

Abstract
AAA+ proteolytic machines unfold proteins before degrading them. Here, we present cryoEM structures of ClpXP-substrate complexes that reveal a postulated but heretofore unseen intermediate in substrate unfolding/degradation. A ClpX hexamer draws natively folded substrates tightly against its axial channel via interactions with a fused C-terminal degron tail and ClpX-RKH loops that flexibly conform to the globular substrate. The specific ClpX-substrate contacts observed vary depending on the substrate degron and affinity tags, helping to explain ClpXP’s ability to unfold/degrade a wide array of different cellular substrates. Some ClpX contacts with native substrates are enabled by upward movement of the seam subunit in the AAA+ spiral, a motion coupled to a rearrangement of contacts between the ClpX unfoldase and ClpP peptidase. Our structures additionally highlight ClpX’s ability to translocate a diverse array of substrate topologies, including the co-translocation of two polypeptide chains.
Subject terms: Cryoelectron microscopy, Proteases, Cryoelectron microscopy
AAA proteases must unfold substrates before degradation. Here the authors report cryo-EM structures to visualize how these machines achieve this by pulling the substrate tightly against their narrow axial channel and encircling the substrate’s folded domain using only the unfoldase’s flexible loops.
Introduction
From bacteria to mammals, ATP-fueled AAA+ proteases degrade regulatory, unneeded, or damaged intracellular proteins1–3. For target proteins with stable three-dimensional structures, ClpXP and other AAA+ proteases harness the energy of ATP hydrolysis to unfold this structure before translocating the denatured polypeptide through a narrow axial channel and into a self-compartmentalized peptidase chamber for degradation. Within the ClpXP complex, the ClpX unfoldase mediates substrate specificity by recognizing short ‘degron’ peptides, such as the ssrA tag, within target substrates. Unfolding is thought to occur when ClpX translocates a disordered substrate segment containing a degron, until the attached native structure is pulled against the axial channel. Subsequent ATP-fueled pulling events then endeavor to translocate the substrate polypeptide further through the channel, resulting in repeated application of a force that increases the cumulative probability of unfolding. Once the native protein is eventually denatured, sequence-independent translocation of the unfolded polypeptide into the ClpP peptidase chamber results in proteolysis (Fig. 1A).
Fig. 1. ClpXP bound to degron-tagged DHFR.

A Cartoon of steps in ClpXP degradation of a protein substrate. B Overlay of cryoEM map and model of ClpXP bound to the branched-degron-DHFR substrate (left). Density for the degron tail could be modeled extending from the folded domain of DHFR to the ClpP entry portal (center), with the maleimide-cysteine branchpoint marked by a yellow sphere. The insets (right) display density for methotrexate and the DHFR Trp30 and Phe31 side chains (top), and the last two residues of native DHFR, Arg158, and Arg159, which were contacted by the pore-1 loop of ClpX subunit A, which includes Tyr153 (bottom).
This model accounts for many experimental studies4. Moreover, X-ray crystallography and cryoEM have revealed architectural features of ClpX ring hexamers, double-ring ClpP tetradecamers, ClpX-ClpP docking, ClpX recognition of the ssrA degron with and without adaptor assistance, and ClpX interactions with the polypeptides of translocating substrates5–11. Notably, visualization at near-atomic resolution of a native substrate being pulled against the axial channel of any AAA+ protease has not to the best of our knowledge, until now, been realized.
Results and discussion
Substrate design
For structural studies, we used Escherichia coli ClpX∆N, an active variant lacking the N-terminal domain12, E. coli ClpP, and a protein substrate consisting of E. coli dihydrofolate reductase (DHFR) with different C-terminal degrons. One of our DHFR substrates had a branched degron tail, created by crosslinking a peptide with a C-terminal ssrA tag to a cysteine in a DHFR extension (Supplementary Fig. 1A, B). As expected from studies with another branched protein13, ClpXP degraded the branched-degron DHFR substrate (Supplementary Fig. 2), highlighting ClpX’s ability to translocate two chains simultaneously. Addition of methotrexate (MTX), a small molecule that binds and stabilizes DHFR, prevented ClpXP degradation of this substrate (Supplementary Fig. 2), as shown previously for different ssrA-tagged DHFR substrates14. We designed a second substrate, in which DHFR had an N-terminal H6-TEV tag and a linear C-terminal degron that terminated with the ssrA tag (Supplementary Fig. 1C).
ClpXP-DHFR structures
To visualize “engaged” complexes with DHFR pulled against the axial channel, we prepared cryoEM grids using samples containing ClpX∆N, ClpP, one of the DHFR substrates, ATP/Mg++, and MTX. As a control, we additionally imaged a sample lacking MTX. After grid screening, data collection, and image processing (see Methods), we determined two structures with native DHFR•MTX closely associated with the axial channel of ClpXP. Another structure, determined in the absence of MTX, did not contain native DHFR but did have substrate polypeptide in the ClpX channel. The resolutions of these structures ranged from ~2.6 to 3.7 Å (Table 1; Supplementary Figs. 3–14).
Table 1.
