

Abstract
PURPOSE
We introduce a novel algorithmic approach to design phase I trials for oncology drug combinations.
METHODS
Our proposed Toxicity Adaptive Lists Design (TALE) is straightforward to implement, requiring the prespecification of a small number of parameters that define rules governing dose escalation, de-escalation, or reassessment of previously explored dose levels. These rules effectively regulate dose exploration and control the number of toxicities. A key feature of TALE is the possibility of simultaneous assignment of multiple-dose combinations that are deemed safe by previously accrued data.
RESULTS
A numerical study shows that TALE shares comparable operative characteristics, in terms of identification of the maximum tolerated dose (MTD), to alternative approaches such as the Bayesian optimal interval design, the COPULA, the product of independent beta probabilities escalation, and the continual reassessment method for partial ordering designs while reducing the risk of overdosing patients.
CONCLUSION
The proposed TALE design provides a favorable balance between maintaining patient safety and accurately identifying the MTD. To facilitate the use of TALE, we provide a user-friendly R Shiny application and an R package for computing relevant operating characteristics, such as the risk of assigning highly toxic dose combinations.
INTRODUCTION
Phase I clinical trials typically aim to identify the maximum tolerated dose (MTD) of a therapy. In oncology, the therapy often combines two or more agents or drugs that can be administered at different dose levels (DLs).1 Simple, efficient, and practical phase I trial designs are needed to identify safe and effective dose combinations and to assess their toxicity (Fig 1). These designs need to be simple to describe and implement. Additionally, they must control the risk of severe toxicities during the study while generating data that allow investigators to estimate the relationship between DLs of the therapy and toxicities.
FIG 1.
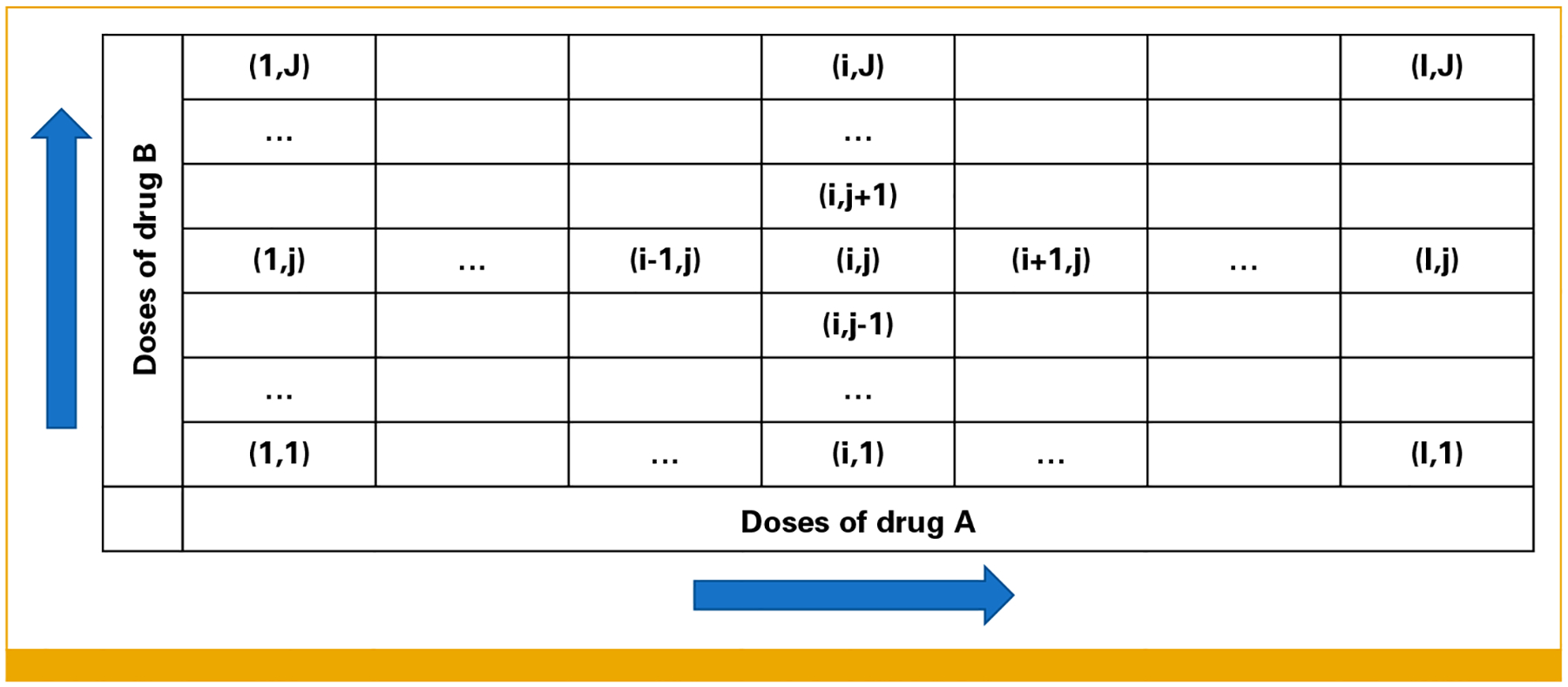
Example of a grid in which DLs of drug A (rows) and drug B (columns) are organized. Doses for drug A are increasing from left to right and for drug B from bottom to top. DLs, dose levels.
