

Abstract
Energy flow within cellular elements of the brain is a well-orchestrated, tightly regulated process, however, details underlying these functions at the single-cell level are still poorly understood. Studying hypometabolism in aging and neurodegenerative diseases may benefit from experimentation on unicellular bioenergetics. Here, we examined energy status in neurons and astrocytes using mixed hippocampal cultures and PercevalHR, an ATP:ADP nanosensor. We assessed exposures of several compounds including KCl, glutamate, FCCP, insulin, and glucose. A mitochondrial stress test was performed, and PercevalHR’s fluorescence was corrected for pH using pHrodo. Results demonstrate that PercevalHR can reliably report on the energetic status of two cell types that communicate in a mixed-culture setting. While KCl, glutamate, and FCCP showed clear changes in PercevalHR fluorescence, insulin and glucose responses were found to be more subtle and sensitive to extracellular glucose. These results may highlight mechanisms that mediate insulin sensitivity in the brain.
Subject terms: Cell biology, Metabolism
Introduction
The flow of energy substrates in the brain is shared among numerous cell types, each with different energy requirements, transport mechanisms, and metabolic enzymes, ultimately giving rise to a complex, yet tightly orchestrated system. This homeostasis must be sustained, particularly during periods of high cellular activity, where energy consumption is elevated and often restricted to specific substructures within an area of the brain or a cell1–3. High flux energy demands for ATP production have been identified in multiple cell types. These points are particularly true in neurons and astrocytes, where maintenance of the resting membrane potential via the stimulation of multiple ATPases (e.g., Na+/K+-ATPases, plasma membrane Ca2+ ATPases, vesicular H+-ATPases) must be engaged during synaptic events3–6. Irrespective of the conditions and demands on the brain region, homeostatic equilibrium is usually well-maintained, and chronic reductions in glucose metabolism are seen mostly under conditions associated with normal aging, diabetes, and Alzheimer’s Disease (AD)7,8 reviewed in ref. 9 reviewed in ref. 10. This decline in cerebral rates of glucose utilization can be explained by multiple processes, including decreases in synaptic neuronal/astrocytic activity, the state of astrocytic glutamate transporters, alterations in enzymatic function, reduced numbers or function of insulin receptors or glucose transporters, or glucose levels11 reviewed in ref. 12 reviewed in refs. 13–15. Irrespective of the source of the dysregulation, the degree of decline is often associated with the severity of cognitive impairment reviewed in refs. 10,16. Importantly, recent work has reported on the significant increase in glucose concentrations in key areas of the brain affected most by AD pathology, implicating reduced utilization and transport as potential mechanisms for the neuropathology reviewed in refs. 13,17.
In addition to the well-established role of glucose metabolism in the CNS, insulin has also been recognized to play an essential role in the regulation of cognitive function, particularly in the hippocampus, where it can ameliorate spatial memory recall18–20. Type 2 diabetes mellitus is associated with deficient insulin sensitivity, certainly in the periphery, but also perhaps in the brain reviewed in ref. 21 reviewed in refs. 22,23 reviewed in ref. 24. In alignment with this point, work from multiple labs measuring the impact of insulin delivery to the brain (intranasal or otherwise) in animal models of aging or AD25–27, as well as in the clinic reviewed in refs. 22,23,28, has provided supportive evidence for offsetting cognitive decline. Characterizing sensitivity to insulin and understanding the impact of this hormone on neuronal or astrocytic bioenergetics would represent significant improvements in the search for therapeutics to limit cognitive decline or age-related alterations in cognitive function.
Over the past decade, dynamic measures of metabolism at the single-cell level have become available. The development of fluorescent biosensors for measuring key intracellular energetic substrates including glucose, NADH, NADPH, lactate, pyruvate, ATP, and ADP has helped define responses to network activation at the single-cell level reviewed in ref. 2 reviewed in ref. 29 reviewed in ref. 30. Measures that can infer on energy status across large areas of the brain and define structural boundaries around high or low-activity foci, such as magnetic resonance imaging (MRI) and blood oxygenation level-dependent (BOLD) imaging, are less useful for investigating unicellular bioenergetics; however, techniques with high spatial resolution, when combined with a simplified system where few cell types interact (e.g., neurons and astrocytes in primary mixed cultures or acute tissue slices used in tandem with live-cell nanosensors), help provide a clearer picture of bioenergetic activity and infer on the dynamic interplay within and between cells reviewed in ref. 29 reviewed in refs. 31–36. Literature utilizing PercevalHR, an ATP:ADP biosensor, reports on the use of multiple species, including yeast, C. elegans37–40, chickens41, mice42,43, and D. melanogaster44,45. Some studies have also been conducted across numerous cell types: coronary artery smooth muscle cells46,47, neurons36,41,48,49, astrocytes36,48, pancreatic beta cells50,51, and immune cells42. At the time of writing, we are unaware of any reports using this indicator in the context of aging or age-related diseases in vivo. Overall, it is clear there is a necessity for further investigations using in vivo probes to measure multiple variables including mitochondrial function, glycolytic activity, high-energy intermediates, and energy metabolism in the brain reviewed in ref. 52.
Here, we present results associated with the use of PercevalHR in a mixed primary hippocampal culture setting using a lentiviral vector for PercevalHR delivery. The hypothesis tested that PercevalHR fluorescence ratios (PFRs) in neurons and astrocytes are sensitive to potassium chloride (KCl), glutamate, carbonyl cyanide-p-trifluoromethoxy phenylhydrazone (FCCP), insulin, and changes in glucose concentrations. We used pHrodo fluorescence in the same cells expressing PercevalHR to correct for the pH sensitivity of PercevalHR. As expected, exposures to KCl, glutamate, and FCCP reduced PFRs. We show that cellular responses to insulin and glucose are sensitive to pretreatments of extracellular glucose and the duration of insulin exposure.
Results
Our experiments assessed compounds thought to decrease ATP:ADP (e.g., depolarization or mitochondrial inhibition) and investigated the impact of extracellular glucose concentrations and insulin exposure on PFRs. Independent analyses of OCR and ECAR assays were used to corroborate the results of PercevalHR imaging with an alternative method and to address measures of basal respiration, maximal respiration, ATP production, and glycolytic rate in response to insulin exposures. The data presented here were collected from a total of approximately 164 culture dishes obtained from 16 dams. Measurements of PFRs were obtained across ~475 cells during imaging experiments.
Perceval imaging pH correction
In cells expressing both PercevalHR and pHrodo, we assessed the impact of intracellular pH changes on PercevalHR fluorescence53 with excursions across 1 pH unit (i.e., 6.5–7.5 or 7.5–6.5). After normalization to baseline fluorescence, the average change in pHrodo fluorescence for a single unit of pH change was determined to be 80.2% ± 8.22% (n = 51; both cell types combined, see below), at the same time the mean change in PFRs for 1-unit change in pH was 0.669 ± 0.067 (n = 50; both cell types combined, see below) (see Supplementary Fig. 1). In cells where pHrodo was not available, we corrected for pH changes using the average pH change measured across a new group of cells exposed to pHrodo and imaged under identical conditions. Therefore, on a cell-by-cell basis, using pHrodo data, we corrected ratiometric PercevalHR measures for changes in pH caused by exposure to different experimental conditions (i.e., KCl, glutamate, ethanol (EtOH), insulin, FCCP, and glucose excursions). An initial analysis was conducted to address if the percent change in pHrodo fluorescence and the ratiometric PercevalHR change were different between neurons and astrocytes during pH challenges and no differences were found (pHrodo: unpaired t-test, two-tailed, p = 0.8112, t = 0.168, df = 49; PercevalHR: unpaired t-test, two-tailed, p = 0.985, t = 0.019, df = 48). Thus, all PFRs presented in the current study were corrected for the sensitivity of PercevalHR to pH.
