition of the interaction of the VWF A1 domain with platelets prevented thrombosis in wild-type mice [16]. Together, these findings suggest that VWF-platelet interactions play a role in DVT pathogenesis. Subsequently, Michels et al. [18] investigated whether VWF contributed to VTE in a murine model of diet-induced obesity. Obesity is associated with chronic proinflammatory activity and a significantly increased thrombotic risk. Using the IVC-stenosis model of DVT, they demonstrated that obese mice developed larger venous thrombi. Moreover, thrombi from obese mice contained substantially more VWF and leucocytes than those from nonobese littermate controls. In addition, immunofluorescent studies of the thrombi confirmed that leucocytes were abundant in VWF-rich areas of the murine thrombi. Finally, infusion of a polyclonal anti-VWF antibody or an anti-A1 domain nanobody was again protective against DVT in this model [18].
To further elucidate the role of VWF in regulating immuno-thrombosis in vivo, Choi et al. [1] combined the established murine IVC-stenosis model of DVT with the LPS-mediated model of endotoxemia. They observed a significant increase in venous thrombosis in the presence of LPS (76% of LPS-treated mice developed thrombosis compared with only 40% of untreated controls; p = .045) (Figure). In contrast to the findings observed in obese mice, there was no increase in thrombus burden in LPS-treated mice. In keeping with previous studies [17,18], the increased risk of thrombosis in LPS-treated mice was significantly attenuated in the presence of a polyclonal anti-VWF antibody. Because thrombus weight was not altered in the presence or absence of anti-VWF, the authors proposed that VWF may be more important in thrombus initiation than clot propagation. However, the mechanistic basis for this still needs to be clarified. The infusion of recombinant human ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) had no significant effect on clot formation, suggesting that the role played by VWF in venous thrombus clot initiation is multimer independent, at least in this murine model. Finally, to better understand cellular interactions within thrombi, Choi et al. [1] proceeded to use imaging mass cytometry to analyze clot architecture in the presence or absence of LPS. They identified distinct regions of red thrombus (consisting mainly of erythrocytes