CryoEM data collection, processing, model building, and validation statistics
| Sample and data deposition information | |||
|---|---|---|---|
| Sample name | ClpX∆N•ClpP•branched-DHFR•MTX | ClpX∆N•ClpP•linear-DHFR•MTX | ClpX∆N•ClpP•branched-DHFR |
| Nucleotide added | ATP | ATP | ATP |
| PDB code | 8V9R | 9C87 | 9C88 |
| EMDB ID | 43081 | 45299 | 45300 |
| Data collection and image processing | |||
| Microscope | Titan Krios G3i | Titan Krios G2 | Titan Krios G2 |
| Camera | Gatan K3 (counting mode) | Gatan K3 (counting mode) | Gatan K3 (counting mode) |
| Magnification (nominal) | 130,000 X | 130,000 X | 130,000 X |
| Accelerating voltage (kV) | 300 | 300 | 300 |
| Total electron dose (e-/Å2) | 51.94 | 47.54 | 47.54 |
| Defocus range (µm) | -0.5 to -1.8 | -0.5 to -1.75 | -0.5 to -1.75 |
| Micrographs collected | 15,920 | 15,864 | 14,574 |
| Pixel size (Å) initial/calibrated | 0.679 / 0.654 | 0.65 | 0.65 |
| Map reconstruction | |||
| Image processing package | cryoSPARC | ||
| Total extracted ClpXP particles | 1,005,797 | 355,300 | 434,826 |
| Final particle count | 449,424 | 71,203 | 178,399 |
| Symmetry imposed | C1 | C1 | C1 |
| Resolution (Å) | |||
| 0.143 GSFSC unmasked | 3.3 | 4.4 | 3.3 |
| 0.143 GSFSC spherical mask | 3.1 | 4.1 | 3.0 |
| 0.143 GSFSC tight/local mask | 2.8 | 3.7 | 2.6 |
| 3DFSC sphericity (out of 1.0) | 0.986 | 0.931 | 0.972 |
| Model composition | |||
| Number of atoms | 72,633 | 77,422 | 74,036 |
| Protein residues | 4,955 | 4,943 | 4,733 |
| Ligands | ATP (3); ADP (3) Mg (4); MTX (1) | ATP (3); ADP (3) Mg (4); MTX (1) | ATP (3); ADP (3) Mg (4) |
| Model refinement | |||
| Refinement package | Phenix and Coot | Phenix and Coot | |
| Map-to-model cross correlation | |||
| masked | 0.81 | 0.75 | 0.81 |
| unmasked | 0.83 | 0.84 | 0.78 |
| RMSD bond lengths (Å) [#>4σ] | 0.003 [0] | 0.002 [0] | 0.002 [0] |
| RMSD bond angles (°) [#>4σ] | 0.666 [0] | 0.653 [0] | 0.501 [0] |
| Model validation | |||
| MolProbity score | 0.94 | 1.03 | 0.89 |
| Clash score | 1.80 | 2.48 | 1.04 |
| C-beta outliers (%) | 0.02 | 0.02 | 0.0 |
| Rotamer outliers (%) | 0.0 | 0.0 | 0.003 |
| Ramachandran favored (%) | 99.5 | 99.9 | 97.6 |
| Q-Score [mean] | |||
| Global / expected from resolution | 0.62 / 0.61 | 0.47 / 0.45 | 0.63 / 0.65 |
| ClpX | 0.61 | 0.47 | 0.54 |
| ClpPcis | 0.71 | 0.56 | 0.66 |
| DHFR | 0.60 | 0.22 | 0.72 |
The highest-resolution structure with density for folded DHFR•MTX (~2.8 Å) was obtained with the branched-degron substrate (Table 1; Supplementary Figs. 3, 6, 9) and showed native DHFR pulled against the ClpX ring (Fig. 1B). In this structure, 19 degron residues were modeled in a β-ribbon conformation that extended ~50 Å through the channel of ClpX and into the entry portal of ClpP. The degron branchpoint was within the ClpP portal, suggesting that remaining parts of the degron extended into the ClpP chamber, but the resolution in this region of the structure was insufficient to model either the branch or the degron termini. Density for residues 1–159 of native DHFR and MTX was present. DHFR in this structure aligned to a crystal structure of DHFR•MTX (PDB code 1RG7)15 with a Cα RMSD of 1.3 Å, indicating that ClpX binding does not cause major changes in the native conformation of MTX-bound DHFR.
We obtained a structure of ClpXP bound to linear-degron DHFR•MTX at ~3.7-Å resolution (Table 1, Supplementary Figs. 4,7,10). Notably, the orientation of DHFR with respect to ClpX observed for the branched-degron substrate fit poorly into this map, and the best rigid-body fit of the density was for a ~180° rotation of the native portion of DHFR compared to its position in the branched-degron structure (Fig. 2A, B). This rotation may be caused by the N-terminal H6-TEV tag on this DHFR construct, which would clash with ClpX in the DHFR orientation in the branched-degron structure. Local resolution of native DHFR in the linear-degron structure was low compared to most parts of ClpX and ClpP (Supplementary Fig. 7), suggesting that it may be bound less tightly and/or in an ensemble of slightly different conformations. The first nine residues of the linear degron of the DHFR substrate were clearly resolved and extended ~25 Å into the axial channel of ClpX. The linear degron tail immediately C-terminal to DHFR followed a similar trajectory to that of the branched degron substrate.
Fig. 2. Fully engaged DHFR can assume distinct orientations with respect to ClpX6 and its RKH loops.

ClpX is shown in outline representation with the positions of subunits A1, B2, C3, and D4 marked and the RKH loops of these subunits shown as spheres in different colors. The DHFR substrate is depicted in cartoon/outline representation in a rainbow-color scheme, with blue representing the N-terminus and red the C-terminus. A DHFR•MTX positioning in the branched-degron structure. B DHFR•MTX positioning in the linear-degron structure.
A peripheral collar of six RKH loops in the ClpX hexamer interacted with native DHFR in both engaged structures, albeit in different orientations made possible by RKH-loop flexibility (Fig. 2A). Indeed, aligning the RKH loops in our engaged DHFR complexes with the RKH loops in an ssrA-degron complex, some of which stabilize recognition of the ssrA tag6 (PDB code 6WRF), revealed movements of as much as 30 Å (Supplementary Fig. 12). This RKH-loop flexibility is likely to allow ClpXP to engage the divergent array of native cellular proteins it must unfold and degrade, as well as assisting in recognition of specific degrons present in these proteins6,16–20.
When MTX was omitted from the branched-degron substrate sample, we obtained a cryoEM structure (~2.6-Å resolution) containing ClpXP and a decapeptide in the channel of ClpX, presumably from the DHFR substrate (Table 1; Supplementary Figs. 5, 8, 11). Side-chain density for this peptide was not sufficient to identify a specific sequence from the degron or DHFR, and it was modeled as polyalanine. This structure appears to be a translocation complex following DHFR denaturation, as it aligned well with a structure containing a partially translocated ssrA tag (PDB code 6WSG)7 with an RMSD of 1.3 Å for all Cα atoms of ClpX (Supplementary Fig. 13).