There are several phase I trial designs for drug combinations. Some convert the study of dose combinations into a series of single-drug subtrials, often requiring a large sample size. In each subtrial, the goal is to identify the MTD of one drug while keeping the other drug at a fixed dose. Others investigate combinations of DLs from a prespecified grid of doses. Designs can be broadly classified as algorithm-based and model-based.2 Algorithm-based methods, such as the 3 + 3 design and extensions,3–6 are commonly used for their simplicity but might have suboptimal operating characteristics. Model-based designs use parametric models to describe the relationship between DLs and toxicities and can incorporate data from previous studies on single drugs.7–21 We also mention model-assisted designs that follow easy-to-interpret rules for sequential decision making, which in turn are selected using dose-toxicity models. Notable examples include the Bayesian optimal interval (BOIN) design,22 the modified toxicity probability interval design,23,24 and the Keyboard design.4 Comprehensive comparisons of model-based and model-assisted designs for drug combinations have been previously published.1,8,25–27
Previous studies comparing phase I trial designs suggest that model-based designs tend to outperform algorithm-based designs.2,3,8,9 However, stakeholders (eg, sponsors and investigators) might perceive model-based designs as black boxes because the criteria underlying the decisions to escalate and de-escalate DLs can be difficult to interpret.3–5 Additionally, they require sequential inference on the relation between DLs and toxicities and rely on parametric models whose assumptions might be violated.1,26
We introduce an algorithm-based approach for phase I drug combination trials: the Toxicity Adaptive Lists Design (TALE). TALE is a multistage design that, at each stage, defines a list of explorable DLs of the combination therapy. DLs are included in the list only if lower doses have been previously explored and the available data suggest acceptable toxicity rates in the study population. The sequential rules defining the study design control the risk of assigning patients to toxic DLs and generate data for rapid characterization of the relationship between DLs and toxicities. A distinctive feature of TALE is that patients enrolled during a phase of the study might be assigned to multiple DLs. When patients’ accrual is sufficiently rapid, this can lead to a considerable reduction in the trial duration compared with other phase I designs that assign a single cohort per phase. Moreover, simultaneous exploration of DLs tends to reduce the number of times the data are analyzed for sequential decision making during the study.
TALE is straightforward to interpret and practical to implement. We illustrate TALE operating characteristics and discuss comparisons with more complex model-based and model-assisted designs.
A Phase I Study of PK and ATR Inhibitors
We used TALE in a recent phase I clinical trial (Clinical-Trials.gov identifier: NCT05687136; CTEP 10527). In this study, we explore the combination of two anticancer drugs: the DNA-PK inhibitor peposertib (M3814) and the ATR inhibitor tuvusertib (M1774). DNA-PK and ATR are critical proteins involved in DNA repair and in the cellular response to replicative stress. In preclinical models, the combination demonstrates synergistic lethality to cancer cells dependent on the ATR pathway, such as those with mutations in ATM or those with a high degree of replicative stress.28 Both agents have been studied as monotherapies and in combinations with other agents; however, this is the first trial evaluating this specific combination. It will start accruing patients in 2024.
The same strategy is being used in a phase I trial combining gemcitabine with the ATR inhibitor elimusertib (Clinical-Trials.gov identifier: NCT04616534; CTEP 10403).
TALE Design
We consider a design to investigate the toxicity of the combination of two drugs, A and B. The design components are defined in the Data Supplement. DLs for the drugs are included in a grid and labeled as (i, j), where i and j represent the DLs of drugs A and B, respectively (Fig 1). TALE proceeds in stages, and at each stage k, a cohort of n (eg, 3) patients is assigned to each DL in an explorable list. Initially (stage I), the list includes only the lowest dose in the grid. It is then updated sequentially after each stage on the basis of the observed number of dose-limiting toxicities (DLTs) and a set of prespecified rules (Figs 2A and 2B). The trial ends when a sample size of N patients is reached or the list of explorable DLs is empty.
FIG 2.

Illustration of how the list of explorable DLs for the TALE design is updated throughout the trial. The figure includes two components (A and B) to summarize the available data up to stage k and two more components (C and D) translating available data into the decision of which DLs will be assigned during the next stage (k+1). The set Lk includes all the (i, j) dose combinations assigned during the k-th stage. For example, L3 = {(1, 3), (2, 1)} indicates that during the third stage, two distinct dose combinations are explored (1, 3) and (2, 1). DLs, dose levels; TALE, Toxicity Adaptive Lists Design.
A maximum of nmax patients can be assigned to a DL. We indicate with pij the observed toxicity rate at DL (i, j), that is:
After the k-th stage, the pij rates are updated, and TALE identifies the list Lk + 1 of explorable DLs for the next stage. The list is obtained comparing the values of pij with prespecified parameters, λe, λd, and λr (Fig 2B), which categorize the observed toxicity rate of a DL (i, j):
pij ≤ λe low toxicity.
λe < pij ≤ λd intermediate-low toxicity.
λd < pij ≤ λr intermediate toxicity.
pij ≥ λr high toxicity.
Figure 2B associates colors with toxicity levels: green for low toxicity, blue for intermediate-low toxicity, orange for intermediate toxicity, and red for high toxicity. Thresholds λe, λd, and λr are trial-specific (possible values are in the Data Supplement, Table S1). The use of the three parameters, λe, λd, and λr, provides flexibility in defining how many toxicity events suggest escalation, de-escalation, and revisiting of the DLs. TALE can, however, be simplified by choosing the escalation parameters (λe) and letting λd = cλe and λr = cλd for some c > 0. For example, one can choose λe = 0.16 and c = 1.6 corresponding to (λe, λd, λe) = (0.16, 0.272, 0.4624), which, for nmax = 3, defines the same decisions as the vector (λe, λd, λe) = (0.16, 0.24, 0.45) in the Data Supplement (Table S1).
Next, TALE provides the definition of Lk + 1, that is, which DLs can be explored in the stage k + 1. As summarized in Figures 2C and 2D, previous data from the explored doses trigger specific actions: escalation, de-escalation, stay, and reconsider.
Figure 2 provides a complete description of how the available data are sequentially translated into decisions for the subsequent stage of the trial. The figure includes two components (boxes A and B) to summarize the available data up to stage k and two more components (boxes C and D) translating available data into the decision of which DLs will be assigned during the next stage (k + 1).