KCl exposures with pH buffers
Because previous work has shown that high KCl concentrations increase Ca2+ influx during depolarization in neurons and astrocytes54–56, we used a 2-min 50 mM KCl exposure to challenge the cells and induce ATP consumption. A significant decrease of PFRs with time was observed following exposure to 50 mM KCl in both cell types (Two-way RM ANOVA: time: F(16, 592) = 17.13, P < 0.0001; cell type: F(1,37) = 0.2899, P = 0.5935; Fig. 1a). To investigate whether the inclusion of bicarbonate in our buffering system may have modulated glycolytic activity in our cells and altered responses to KCl depolarization (i.e., the Crabtree effect)57, we conducted a second set of experiments without bicarbonate buffer. Under these conditions, a significant decrease of PFRs was again detected in response to KCl in both cell types (Two-way RM ANOVA: time: F(1.288, 24.47) = 16.67, P = 0.0002; cell type: F(1,19) = 1.535, P = 0.2305), comparable to that observed in experiments tested with bicarbonate buffer (Fig. 1b).
Fig. 1. PFR reductions with 50 mM potassium chloride and 20 µM glutamate exposures.

a shows neuronal (n = 30) and astrocytic (n = 9) PFR reductions in response to a 50 mM KCl exposure perfused in a solution containing bicarbonate buffer (left, shaded region). b displays a similar bioenergetic response across KCl exposures perfused without bicarbonate buffer (12 neurons; 9 astrocytes). c displays the differential responses of neurons and astrocytes to glutamate (16–21 neurons; 7–9 astrocytes). The spread of the data is displayed as averages of data collected at different time points before, during, and after compound exposure for each respective set of imaging experiments (rightmost graphs). Asterisks represent a significant main effect of exposure. Error bars represent the standard error of the mean.
Glutamate exposures
Prior studies have shown that glutamate exposure increases intracellular Ca2+ and depolarizes cells54,55,58,59, likely causing a reduction in ATP and, in response, a concomitant increase in glycolytic activity3,53,60–62. As predicted, a significant decrease in PFRs in response to glutamate was seen (Two-way ANOVA: F(17, 454) = 30.7, P < 0.0001; Fig. 1c), again, likely due to the energetic demand caused by an influx of cations. The responses to glutamate exposures were similar in astrocytes and neurons (Two-way ANOVA: F(1, 28) = 4.128, P = 0.2973); however, a significant interaction term between cells was detected (Two-way ANOVA; F(17, 454) = 30.7, P = 0.0005). Specifically, neurons responded more robustly to glutamate than astrocytes. These initial experiments replicated the work of Tantama and others well53,63,64, and in the next set of experiments, we investigated the mitochondrial activity of neurons and astrocytes in the same imaging conditions.
FCCP and EtOH exposures
To address mitochondrial engagement in the production of ATP at rest, a subset of cells was exposed to 10 µM FCCP. At concentrations of 1 nM–10 µM, FCCP collapses the membrane potential established by the electron transport chain and is typically used to determine maximal respiration and glycolytic activity65. PFRs decreased significantly with FCCP exposure (Mixed-effects model (REML): time: F(2.394, 69.10) = 20.36, P < 0.0001, Geisser-Greenhouse’s ∑ = 0.0798; cell type: F(1,29) = 0.0500, P = 0.8246), suggesting that the cells were metabolically active and producing ATP via oxidative phosphorylation (Fig. 2a). A time control experiment was used to investigate if the vehicle used for FCCP exposures (0.12% EtOH) contributed to the PFR decrease. A small but significant effect of EtOH was detected under these conditions (Mixed-effects model (REML): time: F(28, 1347) = 4.789, P < 0.0001, Geisser-Greenhouse’s ∑ = 0.0577; cell type: F(1,49) = 4.933, P = 0.0310), suggesting that a small portion of the FCCP response was mediated by the vehicle (~65% reduction with FCCP versus ~13% reduction with EtOH, seen primarily in astrocytes) (Fig. 2b).
Fig. 2. PFR responses to 10 µM FCCP and 0.12% EtOH.

a shows large significant decreases in neuronal (n = 6–10) and astrocytic (n = 21) PFRs in response to a 10 µM FCCP exposure (dotted line). b shows the response to 0.12% EtOH (dotted line), which was used as the vehicle for FCCP (24–30 neurons; 16–23 astrocytes). It appears that EtOH contributes somewhat to the reduction of PFRs found in FCCP exposures, however the impact is relatively small. The spread of the data is displayed as averages of data collected at different time points before and after compound exposure for each respective set of imaging experiments (rightmost graphs). Asterisks represent a significant main effect of exposure or cell type. Error bars represent the standard error of the mean.
Glucose and insulin exposure
To attempt to normalize bioenergetic conditions in cultures exposed chronically to high glucose levels prior to experimentation, all cultures received a half-medium exchange using MEM (no added glucose) 24 h prior to experimentation, and cells also underwent a 30-min pretreatment exposure in either high (5–10 mM) or more physiologically relevant (2 mM) extracellular glucose concentrations. Note that the concentrations used (i.e., low or high glucose) in the pretreatment exposures were continued in the imaging phase of the experiment.
Under high glucose pretreatment conditions, Insulin exposures (10 nM) elicited a small, but significant decrease in PFRs in both cell types, and also revealed a potential cell type difference (trend), whereby neurons displayed slightly elevated PFRs and appeared more responsive to insulin than astrocytes (Mixed-effects model (REML): time: F(2.281, 201.7) = 3.069, P = 0.0420, Geisser-Greenhouse’s ∑ = 0.0992; cell type: F(1,91) = 3.018, P = 0.0857; Fig. 3a). Glucose excursions (0.1–10 mM) did not alter PFRs in either cell type (Mixed-effects model (REML): time: F(1.797, 8485) = 1.674, Geisser-Greenhouse’s ∑ = 0.7104; cell type: F(1,48) = 1.969, P = 0.1670; Fig. 3b). To control for time-dependent changes across the duration of insulin exposure experiment, time control experiments were conducted in a subset of cells and no significant change in PFRs were detected (Mixed-effects model (REML): time: F(1.801, 83.21) = 16.80, P = 0.1952, Geisser-Greenhouse’s ∑ = 0.0530; cell type: F(1, 47) = 1.651, P = 0.2051; Fig. 3c). While these results seem to indicate that there is no impact of extracellular glucose and little impact of insulin on PFRs, studies investigating chronic insulin are necessary, particularly given prior evidence using the same mixed-culture system that constitutive activity at the receptor increases glucose uptake and utilization only in neurons66,67.
Fig. 3. Effects of high extracellular glucose pretreatment (30 min) on insulin exposures, glucose excursions, and time controls.

a displays PFRs measured during 12-min imaging experiments in neurons (n = 67–75) and astrocytes (n = 14–18) with insulin exposures (shaded region). A small but significant reduction in PFRs was detected following insulin exposures. Panel (b) shows the lack of response in both neurons (n = 33 – 35) and astrocytes (n = 8–12) with glucose excursions. c represents time control experiments in neurons (n = 10), and astrocytes (n = 5). The spread of the data is displayed as averages of data collected at different time points before, during, and after compound exposure for each respective set of imaging experiments (rightmost graphs). Asterisks represent a significant main effect of exposure. Error bars represent the standard error of the mean.
No significant differences were found in either cell type in response to insulin exposures under low glucose pretreatment conditions (Mixed-effects model (REML): time: F(2.467, 122.8) = 2.096, P = 0.116, Geisser-Greenhouse’s ∑ = 0.0949; cell type: F(1,52) = 0.0260, P = 0.8725; Fig. 4a). However, as opposed to high glucose pretreatment conditions, where glucose excursions elicited no significant PFR alterations, glucose excursions did show subtle, but significant, PFR reductions to extracellular glucose challenges in both cell types (Mixed-effects model (REML): time: F(2.312, 191.2) = 4.990, P = 0.0053, Geisser-Greenhouse’s ∑ = 0.7104; cell type: F(1,85) = 6.260, P = 0.0143; Fig. 4b). Post-hoc analyses (Tukey’s) showed the main effect of extracellular glucose concentrations is primarily carried by the initial shift from 5.5 mM to 0.1 mM extracellular glucose in both cell types (neurons: 5.5 mM vs. 0.1 mM, P = 0.0013; astrocytes: 5.5 vs. 0.1 mM, P = 0.0179). To account for time-dependent changes during the insulin experiments, a subset of cultures was imaged without hormone treatment. A small time-dependent decrease in PFRs was noted in this experiment, which was absent during 10 mM glucose incubations, perhaps suggesting that both cell types are more energetically engaged in physiologically relevant glucose levels (Mixed-effects model (REML): time: F(1.554, 45.30) = 4.018, P = 0.0340, Geisser-Greenhouse’s ∑ = 0.0598; cell type: F(1,31) = 0.0144, P = 0.9053; Fig. 4c).