Implications for substrate unfolding
The C-terminal β-strand of native DHFR, which is embedded in a β-sheet, connected to the degron tails in both engaged structures without slack but at different angles (Fig. 2). We posit that the absence of slack would ensure that a pulling force from an attempted power stroke is directly applied to the native portion of the substrate. The contact angle between the substrate and unfoldase may also influence unfolding efficiency, as some angles would optimize peeling of secondary-structure elements, whereas others would require shearing of multiple hydrogen bonds14,21. In this regard, we note that ClpXP unfolds the native titinI27 domain at rates differing by ~50-fold depending on whether an ssrA degron is attached to its N- or C-terminus22, and this impact likely results from a combination of the inherent stability of the local structure and the angle at which force is applied.
In the axial channels of our here reported structures as well as in previous structures of ClpXP and related AAA+ proteases and protein-remodeling machines, the pore-1 loops of five subunits of the AAA+ enzyme interact in a spiral conformation with every two residues of the backbone of the substrate polypeptide6–10,23. Conventions for naming ClpX subunits differ. For example, in our here reported structures, as well as some previous ClpXP structures, subunits A-E make these spiral interactions. In some ClpXP structures, however, subunits B-F make these contacts. We define subunits 1 and 5 as those whose pore-1 loops are positioned at the top and bottom of the ClpX spiral, respectively. The remaining ‘seam’ subunit7,8,23 connects the flanking subunits in the hexameric ring but its pore-1 loop is disengaged from the substrate backbone. In our DHFR-engaged structures, the pore-1 loops of ClpX subunits A1, B2, C3, D4, and E5 (where the superscript represents position in the spiral), and the pore-2 loops of subunits A1, B2, C3, D4 contacted degron residues in the channel (Fig. 3), with many interactions involving large side chains of the degron tail. Grip between the channel loops of ClpX and a degron tail affects unfolding efficiency, with larger non-polar side chains in the tail providing superior grip and faster unfolding1,4,24. Hundreds of ATP-hydrolysis events can be needed on average for ClpXP to unfold and degrade a stable substrate25,26. The extensive contacts observed between ClpX and the degron tail and native domain of DHFR in our engaged complexes (Fig. 3) should maximize grip and minimize substrate dissociation after unsuccessful denaturation attempts, increasing the cumulative probability of unfolding.
Fig. 3. Interactions between ClpX axial pore-loops and DHFR-degron tails.

Pore-1 loops (residues 150-155) are shown as spheres, pore-2 loops (residue 198-205) are shown as cartoons, and DHFR is shown in red surface representation. A Front and back views for the branched-degron substrate. B Front and back views for the linear degron substrate. Note that in each structure, the pore-1 loop from the Fseam subunit is not engaged with the substrate.
Distinct hexamer conformations
In some ClpX hexamers (e.g., pdb codes 6PP5, 6PP6, 6WRF, and 8E91), the pore-1 loop of the Fseam subunit is closest to the corresponding loop of subunit E5 near the bottom of the spiral. In all of our here reported structures, by contrast, Fseam moved higher with its pore-1 loop closest to the pore-1 loop of subunit A1 (Supplementary Fig. 14, Supplementary Movie 1).This upward movement of Fseam has been noted previously7–9. In Fseam ‘up’ structures, the ClpP-binding IGF loop of subunit 5 hops into a cleft in the ClpP7 ring that is unoccupied in Fseam ‘down’ structures (Fig. 4). This rearrangement of ClpX-ClpP contacts, which maintains a full complement ClpX IGF loops bound to ClpP clefts, should help to maintain tight binding in both Fseam ‘up’ and ‘down’ conformations. ClpP binding may stabilize conformations of ClpX that are more effective in protein unfolding as ClpX binds ClpP with ~5-fold different affinities depending on whether it is degrading a stable native substrate or an unfolded substrate27. Additionally, ClpP binding suppresses the unfolding defects of certain ClpX mutants and enables ClpXP to unfold GFP-ssrA approximately 3-fold faster than ClpX alone27,28.
Fig. 4. Rearrangement of ClpX-ClpP contacts correlates with upward movement of the seam subunit.

In many ClpXP complexes, the empty binding cleft on a ClpP7 ring lies between the clefts occupied by the IGF loops of ClpX subunits E5 and Fseam (A; PDB code 6WRF). In the structures presented here and some other ClpXP structures (B; PDB code 8V9R), the IGF loop of chain E5 moves into a binding cleft that is unoccupied in structures similar to that in (A). These movements correlate with Fseam upward movement, as shown in (C, D).
We used cryoDRGN29–31 to test for minor populations of ClpX in our ClpXP dataset with branched-degron DHFR•MTX, specifically looking for structures in which Fseam moved ‘down’ or the IGF-loop of subunit E5 ‘hopped’. Neither structure was detected. Instead, we observed DHFR moving subtly or becoming unresolved above the channel (Supplementary Movie 2). ClpXP continues to hydrolyze ATP as it attempts to unfold DHFR•MTX (Fig. 5), and the movements revealed by cryoDRGN may result from ATP-hydrolysis events that do not result in a productive power stroke because they are resisted by DHFR•MTX. This dynamic view of the structure suggests that ClpX attempts to denature DHFR•MTX, even when this outcome is highly improbable.
Fig. 5. Rates of ATP hydrolysis by ClpXP in the absence and presence of a degron-tagged DHFR•MTX substrate.