A DL (i, j) can enter in Lk only if safety is suggested by lower adjacent neighbors that are the DLs (i − 1, j) and (i, j − 1). Figure 2C contains a scheme that uses four questions to determine whether a DL can enter in Lk + 1 on the basis of the available data.
After the list Lk + 1 has been created (Fig 2C), some DLs are excluded (Fig 2D). We remove (1) overly toxic DLs, (2) DLs with nmax previously assigned patients (low uncertainty), and (3) DLs defined by lower or identical doses for both agents as other combinations in the list of explorable DLs. In successive stages, some DLs could be re-explored. A DL with an intermediate toxicity rate at completion of the k-th stage does not continue to enroll patients in the (k + 1)-th stage, but it might be revisited later in the study (Fig 2C). TALE can be described in three steps:
For the first stage k = 1, the list of explorable DLs L1 includes only the lowest DL (1, 1).
During the k-th stage of the trial, the design assigns a cohort of n patients to each DL in Lk. After the k-th stage, the toxicity rates pij are updated, on the basis of the available data, and a new list of explorable DLs Lk + 1 is provided (Figs 2C and 2D).
The trial completes the enrollment when the maximum sample size N is reached or if the list of explorable DLs Lk + 1 for the next stage is empty.
In the final stage, the sample size N could be insufficient to investigate all DLs in Lk + 1. For example, with N = 30 and a cohort size of three patients, after the enrollment of 27 patients during the initial k stages, the data might indicate two explorable DLs for the (k + 1)-th stage. Two cohorts require six patients, but only 30–27 = 3 patients can be enrolled. In this case, the remaining three patients are assigned to one of the explorable DLs, giving priority to the DL with the lowest number of enrolled patients. When patient accrual is completed, the MTD is estimated via isotonic regression.29
Example of TALE Trial
Figure 3 provides a stylized example to illustrate the rules governing the escalation and de-escalation decisions in TALE. This example illustrates how the escalation and de-escalation rules work and does not refer to a specific clinical context. Both drugs A and B have three DLs, and the trial can enroll up to N = 30 patients in cohorts of n = 3 patients. The design parameters are λe = 0.16, λd = 0.24, and λr = 0.45. The Data Supplement (Table S2) indicates the number of DLTs during the course of the study that dictate escalation (Q1 in Fig 2C) and de-escalation (Q4 in Fig 2C) decisions for the subsequent stage of the trial. The maximum number of patients that can be assigned to a single DL is set at nmax = 9. The trial results are illustrated in Figure 3, and the key events are as follows:
Stage I: Three patients receive DL (1, 1) and 0/3 DLTs are reported. Escalation to DLs (1, 2) and (2, 1) is allowed.
Stage II: Two cohorts of patients are assigned to DLs (1, 2) and (2, 1). At DL (1, 2), 0/3 patients experience DLTs; therefore, escalation to DL (1, 3) is allowed. At DL (2, 1), 1/3 patients experience a DLT; therefore, de-escalation to DL (1, 1) is suggested. However, DL (1, 1) will not be explored in stage III since (1, 3) is in the list of explorable DLs.
Stage III: A cohort receives DL (1, 3) and 1/3 patients experience a DLT; therefore, de-escalation to DL (1, 2) is suggested. Moreover, revisit is allowed at DL (2, 1) since p21 ≤ λr, which re-enters the list of explorable DLs.
Stage IV: Two cohorts of patients are assigned to DLs (1, 2) and (2, 1). For both levels, 0/3 patients experience DLTs. In total, 1/6 patients assigned to DL (2, 1) experience a DLT and stay/continue assigning patients to this DL is suggested. At DL (1, 2), 0/6 patients experience DLTs and escalation to DL (1, 3) is allowed.
Stage V: Two cohorts of patients are assigned to DLs (1, 3) and (2, 1). For both DLs, 0/3 patients experience DLTs. On the basis of these outcomes, escalation is allowed to DLs (2, 2) and (3, 1) and stay/continue at DL (1, 3) is suggested.
Stage VI: Three cohorts of patients are assigned to each of the DLs (1, 3), (2, 2), (3, 1). At DL (1, 3) and (2, 2) 1/3 patients experience a DLT, whereas at DL (1, 3) 2/9 patients experience DLTs. The maximum sample size is reached, and the trial stops.
FIG 3.