Fig. 4. Effects of low extracellular glucose pretreatment (30 min) on insulin exposures, glucose excursions, and time controls.

a displays no change in PFRs with a 5-min exposure to insulin during a 14-min imaging experiment (27–34 neurons; 15–20 astrocytes). b shows significant alterations in PFRs with glucose in both neurons (n = 49–57) and astrocytes (n = 25–30), which is dependent on cell type. Note that the largest change in PFRs was recorded in response to switching from 5.5 mM–0.1 mM. c shows the baseline activity of neurons (n = 17–21) and astrocytes (n = 6–11) during time control experiments. The spread of the data is displayed as averages of data collected at different time points before, during, and after compound exposure for each respective set of imaging experiments (rightmost graphs). Asterisks represent a significant main effect of exposure, time, or cell type. Error bars represent the standard error of the mean.
SeahorseXFe96 validation
Although confident in the health of our cultures [low resting calcium levels near ~100 nM54,58, significant baseline glycolytic activity54, hyperpolarized resting membrane potentials59, and low cell death during extended culture periods68–70], we sought to corroborate the PercevalHR-derived imaging results with an alternative method. Thus, we used a SeahorseXFe96 assay to evaluate the combined neuronal and astrocytic responses to acute and 1 h insulin exposures in 2 mM and 10 mM extracellular glucose concentrations. OCR measures were obtained to assess mitochondrial bioenergetics (basal respiration, maximal respiration, and ATP production), and ECAR data were used to determine glycolytic rates.
Acute insulin exposures, whether in 2 mM or 10 mM glucose, significantly increased basal respiration (F(2,49) = 5.023, P = 0.0104; Fig. 5d) and maximal respiration (F(2,49) = 5.833, P = 0.0053; Fig. 5e), but reduced coupling efficiency (F(2,48) = 4.133, P = 0.0221; Fig. 5c). A significant main effect of glucose concentration was found only for measures of ATP production (F(1,48) = 5.986, P = 0.0181; Fig. 5f), where ATP production was increased with elevated glucose concentrations. Using Tukey’s multiple comparisons tests, basal respiration was significantly increased with acute insulin treatment in both 2- and 10 mM glucose, which was maintained only in the 10 mM glucose condition (2 mM glucose: acute insulin vs. insulin 1 h: P = 0.0143; insulin 1 h: 2 mM vs. 10 mM glucose: P = 0.0046; Fig. 5d, hashtags). Comparable results were detected in measures of ATP production (2 mM glucose: acute insulin vs. insulin 1 h: P = 0.0278; insulin 1 h: 2 mM vs. 10 mM glucose: P = 0.0006; Fig. 5f, hashtags). Of note, while coupling efficiency showed reduction with insulin treatments, longer insulin exposures reduce coupling efficiency further in higher glucose concentrations (10 mM glucose: control vs. insulin 1 h: P = 0.0357; Fig. 5c, hashtag). Clearly, a significant interaction between insulin effects, the duration of its exposure, and extracellular glucose concentrations on measures of basal respiration and ATP production was seen.
Fig. 5. SeahorseXFe96-based OCR and ECAR data.

a and b shows the oxygen consumption rate normalized to 1000 cells in mixed hippocampal cultures treated with several selective mitochondrial substrates/inhibitors in 2 mM and 10 mM extracellular glucose, respectively, as well as different durations of insulin exposure (control, acute insulin, and insulin 1 h). Variables extracted from this dataset include coupling efficiency (c), basal respiration (d), maximal respiration (e), and ATP production (f). g and h highlight the extracellular acidification rate normalized to 1000 cells in 2 mM and 10 mM glucose conditions, respectively, as well as insulin exposures. Variables extracted from these measurements include glycolytic capacity (i), glycolytic reserve (j), and glycolysis (k). Asterisks represent a significant main effect of exposure. Hashtags denote significance derived from post-hoc testing. Error bars represent the standard error of the mean.
Glycolytic capacity, glycolytic reserve, and glycolysis were quantified, and a main effect of glucose concentration was detected for measures of both glycolytic capacity (F(1,44) = 5.166, P = 0.0280; Fig. 5i) and reserve (F(1,40) = 19.80, P < 0.0001; Fig. 5j), whereby elevations in extracellular glucose increased these values. Using Tukey’s multiple comparisons tests, significant differences within measures were revealed. Compared to 2 mM glucose conditions, in 10 mM, both acute and chronic insulin increased glycolytic capacity (acute insulin, 2 mM vs. 10 mM glucose: P = 0.0395; insulin 1 h, 2 mM vs. 10 mM glucose: P = 0.0164, Fig. 5i, hashtags). Similar findings were identified on measures of the glycolytic reserve, where acute insulin significantly raised values only in the 10 mM condition (acute insulin, 2 mM vs. 10 mM glucose: P = 0.0017; insulin 1 h, 2 mM vs. 10 mM glucose: P = 0.0011; 10 mM glucose, control v. acute insulin: P = 0.0339; Fig. 5k, hashtags). Interestingly, glycolysis did not appear to be altered by extracellular glucose or insulin exposures (insulin: F(2,44) = 0.6003, P = 0.5531; glucose: F(1,44) = 0.4890, P = 0.4881; Fig. 5k). Again, here, clear associations are seen between extracellular glucose concentrations and insulin exposure on potential glycolytic engagement.
Discussion
Discrepancies in energetic measures obtained using traditional biochemical approaches are difficult to reconcile with those obtained using alternative methods. Traditional methods of quantifying metabolic processes in the brain, [e.g., mass spectroscopy, biochemical, chemiluminescent or bioluminescent assays, radioactively labeling nucleotides, etc. reviewed in ref. 71] lose the nuance of cell-to-cell interactions found in unicellular imaging. Furthermore, it is important to note that measurements are often reported as calibrated, uncalibrated, occupancy measures, or percent change from baseline, and for this reason it is difficult to compare between experiments. Indeed, ATP:ADP of about 10 has been measured in the murine brain during stimulated activity72. However, in other cell types, such as the rat liver, ratios span from 5 to 100+, although the rate of ATP synthesis from the mitochondria appears to be negligible73. Using a modified luminescence assay, ATP:ADP measures of 20 and 1000 have been recorded in E. coli74. Directly addressing calibrated ATP:ADP measures, the Yellen group proposes that healthy mammalian cultures display values between 1 and 100, with baseline values for individual cells nearing 548; likewise, in mouse cortical neurons, measures of PercevalHR occupancy of approximately 1 have been published49. Several others describe ATP:ADP as a percent change from baseline, precluding us from addressing nucleotide ratios36,45,47,75,76. While we do not measure occupancy in this study, nor do we report on calibrated [ATP:ADP] values, we describe baseline PFRs that fall within a range of 2–30, with an average of ~5, which aligns well with prior work from the Yellen lab Figs.1b, 3b53. We also report a very similar pH calibration value for PercevalHR fluorescence correction based on this work (~0.67 F475/F427 change for every unit of pH change) with a different pH indicator. Overall, the large number of methods available to measure and report on ATP:ADP seems to contribute to the significant variations apparent throughout the literature. Additionally, the use of different cell types as well as different conditions for growth [mixed cultures, media, and experimentation (e.g., temperature, oxygenation, pH buffers)] also clearly influences metabolic status.