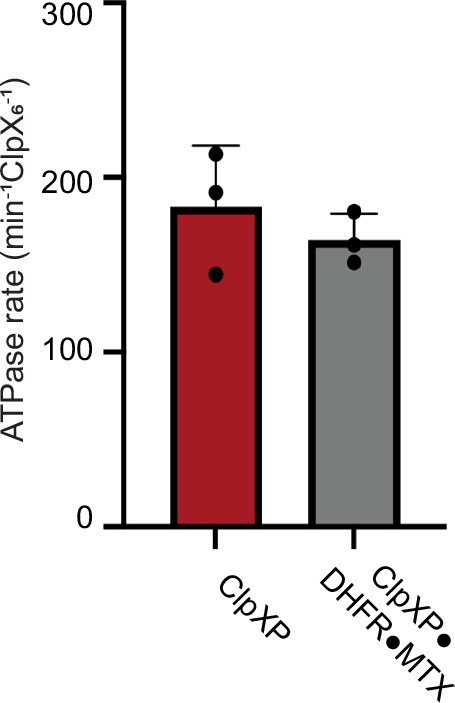
Bars show mean rates of ATP hydrolysis for ClpXP, or ClpXP in the presence of branched-degron tagged DHFR•MTX ± 1 SD, with symbols denoting replicate (n = 3) assays.
Models of ATP hydrolysis, translocation, and unfolding
In previous ClpXP structures, four or five ClpX subunits had ATP/ATPγS bound and one or two subunits had ADP bound6–10. By contrast, in the three Fseam ‘up’ structures presented here, subunits A1, B2, and C3 were ATP bound, whereas D4, E5, and Fseam were ADP bound (Supplementary Fig. 15, Table 2). Given the high prevalence of this confomational state during the processes of substrate unfolding and translocation, as judged by the number of cryoEM particles in our data sets, why might it have been overlooked previously? It is possible that this observed configuration of ClpX-bound nucleotides was underpopulated in prior structures due to the use of ATPγS, which ClpX hydrolyzes slowly9,32, and/or the use of a E185Q Walker-B mutation that slows ATP hydrolysis dramatically6–8,33. Specifically, we posit that our here reported ATP3/ADP3 structures would be similar to some ATP4/ADP2 structures if ATP/ATPγS was hydrolyzed slowly and a rate-limiting post-hydrolysis conformational change was slow. Indeed, ClpX in our ATP3/ADP3 translocation structure (PDB code 9C88), which was obtained using the branched-DHFR substrate in the absence of MTX, aligned with ClpX in a ATP4/ADP2 structure (PDB code 6WSG) with a Cα RMSD of 1.3 Å.
Table 2.
Nucleotide occupancy of subunits in ClpXP structures
| Structure | Subunit position in ClpX spiral | Seam subunit confirmation | ClpX state | ATPγS | Walker-B mutation | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | seam | |||||
| 8V9R | Ta | Tb | Tc | Dd | De | Df | Up | Substrate-engaged | No | No |
| 9C87 | Ta | Tb | Tc | Dd | De | Df | Up | Substrate-engaged | No | No |
| 9C88 | Ta | Tb | Tc | Dd | De | Df | Up | Translocation | No | No |
| 6PP7/6POD | Tb | Tc | Td | Te | tf | Da | Up | Translocation | Yes | Yes |
| 6WSG | Tb | Tc | Td | Te | tf | Da | Up | Translocation pre-engagment | Yes | No |
| 6VFS | Tb | Tc | Td | Te | Df | Da | Up | Translocation | No | Yes |
| 8ET3 | Ta | Tb | Tc | Td | De | Df | Up | Adaptor substrate recognition | Yes | No |
| 6PP8/6POS | Ta | Tb | Tc | Td | Te | Df | Down | Substrate-bound* | Yes | Yes |
| 6PP6/6PO3 | Ta | Tb | Tc | Td | Te | Df | Down | Substrate-bound* | Yes | Yes |
| 6PP5/6PO1 | Ta | Tb | Tc | Td | Te | Df | Down | Translocation | Yes | Yes |
| 6WRF | Ta | Tb | Tc | Td | Te | Df | Down | ssrA-tag recognition | Yes | No |
| 6VFX | Ta | Tb | Tc | Td | Te | Df | Down | Translocation | No | Yes |
| 8E91 | Ta | Tb | Tc | Td | De | Df | Down | Substrate-free | Yes | No |
| 8E8Q | Ta | Tb | Tc | Td | De | Df | Down | Substrate-free | No | No |
| 8E7V | Ta | Tb | Tc | Td | De | Df | Down | Substrate-free | No | No |
‘T’ denotes a nucleoside triphosphate (ATP or ATPγS) in which the ‘Arg-finger’ side chain of residue 307 from an adjacent subunit appears to be in a conformation that would support hydrolysis. ‘t’ denotes a nucleoside triphosphate in a site in which Arg307 is disengaged. ‘D’ represents ADP. Subscripts following the nucleotide designation are the chain identity of the subunit in the PDB file. In structures with PDB codes 6PP8/6POS and 6PP6/6PO3, native structure of an unknown substrate/adaptor or ensemble of substrates was present above the ClpX channel, but the resolution was too poor to determine if there was a direct connection to polypeptide density in the axial channel. In this instance, the ClpX state is noted as substrate-bound*. The remaining ClpX states were defined as follows. In the ssrA-tag recognition state, the axial channel of ClpX is closed and is interacting with the C-terminus of the degron tag, which is above this point of closure. In contrast, structures of the substrate-engaged and translocation states all exhibit an open axial channel of ClpX, with either the degron tag or the unfolded substrate threaded through the opening. In the substrate-engaged-state (reported here), the folded domain of the substrate is resolved and observed to make contact with the axial channel. In translocation state, the substrate is unresolved and sidechains of the substrate polypeptide in the axial channel cannot be assigned.
