

Abstract
Non-typhoidal Salmonella (NTS) is one of the leading causes of foodborne outbreaks worldwide, especially in low- and middle-income countries such as Peru. To understand the dynamics of NTS serotypes circulating in the country, the whole genomes of 1122 NTS strains from 1998 to 2018 were analyzed using phylogenomic and comparative genomics tools. A total of 40 different Sequences Type (STs) were identified, the five most frequent being ST-32 (S. Infantis, 37.25%), ST-11 (S. Enteritidis, 23.8%), ST-19 (S. Typhimurium, 14.17%), ST-31 (S. Newport, 6.77%), and ST-413 (S. Mbandaka, 4.72%). Furthermore, the maximum likelihood phylogeny showed high clonality between strains from the same ST recovered from different isolation sources, as well as a variable recombination rate, when comparing each ST individually. Moreover, several virulence factors involved in adherence and invasion, as well as plasmids and prophages, are strongly associated with the most frequent STs, while multidrug resistance markers are mostly linked to ST-32. This work provides an overview of the main genomic characteristics linked to the high-frequency ST, which have undergone few genetic modifications over time, suggesting a high adaptation of these NTS circulating clones in Peru.
Keywords: Salmonellosis, Next-generation sequencing, Molecular epidemiology, Public health
Subject terms: Molecular biology, Microbiology, Clinical microbiology, Microbial genetics, Pathogens
Introduction
Acute diarrheal diseases constitute one of the main public health problems in developing countries, where deficiencies in sanitation and limited access to drinking water in rural areas facilitate their rapid spread, leading to high rates of morbidity and mortality1. Among the most recurrent diarrheic pathogens, non-typhoidal Salmonella (NTS) constitutes a bigger concern around several countries2. This group is made up of more than 2,500 serotypes, with particular antigenic and genotypic characteristics that make their molecular study more complex than other bacteria frequently recovered in the first level of care3.
NTS can colonize several human and animal hosts, frequently causing mild gastrointestinal infections in humans; however, some strains are capable of causing bloodstream infections and compromise of other organisms, known as invasive non-typhoidal Salmonella (iNTS)4. NTS cause 93.8 million cases and around 155,000 deaths each year worldwide3, which are frequently attributed to therapeutic failure caused by the increase in antimicrobial resistance in the recent years5.
Surveillance of acute diarrheal diseases in Peru reveals that each year there is an increase in cases, which are especially accentuated in the summer months6. Although specific information regarding the epidemiology of NTS and the dynamics of serotypes is limited7, some outbreaks have been reported in the country8,9. This scenario creates the need to reinforce epidemiological surveillance, especially with the high frequency of antimicrobial-resistant strains in some of the serotypes circulating in Peru10.
The incorporation of whole genome sequencing (WGS) into diarrheal disease surveillance systems worldwide has improved the understanding of the virulence, pathogenicity, transmission, and dynamics of gastrointestinal pathogens such as NTS, efficiently using genomic information for the control and prevention of outbreaks2. Furthermore, it has allowed the determination of the emergence and spread of more virulent clones or those with multidrug-resistant profiles (MDR) worldwide3. The application of WGS in the analysis of NTS in Peru is limited, particularly restricted to a single serotype of Salmonella, such as S. Infantis10,11, S. Enteritidis9, and S. Typhimurium12,13. Therefore, this study used WGS to analyze the epidemiological trends, antimicrobial resistance (AMR), and genome dynamics of NTS in Peru based on a comprehensive dataset of strains recovered through national surveillance over a period of 21 years.
Results
Temporal distribution of Peruvian NTS
The distribution of the NTS population (N = 2,244) remitted to the Instituto Nacional de Salud (INS) revealed the predominance of S. Enteritidis, S. Typhimurium and S. Infantis over the other NTS serotypes reported (Fig. 1, see Suppl. Table S1). Analyzing all the period (1998–2018), the case trend showed a behavior of non-significant reduction (APC: −3.6; 95% CI: −16.2; 11.0), identifying four periods, as well as a significant reduction in the period 1998–2003 (APC: −23.2, 95% CI: −48.1; −11.4). Additionally, when analyzed by serotypes, only a significant reduction trend was found for S. Enteritidis (APC: −6.1; 95% CI: −9.5; −2.5). In contrast, in the evaluation of the period 2008–2018 (since the first report of S. Infantis), only one trend of cases with a non-significant reduction behavior was found (APC: 2.5, 95% CI: -3.8; 9.1). For this period, a significant increasing trend was found for S. Infantis (period 2008–2010; APC: 51.0; 95% CI: 9.9; 107.6) as well as a significant reduction trend for S. Enteritidis (period 2008–2014; APC: -20.9, 95% CI: -37.2; -0.3) (see Suppl. Table S2, Suppl. Fig S1).
Fig. 1.