In vitro investigations of metabolic fluxes are useful for elucidating molecular processes in meticulously controlled conditions. For this purpose, the most common neuronal and glial cell culture methods can be categorized into three types: unicellular cultures of separated neurons and astrocytes, co-cultures (astrocyte-enriched neuronal cultures, seeded separately), and mixed cultures (neurons and astrocytes jointly seeded). A subtle, but key component underlying this study is the use of a mixed-culture system. Neurons and astrocytes independently maintained for imaging are not able to reflect on the interactions known to occur in the brain, given the proximity between cells and the sharing of metabolic intermediates. This concept is essential when evaluating energy production and consumption, as predicted by the astrocyte-neuron lactate shuttle hypothesis (ANLSH)32,77. As compared to the Yellen group, who appears to use unicellular cultures, here a mixed-culture system was used, which may have led to the differences from the results previously published, particularly with respect to the sensitivity of glucose challenges53. Indeed, unicellular neuronal cultures exposed to bradykinin to stimulate glutamate release show no alterations in Ca2+ levels; however, astrocyte-enriched neuronal cultures respond to the exposure with increases in Ca2+, suggesting that neuronal Ca2+ signaling depends on the presence of astrocytes78. Likewise, co-cultured neurons and astrocytes may suffer from a lack of communication due to the absence of an initial seeding period when cells develop together. Our use of a mixed-culture system was an attempt to address some of these limitations. It is possible that the lack of a robust glucose response when compared to Tantama and colleagues53 may be due to the proximity of neurons and astrocytes, where glutamate-derived, astrocyte-produced lactate delivers energy to neurons, thus reducing the need for glycolysis in neurons; however, this hypothesis will require further investigation. Furthermore, early mixed-culture work of neurons and astrocytes has shown that the propagation of Ca2+ signaling is indeed mediated by connections between the cell types79. In this study, only neurons in direct contact with astrocytes displayed increases in Ca2+ with stimulation. Furthermore, it is important to note that using the FLIP nanosensor in a mixed-culture environment, prior work has demonstrated that compared to younger cultures, more mature cultures, as those used here (>14 DIV) show decreases in neuronal glycolytic rate (despite increases in astrocytes)36 reviewed in ref. 77.
It does not appear that the use of sera in culture maintenance altered the results presented in this report, as the Tantama manuscript reports on the reliability of the PercevalHR indicator in experiments with 10% FBS (HEK293 and astrocytic cultures), as well as in experiments with cells unexposed to serum (neuronal cultures). Furthermore, the initial work of Takahashi et al.80 was conducted in the presence of FBS, where co-cultures of astrocytes and neurons responded to K+-mediated depolarization with increases in metabolism after 15 min. While astroglia exposed to physiological levels of glucose depend on oxidative metabolism more than cultures grown in high glucose solutions, these differences were obtained in the presence of FBS in the growth medium34, suggesting that the use of FBS does not invalidate results. Overall, the culture type, growing conditions, and stage of development are all essential elements to consider when interpreting results.
Cells with resting membrane potentials (hyperpolarized) that also have voltage-gated calcium channels (VGCCs) will show increases in intracellular Ca2+ when depolarized by extracellular potassium81. Likewise, in acute slices, astrocytes, and oligodendrocytes respond to potassium depolarization with significant increases in Ca2+ through the recruitment of VGCCs82–84. In mixed neuronal and astrocytic cultures, exposures to high potassium have been shown to increase metabolism80. In addition, increases in extracellular potassium activate inward rectifying K+ channels in astrocytes, consequently initiating K+ conductance85. Moreover, in conditions where neurons and astrocytes co-exist, K+ may depolarize presynaptic terminals on neurons, releasing glutamate onto astrocytes83. Therefore, it is not surprising to observe reduced PFRs in response to potassium exposure. Yet, in contrast, some groups note small (~4–5%) increases in ATP in response to K+ in cultured astrocytes and organotypic slices86,87. Using culture and hippocampal slices, elevations in ATP have been linked to an influx of bicarbonate (and subsequent cytosolic alkalinization) during K+-mediated astrocyte depolarization, ultimately stimulating glycolysis (e.g., the Crabtree effect)57. Here, however, we observed mild acidification (~8–20%; Fig. S2) in neurons and astrocytes during imaging in response to KCl. To further validate these results, a separate series of experiments was conducted in the absence of bicarbonate buffer in the perfusion system, and again, we saw similar decreases in PFRs, suggesting that the Crabtree effect is not a robust contributor in our culture. These results corroborate prior work exposing neuronal hippocampal cultures to 50 mM KCl – where cells were shown to initially respond to treatment with acidification, which was followed by alkalinization at much later time points [i.e., 10–50 min later88].
The role of insulin in the mammalian brain is still unclear. Traditionally, it is thought that the brain is insensitive to insulin; however, it is well characterized that insulin receptors are expressed in cultured neurons89, and several labs have shown the localization and expression of IR in both neurons90 and astrocytes91 in vitro. Prior work in our lab investigating the acute actions of insulin and insulin sensitizers on calcium homeostasis has reliably shown alterations in processes such as calcium-induced calcium release, reductions in Ca2+-sensitive K+ voltages, and voltage-gated Ca2+ currents25,55,92. Here, we show that the duration of the insulin exposure in combination with extracellular glucose levels differentially impacts metabolism (see Mitochondrial Stress Test), highlighting the brain’s insulin sensitivity. Although we believe our report is the first to directly test for the impact of insulin on bioenergetics using PercevalHR in neurons and astrocytes, it is essential to note that previous work from McNay and colleagues initially showed that in vivo local insulin application reduced extracellular glucose and elevated lactate levels93. Perhaps the small reductions in PFRs during acute insulin treatment in high glucose conditions are mediated by the recruitment of high-energy phosphates during autophosphorylation of the IR upon insulin binding, thus triggering further ATP utilization through signaling pathways including, but not limited to, Akt, PI3K, IRS-1, and PIP3.
We initially expected PFRs would mirror extracellular glucose levels (ranging from 0.1 mM–10 mM) yet only detected a minor, but significant, decrease in PFRs when changing the solution from 5.5 mM to 0.1 mM glucose under the 2 mM pretreatment condition. Prior work shows astrocytes and Neuro2A cells demonstrate large ATP:ADP changes in response to glucose excursions94, similarly, in primary astrocytic cultures, ATP:ADP appears coupled to available glucose53. Furthermore, work from Bittner and colleagues highlights the presence of dynamic glucose transport systems across multiple cell types, including mixed murine cortical cultures, adipocytes, myoblasts, fibroblasts, HeLa cells, and hippocampal tissue slices, capable of altering glycolytic rate33. Surprisingly, somewhat overshadowed by a large publication record focusing mostly on astrocytes, few papers have conducted dynamic glucose excursions in neurons while looking at bioenergetic changes33,34,36 reviewed in ref. 95. Neuronal and astrocytic cultures are routinely maintained in very high extracellular glucose concentrations (~35 mM), which is known to alter glucose oxidation rates, glycogen levels, and lactate release34. We attempted to address this issue by normalizing glucose levels 24 h prior to experimentation (see “Methods” section) and, while the effects of glucose excursions were not seen in 10 mM pretreatment conditions, we did note a significant reduction, albeit small, with pretreatment in more physiological conditions. Unfortunately, procedural details in many prior manuscripts fail to identify if, and for what pretreatment duration cells were exposed to physiological levels of extracellular glucose prior to experimentation. Further analysis should be conducted to investigate to what extent different durations of glucose pretreatments affect cellular bioenergetics.