and fibrin) and white thrombus (primarily composed of platelets and fibrin). Similar to obese mice [18], the proportion of red thrombus was also significantly increased in LPS-treated mice. VWF was predominantly located within the white thrombus, where it was associated with CD62P (P-selectin) (a marker of platelet activation) and fibrin but not with neutrophil extracellular traps. In the presence of LPS, the amount of VWF and its localization within the clot relative to other constituents were also altered.
FIGURE.
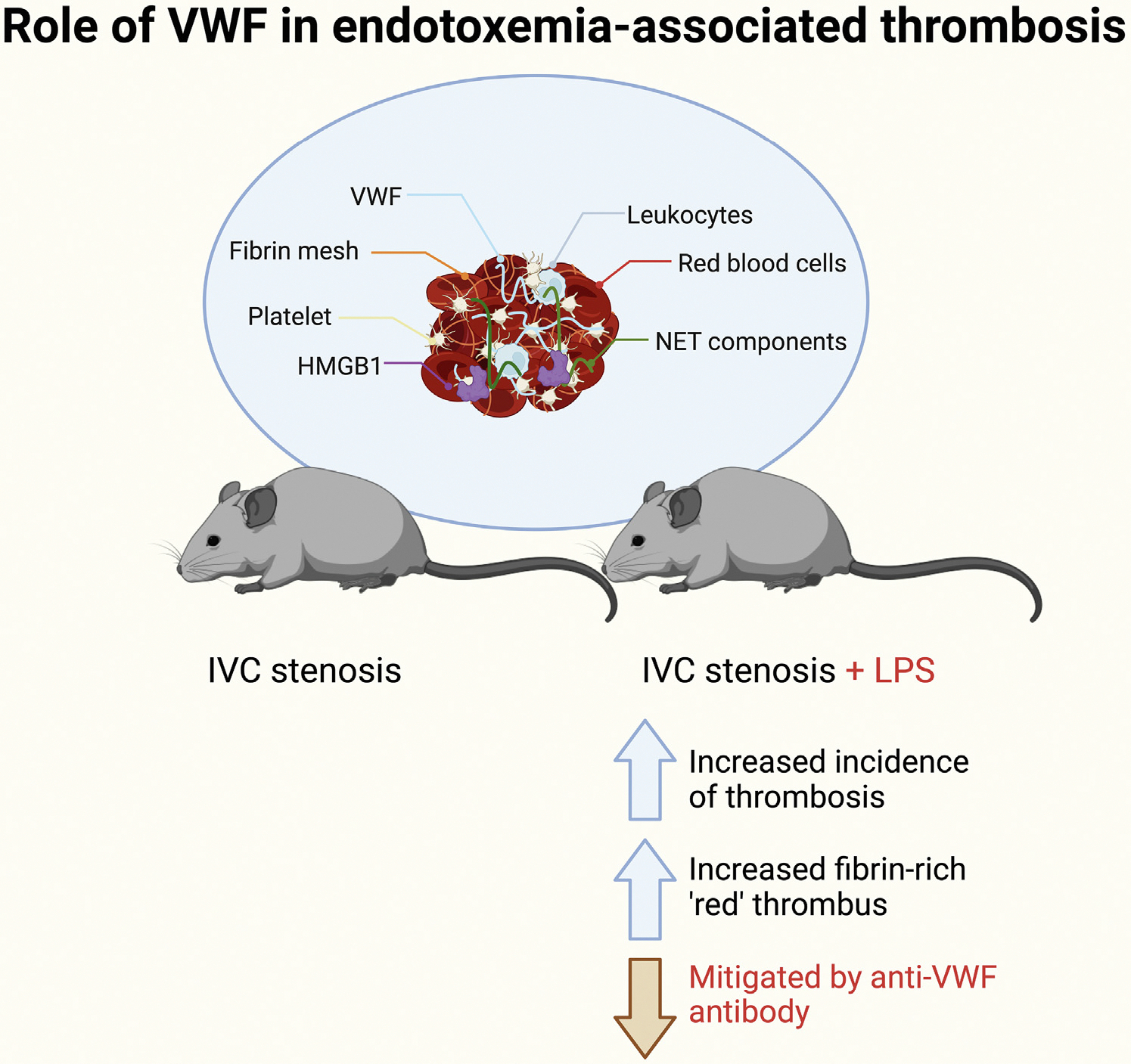
Role of von Willebrand factor in endotoxemia-associated thrombosis. Venous thrombi were significantly increased in endotoxemic mice after IVC stenosis. Endotoxemia also altered thrombus architecture, with increased erythrocyte and fibrin thrombi isolated from lipopolysaccharide (LPS)-treated mice. Importantly, however, this increased the risk of thrombus development in LPS-treated mice, which was mitigated by the inhibition of von Willebrand factor activity. Localization of cellular and protein clot components (fibrin, leukocyte, NETs, HMGB1, and platelets; pictured) within thrombi was not affected by LPS. HMGB1, high mobility group box 1; IVC, inferior vena cava; NET, neutrophil extracellular trap; VWF, von Willebrand factor.
Taken together, these new data provide additional insights into the significance of VWF in DVT pathogenesis. As such, they may be of direct clinical relevance across several different types of sepsis, including, for example, in understanding the mechanisms underlying thrombotic risk in patients with acute COVID-19 [19]. In addition, the findings further support the hypothesis that VWF plays critical roles in modulating crosstalk between inflammation and hemostasis in vivo. Further studies will be necessary to elucidate the biological mechanisms involved and to determine whether VWF-targeted therapies may be beneficial in this context.
ACKNOWLEDGMENTS
J.O.D. was supported by funds from the NIH for the Zimmerman Program (HL081588) and a Science Foundation Ireland Frontiers for the Future Award (20/FFP-A/8952). R.J.S.P. was supported by Science Foundation Ireland (21/FFP-A/8859), the National Children’s Research Centre (PRPG/H/18/315), and the Health Research Board (ILP-POR-2022–060).
Footnotes
DECLARATION OF COMPETING INTERESTS
There are no competing interests to disclose.