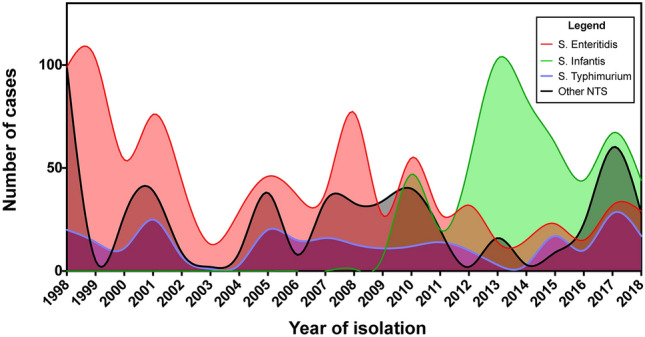
Number of NTS cases from 1998 to 2018 in Peru. Each color indicates a different serotype. X axis indicates the year of isolation, while Y axis indicates the number of NTS cases.
Regarding the behavior of trends among serotypes for the period 1998–2018, a parallel distribution of cases of S. Enteritidis and S. Typhimurium relative to other NTS was found, with different behaviors observed only between S. Enteritidis and S. Typhimurium (p > 0.05) (see Suppl. Table S3, Suppl. Fig S2). In contrast, for the period 2008–2018, the number of cases of S. Enteritidis was parallel to those of S. Typhimurium and other NTS, and the number of cases of S. Typhimurium was distributed parallel to those of Other NTS (p > 0.05). However, when comparing S. Typhimurium with S. Infantis, S. Typhimurium showed a unique trend during the period 2008–2018. In contrast, S. Infantis showed a trend characterized by a high APC from 2008–2010 (p < 0.05) followed by a lower APC from 2010–2018 (p > 0.05) (see Suppl. Table S4, Suppl. Fig S3).
Genomic analysis and genetic diversity of NTS
Of the 2,244 available cryopreserved NTS strains, a non-probabilistic convenience sampling of 1,000 viable strains was conducted for whole-genome sequencing. The analysis of raw reads identified 158 genomes as contaminated or of low quality, which were subsequently excluded. This left a total of 842 high-quality genomes for further analysis. The average metrics for these genomes were as follows: 115 contigs, 52% GC content, an L50 of 17 contigs, and a genome size of 4.8 Mb (see Suppl. Table S5). Of these strains, 698 were recovered from clinical samples (human), while the remaining 148 from environmental sources (animal or food). Additionally, to include all genomes sequenced in Peru from 1998 to 2018, NTS genomes retrieved from the NCBI, obtained from other studies conducted in Peru that met the inclusion criteria, were added (n = 280, 260 clinical strains and 20 environmental strains). As a result, a total of 1122 genomes were included in the final analysis (Table 1, see Suppl. Table S6).
Table 1.
Frequency of sequenced Peruvian NTS analyzed in this study.
| Serotype | Sequence Type (ST) | Source | Reference | Total per serotype | Percentage (%) | ||
|---|---|---|---|---|---|---|---|
| Clinical | Environmental | This study | Other studies | ||||
| Infantis | ST-32 | 389 | 29 | 209 | 209 | 418 | 37.25 |
| ST-2937 | 2 | 0 | 0 | 2 | 2 | 0.18 | |
| Enteritidis | ST-11 | 241 | 26 | 242 | 25 | 267 | 23.8 |
| Typhimurium | ST-19 | 138 | 21 | 135 | 24 | 159 | 14.17 |
| Newport | ST-31 | 52 | 24 | 76 | 0 | 76 | 6.77 |
| ST-45 | 18 | 0 | 18 | 0 | 18 | 1.6 | |
| ST-118 | 0 | 1 | 0 | 1 | 1 | 0.09 | |
| Mbandaka | ST-413 | 32 | 21 | 53 | 0 | 53 | 4.72 |
| Corvallis | ST-1541 | 11 | 4 | 15 | 0 | 15 | 1.34 |
| Senftenberg | ST-14 | 1 | 10 | 11 | 0 | 11 | 0.98 |
| ST-210 | 0 | 1 | 1 | 0 | 1 | 0.09 | |
| Agona | ST-13 | 6 | 3 | 8 | 1 | 9 | 0.8 |
| Anatum | ST-64 | 6 | 3 | 8 | 1 | 9 | 0.8 |
| ST-2858 | 1 | 1 | 2 | 0 | 2 | 0.18 | |
| Kentucky | ST-152 | 5 | 4 | 9 | 0 | 9 | 0.8 |
| Braenderup | ST-22 | 7 | 0 | 6 | 1 | 7 | 0.62 |
| Oranienburg | ST-174 | 6 | 0 | 6 | 0 | 6 | 0.53 |
| ST-23 | 2 | 2 | 3 | 1 | 4 | 0.36 | |
| Other | Several (22) | 37 | 18 | 40 | 15 | 55 | 4.92 |
| Total per category | 954 | 168 | 842 | 280 | 1,122 | 100 | |
From the final genome dataset (n = 1122), at least one NTS strain was obtained from 21 Peruvian regions (Fig. 2): Ancash (n = 26), Apurímac (n = 6), Arequipa (n = 10), Ayacucho (n = 2), Cajamarca (n = 19), Callao (n = 25), Cuzco (n = 10), Huánuco (n = 3), Ica (n = 10), Junin (n = 7), La Libertad (n = 34), Lambayeque (n = 38), Lima (n = 638), Loreto (n = 8), Madre de Dios (n = 4), Moquegua (n = 7), Piura (n = 1), Puno (n = 3), Tacna (n = 4), Tumbes (n = 6) and Ucayali (n = 1), in addition to a group of strains without an exact record of origin (n = 260). Conversely, 40 different STs were identified. The five most frequently STs were ST-32 (S. Infantis, n = 418, 37.25%), followed by ST-11 (S. Enteritidis, n = 267, 23.8%), ST-19 (S. Typhimurium, n = 159, 14.17%), ST-31 (S. Newport, n = 76, 6.77%) and ST-413 (S. Mbandaka, n = 53, 4.72%) (Table 1, see Suppl. Fig S4). Based on the year of isolation, the highest frequency of sequenced S. Enteritidis, S. Typhimurium, S. Mbandaka, and S. Newport ST-31 strains occurred from 1998 to 2009. From 2010 to 2018, however, the highest frequency was observed for S. Infantis ST-32 (see Suppl. Fig S5).