The Seahorse data reported here indicates neurons and astrocytes are sensitive to insulin exposure and/or extracellular glucose concentrations. Significant elevations with either or both treatments are seen in measures of basal respiration, maximal respiration, ATP production, glycolytic capacity, and glycolytic reserve, whereby only cells exposed to acute insulin under the 10 mM glucose condition show increases in glycolytic reserve and capacity. These results align well with PercevalHR imaging data, showing insulin exposure alters PFRs only in the 10 mM condition (see Fig. 3a); however, clear differences are also noted, where elevations in glycolytic capacity and reserve are seen despite reductions in PFRs. Here, we show that insulin exposures do not increase ATP production (see Fig. 5f) but increase oxidative phosphorylation, as evidenced by elevations in basal and maximal respiration (see Fig. 5d, e, respectively). Along with this, insulin exposures reduce coupling efficiency (see Fig. 5c) and increase proton leak (Fig. S3), perhaps shifting bioenergetics to the well-known anabolic effects of the hormone or synthesis of glycogen, which may explain the subtle impact of insulin on PFRs. Alternatively, it is important to consider the difference in hormone exposure duration between imaging and Seahorse experiments. Indeed, the high temporal resolution obtained during PercevalHR imaging (i.e., minutes) captures transient changes across short periods of time after exposures to insulin, whereas the data derived from the Seahorse assay are delayed and collected approximately 20–100 min following the introduction of insulin into the medium. However, both methods provide evidence for significant associations between insulin action (or lack thereof) and extracellular glucose levels in neurons and astrocytes. These results emphasize potential bioenergetic mechanisms that modulate insulin action on CNS metabolism and may inform on homeostatic processes in both healthy and pathological states, particularly given the evidence of elevated extracellular glucose levels in the brains of AD patients17.
Although useful for studying molecular mechanisms, the cellular model presented here is not representative of the systems-level mechanisms at work in the brain, making it difficult to infer on in vivo physiology and the impact of insulin or glucose changes in the brain. In vivo approaches for evaluating brain metabolism using high-resolution biosensors have been recently published96, demonstrating that, when combined with imaging techniques that allow for awake and even ambulating animals to be studied (e.g., 2-photon), nanosensors such as PercevalHR are critical for addressing the long-contested points of brain metabolism and bioenergetics. That is, this integrative approach is powerful and may be able to directly tackle discrepancies in results seen across traditional whole-animal imaging and biochemical methods. Thus, we recognize the limitations of this report and propose further investigations of unicellular bioenergetics using PercevalHR in vivo. Furthermore, the work presented depicts clear interactions between insulin actions on metabolism and extracellular glucose levels in the brain, inferring on potential mechanisms of central insulin sensitivity.
Material and methods
Cell culture preparation and maintenance
All animals used in this study were maintained according to an Institutional Animal Care and Use Committee protocol approved by the University of Kentucky (IACUC# 2020-3555). Mixed primary neuronal and astrocytes hippocampal cultures were established from Sprague Dawley rat pups (E18), as described in previous work54,55,66,68,97. Briefly, following deep anesthesia (5% isoflurane) and euthanasia, pups were removed and hippocampi were isolated in Hank’s balanced salt solution (HBSS) (all salts were obtained from ThermoFisher Scientific; Waltham, MA, United States), supplemented with 4.2 mM NaHCO3 and 12 mM HEPES, over ice. The hippocampi were subsequently added to a 50 mL conical tube containing 10 mL 0.25% trypsin-EDTA and incubated for 11 min at room temperature (~23 °C). After drawing off the trypsin-EDTA solution, the hippocampi were washed 3 times with warm (37 °C) Minimum Essential Medium, supplemented with 2 mM L-glutamine and 30 mM D-glucose (SMEM), then triturated in 10 mL of SMEM1 mL of this solution was dispensed into individual 35 mm glass-bottom dishes (Matsunami Glass Ind., Ltd., Osaka, Japan) already containing 1 mL of plating media (SMEM supplemented with horse serum and fetal bovine serum). A hemocytometer was used to count the number of cells per 100 nL of solution for diluting to a final plating density of approximately 450,000 cells per dish using SMEM. Dishes were either coated with 0.5% poly-L-lysine and etched with HCl the night before or treated with polyethyleneimine (PEI) an hour prior to culturing. To account for evaporation, an uncovered 35 mm plastic dish filled with 2 mL of sterile water was placed in the center of a 100 mm dish housing the cultures. Cultures were maintained in an incubator at 37 °C, in a humidified environment of 5% CO2 and 95% air. A half-medium exchange was conducted on day 1 in vitro (DIV 1). On DIV 4, a half-medium exchange of SMEM supplemented with FUDR and uridine was conducted to stall glial cell growth. In attempts to normalize glucose transporter function following 12–15 days in high glucose concentration, cells underwent a last half-medium exchange into a serum-free glucose solution (approximately 5.5 mM)~24 h prior to imaging experiments.
PercevalHR lentiviral construction and transduction
The FUGW-PercevalHR plasmid (plasmid #49083; RRID: Addgene 49083), containing a Human Ubiquitin C promoter, was purchased from Addgene (Watertown, MA, United States), purified, and further packaged into a lentivirus by the Cincinnati Children’s Hospital Viral Vector Core (Cincinnati, OH, United States) at a titer of ~1 × 109 TU/mL. Cultures were transduced with the lentivirus on DIVs 10–12 at a multiplicity of infection (MOI) of 3–10.
Image acquisition
Epifluorescence microscopy was used to sequentially image multiple excitation wavelengths (i.e., 475 nm, 427 nm, 545 nm), and obtain signals from both the ratiometric (PercevalHR) and single-wavelength (pHrodo) indicators (Fig. 6b), across two cell types in the same FOV with unicellular resolution (a; Fig. 7b).
Fig. 6. Imaging timeline schematics.
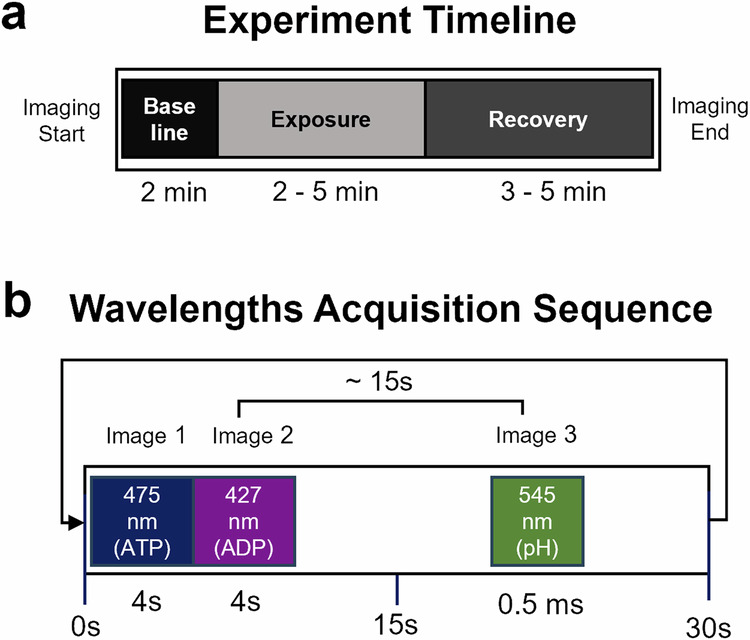
a shows a sample timeline for a typical experiment where baseline imaging data are acquired for 2 min, followed by 2–5 min of drug or solution exchange, and 3–5 min of recovery. b shows wavelength excitations for PercevalHR (475 and 427 nm, Image 1 and 2, respectively), and 15 s later, pHrodo was excited at 545 nm (Image 3).
Fig. 7. Representative images of astrocytes and neurons during exposure to 50 mM KCl.

a shows an astrocyte imaged using both wavelengths (grayscale) and the resulting ratio (right) using pseudocolor imaging (left, scale, where “L” indicates low PFRs and “H” indicates high PFRs). b highlights a single field of view taken at baseline (left column), during the response to KCl (middle column), and during recovery (right column). Both ATP, ADP, and ratioed images are shown, as well as the pHrodo images (bottom). Sample regions of interest are shown in red on the baseline 475 nm image.