There are several models for how ClpX and other AAA+ machines translocate protein substrates and drive substrate unfolding4,8,23. The ‘hand-over-hand’ model posits that ATP hydrolysis generates a power stroke that moves two amino acids through the axial channel via a structural transformation in the AAA+ hexamer that results in subunits 1–4 moving down one position in the spiral, subunit 5 becoming the new seam subunit, and the old seam subunit becoming subunit 1 at the top of the spiral23. If ClpX operates by this mechanism, then the Fseam ‘up’ conformation might represent an intermediate in the movement of the seam subunit to the top of the spiral. An alternative ‘large-step’ model posits that a subunit near the top of the ClpX spiral drags the substrate down to near the bottom of the spiral during a power stroke, generating a translocation step of ~6 residues7. This ‘large-step’ model is consistent with single-molecule measurements of translocation step size and with the movement of an ssrA-like degron between ClpXP structures 6WRF and 6WSG4,6,22,26,34–36. In one variation of a ‘large-step’ model, the Fseam ‘up’ conformation might be a good candidate for engaging the substrate if its pore-1 loop moved to contact the substrate polypeptide in the channel and then dragged it down via a conformational change to the Fseam ‘down’ conformation. Supplemental Movie 3 shows a morph of a potential movement of this type, which could be part of a sequential or reciprocating mechanism. The ‘large step’ model is attractive in that it accounts for measured translocation step sizes. In contrast, the ‘hand-over-hand’ model would require ~3 sequential hydrolysis events to occur in less than 0.1 s to account for the observed kinetics of translocation steps in single-molecule experiments, whereas bulk assays of ClpX-mediated hydrolysis indicate that 3 sequential hydrolysis events would require ~0.8 s (Fei, Bell et al. 2020, and references therein). Notably however, unlike the ‘hand-over-hand’ model, the ‘large step’ model requires coordinated substrate release by pore loops of subunits A1-E5. As translocation step size and the number of substrate-pore loop contacts maintained during a power stroke are the primary discriminating features between the ‘hand-over-hand’ and the ‘large step’ models, biophysical experiments aimed at assessing these features hold the greatest promise in discriminating between these or additional models.
Methods
Protein purification
E. coli ClpP-His6 and a C-terminally His6-tagged ClpX∆N variant consisting of three ClpX∆N subunits connected by genetically encoded peptide linkers were expressed separately in E. coli and purified by Ni++-NTA, ion-exchange, and gel-filtration chromatography12.
For the branched-degron substrate, a gene encoding E. coli DHFR gene was modified to encode a C-terminal GSHLGLIEVEKPLYCVEPFVGETAHFEIELSEPDVHGQWKLTSH6 peptide tail and cloned into a pETDuet expression vector. We used this branched-degron DHFR substrate in hopes of visualizing how ClpX accommodates multiple polypeptides in its axial channel. However, the branch was not well-ordered and could not be modeled in the structure. This tagged DHFR protein was expressed in BL21 (DE3) cells at 18 °C overnight and purified using Ni++-NTA affinity chromatography by loading and washing in buffer N1 [20 mM HEPES (pH 7.8), 400 mM NaCl, 100 mM KCl, 10% glycerol, 1 mM DTT, 15 mM imidazole] and eluting in buffer N1 containing 250 mM imidazole. The eluate was concentrated and then subjected to gel-filtration chromatography in buffer GF [20 mM HEPES (pH 7.5), 300 mM KCl, 10% glycerol, and 1 mM TCEP]. A synthetic degron-tag peptide consisting of maleimide-GSGSWSHPQFEKAANDENYALAA (21st Century Biochemicals, Inc.), where the underlined sequence is the ssrA tag, was crosslinked to the tagged DHFR protein by reacting ~20 equivalents of the peptide with one equivalent of tagged DHFR for 2 h at room temperature in buffer CL [100 mM HEPES (pH 7.5), 500 mM NaCl, 10% glycerol, 1 mM TCEP] that had been degassed using argon. Unreacted peptide was quenched using 20 mM DTT, removed using a PD-10 desalting column, and the crosslinked DHFR-C15-ssrA protein was concentrated and flash-frozen in liquid nitrogen for storage.
For the linear-degron substrate, the DHFR gene was modified to encode a His6-TEV tag at the N-terminus and GSHLGLIEVEKPLYCGSGSWSHPQFEKAANDENYALAA at the C-terminus (ssrA tag underlined). Expression and purification of this protein by Ni++-NTA affinity and gel filtration was performed as described for the DHFR variant described above prior to crosslinking.
CryoEM single particle analysis
ClpX∆N (5.7 µM pseudohexamer) and ClpP (1.5 µM tetradecamer) were incubated with ATP (5 mM), MTX (0.5 mM), and branched-degron DHFR (20 µM) in buffer R1 [20 mM HEPES (pH 7.5), 100 mM KCl, and 25 mM MgCl2] at room temperature for 5 min. Prior to vitrification, 2.5 µL sample aliquots were placed on 200-mesh Quantifoil 2/1 copper grids, which had been glow-discharged at –15 mA for 60 s using an easiGlow discharger (Pelco), and samples were blotted using a FEI Vitrobot Mk IV with a 0 blot force at 6 °C and 95% relative humidity for 4 s. Essentially the same conditions were used for linear-degron DHFR•MTX and the branched-degron DHFR in the absence of MTX, except these samples were frozen 3 min after the incubation of the substrate with the pre-formed ClpXP complex.
For the branched-degron DHFR with MTX complex, movies were collected with EPU (Thermo Fisher Scientific) using aberration-free image shift (AFIS) and hole-clustering methods on a Titan Krios G3i (Thermo Fisher Scientific) with an acceleration voltage of 300 kV and magnification of 130,000X and detected in super-resolution mode on a K3 detector (Gatan). Movies were collected over 40 frames with total exposures per specimen and defocus ranges listed in Table 1. Data processing was performed in cryoSPARC (v.3.3.1)37 as depicted in Supplementary Fig. 3. Raw movies (15,290) were pre-processed using ‘patch motion correction’, and ‘patch CTF estimation’. Particles (~47,000) picked using the blob-picker tool from 1000 random micrographs were extracted (box size 440px, Fourier cropped to 256px) and subjected to ‘2D classification’. A set of four well-resolved 2D classes composed of side and shoulder views were provided to the ‘Template picker’ tool and applied to the full dataset. The particles and micrographs were subjected to ‘manually curate exposures’, filtering out low resolution and low particle count exposures. Particles were extracted (box size 440px, Fourier cropped to 256px); after two rounds of 2D classification, the preliminary stack included 1,005,797 particles.