Fig. 2.

(a) Map of Peru indicating the geographical origin of the NTS genomes used in this study (n = 1122). The size of the pie charts indicates the number of strains for each region, with each pie chart colored based on the STs detected per region. (b) Frequency of NTS genomes sequenced throughout the period 1998–2018. Each portion of the stacked bars is colored based on the STs detected per year. Map created using the Free and Open Source QGIS Desktop v3.26.3 (http://www.qgis.org).
Additionally, high-quality assembled genomes were recovered from Brazil (n = 2644), Chile (n = 2461), Ecuador (n = 585), Colombia (n = 207), Argentina (n = 139), Venezuela (n = 42), Guyana (n = 30), Paraguay (n = 29), Suriname (n = 15), Bolivia (n = 13) and Uruguay (n = 2). Most of these non-Peruvian strains (n = 6167) were recovered from environmental sources (n = 5685) (see Suppl. Table S7). These genomes were then compared to the Peruvian strains (n = 1122) using an MST based on MLST profiles A total of 211 different genotypes were detected from studies conducted in South America, with Brazil reporting the highest number of STs (140), followed by Chile (70), Peru (40), Ecuador (39), and Colombia (38). Peru was observed to share the circulation of most of the frequent STs with the other South American countries, displaying a similar frequency of sequenced S. Enteritidis and S. Typhimurium as Chile and Brazil, as well as a similar frequency of sequenced S. Infantis ST-32 as Chile and Ecuador. In contrast, S. Heidelberg ST-45, with a high frequency of sequenced strains particularly in Brazil and Chile, was absent in Peru. (Fig. 3, see Suppl. Fig S6).
Fig. 3.

Minimum spanning tree of NTS strains sequenced in South America and its comparison with strains sequenced in Peru. Each node represents a different ST and is proportional to the number of strains assigned to that ST. The Peruvian strains (n = 1122) in each node are colored according to the legend, while the strains from other countries are indicated in white.
Phylogenetic analysis of NTS
The phylogenetic analysis conducted using the core genome revealed high clonality among the identified STs, grouping them into distinct clades corresponding to individual STs, with bootstrap values approaching 100%. Notably, significant genetic distances between different STs were observed, evident in the elongated branches within the phylogenetic tree. Additionally, there was no discernible association between the STs and the origin of the strains. Furthermore, the analysis of the population structure, as determined by Bayesian methods, identified the existence of 15 subpopulations labeled from Pop 1 to 15. The larger subpopulations aligned with the five most prevalent STs, without identifying any subpopulations within those STs, while the smaller ones resulted from the union of two or more less common STs (Fig. 4).
Fig. 4.

Maximum likelihood phylogenetic tree of 1122 NTS strains isolated in Peru inferred using the GTR + G model, with support nodes calculated through a bootstrap of 1000. The inner circle denotes the source of isolation of the strains, the middle circle specifies the ST to which each strain belongs, and the outer circle represents the subpopulations according to hierBAPS.
Conversely, ClonalFrameML provided values for the relative rate of recombination to mutation (ρ/θ), the inverse of the mean length of recombined DNA (1/δ), the mean divergence of imported DNA (ν), and the relative contribution of recombination versus mutation to the overall genomic diversity (r/m) for all the genomes included in the phylogeny and for each of the most frequent STs. These values indicated that all Peruvian NTS present moderate recombination events (r/m = 1.84), while Peruvian S. Enteritidis, S. Typhimurium, and S. Infantis ST-32 have low recombination events, similar to what was observed in genomes of these serotypes sequenced in Chile and Brazil. Moreover, Peruvian S. Newport ST-31 shows moderate recombination events, with a slightly higher value than that observed in genomes of this serotype in Chile. Furthermore, Peruvian S. Mbandaka has high recombination events, which contrasts with genomes of this serotype in Chile and Brazil, where low recombination events were observed (see Suppl. Table S8).