Live-cell hippocampal neuronal and astrocytic fluorescence imaging
All cells were imaged between DIVs 12–16, ~72 h after PercevalHR transduction, using an epifluorescence microscope (E600FN; Nikon Inc., Melville, NY, United States) equipped with a heated objective, a flow-through chamber (RC22 Warner Instruments; Holliston, MA, United States), and a preheater (HPRE2 Cell Microcontrols; Norfolk, VA, United States) (35–37 °C). Cultured dishes were perfused with a peristaltic pump (PPS2, Multichannel Systems; Kusterdingen, Germany) at 1 mL/min or 2 mL/min using a standard HEPES and bicarbonate-based oxygenated solution (120 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 0.1–10 mM D-Glucose; 95% air, 5% CO2) unless otherwise noted (i.e., pH calibration assays and bicarbonate-free imaging experiments, see below). Compounds were introduced upstream of the imaging chamber using either a syringe pump (Model 341b, Sage Instruments; Watsonville, CA, United States) or bolus administration (Fig. 6a). Experiments were conducted across conditions of low (2 mM) or high (5–10 mM) extracellular glucose. For those experiments (i.e., insulin and glucose excursions), cells were pretreated for 30 min with either solution.
Imaging Workbench 5.0 (Indec Biosystems; Los Altos, CA, United States) was used to acquire all fluorescence signals and to control the fluorescence excitation filter device (Mercury lamp; Lambda DG4, Sutter Instruments; Novato, CA, United States) using a Transistor-Transistor-Logic (TTL) signal. PercevalHR fluorescence was captured through an emission filter (535 ± 45 nm) using an Andor Technology DV887 iXon camera (Belfast, United Kingdom). A dichroic mirror centered at 505 nm and two exciter filters (ATP: 475 ± 40 nm; ADP: 427 ± 40 nm) were used to excite the PercevalHR nanosensor using 2–4 s exposures for each channel. To correct PercevalHR’s pH sensitivity, experiments were conducted concomitantly with the intracellular pH indicator pHrodo (Catalog number: P35372; ThermoFisher Scientific). Cells were exposed to pHrodo AM, according to manufacturer instructions (in either low or high glucose conditions, see paragraph above). pHrodo fluorescence was excited through a 545 ± 25 nm filter and emitted light was passed through a 565 nm dichroic filter and a 605 ± 70 nm emission filter (Chroma Technology Corp.; Bellows Falls, VT, United States). A phase image was taken of a field of view (FOV) to distinguish morphological features of neurons and astrocytes, thus allowing us to separate data by cell type. For most experiments, a 2-min baseline was imaged prior to exposure to one of several compounds for 2–5 min [50 mM KCl; 20 µM glutamate; 10 µM FCCP; 10 nM Apidra (zinc-free glulisine); 0.1 mM, 5.5 mM, and 10 mM glucose].
Fluorescence quantification and statistical treatment
Regions of interest (ROIs) were hand-drawn around the soma of neurons and astrocytes with Imaging Workbench 5.0 and used to extract all signals (phase, pHrodo, PercevalHR). Excel, GraphPad Prism 9.5.1 (GraphPad Software Inc.; San Diego, CA, United States), and MATLAB (MathWorks; Natick MA, United States; version R2023b) code written in-house were used to process and analyze the data. An area free of cells was selected for background subtraction on all wavelengths acquired. Fluorescence values were averaged between cells and dishes for corresponding fluorophores to determine mean PFRs across experiments. Data were tested for significance, using a two-way ANOVA with repeated measures using time and cell type as main factors. When frames were inadvertently skipped during imaging, the data lost prevented the use of repeated measures ANOVA; thus, in these cases, a mixed-effects model (REML) was used, followed by post-hoc testing (Šídák’s or Tukey’s multiple comparison tests). We used Geisser-Greenhouse correction factors for one-way repeated measures ANOVAs, as we could not assume the sphericity of the datasets. On occasion, this correction can report on a non-whole degree of freedom. Significance was defined as p < 0.05, and all data is presented as means ± standard error of the mean (SEM). Cells displaying resting baseline PFRs <1.5 were removed from the analysis (9 neurons, 3 astrocytes) to represent a population of healthy cells. Additionally, data was filtered using a root mean square (RMS) method to eliminate exceptionally noisy PFRs during baseline.
Mitochondrial and glycolytic function
Using similar techniques to those described above, astrocytes and neurons were plated at 90,000 cells per well in a Seahorse 96-well plate. Media exchanges were conducted as described previously on DIVs 1 and 4. The day prior to experimentation (DIV 12), the sensor cartridge from the Extracellular Flux kit was filled with standard XF calibrant solution (Agilent Technologies, Santa Clara, CA, USA) and incubated at 37 °C overnight. The following morning, injection ports solutions were prepared with mitochondrial and glycolytic substrates/inhibitors to measure oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs), respectively. The Mito stress test and Glycolytic stress test assays were carried out using the SeahorseXFe96 Flux Analyzer (Agilent Technologies), as described previously98. An hour before the assays, some wells received 10 nM insulin (Insulin 1 h), while other wells were exposed acutely to insulin (Acute Insulin) prior to the start of the OCR assay (see Fig. 6). All wells were filled to 175 µL with XF DMEM assay media reconstituted with 10 mM or 2 mM glucose, 2 mM glutamine and 1 mM pyruvate. We diluted chemical stocks in the reconstituted assay media. After all injections, the final concentration of each compound was as follows: 2.5 µM oligomycin (Port A), 3 µM FCCP (Port B), 0.1 µM rotenone and 1 µM antimycin A (Port C) and 2-DG (Port D). The OCR and ECAR measurements were conducted over a period of approximately 70 min. The mitochondrial stress test report was generated using the standard export protocol. The glycolytic rates were derived manually from the ECAR data obtained during the assay using measures after 2-DG injection as the baseline. Following the assay, the cells per well were counted using a Biotek Cytation-5 Multimode Plate Reader (Agilent Technologies). All data collected during OCR and ECAR assays were normalized to 1000 cells.
pHrodo calibration and PercevalHR pH correction
Using HEPES-based solutions with a low pH (6.5) and a high pH (7.5), we quantified the change in PFRs as well as pHrodo fluorescence in 23 dishes. Briefly, to equilibrate intracellular and extracellular pH during the solution exchanges, dishes were pretreated with 10 µM valinomycin/DMSO and 10 µM nigericin/DMSO using an intracellular pH calibration buffer kit (Catalog #: P35379; ThermoFisher Scientific). A calibration assay was performed over a period of up to 40 min. For the first 2.5–5 min, cells were perfused with either the low or high pH solution, then switched to the alternative pH solution and imaged until a plateau was reached (approximately 15–30 min). Because pHrodo is a single-wavelength indicator, fluorescence levels were normalized to the baseline measures obtained prior to solution exchange and were expressed as a percent change from baseline. Alterations in pHrodo fluorescence across the 1-unit pH change were used to determine the average corresponding PercevalHR fluorescence change during pH excursions. Thus, based on this relationship, we determined that a 1 pH unit excursion yielded a 0.67 change in PFRs, and all data were corrected for measured changes in pHrodo fluorescence acquired across individual experiments.
Supplementary information
Acknowledgements
We thank Dr. Susan Kraner, Mr. Grant Fox, Mrs. Irina Artiushin, and Ms. Jacquelyn Rhinehart for their helpful technical contributions. This work was made possible by the National Institutes of Health (P01AG078116 and R01AG033649 to OT; NIGMS, P20 GM148326 to PS) and the University of Kentucky (Neuroscience Research Priority Area Pilot Grant to OT; Diabetes and Obesity Research Priority Area funded by the Office of the Vice-President for Research to JT; CNS Metabolism COBRE to PS, supported by a grant from the National Institute of General Medical Sciences).
Author contributions
S.S., H.F., and S.C. conducted and analyzed all culture imaging experiments. R.-L.L. helped with aspects of analysis and presentation. J.T. helped with some of the initial FCCP experiments. H.V. and P.S. conducted and analyzed all SeaHorse experiments. O.T. conceived experiments and helped analyze, interpret, and write the final document with S.S.
Data availability
The datasets generated during the course of these studies are available upon reasonable request.