For the linear-degron DHFR with MTX and branched-degron DHFR without MTX complexes, movies were collected on a Titan Krios G2 equipped with a K3 Gatan detector and a GIF energy filter and operatued using SerialEM (4.1.6). Imaging parameters included: an acceleration voltage of 300 kV; magnification of 130,000X magnification, resulting in a final pixel size of 0.65 Å; target defocus range of −0.5 µm to −1.7 µm; and a targeted total exposure dose per specimen of 47.54 e − /Ų, collected over 40 frames.
For the linear-degron DHFR•MTX complex, data processing was performed in cryoSPARC (v.4.4.1) as outlined in Supplementary Fig. 4. 15,864 raw movies were pre-processed using ‘patch motion correction’ and ‘patch CTF estimation’. Particles (499,567) were picked using the blob-picker tool from 4,000 random micrographs and extracted with a box size of 440px, then Fourier cropped to 256px, and subjected to ‘2D classification’. A set of three well-resolved 2D classes, composed of side, top, and shoulder views, were provided to the ‘Template picker’ tool and applied to the full dataset. Particles were extracted again with a box size of 440px and Fourier cropped to 128px. After two rounds of 2D classification, the preliminary stack, which included 355,300 particles extracted with a box size of 440px (Fourier cropped to 256px), was used for further processing.
For the branched-degron DHFR without MTX complex, data processing in cryoSPARC (v.4.4.1) is outlined in Supplementary Fig. 5. 14,574 raw movies were pre-processed using ‘patch motion correction’ and ‘patch CTF estimation’. Particles (95,106), picked using the blob-picker tool from 1000 random micrographs, were extracted with a box size of 440px and Fourier cropped to 256px, then subjected to ‘2D classification’. Two well-resolved 2D classes, consisting of side and shoulder views, were used in the ‘Template picker’ tool and applied to the complete dataset. Particles were extracted again with a box size of 440px and Fourier cropped to 128px. The preliminary stack had 434,826 particles after two rounds of 2D classification, which were extracted with a box size of 440px, Fourier cropped to 256px, and used for further processing.
Ab-initio reconstruction, global refinement, and model building
For ClpXP structure with branched-degron DHFR•MTX (PDB code 8V9R), multiclass ‘ab-initio’ reconstruction was performed using three classes. One class consisted of 629,143 particles (group I) and another of 251,567 particles (group II). Inspection of the remaining class via 2D classification revealed a mixture of free ClpP, a low-resolution ClpXP complex, and ‘junk’ particles, likely corresponding to errantly picked particles; these particles were not further considered. After separate homogeneous refinements of groups I and II, heterogeneous refinement was performed using six sub-classes of group I and four sub-classes of groups II. The resulting maps were visually inspected, and four classes from group I and three classes from group II were selected for another round of homogeneous refinement (801,567 particles) that used an ab initio map from group I as an initial model. These aligned particles were then recentered on ClpX using ‘volume tools’ followed by homogeneous refinement with per-particle defocus estimation enabled and subjected to another round of heterogeneous refinement (four classes) that identified the 449,424 particles used for final reconstructions. The final map was obtained through homogeneous refinement, followed by local refinement employing a mask focused on ClpX and the cis ClpP ring. The final map was rescaled using a calibrated pixel size of 0.654 Å/px10 in cryoSPARC and had a GSFSC of ~2.8 Å after FSC-mask auto-tightening. After centering particles on the ClpP equatorial ring and extracting the centered particles with a larger box size, we also visualized a second ClpX•DHFR complex bound to the second heptameric ring of ClpP14, albeit at lower resolution (Supplementary Fig. 16).
Local resolution was estimated using MonoRes38 within cryoSPARC; angular FSCs were calculated using the 3DFSC server39; and Q-scores were calculated using a ChimeraX Q-score plugin40. Model building was performed using a combination of ChimeraX-1.341, Coot-0.9.442, and Phenix-1.1443. The final map was sharpened using CryoSPARC with a B-factor of -50.
For the ClpXP structure with linear-degron DHFR•MTX (PDB code 9C87), multiclass ‘ab initio’ reconstruction was performed using two classes. One class, consisting of 158,892 particles, was selected for further processing. The second class, which corresponded to free ClpP, was not considered further. Homogeneous refinement was performed, followed by heterogeneous refinement using two classes. Of these classes, 105,660 particles were selected for homogeneous refinement as the other class yielded a low-resolution structure. The particles were recentered on ClpX using ‘volume tools’, followed by local refinement using a mask around ClpX and cis-ClpP (mask a). To separate the classes that contained substrate from substrate-free ClpXP, a new mask (mask b) was generated that included only DHFR, followed by a subsequent 3D classification using six classes. Four classes that showed a good DHFR density were selected and separately refined using mask a through local refinement. Because all four structures appeared similar by visual inspection, particles were merged for another round of local refinement using mask a. Following global and local CTF refinements, an additional local refinement was performed using mask a.
For the ClpXP structure containing branched-degron DHFR without MTX (PDB code 9C88), multiclass ‘ab-initio’ reconstruction was performed using two classes. One class, consisting of 264,996 particles, was kept and another class, corresponding to free ClpP, was discarded. Particles were extracted with a box size of 440px (Fourier cropped to 256px), followed by a second multiclass ‘ab-initio’ reconstruction using two classes. Homogeneous refinement was performed by selecting both classes and choosing one of the volumes that showed a better initial model for the ClpXP complex. Heterogeneous refinement was performed using three classes. Out of these classes, 178,399 particles were selected for homogeneous refinement as the other classes yielded low-resolution structures. Because substrate density in the ClpX axial channel was not visible, perhaps due to residual heterogeneity, another round of heterogeneous refinement was performed using two classes (87,656 and 90,743). Each class was separately recentered on ClpX using ‘volume tools,’ followed by local refinement using a mask around ClpX and cis-ClpP. Subsequently, global and local CTF refinements were performed on each particle stack (87,656 and 90,743) separately, followed by local refinement. The particles were then merged to generate the final map using the local refinement using a mask around ClpX and cis-ClpP.