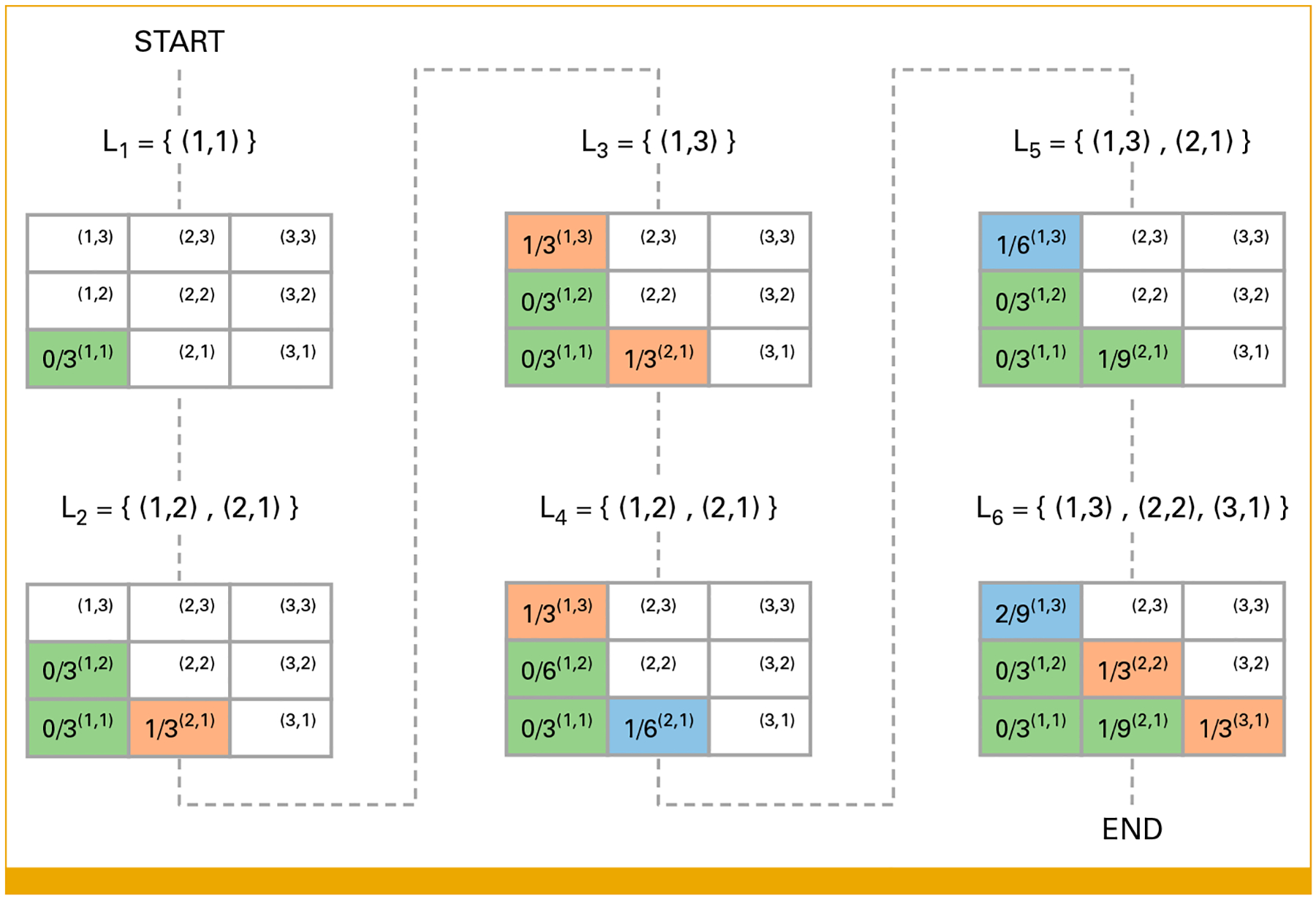
A hypothetical phase I clinical trial using the TALE design. Each table represents a stage of the trial, and the number in each cell represents the number of DLTs/number of enrolled patients. The set Lk includes all dose combinations that are assigned at stage k. DLTs, dose-limiting toxicities; TALE, Toxicity Adaptive Lists Design.
Numerical Study
We compare TALE, the dual-agent BOIN,22 the model-based COPULA,14 the continual reassessment method for partial ordering (POCRM),21,30 and the product of independent beta probabilities escalation (PIPE)15 designs across eight simulation scenarios (Table 1). Each scenario considers five DLs for drugs A and B, resulting in 25 combinations. The maximum sample size is N = 42 patients, enrolled in 14 cohorts of n = 3 patients. For each scenario and design, we simulate 1,000 independent trials.
TABLE 1.
Toxicity Scenarios for Two-Drug Combinations
| Drug A | |||||
|---|---|---|---|---|---|
| Level | 1 | 2 | 3 | 4 | 5 |
| Drug B | Scenario 1 | ||||
| 5 | 0.32 | 0.37 | 0.42 | 0.43 | 0.46 |
| 4 | 0.27 | 0.33 | 0.37 | 0.40 | 0.43 |
| 3 | 0.24 | 0.29 | 0.34 | 0.37 | 0.42 |
| 2 | 0.20 | 0.23 | 0.29 | 0.33 | 0.37 |
| 1 | 0.11 | 0.20 | 0.24 | 0.28 | 0.32 |
| Drug B | Scenario 2 | ||||
| 5 | 0.26 | 0.32 | 0.38 | 0.41 | 0.46 |
| 4 | 0.24 | 0.27 | 0.34 | 0.38 | 0.41 |
| 3 | 0.20 | 0.24 | 0.29 | 0.34 | 0.38 |
| 2 | 0.12 | 0.20 | 0.22 | 0.29 | 0.32 |
| 1 | 0.09 | 0.13 | 0.20 | 0.23 | 0.26 |
| Drug B | Scenario 3 | ||||
| 5 | 0.12 | 0.15 | 0.22 | 0.40 | 0.42 |
| 4 | 0.12 | 0.15 | 0.20 | 0.35 | 0.42 |
| 3 | 0.10 | 0.12 | 0.18 | 0.33 | 0.40 |
| 2 | 0.10 | 0.12 | 0.15 | 0.26 | 0.27 |
| 1 | 0.00 | 0.10 | 0.10 | 0.20 | 0.22 |
| Drug B | Scenario 4 | ||||
| 5 | 0.23 | 0.33 | 0.35 | 0.46 | 0.48 |
| 4 | 0.13 | 0.23 | 0.34 | 0.36 | 0.43 |
| 3 | 0.05 | 0.20 | 0.27 | 0.34 | 0.35 |
| 2 | 0.02 | 0.05 | 0.08 | 0.27 | 0.33 |
| 1 | 0.00 | 0.01 | 0.02 | 0.10 | 0.30 |
| Drug B | Scenario 5 | ||||
| 5 | 0.29 | 0.53 | 0.60 | 0.61 | 0.92 |
| 4 | 0.20 | 0.39 | 0.45 | 0.57 | 0.79 |
| 3 | 0.04 | 0.14 | 0.15 | 0.18 | 0.32 |
| 2 | 0.03 | 0.09 | 0.13 | 0.15 | 0.23 |
| 1 | 0.02 | 0.02 | 0.05 | 0.09 | 0.13 |
| Drug B | Scenario 6 | ||||
| 5 | 0.21 | 0.45 | 0.77 | 0.87 | 0.99 |
| 4 | 0.08 | 0.35 | 0.48 | 0.83 | 0.97 |
| 3 | 0.07 | 0.31 | 0.46 | 0.82 | 0.97 |
| 2 | 0.04 | 0.08 | 0.20 | 0.51 | 0.94 |
| 1 | 0.02 | 0.06 | 0.15 | 0.17 | 0.93 |
| Drug B | Scenario 7 | ||||
| 5 | 0.39 | 0.49 | 0.53 | 0.55 | 0.58 |
| 4 | 0.20 | 0.40 | 0.44 | 0.45 | 0.56 |
| 3 | 0.19 | 0.33 | 0.42 | 0.44 | 0.54 |
| 2 | 0.02 | 0.20 | 0.31 | 0.33 | 0.43 |
| 1 | 0.01 | 0.06 | 0.20 | 0.32 | 0.32 |
| Drug B | Scenario 8 | ||||
| 5 | 0.36 | 0.50 | 0.56 | 0.58 | 0.58 |
| 4 | 0.20 | 0.32 | 0.34 | 0.48 | 0.58 |
| 3 | 0.05 | 0.24 | 0.27 | 0.40 | 0.55 |
| 2 | 0.03 | 0.20 | 0.23 | 0.26 | 0.46 |
| 1 | 0 | 0.07 | 0.17 | 0.20 | 0.34 |
NOTE. The MTDs at 0.20 are in bold.