Virulence genes associated with NTS
The analysis detected the presence of 11 out of the 14 main Salmonella Pathogenicity Islands (SPIs) in the Peruvian strains (see Suppl. Table S9). All strains harbored complete six pathogenicity islands (SPI-1, SPI-2, SPI-3, SPI-9, SPI-11 and SPI-12), while the five most frequent STs shared ten complete pathogenicity islands (SPI-4, SPI-5, SPI-13, and SPI-14, in addition to the six aforementioned). Furthermore, S. Enteritidis exhibited the presence of SPI-10 (Fig. 5, with the presence of SPIs highlighted in blue). Moreover, all strains carried genes encoding the type 3 secretion system (T3SS), and the majority of STs possessed genes for the type 6 secretion system (T6SS, see Suppl. Table S10), except for S. Enteritidis (Fig. 5, with the presence of T6SS genes highlighted in orange).
Fig. 5.

Phylogenetic relationship of Peruvian NTS (n = 1122) inferred through maximum likelihood and its association with virulence genes. The first column is color-coded to represent the most frequent STs. Blue blocks denotate the presence of pathogenicity islands, orange blocks indicate the presence of genes associated with the T6SS, purple blocks denote the presence of adherence-related genes, and green blocks signify the presence of genes associated with invasion and cytotoxicity. The names of the identified genes are positioned above the corresponding blocks, while a black block indicates the absence of a gene.
In terms of virulence factors associated with adherence (see Suppl. Table S11), the presence of the majority of pefABCD genes was specifically linked to S. Enteritidis and S. Typhimurium, and the sdhA gene was predominantly associated with S. Infantis ST-32. Additionally, the distribution of lpfABCDE genes was mainly connected to the five most frequent STs, whereas the ratB gene was absent in S. Mbandaka strains but present in the other prevalent STs (Fig. 5, with the presence of adherence genes highlighted in purple). Conversely, the spvB exotoxin genes, the superoxide dismutase gene (sodCI), and the macrophage-inducible gene 5 (mig-5) were predominantly detected in S. Enteritidis, S. Typhimurium, and S. Newport ST-31 strains. Furthermore, the resistance to complement-mediated killing gene (rck) seemed to be restricted to S. Enteritidis and S. Typhimurium (Fig. 5, with the presence of genes highlighted in green).
Mobile genetic elements and antimicrobial resistance genes associated with NTS
Among the analyzed genomes, 86.8% (n = 974) harbored at least one plasmid. Specifically, 45.6% (n = 512) contained a single plasmid, 30% (n = 337) possessed two plasmids, and 10.2% (n = 114) carried three plasmids. A total of 29 plasmid types were identified, with the most common being the incompatibility (Inc) group, comprising 23 different plasmids (see Suppl. Table S12). Notably, the distribution of most plasmids did not appear to follow a pattern associated with STs, except for IncFIB(S) linked to S. Enteritidis (261/267, 97.8%) and S. Typhimurium (145/159, 91.2%), IncFII(S) associated with S. Enteritidis (236/267, 88.4%), S. Typhimurium (147/159, 92.5%), and S. Newport ST-31 (65/76, 85.5%), as well as the megaplasmid pESI-like associated with S. Infantis ST-32 (415/418, 99.3%) (Fig. 6, with the presence of plasmids highlighted in green).
Fig. 6.

Phylogenetic relationship of Peruvian NTS (n = 1122) inferred through maximum likelihood and its association with mobile genetic elements and antimicrobial resistance genes. The first column is color-coded to represent the most frequent STs. Green blocks indicate the presence of plasmids, red blocks denote the presence of prophages, and resistance genes are indicated by color blocks based on their spectrum of action: Beta-lactamases (Brown), chloramphenicol (light blue), fosfomycin (purple), aminoglycosides (yellow), trimethoprim (orange), fluoroquinolones (blue), sulfamethoxazole (light purple), and tetracycline (pink). The names of the identified genes are positioned above the corresponding blocks, while a black block indicates the absence of a gene.
Conversely, a 98.7% (n = 1107) of the NTS genomes exhibited at least one complete prophage sequence. Specifically, 5.5% (n = 62) featured one prophage, 43.5% (n = 488) had two prophages, 30.3% (n = 340) possessed three prophages, and 18.4% (n = 206) presented four prophages. A total of 70 complete prophages were identified (see Suppl. Table S13), with the most prevalent being those associated with Enterobacteriaceae (Entero), totaling 24, and Salmonella-associated prophages (Salmon), totaling 22. Notably, certain ST-specific prophages were identified: S. Typhimurium with Edward GF 2 (80/159, 50.3%) and Entero UAB Phi20 (38/159, 23.4%); S. Newport ST-31 with Salmon SP004 (66/76, 86.8%); S. Infantis ST-32 with Entero BP_4795 (154/418, 36.8%), Escher 500465 (216/418, 51.7%), Escher pro483 (51/418, 12.2%), Salmon Fels 1 (124/418, 29.7%), Sxt2 c 1717 (239/418, 57.2%) and Yersin L 413C (360/418, 86.1%); and S. Mbandaka with Entero mEp234 (51/53, 96.2%) and Salmon Fels 2 (48/53, 90.6%). In contrast, common prophages such as Gifsy 2 and Salmon 118970 were detected in S. Enteritidis and S. Typhimurium; Salmon RE 2010 in S. Enteritidis and S. Newport ST-31; and Gifsy 1 in S. Typhimurium, S. Newport ST-31, and S. Infantis ST-32 (Fig. 6, with the presence of prophages highlighted in red).