Local resolution, angular FSCs, Q-scores, and model building were performed using tools described above.
ATPase assays
ATP hydrolysis was measured using a coupled enzymatic reaction44 in which NADH oxidation to NAD+ reduces absorbance at 340 nm (∆ε = 6.22 mM-¹ cm-¹) using a SpectraMax M5 plate reader and a 384-well assay plate (Corning, 3575). A stock ATPase reaction mix (20X) contained 20 µL of a mixture of pyruvate kinase and lactic dehydrogenase from rabbit muscle (P0294, Sigma Aldrich), 10 µL of 200 mM NADH, grade II (CAS# 606688), 15 µL of 1 M phosphoenolpyruvate (Sigma Aldrich, 10108294001) in 25 mM HEPES-KOH (pH 7.6), and 25 µL of 200 mM ATP (pH 6.5). For assays, ClpX6 (1 µM) with or without ClpP14 (3 µM) and DHFR-gsylaalaa45 plus MTX (16 µM each) was present in 10 µL of buffer AB [25 mM HEPES, pH 7.8, 100 mM KCl, 20 mM MgCl2, 10% glycerol]. After 5 min of incubation at 30 °C with 2 mM ATP, the ATPase assay was initiated by addition of an equal volume of 2X ATPase reaction mix in buffer AB. Final reaction concentrations were: 0.5 µM ClpX6, 1.5 µM ClpP14 (if present), 8 µM DHFR-gsylaalaa with MTX (if present), ATPase reaction mix (1X) in a total reaction volume of 20 µL.
Degradation assay
Degradation was assayed by SDS-PAGE. Assays were conducted at 30 °C in 25 mM HEPES (pH 7.5), 5 mM MgCl2, 200 mM KCl, 10% glycerol, with 0.5 µM ClpX6 or ClpX6ΔΝ, 1.5 µM ClpP14, 5 mM ATP, 32 mM creatine phosphate, and 0.08 mg/mL creatine kinase. Assays were initiated by adding a final concentration of 5 µM of the branched-degron DHFR substrate to a preincubated mixture of ClpX and ClpP with ATP in the presence or absence of MTX (20 µM). ClpXP did not degrade DHFR-GSHLGLIEVEKPLYCVEPFVGETAHFEIELSEPDVHGQWKLTSH6 lacking a crosslinked ssrA degron, showing that degradation is specific (Supplementary Fig. 2).
CryoDRGN analysis
CryoDRGN was used to analyze the full set of 449,424 particles where the signal for the trans ring of ClpP had been subtracted in cryoSPARC by: (i) aligning particles on ClpP14 through a local refinement with a mask surrounding ClpP14; (ii) subtracting signal of the trans ClpP7 ring; and (iii) performing a final local refinement using the signal-subtracted particle stack and a mask encompassing DHFR, ClpX6, and the ClpP7 cis ring. These particles were down-sampled to a box size of 254 pixels (1.13 Å/pixel) and used to train an eight-dimensional latent-variable model in cryoDRGN v2.3.0 using 1024×3 encoder and decoder architectures. The poses and CTF parameters for cryoDRGN training were supplied from the aforementioned local refinement. Following 20 epochs of training, 100 volumes were sampled from the k-means cluster centers of latent embeddings. After extensive visual inspection of these volumes using our atomic model as a reference, we observed neither the ‘down’ conformation in F subunit nor any instances of IGF ‘hopping’. However, the sampled volumes did reveal subtle movements of DHFR or instances where it was unresolved (Supplemental Movie 2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Supplementary Movie 1. Movements of the Fseam subunit pore-1 loop.
Supplementary Movie 2. Analysis of structural heterogeneity in the branched-degron DHFR•MTX complex.
Supplementary Movie 3. Fseam subunit conformations.
Description of Additional Supplementary Files
Source data
Acknowledgements
The authors thank Ed Brignole, Laurel Kinman, Barrett Powell, and Andy Martin for helpful discussion and feedback, and the MIT-IBM Satori team and the MIT SuperCloud Supercomputing Center for high performance computing resources and support. This work was supported by NIH grants R01-GM144542 and R35-GM141517, and NSF-CAREER grant 2046778. Samples were prepared at the Automated Cryogenic Electron Microscopy Facility in MIT.nano and screened on a Talos Arctica microscope, which was a gift from the Arnold and Mabel Beckman Foundation.
Author contributions
A.G. purified proteins, performed biochemical assays, prepared samples for EM imaging and collected data. A.G. and J.H.D. performed EM data processing and 3D reconstructions. A.G. and R.T.S. built and refined the atomic models. All authors contributed to writing and editing the manuscript. J.H.D. and R.T.S. supervised the project.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
Cryo-EM maps and associated atomic models are available using the PDB and EMDB codes as follows: 8V9R, 9C87, 9C88 (PDB); 43081, 45299, 45300 (EMDB). Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Robert T. Sauer, Joseph H. Davis.
Contributor Information
Robert T. Sauer, Email: bobsauer@mit.edu
Joseph H. Davis, Email: jhdavis@mit.edu
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-53681-9.