Abbreviation: MTDs, maximum tolerated doses.
In scenarios 1 and 2, drugs A and B share similar toxicity effects; the toxicity probability increases approximately symmetrically with drugs A and B. In scenarios 3 and 4, the toxicity probability increases markedly with the DL of drug A, whereas the toxicity effects of drug B are less pronounced. Scenarios 5–8 are randomly generated using the algorithm provided by Liu et al.27
We use the following parameters for the designs:
TALE: We set nmax = 9, λe = 0.16, λd = 0.24, and λr = 0.45; see the Data Supplement (Table S2) for the corresponding numbers of DLTs. TALE uses isotonic regression and selects the highest DL with an estimated MTD below 0.2.
BOIN: We follow the recommendations of Lin and Yin.22 The trial stops if the number of patients assigned to a single DL reaches nine patients, and the data suggest staying at the same DL.
COPULA: This approach requires specifying prior probabilities of observing a DLT when drugs A and B are administered separately. In our numerical study, drugs A and B share the same single-agent probability of DLT (0, 0.05, 0.10, 0.15, 0.20, for DLs 1, …, 5). The dose escalation and de-escalation thresholds are set (default parameters) at 0.8 and 0.45, respectively.
POCRM: We use the default design settings with three equally probable partial orderings.21,30 The trial stops when nine patients are assigned to the same DL.
PIPE: We set the safety threshold at 0.8 and prior medians equal to the target toxicity rate. Other parameters are set to their default.15
We consider the following metrics to compare trial designs:
The percentage of simulations a design selects an MTD with a toxicity rate of 0.2.
The percentage of simulations a design selects an MTD with a toxicity rate in (0.16, 0.24), a symmetric interval around the target toxicity.
The percentage of patients allocated at a DL with a toxicity rate of 0.
The average number of toxicities per trial.
The percentage of overdose assignments, that is, the percentage of patients receiving dose combinations with toxicity probability higher than 0.3.
The percentage of underdose assignment, that is, the percentage of patients receiving combinations with toxicity probability equal to or below 0.12.
Metrics a-c focus on the MTD selection accuracy, whereas metrics d-f focus on the number of toxicities during the trial.
RESULTS
Accuracy Metrics (a, b, and c)
No design outperforms the others across all scenarios and metrics (Figs 4A and 4B). COPULA performs particularly well in scenario 2, where it selects the true MTD with a frequency of 45.2% simulated trials compared with 29.8% of BOIN, 34.7% of TALE, 26.4% of PIPE, and 31% of POCRM. With metric b, PIPE has the highest accuracy in scenario 3 (30.2%) and COPULA in scenarios 2 and 4 (56.3% and 37.8%, respectively), whereas TALE has a high accuracy in scenario 1 (45.9%). COPULA allocates a higher percentage of patients at the MTDs compared with the other designs in most scenarios (Fig 4C). However, in scenario 5, it has the lowest allocation rate at 2.8%. On average, COPULA has 27.5% of the enrolled patients treated at the MTDs, against 26.9% of BOIN, 27.6% of TALE, 24.9% of POCRM, and 18.6% of PIPE. In scenario 3, TALE shows a smaller percentage of allocation around the true MTD (metric b) than the competitor designs. At the same time, the percentage of accurate selection of the MTD (metric a) and the number of patients allocated to the MTD (metric c) are closer to the other designs. This behavior can be explained by the structure of the toxicity matrix defined in Table 1, having most of the lower adjacent neighbors of the MTDs outside the interval (0.16, 0.24) considered by metric b.
FIG 4.