Similarly, 54 genes of plasmid origin associated with antimicrobial resistance were identified, along with six chromosomal gene mutations conferring resistance to fluoroquinolones (see Suppl. Table S14). The most prevalent genes included aac(3)-Iva (20.9%, n = 234), ant(3'')-Ia (37.6% n = 422), aph(3')-Ia (32.3%, n = 362), aph(4)-Ia (21%, n = 236), blaCTX-M-65 (20.3%, n = 228), dfrA14 (35.6%, n = 399), floR (19.7%, n = 221 ), fosA3 (15.3%, n = 172), sul1 (36.6%, n = 411) and tetA (38.4%, n = 431). Moreover, the most frequently observed chromosomal gene mutation was D87Y in the gyrA gene (37.3%, n = 419). A majority of the antimicrobial resistance genes were associated with S. Infantis ST-32 strains, displaying two distinct groups: the first group, isolated since 2008, features genes ant(3’’)-Ia, aph(3’)-Ia, dfrA14, sul1, tetA and the D87Y mutation in gyrA; and the second group, isolated mostly since 2012, comprises aac(3)-Iva, aph(4)-Ia, blaCTX-M-65, floR and fosA3, in addition to the aforementioned genes (Fig. 6, with the presence of genes highlighted in different colors, according to the spectrum of action).
Pangenome of the most frequent STs in Peru
The pangenome analysis conducted on the five most frequent STs unveiled a core genome comprising 3787 cluster genes (61.5%) and an accessory genome consisting of 2368 cluster genes. Additionally, a high similarity was noted among genomes within the same ST, as evidenced by the presence of accessory genes and the heatmap of the average nucleotide identity (ANI) values (Fig. 7).
Fig. 7.

Comparative genomic analysis conducted on the genomes of the most frequent STs of NTS circulating in Peru. The inner layers represent individual genomes organized based on MLST and their phylogenetic relationships within the maximum likelihood tree, using the previously established color-coding. Dark colors in these layers indicate the presence of a gene cluster, while light colors denote its absence. The outmost layer represents the core and accessory genomes in red and blue, respectively. The blue layers represent the number of genomes contributing to each gene group, GC content of genes, and the number of paralogs. The dark green layers represent the genetic, functional, and combined homogeneity indexes. Additionally, the heatmap of average nucleotide identity (ANI) values is presented, indicating high similarity in dark blue and low similarity in light blue.
In general, the shared accessory genes among strains of the five most frequent STs primarily involve virulence, plasmid, and prophage genes. Notably, S. Infantis ST-32 exhibited the largest number of exclusive genes, totaling 263, predominantly associated with bacterial conjugation, replication, and the recombination of phages and plasmids. Noteworthy among these are the plasmid genes of the transfer operon (tra genes) from the pESI-like plasmid (see Suppl. Table S15). In the case of S. Enteritidis, 151 exclusive genes were identified, with a focus on those related to bacterial invasion processes, fimbriae formation, and fragments associated with prophages (see Suppl. Table S16). S. Mbandaka presented 138 exclusive genes, many of which are linked to recombination processes involving prophages (see Suppl. Table S17). S. Newport ST-31 encoded 95 exclusive genes related to bacterial replication, carbohydrate metabolism, and prophage-derived proteins (see Suppl. Table S18). Finally, S. Typhimurium exhibited the fewest exclusive genes compared to other STs, with a total of 90 genes involved in carbohydrate metabolism, bacterial replication, and prophage recombination, particularly Gifsy 2 (see Suppl. Table S19).
Comparative genomics of most frequent plasmids in NTS
When comparing the IncFIB(S) plasmids of S. Enteritidis and S. Typhimurium to the reference (plasmid pSLT-BT, accession number: FN432031), several differences are observed. Both STs exhibit the absence of a small region of approximately 2 kbp, flanked by the locus tag prefixes SLT-BT0161 and SLT-BT0181, and a larger region of 19 kbp associated with antimicrobial resistance, flanked by two tnpA transposons. In addition, S. Enteritidis lacks three other regions: a small one of approximately 0.5 kbp corresponding to the locus tag prefix SLT-BT0041, a medium one of 4.5 kbp flanked by the locus tag prefixes SLT-BT0651 and SLT-BT0741, and a large one of around 25 kbp flanked by the conjugative transfer genes traR and traX (Fig. 8a).
Fig. 8.