Code availability
The MATLAB codes (MathWorks; Natick MA, United States; version R2023b) generated during this study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s44324-024-00037-y.
References
- 1.Harris, J. J., Jolivet, R. & Attwell, D. Synaptic energy use and supply. Neuron75, 762–777 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Yellen, G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol.217, 2235–2246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attwell, D. & Laughlin, S. B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow. Metab.21, 1133–1145 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Pellerin, L. & Magistretti, P. J. Glutamate uptake stimulates Na+,K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J. Neurochem.69, 2132–2137 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Hertz, L., Song, D., Xu, J. N., Peng, L. & Gibbs, M. E. Role of the astrocytic Na+, K+- ATPase in K+ homeostasis in brain: K+ uptake, signaling pathways and substrate utilization. Neurochem. Res.40, 2505–2516 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res.24, 321–329 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Yaffe, K. et al. Diabetes, glucose control, and 9-year cognitive decline among older adults without dementia. Arch. Neurol.69, 1170–1175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croteau, E. et al. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol.107, 18–26 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Schwartz, M. W. & Porte, D. Jr. Diabetes, obesity, and the brain. Science307, 375–379 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging10.1007/s40336-013-0026-y (2013). [DOI] [PMC free article] [PubMed]
- 11.Griffith, C. M., Eid, T., Rose, G. M. & Patrylo, P. R. Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology136, 202–215 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Frazier, H. N. et al. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp. Neurol.313, 79–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang, Q. et al. Glucose Metabolism, Neural Cell Senescence and Alzheimer’s Disease. Int. J. Mol. Sci.10.3390/ijms23084351 (2022). [DOI] [PMC free article] [PubMed]
- 14.Frolich, L. et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. (Vienna)105, 423–438 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Hoyer, S. Senile dementia and Alzheimer’s disease. Brain blood flow and metabolism. Prog. Neuropsychopharmacol. Biol. Psychiatry10, 447–478 (1986). [DOI] [PubMed] [Google Scholar]
- 16.Mayeda, E. R. et al. Type 2 diabetes and cognitive decline over 14 years in middle-aged African Americans and whites: the ARIC Brain MRI Study. Neuroepidemiology43, 220–227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.An, Y. et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement.14, 318–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adzovic, L. et al. Insulin improves memory and reduces chronic neuroinflammation in the hippocampus of young but not aged brains. J. Neuroinflamm.12, 63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunner, Y. F., Kofoet, A., Benedict, C. & Freiherr, J. Central insulin administration improves odor-cued reactivation of spatial memory in young men. J. Clin. Endocrinol. Metab.100, 212–219 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Pearson-Leary, J., Jahagirdar, V., Sage, J. & McNay, E. C. Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res.338, 32–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Talbot, K. Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener. Dis. Manag.4, 31–40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benedict, C. et al. Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology32, 239–243 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Reger, M. A. et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis.13, 323–331 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hallschmid, M. Intranasal insulin. J. Neuroendocrinol.33, e12934 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Maimaiti, S. et al. Intranasal insulin improves age-related cognitive deficits and reverses electrophysiological correlates of brain aging. J. Gerontol. A Biol. Sci. Med Sci.71, 30–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frazier, H. N. et al. Long-term intranasal insulin aspart: a profile of gene expression, memory, and insulin receptors in aged F344 rats. J. Gerontol. A Biol. Sci. Med. Sci.75, 1021–1030 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo, Z. et al. Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Sci. Rep.7, 45971 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nijssen, K. M. R., Mensink, R. P. & Joris, P. J. Effects of intranasal insulin administration on cerebral blood flow and cognitive performance in adults: a systematic review of randomized, placebo-controlled intervention studies. Neuroendocrinology113, 1–13 (2023). [DOI] [PubMed] [Google Scholar]
- 29.San Martin, A. et al. Single-cell imaging tools for brain energy metabolism: a review. Neurophotonics1, 011004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koveal, D., Diaz-Garcia, C. M. & Yellen, G. Fluorescent biosensors for neuronal metabolism and the challenges of quantitation. Curr. Opin. Neurobiol.63, 111–121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barros, L. F., Ruminot, I., Sandoval, P. Y. & San Martin, A. Enlightening brain energy metabolism. Neurobiol. Dis. 10.1016/j.nbd.2023.106211 (2023). [DOI] [PubMed]
- 32.Pellerin, L. & Magistretti, P. J. Glutamate uptake into astrocytes stimulates aerobic glycolysis - a mechanism coupling neuronal-activity to glucose-utilization. Proc. Natl Acad. Sci. USA91, 10625–10629 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bittner, C. X. et al. High resolution measurement of the glycolytic rate. Front. Neuroenerget.10.3389/fnene.2010.00026 (2010). [DOI] [PMC free article] [PubMed]
- 34.Abe, T., Takahashi, S. & Suzuki, N. Oxidative metabolism in cultured rat astroglia: effects of reducing the glucose concentration in the culture medium and of D-aspartate or potassium stimulation. J. Cereb. Blood Flow. Metab.26, 153–160 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Porras, O. H., Loaiza, A. & Barros, L. F. Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J. Neurosci.24, 9669–9673 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baeza-Lehnert, F. et al. Non-canonical control of neuronal energy status by the Na(+) pump. Cell Metab.29, 668–680.e664 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Garde, A. & Sherwood, D. R. Visualizing cytoplasmic ATP in C. elegans larvae using PercevalHR. STAR Protoc.3, 101429 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garde, A. et al. Localized glucose import, glycolytic processing, and mitochondria generate a focused ATP burst to power basement-membrane invasion. Dev. Cell57, 732–749.e7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bohnert, K. A. & Kenyon, C. A lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature551, 629–633 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goudeau, J., Samaddar, M., Bohnert, K. A. & Kenyon, C. Addendum: a lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature580, E5 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Ketschek, A., Sainath, R., Holland, S. & Gallo, G. The axonal glycolytic pathway contributes to sensory axon extension and growth cone dynamics. J. Neurosci.41, 6637–6651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russo, E. et al. SPICE-Met: profiling and imaging energy metabolism at the single-cell level using a fluorescent reporter mouse. EMBO J.41, e111528 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plotegher, N. et al. Impaired cellular bioenergetics caused by GBA1 depletion sensitizes neurons to calcium overload. Cell Death Differ.27, 1588–1603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karagas, N. E. et al. Loss of activity-induced mitochondrial ATP production underlies the synaptic defects in a Drosophila model of ALS. J. Neurosci.42, 8019–8037 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris, O., Deng, H., Tam, C. & Jasper, H. Warburg-like metabolic reprogramming in aging intestinal stem cells contributes to tissue hyperplasia. Cell Rep.33, 108423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang, M., Chadwick, A. E., Dart, C., Kamishima, T. & Quayle, J. M. Bioenergetic profile of human coronary artery smooth muscle cells and effect of metabolic intervention. PLoS ONE12, e0177951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang, M., Dart, C., Kamishima, T. & Quayle, J. M. Hypoxia and metabolic inhibitors alter the intracellular ATP:ADP ratio and membrane potential in human coronary artery smooth muscle cells. PeerJ8, e10344 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tantama, M. & Yellen, G. Imaging changes in the cytosolic ATP-to-ADP ratio. Methods Enzymol.547, 355–371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rueda, C. B. et al. Mitochondrial ATP-Mg/Pi carrier SCaMC-3/Slc25a23 counteracts PARP-1-dependent fall in mitochondrial ATP caused by excitotoxic insults in neurons. J. Neurosci.35, 3566–3581 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ofori, J. K. et al. Elevated miR-130a/miR130b/miR-152 expression reduces intracellular ATP levels in the pancreatic beta cell. Sci. Rep.7, 44986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez-Sanchez, A. et al. Disallowance of Acot7 in beta-cells is required for normal glucose tolerance and insulin secretion. Diabetes65, 1268–1282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jett, S. et al. Systematic review of (31)P-magnetic resonance spectroscopy studies of brain high energy phosphates and membrane phospholipids in aging and Alzheimer’s disease. Front. Aging Neurosci.15, 1183228 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tantama, M., Martinez-Francois, J. R., Mongeon, R. & Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun.4, 2550 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pancani, T., Anderson, K. L., Porter, N. M. & Thibault, O. Imaging of a glucose analog, calcium and NADH in neurons and astrocytes: dynamic responses to depolarization and sensitivity to pioglitazone. Cell Calcium50, 548–558 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maimaiti, S. et al. Novel calcium-related targets of insulin in hippocampal neurons. Neuroscience364, 130–142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahlert, S., Zundorf, G. & Reiser, G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J. Neurosci. Res.79, 262–271 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Fernandez-Moncada, I. et al. Neuronal control of astrocytic respiration through a variant of the Crabtree effect. Proc. Natl Acad. Sci. USA115, 1623–1628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attucci, S. et al. Group I metabotropic glutamate receptor inhibition selectively blocks a prolonged Ca(2+) elevation associated with age-dependent excitotoxicity. Neuroscience112, 183–194 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Norris, C. M. et al. Electrophysiological mechanisms of delayed excitotoxicity: positive feedback loop between NMDA receptor current and depolarization-mediated glutamate release. J. Neurophysiol.96, 2488–2500 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kinoshita, P. F. et al. The Influence of Na(+), K(+)-ATPase on glutamate signaling in neurodegenerative diseases and senescence. Front. Physiol.7, 195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lange, S. C. et al. Dynamic changes in cytosolic ATP levels in cultured glutamatergic neurons during NMDA-induced synaptic activity supported by glucose or lactate. Neurochem. Res.40, 2517–2526 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Bak, L. K. et al. Novel model of neuronal bioenergetics: postsynaptic utilization of glucose but not lactate correlates positively with Ca2+ signalling in cultured mouse glutamatergic neurons. ASN Neuro4, AN20120004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Magistretti, P. J., Pellerin, L., Rothman, D. L. & Shulman, R. G. Energy on demand. Science283, 496–497 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Hyder, F. et al. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J. Cereb. Blood Flow. Metab.26, 865–877 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Nicholls, D. G. & Ferguson, S. J. Bioenergetics 4th edn (Elsevier (Academic Press), 2013).