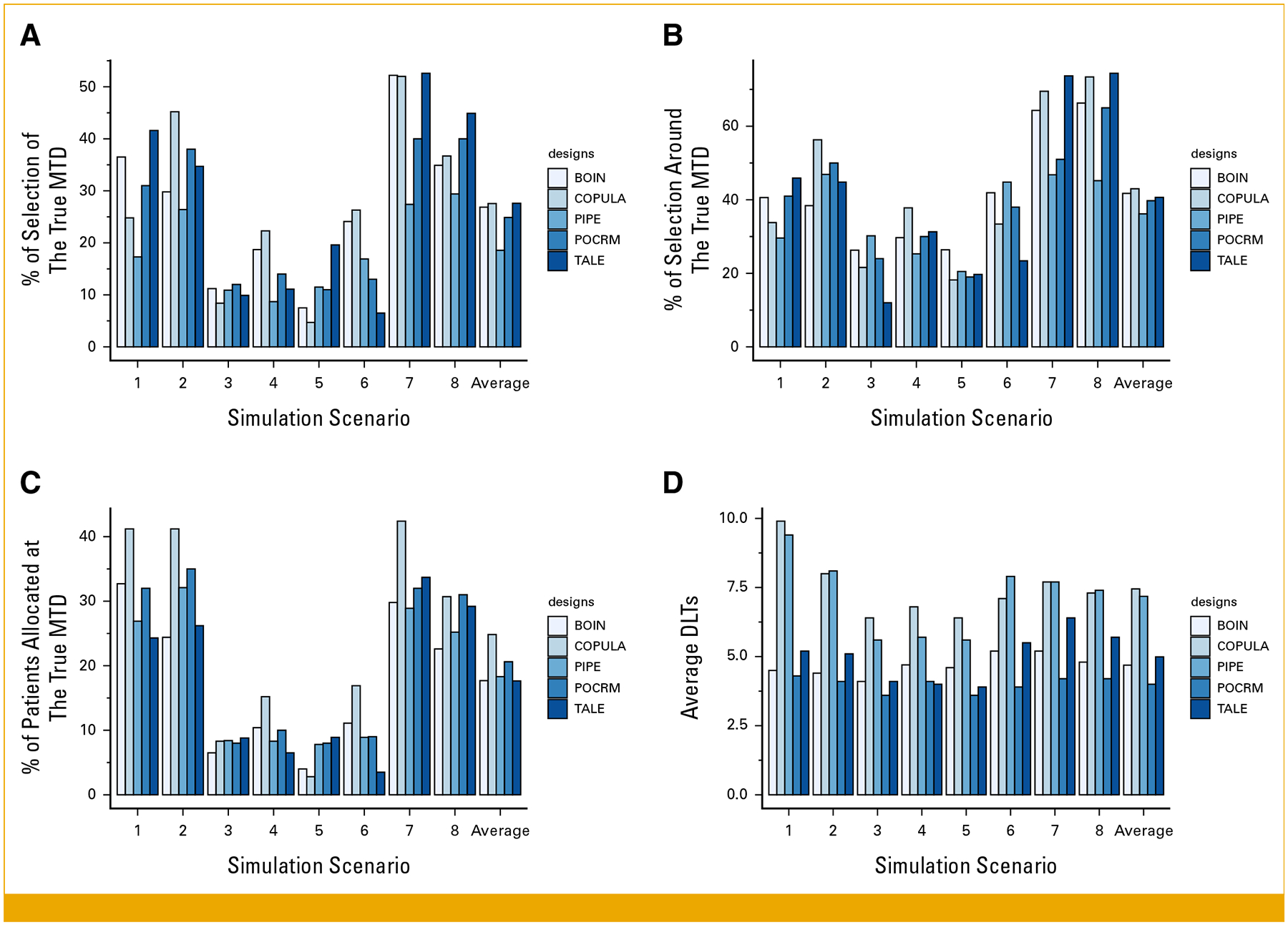
Comparison of the proposed TALE design and existing model-based or model-assisted methods. We focus on metrics a-d evaluated in scenarios 1–8. BOIN, Bayesian optimal interval design; DLTs, dose-limiting toxicities; MTD, maximum tolerated dose; PIPE, product of independent beta probabilities escalation; POCRM, continual reassessment method for partial ordering; TALE, Toxicity Adaptive Lists Design.
Toxicity Metrics (d, e, and f)
On average, COPULA and PIPE produce 3 DLTs more than BOIN, TALE, and POCRM (Fig 4D). Table 2 shows that TALE results in fewer overdose assignments (4.8% on average) than COPULA (10.8%), BOIN (11.6%), POCRM (6%), and PIPE (8.3%). In most scenarios, COPULA has the lowest percentage of patients assigned to a dose lower than 0.12 (Table 2). In scenarios 5, 6, and 7, the lowest percentage of patients assigned to a dose lower than 0.12 is provided by BOIN (14.1% of patients underdosed in scenario 5), PIPE (46.5% of patients underdosed in scenario 7), and POCRM (15% of patients underdosed in scenario 7). On average, TALE results in approximately 44% of patients being assigned to a dose with toxicity probability below 0.12, compared with 39% using BOIN, 27% with COPULA, and 32% with POCRM and PIPE.
TABLE 2.
Comparison of the TALE With Existing Model-Based and Model-Assisted Methods in Terms of the Percentage of Overdose Assignment and Percentage of Underdose Assignment (metrics e and f)
| Scenario | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Average |
|---|---|---|---|---|---|---|---|---|---|
| Percentage of overdose assignment | |||||||||
| TALE | 1.5 | 0.05 | 0 | 5.2 | 1.5 | 9.8 | 14.3 | 6.3 | 4.8 |
| BOIN | 4.8 | 2.8 | 4.7 | 12.7 | 11.8 | 22.1 | 24.4 | 9.5 | 11.6 |
| COPULA | 9.3 | 2.4 | 3.8 | 11.2 | 12.4 | 21.4 | 19 | 6.7 | 10.8 |
| POCRM | 5 | 0 | 1 | 11 | 6 | 13 | 5 | 7 | 6 |
| PIPE | 6.8 | 0.8 | 1.5 | 8.6 | 6.6 | 22.8 | 7.8 | 11.7 | 8.3 |
| Percentage of underdose assignment | |||||||||
| TALE | 15.2 | 12.0 | 54.8 | 70.1 | 65.6 | 60 | 34 | 42.7 | 44.3 |
| BOIN | 33.3 | 37.1 | 44.9 | 52.9 | 14.1 | 53.2 | 37.2 | 40 | 39.1 |
| COPULA | 1.9 | 0.24 | 30.0 | 45.9 | 36.2 | 47.6 | 25 | 28.3 | 26.9 |
| POCRM | 19 | 12 | 40 | 47 | 45 | 49 | 15 | 30 | 32.1 |
| PIPE | 15.7 | 11.4 | 37.7 | 53.9 | 46.2 | 46.5 | 16.5 | 31.7 | 32.4 |
Abbreviations: BOIN, Bayesian optimal interval design; PIPE, product of independent beta probabilities escalation; POCRM, continual reassessment method for partial ordering; TALE, Toxicity Adaptive Lists Design.