REFERENCES
- [1].Choi SJ, Wyer CN, Rapkin L, Cormier M, Hindmarch CC, Nesbitt K, Michels A, Hopman W, Swystun LL, Lillicrap D. The mechanistic and structural role of von Willebrand factor in endotoxemia-enhanced deep vein thrombosis in mice. J Thromb Haemost. 2023;21:586–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lenting PJ, Christophe OD, Denis CV. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015;125:2019–28. [DOI] [PubMed] [Google Scholar]
- [3].Turecek PL, Johnsen JM, Pipe SW, O’Donnell JS, iPATH Study Group. Biological mechanisms underlying inter-individual variation in factor VIII clearance in haemophilia. Haemophilia. 2020;26:575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, Payne EM, Haskard DO, Hughes AD, Cutler DF, Laffan MA, Randi AM. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ishihara J, Ishihara A, Starke RD, Peghaire CR, Smith KE, McKinnon TA, Tabata Y, Sasaki K, White MJ, Fukunaga K, Laffan MA, Lutolf MP, Randi AM, Hubbell JA. The heparin binding domain of von Willebrand factor binds to growth factors and promotes angiogenesis in wound healing. Blood. 2019;133:2559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].O’Sullivan JM, Preston RJ, Robson T, O’Donnell JS. Emerging roles for von Willebrand factor in cancer cell biology. Semin Thromb Hemost. 2018;44:159–66. [DOI] [PubMed] [Google Scholar]
- [7].Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15:1285–94. [DOI] [PubMed] [Google Scholar]
- [8].O’Sullivan JM, Preston RJ, O’Regan N, O’Donnell JS. Emerging roles for hemostatic dysfunction in malaria pathogenesis. Blood. 2016;127:2281–8. [DOI] [PubMed] [Google Scholar]
- [9].Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, Denis CV. P-selectin glycoprotein ligand 1 and beta2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108:3746–52. [DOI] [PubMed] [Google Scholar]
- [10].O’Sullivan JM, Ward S, Lavin M, O’Donnell JS. von Willebrand factor clearance–biological mechanisms and clinical significance. Br J Haematol. 2018;183:185–95. [DOI] [PubMed] [Google Scholar]
- [11].Rastegarlari G, Pegon JN, Casari C, Odouard S, Navarrete AM, Saint-Lu N, van Vlijmen BJ, Legendre P, Christophe OD, Denis CV, Lenting PJ. Macrophage LRP1 contributes to the clearance of von Willebrand factor. Blood. 2012;119:2126–34. [DOI] [PubMed] [Google Scholar]
- [12].Wohner N, Muczynski V, Mohamadi A, Legendre P, Proulle V, Ayme G, Christophe OD, Lenting PJ, Denis CV, Casari C. Macrophage scavenger receptor SR-AI contributes to the clearance of von Willebrand factor. Haematologica. 2018;103:728–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ward SE, O’Sullivan JM, Drakeford C, Aguila S, Jondle CN, Sharma J, Fallon PG, Brophy TM, Preston RJS, Smyth P, Sheils O, Chion A, O’Donnell JS. A novel role for the macrophage galactose-type lectin receptor in mediating von Willebrand factor clearance. Blood. 2018;131:911–6. [DOI] [PubMed] [Google Scholar]
- [14].Drakeford C, Aguila S, Roche F, Hokamp K, Fazavana J, Cervantes MP, Curtis AM, Hawerkamp HC, Dhami SP, Charles-Messance H, Hackett EE, Chion A, Ward S, Ahmad A, Schoen I, Breen E, Keane J, Murphy R, Preston RJ, O’Sullivan JM, et al. von Willebrand factor links primary hemostasis to innate immunity. Nat Commun. 2022;13:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].O’Donnell J, Laffan M. Elevated plasma factor VIII levels—a novel risk factor for venous thromboembolism. Clin Lab. 2001;47:1–6. [PubMed] [Google Scholar]
- [16].Ward SE, O’Sullivan JM, O’Donnell JS. The relationship between ABO blood group, von Willebrand factor, and primary hemostasis. Blood. 2020;136:2864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, Wakefield TW, Lammle B, Massberg S, Wagner DD. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Michels A, Dwyer CN, Mewburn J, Nesbitt K, Kawecki C, Lenting P, Swystun LL, Lillicrap D. Von Willebrand factor is a critical mediator of deep vein thrombosis in a mouse model of diet-induced obesity. Arterioscler Thromb Vasc Biol. 2020;40:2860–74. [DOI] [PubMed] [Google Scholar]
- [19].O’Donnell JS, Peyvandi F, Martin-Loeches I. Pulmonary immuno-thrombosis in COVID-19 ARDS pathogenesis. Intensive Care Med. 2021;47:899–902. [DOI] [PMC free article] [PubMed] [Google Scholar]