- 66.Frazier, H. N. et al. Elevating insulin signaling using a constitutively active insulin receptor increases glucose metabolism and expression of GLUT3 in hippocampal neurons. Front. Neurosci.14, 668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frazier, H. N. et al. Expression of a constitutively active human insulin receptor in hippocampal neurons does not alter VGCC currents. Neurochem. Res.44, 269–280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Porter, N. M., Thibault, O., Thibault, V., Chen, K. C. & Landfield, P. W. Calcium channel density and hippocampal cell death with age in long-term culture. J. Neurosci.17, 5629–5639 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blalock, E. M., Porter, N. M. & Landfield, P. W. Decreased G-protein-mediated regulation and shift in calcium channel types with age in hippocampal cultures. J. Neurosci.19, 8674–8684 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Norris, C. M., Blalock, E. M., Chen, K. C., Porter, N. M. & Landfield, P. W. Calcineurin enhances L-type Ca(2+) channel activity in hippocampal neurons: increased effect with age in culture. Neuroscience110, 213–225 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rajendran, M., Dane, E., Conley, J. & Tantama, M. Imaging adenosine triphosphate (ATP). Biol. Bull.231, 73–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Erecinska, M. & Silver, I. A. Ions and energy in mammalian brain. Prog. Neurobiol.43, 37–71 (1994). [DOI] [PubMed] [Google Scholar]
- 73.Morikofer-Zwez, S. & Walter, P. Binding of ADP to rat liver cytosolic proteins and its influence on the ratio of free ATP/free ADP. Biochem. J.259, 117–124 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meyrat, A. & von Ballmoos, C. ATP synthesis at physiological nucleotide concentrations. Sci. Rep.9, 3070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghosh Dastidar, S. et al. Distinct regulation of bioenergetics and translation by group I mGluR and NMDAR. EMBO Rep.23, e54501 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bressan, C. et al. The dynamic interplay between ATP/ADP levels and autophagy sustain neuronal migration in vivo. eLife10.7554/eLife.56006 (2020). [DOI] [PMC free article] [PubMed]
- 77.Diaz-Garcia, C. M. et al. Quantitative in vivo imaging of neuronal glucose concentrations with a genetically encoded fluorescence lifetime sensor. J. Neurosci. Res.97, 946–960 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parpura, V. et al. Glutamate-mediated astrocyte-neuron signalling. Nature369, 744–747 (1994). [DOI] [PubMed] [Google Scholar]
- 79.Nedergaard, M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science263, 1768–1771 (1994). [DOI] [PubMed] [Google Scholar]
- 80.Takahashi, S., Driscoll, B. F., Law, M. J. & Sokoloff, L. Role of sodium and potassium ions in regulation of glucose metabolism in cultured astroglia. Proc. Natl Acad. Sci. USA92, 4616–4620 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Verkhratsky, A. & Nedergaard, M. Physiology of astroglia. Physiol. Rev.98, 239–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barron, T. & Kim, J. H. Neuronal input triggers Ca(2+) influx through AMPA receptors and voltage-gated Ca(2+) channels in oligodendrocytes. Glia67, 1922–1932 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carmignoto, G., Pasti, L. & Pozzan, T. On the role of voltage-dependent calcium channels in calcium signaling of astrocytes in situ. J. Neurosci.18, 4637–4645 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pasti, L., Volterra, A., Pozzan, T. & Carmignoto, G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci.17, 7817–7830 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ransom, C. B. & Sontheimer, H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J. Neurophysiol.73, 333–346 (1995). [DOI] [PubMed] [Google Scholar]
- 86.Kohler, S. et al. Gray and white matter astrocytes differ in basal metabolism but respond similarly to neuronal activity. Glia71, 229–244 (2023). [DOI] [PubMed] [Google Scholar]
- 87.Lerchundi, R. et al. FRET-based imaging of intracellular ATP in organotypic brain slices. J. Neurosci. Res.97, 933–945 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Hartley, Z. & Dubinsky, J. M. Changes in intracellular pH associated with glutamate excitotoxicity. J. Neurosci.13, 4690–4699 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dakic, T. et al. The Expression of Insulin in the Central Nervous System: What Have We Learned So Far? Int. J. Mol. Sci. 24, 10.3390/ijms24076586 (2023). [DOI] [PMC free article] [PubMed]
- 90.Abbott, M. A., Wells, D. G. & Fallon, J. R. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci.19, 7300–7308 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garwood, C. J. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain8, 51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pancani, T. et al. Effect of high-fat diet on metabolic indices, cognition, and neuronal physiology in aging F344 rats. Neurobiol. Aging34, 1977–1987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McNay, E. C. et al. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn Mem.93, 546–553 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tantama, M., Hung, Y. P. & Yellen, G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J. Am. Chem. Soc.133, 10034–10037 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deitmer, J. W., Theparambil, S. M., Ruminot, I., Noor, S. I. & Becker, H. M. Energy dynamics in the brain: contributions of astrocytes to metabolism and pH homeostasis. Front. Neurosci.13, 1301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Diaz-Garcia, C. M., Nathwani, N., Martinez-Francois, J. R. & Yellen, G. Delivery of AAV for expression of fluorescent biosensors in juvenile mouse hippocampus. Bio Protoc.11, e4259 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pancani, T. et al. Distinct modulation of voltage-gated and ligand-gated Ca2+ currents by PPAR-gamma agonists in cultured hippocampal neurons. J. Neurochem.109, 1800–1811 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pandya, C. D. et al. Azithromycin reduces hemoglobin-induced innate neuroimmune activation. Exp. Neurol.372, 114574 (2024). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the course of these studies are available upon reasonable request.
The MATLAB codes (MathWorks; Natick MA, United States; version R2023b) generated during this study are available from the corresponding author on reasonable request.