References
- 1.Sauer, R. T. & Baker, T. A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem80, 587–612 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Mahmoud, S. A. & Chien, P. Regulated proteolysis in bacteria. Annu Rev. Biochem87, 677–696 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olivares, A. O., Baker, T. A. & Sauer, R. T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol.14, 33–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauer, R. T., Fei, X., Bell, T. A. & Baker, T. A. Structure and function of ClpXP, a AAA+ proteolytic machine powered by probabilistic ATP hydrolysis. Crit. Rev. Biochem. Mol. Biol.57, 188–204 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang, J., Hartling, J. A. & Flanagan, J. M. The structure of ClpP at 2.3 A resolution suggests a model for ATP- dependent proteolysis. Cell91, 447–456 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Fei, X., Bell, T. A., Barkow, S. R., Baker, T. A. & Sauer, R. T. Structural basis of ClpXP recognition and unfolding of ssrA-tagged substrates. Elife9, e61496 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fei, X. et al. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife9, e52774 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ripstein, Z. A., Vahidi, S., Houry, W. A., Rubinstein, J. L. & Kay, L. E. A processive rotary mechanism couples substrate unfolding and proteolysis in the ClpXP degradation machinery. Elife9, e52158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghanbarpour, A., Fei, X., Baker, T. A., Davis, J. H. & Sauer, R. T. The SspB adaptor drives structural changes in the AAA+ ClpXP protease during ssrA-tagged substrate delivery. Proc. Natl Acad. Sci. USA120, e2219044120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghanbarpour, A. et al. A closed translocation channel in the substrate-free AAA+ ClpXP protease diminishes rogue degradation. Nat. Commun.14, 7281 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatsogiannis, C., Balogh, D., Merino, F., Sieber, S. A. & Raunser, S. Cryo-EM structure of the ClpXP protein degradation machinery. Nat. Struct. Mol. Biol.26, 946–954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin, A., Baker, T. A. & Sauer, R. T. RebuiltA. A. A. motors reveal operating principles for ATP-fuelled machines. Nature437, 1115–1120 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Bolon, D. N., Grant, R. A., Baker, T. A. & Sauer, R. T. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol. Cell16, 343–350 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Lee, C., Schwartz, M. P., Prakash, S., Iwakura, M. & Matouschek, A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell7, 627–637 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Sawaya, M. R. & Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry36, 586–603 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Roche, E. D. & Sauer, R. T. Identification of endogenous SsrA-tagged proteins reveals tagging at positions corresponding to stop codons. J. Biol. Chem.276, 28509–28515 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Flynn, J. M., Neher, S. B., Kim, Y. I., Sauer, R. T. & Baker, T. A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell11, 671–683 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Neher, S. B. et al. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol. Cell22, 193–204 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Farrell, C. M., Baker, T. A. & Sauer, R. T. Altered specificity of a AAA+ protease. Mol. Cell25, 161–166 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin, A., Baker, T. A. & Sauer, R. T. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol. Cell29, 441–450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohs, R., Etchebest, C. & Lavery, R. Unraveling proteins: a molecular mechanics study. Biophys. J.76, 2760–2768 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olivares, A. O., Kotamarthi, H. C., Stein, B. J., Sauer, R. T. & Baker, T. A. Effect of directional pulling on mechanical protein degradation by ATP-dependent proteolytic machines. Proc. Natl Acad. Sci. USA114, E6306–E6313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puchades, C., Sandate, C. R. & Lander, G. C. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat. Rev. Mol. Cell Biol.21, 43–58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell, T. A., Baker, T. A. & Sauer, R. T. Interactions between a subset of substrate side chains and AAA+ motor pore loops determine grip during protein unfolding. Elife8, e46808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenniston, J. A., Baker, T. A., Fernandez, J. M. & Sauer, R. T. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell114, 511–520 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Cordova, J. C. et al. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell158, 647–658 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi, S. A., Hersch, G. L., Baker, T. A. & Sauer, R. T. Communication between ClpX and ClpP during substrate processing and degradation. Nat. Struct. Mol. Biol.11, 404–411 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Martin, A., Baker, T. A. & Sauer, R. T. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol. Cell27, 41–52 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinman, L. F., Powell, B. M., Zhong, E. D., Berger, B. & Davis, J. H. Uncovering structural ensembles from single-particle cryo-EM data using cryoDRGN. Nat. Protoc.18, 319–339 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods18, 176–185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun, J., Kinman, L. F., Jahagirdar, D., Ortega, J. & Davis, J. H. KsgA facilitates ribosomal small subunit maturation by proofreading a key structural lesion. Nat. Struct. Mol. Biol.30, 1468–1480 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burton, R. E., Baker, T. A. & Sauer, R. T. Energy-dependent degradation: Linkage between ClpX-catalyzed nucleotide hydrolysis and protein-substrate processing. Protein Sci.12, 893–902 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hersch, G. L., Burton, R. E., Bolon, D. N., Baker, T. A. & Sauer, R. T. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell121, 1017–1027 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Aubin-Tam, M. E., Olivares, A. O., Sauer, R. T., Baker, T. A. & Lang, M. J. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell145, 257–267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maillard, R. A. et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell145, 459–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sen, M. et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell155, 636–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Vilas, J. L. et al. MonoRes: automatic and accurate estimation of local resolution for electron microscopy maps. Structure26, 337–344.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pintilie, G. et al. Measurement of atom resolvability in cryo-EM maps with Q-scores. Nat. Methods17, 328–334 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci.30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casanal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci.29, 1069–1078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D: Struct. Biol.75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Norby, J. G. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol.156, 116–119 (1988). [DOI] [PubMed] [Google Scholar]
- 45.Bell, T. A. Intersubunit Communication and Coordinated Mechanical Activity in the AAA+ Protease ClpXP. PhD thesis, Massachusetts Institute of Technology (2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1. Movements of the Fseam subunit pore-1 loop.
Supplementary Movie 2. Analysis of structural heterogeneity in the branched-degron DHFR•MTX complex.
Supplementary Movie 3. Fseam subunit conformations.
Description of Additional Supplementary Files
Data Availability Statement
Cryo-EM maps and associated atomic models are available using the PDB and EMDB codes as follows: 8V9R, 9C87, 9C88 (PDB); 43081, 45299, 45300 (EMDB). Source data are provided with this paper.