Comparative genomic analysis conducted on the three most prevalent plasmids of NTS circulating in Peru: (a) IncFIB(S) plasmid, (b) IncFII(S) plasmid, and (c) pESI-like megaplasmid. The inner layer of each figure represents the average GC content of the reference plasmids. The middle layers represent individual plasmid genomes organized according to MLST and their phylogenetic relationships within the maximum likelihood tree, using the previously established color-coding. In these layers, dark colors indicate the presence of a gene, while light colors denote its absence. The outer layer represents the annotation of the genes of the reference plasmids, alternating in red and blue.
Conversely, upon comparing the IncFII(S) plasmids of S. Enteritidis, S. Typhimurium, and S. Newport ST-31 genomes with the reference (plasmid pSPCV, accession number: CP000858), significant differences are observed. All three STs exhibit the absence of a region of approximately 3 kbp, flanked by locus tag prefixes SPC_p008 and SPC_p012, and variations in a region of about 1 kbp, flanked by locus tag prefixes SPC_p047 and SPC_p049. Furthermore, S. Enteritidis lacks an 11 kbp region flanked by the conjugative transfer genes traX and traT, while S. Newport ST-31 lacks a 9 kbp region that includes, among other genes, the pefABCD operon (Fig. 8b).
Finally, the analysis of the pESI-like megaplasmid in S. Infantis ST-32 was conducted based on the two patterns of antimicrobial resistance genes observed within this ST. The most notable disparity was observed in a subgroup of strains, where a region of approximately 40 kbp flanked by the parA and tnp genes was absent. This region encodes a significant portion of the resistance determinants of the plasmid, with noteworthy genes such as aac(3)-Iva, aph(4)-Ia, blaCTX-M-65, floR, and fosA3 being prominent (Fig. 8c).
Discussion
The prevalence of foodborne diseases has seen a notable increase in recent decades, attributed to environmental variations, the adaptability of pathogens to new ecological niches, and the escalating challenge of antimicrobial resistance14. The rise of molecular surveillance, primarily through WGS, has allowed for the development of tools to detect and monitor highly virulent clones or those with multiple resistance determinants in real-time at local and/or regional levels. This leads to better decision-making to curb their global dissemination15. While most molecular surveillance studies provide significant insights into the pathogenicity, population genetics, and evolutionary history of some virulent NTS serotypes2,16,17, very few of them provide information on the dynamics of all circulating serotypes within a specific timeframe in a given region18–20. Hence, this study focused on sequencing a large number of NTS strains to analyze and understand the spatial and temporal dynamics of circulating serotypes in Peru.
A wide diversity of STs circulating in Peru was identified, with particular emphasis on the high frequency of some, including ST-32 (S. Infantis), ST-11 (S. Enteritidis), ST-19 (S. Typhimurium), ST-31 (S. Newport), and ST-413 (S. Mbandaka). Previous studies conducted in Peru using serological and molecular methods have consistently reported the presence of these serotypes7,9–11,13. However, due to the limited number of strains analyzed in those studies, there was no clear understanding of changes in the dynamics of NTS serotypes over time. Therefore, this study reveals, for the first time, the real dynamics of NTS serotypes in Peru. The dynamics of the serotypes, as revealed by both phenotypic and molecular methods, indicate that S. Enteritidis and S. Typhimurium have been responsible for the majority of NTS infections in the country, with S. Infantis emerging prominently since 2012. Although the dynamics of serotypes vary among continents and countries, most studies indicate that S. Enteritidis and S. Typhimurium are the most prevalent globally21,22, a trend that aligns with the findings of this study. However, it is interesting how S. Infantis displaces these two serotypes, a rising trend that has been previously observed in other countries in the Americas16. Peru is one of the countries where S. Infantis currently holds the first position in terms of isolation frequency, as observed in this study.
The elevated prevalence of S. Enteritidis, S. Typhimurium, and S. Infantis is a shared characteristic with other South American countries such as Brazil23,24 and Chile25, irrespective of the source of isolation. In contrast, the frequencies of S. Newport ST-31 and S. Mbandaka vary among countries in the region17,26. However, despite the substantial number of genomes included in this study, the multidrug-resistant genotype ST-15 (S. Heidelberg), acknowledged as one of the ten most prevalent NTS serotypes in South America and globally27, was not identified. While the presence of this ST has not been reported in Peru, it is imperative to implement effective molecular epidemiological surveillance and remain vigilant for the potential introduction of this serotype, considering its dissemination in neighboring countries.
At the phylogenetic level, most NTS exhibited high clonality, forming monophyletic clusters corresponding to strains of the same ST, without showing an association with the source of isolation or the region. The phylogenetic proximity between clinical and environmental strains suggests a continuous transmission link between humans and the infectious source, as previously described28,29. In contrast, recombination-related parameters indicated that each ST presented a different r/m, and while the majority of STs exhibited low recombination events, some genotypes, such as S. Newport ST-31 and S. Mbandaka, showed unusually high or moderate values. This characteristic appears to be linked to these genotypes according to some investigations17,30. Despite this, when analyzing all STs together, a low recombination frequency typical of NTS is obtained, as reported in other studies31,32.