The five designs show comparable operative characteristics in terms of accurate identification of the MTDs. TALE tends to reduce the number of DLTs and the number of patients that are assigned to DLs with toxicity above 0.3. Also, TALE allows for multiple cohorts of patients to be assigned to DLs at the same time, potentially reducing the duration of the trial.
Practical Implementation (software)
An R Shiny application31 that allows users to select design parameters, conduct simulation studies, and compute relevant operative characteristics is available at TALE design.32 We also provide an R package33 at GitHub.34
DISCUSSION
We introduced TALE, an algorithm-based design for oncology phase I drug combination. TALE is straightforward to implement and uses escalation and de-escalation rules that account for patients’ safety. It is extremely adaptable: one can define the size of cohorts, the target DLT rate of MTDs, the maximum number of patients treated at each DL, and the parameters that control the dose escalation and de-escalation processes. A key feature of TALE is exploring multiple DLs simultaneously, ultimately reducing the duration of the trial.
TALE categorizes DL toxicity levels using a color scale from green to red (Fig 2). From our experience (Clinical-Trials.gov identifiers: NCT05687136 and NCT04616534), these easy-to-understand categories clarify how sequential decisions are made during the trial on the basis of available data for stakeholders such as clinicians and protocol reviewers.
Our software lets users select the design parameters with simulation studies. Notably, choosing λd = λr removes the intermediate-low category. The software lets users explore different configurations of the design parameters and summarize the operating characteristics across plausible scenarios. Selecting the design parameters should account for relevant operating characteristics and other context-specific considerations. For example, users should examine the comprehensive list of data configurations that make a DL (i, j) explorable for the next stage. Importantly, TALE simplifies this task because the set of data configurations that make DL (i, j) explorable is defined only by pij and the toxicity rates at the neighbor DLs (Fig 2).
TALE also has some limitations. Simultaneous exploration of multiple DLs might be problematic in settings with a slow patient accrual rate. We also mention the restricted number of simulation scenarios that are illustrated for ease of presentation.
Previous studies suggested that model-based or assisted designs tend to outperform algorithm-based designs,2,7–9 especially in terms of MTD selection accuracy. However, in our simulations, TALE demonstrated a similar accuracy in selecting MTDs as the other designs with a reduced number of patients exposed to toxic DLs.
Importantly, TALE reduces the assignment of toxic DLs and the occurrence of DLTs, resulting in an improved balance between patient safety and MTD identification accuracy and providing an attractive option for the statistical design of phase I oncology combination studies.
Supplementary Material
CONTEXT.
Key Objective
To propose a novel algorithmic approach to design phase I clinical trials for drug combinations: the Toxicity Adaptive Lists Design (TALE).
Knowledge Generated
TALE controls patients’ assignment to toxic combinations and adverse events related to therapy, providing a good balance between patient safety and accurate assessment of candidate dose levels (DLs).
Relevance
TALE is simple to implement and can simultaneously explore multiple DLs. In settings where patients’ accrual is sufficiently rapid, this translates into a shorter duration of the phase I trial compared with popular designs that explore only a single DL at the time. The design proceeds in stages, and to simplify decision making, it categorizes both DLs and toxicity rates into four groups on the basis of the observed data. At each stage of the trial, these categories are used to make decisions following an intuitive scheme.
SUPPORT
Supported in part by NIH grant UM1 CA186709 (J.M.C., G.I.S., G.M.C. and L.T., with G.I.S., Contact PI) and NIH 5R01LM013352 (L.T.).
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Francesco Mariani
Research Funding: Novartis
James M. Cleary
Honoraria: Blueprint Medicines, Syros Pharmaceuticals, Incyte, AstraZeneca
Research Funding: Merck, Tesaro, AstraZeneca, Esperas Pharma, Merus (Inst), Roche/Genentech (Inst), BMS (Inst), Bayer, Apexigen, Arcus Biosciences, Servier (Inst), Amgen
Travel, Accommodations, Expenses: Roche, Agios, Bristol Myers Squibb, Incyte
Geoffrey I. Shapiro
Consulting or Advisory Role: Merck Serono, Bicycle Therapeutics, Xinthera
Research Funding: Pfizer (Inst), Lilly (Inst), Merck Serono (Inst), Merck (Inst), Tango Therapeutics (Inst), Bristol Myers Squibb/Medarex (Inst)
Patents, Royalties, Other Intellectual Property: Patent: 9872874 Title: Dosage regimen for sapacitabine and seliciclib Issue Date: 1/23/2018, Patent: 10,934,593 B2 Compositions and methods for predicting response and resistance to CDK4/6 inhibition
Gregory M. Coté
Honoraria: BioAtla, Gilead Sciences
Consulting or Advisory Role: Ikena Oncology, Daiichi Sankyo/UCB Japan, Sonata, C4 Therapeutics
Research Funding: Macrogenics (Inst), PharmaMar (Inst), Epizyme (Inst), Eisai (Inst), Merck KGaA (Inst), Bavarian Nordic, Bayer (Inst), Springworks Therapeutics (Inst), Sumitomo Dainippon Pharma Oncology (Inst), Foghorn Therapeutics (Inst), Repare Therapeutics (Inst), Jazz Pharmaceuticals (Inst), C4 Therapeutics (Inst), Servier (Inst), Rain Therapeutics (Inst), BioAtla (Inst), Ikena Oncology (Inst), Kronos Bio (Inst), Pyxis (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Gasparini M: General classes of multiple binary regression models in dose finding problems for combination therapies: Dose finding problems for combination therapies. J R Stat Soc Ser C: Appl Stat 62:115–133, 2013 [Google Scholar]