The phylogenetic structure of NTS can be explained not only by the STs but also by the genes involved in virulence, particularly the SPIs, which are predominantly linked to the most frequent STs in Peru. The presence of multiple SPIs elucidates their success in survival, colonization, and invasion of several hosts33. The primary virulence machinery consists of two T3SS, present in all analyzed NTS, as previously described34. Conversely, the genes encoding the T6SS are present in the majority of serotypes, with the notable exception of S. Enteritidis. Some authors suggest that this genotype lost the majority of genes encoding T6SS after the divergence of S. Gallinarum, since both serotypes share the same common ancestor. However, the loss of these genes, instead of impairing its virulence, promotes bacterial survival in the host by reducing its antigenicity35,36.
Furthermore, several patterns were observed that indicate a possible relationship between virulence genes, phages, and plasmids with high-frequency STs. The patterns found are similar to those described in previous studies, with minor differences that can be attributed to regional variations resulting from the dissemination and evolution of these microorganisms15,29,37. Virulence genes linked to adherence, invasion, and cytotoxicity are those that showed a clearer contrast, suggesting significant differences in evolutionary pressures regarding pathogenicity for different STs38,39. Conversely, despite the high diversity of phages detected in this study, only a small group seems to indicate evolutionarily important patterns of horizontal gene transfer (HGT) between different NTS genotypes, such as Gifsy-1, Gifsy-2, and Fels-129,40.
The observations made with phages are repeated when analyzing the plasmids. A large number of plasmid replicons were detected within the NTS, although only a few, such as IncFIB and IncFII, were common among strains of the same ST41. IncF-type plasmids are the most frequent among NTS, conferring multiple characteristics that enhance bacterial pathogenicity and virulence42. Comparative genomic analysis of IncFIB and IncFII revealed notable differences in genetic content depending on the STs, which is consistent with previous reports29,43. Interestingly, although multiple reports indicate their involvement in antimicrobial resistance43,44, the IncF-type plasmids detected in Peruvian strains lacked these regions. However, the possibility that these plasmids may recombine and acquire these determinants in the future remains present.
In contrast to the aforementioned, the pESI-like plasmid linked to the circulating S. Infantis ST-32 in Peru exhibited antimicrobial resistance genes against several families such as tetracycline, trimethoprim, sulfamethoxazole, and beta-lactams, with patterns similar to those described previously45,46. These properties, combined with the presence of virulence-associated genes that enhance bacterial colonization capacity and fitness, are pointed out as the main cause of the emergence of this serotype worldwide47. Notably, two different antimicrobial resistance genotypes circulate within the same ST in Peru, as revealed by both phylogenetic analysis and comparative genomics. This is because resistance genes are integrated within two specific regions of the pESI-like plasmid, generating variable patterns in circulating strains from the same region46.
The emergence of S. Infantis stands out as the most significant epidemiological shift among circulating NTS serotypes in Peru. Local studies first reported the unusual rise in cases of this serotype in 2010, characterized by a multidrug-resistant (MDR) pattern that included resistance to fluoroquinolones, tetracycline, and trimethoprim-sulfamethoxazole8. This pattern aligns with the earliest genomes of this serotype, recovered in 2008 and included in this study. Subsequently, a genomic study of strains isolated between 2014 and 2016 identified a shift in the MDR pattern, which expanded to include beta-lactams and fluoroquinolones in addition to the previously mentioned antibiotic classes10. This second pattern matches the genomes recovered from 2012 onwards, as included in this study. All this evidence points to an expansion of the MDR spectrum of the S. Infantis serotype, likely triggered by the indiscriminate use of antibiotics in the Peruvian poultry industry over the past decade, creating selective pressure on these pathogenic bacteria, as previously described48. The widespread circulation of MDR bacterial strains in a country leads to the emergence of complicated infections, resulting in increased healthcare costs due to therapeutic failure, as well as higher morbidity and mortality rates49.
The study presents the limitation of having a low number of strains recovered from regions other than Lima. This occurs because many of regional laboratories lack the logistical capacity, including supplies and reagents, to properly recover and characterize diarrheal bacterial strains, often leading to the omission of stool culture50. Nonetheless, the study included the highest possible number of strains recovered from regions other than Lima to achieve the best representativeness. Conversely, few strains were recovered from certain time periods, especially between 2003–2004. This was attributed to both the loss of viability of the strains from those years and the low number of strains sent to the INS during that period, as indicated by the analysis of the temporal distribution of NTS in this study. Despite the limitations mentioned, the present study represents the first comprehensive analysis of a set of strains recovered in Peru over a 21-year period.
In conclusion, this study represents a comprehensive analysis of circulating NTS in Peru over a 21-year period, revealing genetic patterns of virulence and resistance that help explain the dynamics and predominance of certain serotypes such as S. Infantis, S. Enteritidis and S. Typhimurium responsible for diarrheal infections in the country. The persistence of clonal strains over the evaluated period suggests that the sanitary conditions in Peru have not changed in the last 21 years. Therefore, it is necessary to improve sanitation so that, together with molecular surveillance tools, the currently applied strategies for monitoring, prevention, and control can be reinforced to curb the increase in cases of emerging pathogens associated with acute diarrheal diseases. Moreover, the information related to virulence genes, resistance, and mobile genetic elements of different serotypes can serve as a starting point for understanding the evolution of NTS in the future. Finally, the use of WGS for future surveillance of circulating NTS in the country could support the implementation of more effective One Health intervention strategies focused on preventing and controlling foodborne pathogens.