- 2.Jaki T, Clive S, Weir CJ: Principles of dose finding studies in cancer: A comparison of trial designs. Cancer Chemother Pharmacol 71:1107–1114, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan Y, Lee JJ, Hilsenbeck SG: Model-assisted designs for early-phase clinical trials: Simplicity meets superiority. JCO Precis Oncol 10.1200/PO.19.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan F, Mandrekar SJ, Yuan Y: Keyboard: A novel Bayesian toxicity probability interval design for phase I clinical trials. Clin Cancer Res 23:3994–4003, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang B, Bycott P, Talukder E: Novel dose-finding designs and considerations on practical implementations in oncology clinical trials. J Biopharm Stat 27:44–55, 2017 [DOI] [PubMed] [Google Scholar]
- 6.Yuan S, Zhou T, Lin Y, et al. : The Ci3+3 design for dual-agent combination dose-finding clinical trials. J Biopharm Stat 31:745–764, 2021 [DOI] [PubMed] [Google Scholar]
- 7.Riviere MK, Dubois F, Zohar S: Competing designs for drug combination in phase I dose-finding clinical trials. Statist Med 34:1–12, 2015 [DOI] [PubMed] [Google Scholar]
- 8.Iasonos A, O’Quigley J: Adaptive dose-finding studies: A review of model-guided phase I clinical trials. J Clin Oncol 32:2505–2511, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Brummelen EMJ, Huitema ADR, van Werkhoven E, et al. : The performance of model-based versus rule-based phase I clinical trials in oncology: A quantitative comparison of the performance of model-based versus rule-based phase I trials with molecularly targeted anticancer drugs over the last 2 years. J Pharmacokinet Pharmacodyn 43:235–242, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Yin G: Escalation with overdose control for phase I drug-combination trials. Statist Med 32:4400–4412, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Sweeting MJ, Mander AP: Escalation strategies for combination therapy phase I trials. Pharmaceut Statist 11:258–266, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thall PF, Millikan RE, Mueller P, et al. : Dose-finding with two agents in phase I oncology trials. Biometrics 59:487–496, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Tighiouart M, Piantadosi S, Rogatko A: Dose finding with drug combinations in cancer phase I clinical trials using conditional escalation with overdose control. Statist Med 33:3815–3829, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin G, Yuan Y: Bayesian dose finding in oncology for drug combinations by copula regression. J R Stat Soc Ser C: Appl Stat 58:211–224, 2009 [Google Scholar]
- 15.Mander AP, Sweeting MJ: A product of independent beta probabilities dose escalation design for dual-agent phase I trials. Statist Med 34:1261–1276, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin G, Yuan Y: A latent contingency table approach to dose finding for combinations of two agents. Biometrics 65:866–875, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Braun TM, Wang S: A hierarchical Bayesian design for phase I trials of novel combinations of cancer therapeutic agents. Biometrics 66:805–812, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Ning J: A Bayesian dose-finding design for drug combination trials with delayed toxicities. Bayesian Anal 8:703–722, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riviere MK, Yuan Y, Dubois F, et al. : A Bayesian dose-finding design for drug combination clinical trials based on the logistic model. Pharmaceut Statist 13:247–257, 2014 [DOI] [PubMed] [Google Scholar]
- 20.Tighiouart M, Li Q, Rogatko A: A Bayesian adaptive design for estimating the maximum tolerated dose curve using drug combinations in cancer phase I clinical trials: Adaptive design for estimating the maximum tolerated dose curve. Statist Med 36:280–290, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wages NA, Conaway MR, O’Quigley J: Continual reassessment method for partial ordering. Biometrics 67:1555–1563, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin R, Yin G: Bayesian optimal interval design for dose finding in drug-combination trials. Stat Methods Med Res 26:2155–2167, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Ji Y, Li Y, Nebiyou Bekele B: Dose-finding in phase I clinical trials based on toxicity probability intervals. Clin Trials 4:235–244, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Ji Y, Wang SJ: Modified toxicity probability interval design: A safer and more reliable method than the 3 + 3 design for practical phase I trials. J Clin Oncol 31:1785–1791, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brock K, Homer V, Soul G, et al. : Is more better? An analysis of toxicity and response outcomes from dose-finding clinical trials in cancer. BMC Cancer 21:777, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mauro G, Bailey S, Neuenschwander B, et al. : Correspondence: Bayesian dose finding in oncology for drug combinations by copula regression. J R Stat Soc Ser C: Appl Stat 59:543–546, 2010 [Google Scholar]
- 27.Liu R, Yuan Y, Sen S, et al. : Accuracy and safety of novel designs for phase I drug-combination oncology trials. Stat Biopharm Res 14:270–282, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buisson R, Boisvert JL, Benes CH, et al. : Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol Cell 59:1011–1024, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bril G, Dykstra R, Pillers C, et al. : Algorithm AS 206: Isotonic regression in two independent variables. Appl Stat 33:352, 1984 [Google Scholar]
- 30.Wages NA, Varhegyi N: pocrm: An R-package for phase I trials of combinations of agents. Computer Methods Programs Biomed 112:211–218, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang W, Cheng J, Allaire JJ, et al. : shiny: Web application framework for R. R package version 1.6.0. https://CRAN.R-project.org/package5shiny
- 32.Shiny app. https://6kp5ow-francesco-mariani.shinyapps.io/TALEdesign/
- 33.Leisch F: Creating R packages: A tutorial. The Comprehensive R Archive Network (CRAN), contributed documentation. 2009. https://cran.r-project.org/doc/contrib/Leisch-CreatingPackages.pdf
- 34.R package. https://github.com/framar1997/TALEdesign/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.