Methods
Study design and population
This is an observational, retrospective and cross-sectional study. Salmonella is routinely investigated in public and private health laboratories in Peru; however, notification is not mandatory. When other laboratories require confirmation of species, serotype, and/or antimicrobial susceptibility, the strains are sent to the Laboratorio de Referencia Nacional de Bacteriología Clínica (LRN-BACLI) of the Instituto Nacional de Salud. Therefore, this study represents a passive surveillance.
A total of 2244 NTS strains were received by the LRN-BACLI from 1998 to 2018, under the framework of the National Surveillance of Enteric Pathogens. The records of these strains were loaded into the Prism software v9.3.1 (GraphPad, USA) to generate a line graph evaluating the dynamics of NTS strains over the study period. Moreover, the characterization of periods of change in the trend of case counts per year was conducted both in general and according to serotype, using annual percentage change (APC) via Joinpoint Desktop software v.5.1.0.0. Additionally, the determination of case incidence adjusted for the interaction of serotype and the evaluated period was performed with Stata v17.0. The sample for this study consisted of 1000 NTS strains recovered through microbial cultivation.
Ethical statement
This study was conducted within the framework of the project "1000 Genomes: Phylogeography and population structure of Salmonella enterica in Peru 1999–2017", approved by D.R. No. 101-2019-OGITT-OPE/INS. All procedures and methods were performed in accordance with ethical standards outlined in the Declaration of Helsinki or comparable relevant guidelines and regulations. Informed consent was waived due to the retrospective nature of this study by the Institutional Committee of Research and Ethics (IRB) of the Instituto Nacional de Salud of Peru, in accordance with national legislation and institutional requirements for Public Health Surveillance. Patient information was anonymized completely in this study.
Culture and typing of Salmonellaenterica
Salmonella strains were enriched in trypticase soy broth (Oxoid, England) at 37 °C for 6 to 8 h. Each strain was then cultured on Salmonella-Shigella selective agar (Oxoid, England) and incubated at 37 °C from 18 to 24 h. Presumptive identification was carried out using conventional biochemical tests such as three sugar iron agar, lysine iron agar, sulfide indole motility agar and Simmons citrate agar (Oxoid, England). Serotyping was performed based on the White-Kaufmann-Le Minor scheme, using commercially available monovalent and polyvalent antisera (Denka Seiken Co., Ltd, Japan).
Library preparation and whole genome sequencing
DNA extraction was performed using the DNeasy Blood & Tissue kit (Qiagen, Germany) following the manufacturer’s instructions. The genomic DNA obtained was quantified using fluorometry (Qubit 3.0 Invitrogen, Malaysia). Sequencing libraries were prepared using the Nextera XT library preparation kit (Illumina, USA), and genomic sequencing was performed using a MiSeq benchtop sequencer (Illumina, USA) with 2 × 250 bp paired-end chemistry.
Genomic data analysis
Quality control of each sequence was assessed using Fastqc v0.11.551. Adapters and low-quality nucleobases were removed using Trimmomatic v0.3852. Raw reads were assembled de novo using the A5-miseq v20160825 pipeline53. The quality metrics of each assembled genome were calculated using QUAST54. Gender confirmation and detection of possible contaminated contigs were performed using Kraken v1.01555. The inclusion criteria applied were genomes with fewer than 500 contigs, with 90% of them belonging to Salmonella spp.
In silico serotyping of each sequence was performed using SISTR v1.1.156, while the sequence type (ST) was determined using multilocus sequence typing (MLST) v2.10 according to the scheme of Achtman et al.57. Furthermore, to analyze the frequency of STs detected in this work in relation to others reported in South America, a minimum spanning tree (MST) was inferred using the MLST profiles in the Bionumerics software (Applied Maths, bioMérieux).
Phylogenetic analysis
In addition to the genomes sequenced in this study, other genomes from previous studies of Salmonella enterica carried out in Peru were recovered from the NCBI and included for phylogenetic inference. An ANI analysis was conducted using Bionumerics software (Applied Maths, bioMérieux) to confirm that none of the genomes from previous studies were identical to those obtained in this study. Genome core alignment was performed using Parsnp v1.2, and the locally collinear blocks (LCB) containing the SNPs were submitted to HarvestTools v1.2 to extract the alignment58.
A maximum likelihood phylogeny was inferred using RaxML v8.059, using the GTR + G model and supporting nodes calculated with 1000 bootstrap replicates, where clades were defined by branch length. Also, ClonalFrameML v1.11-3-g4f13f23 was used to detect and remove recombinant regions60. Moreover, a hierarchical clustering method to identify and visualize sub-populations and genetic relationships within the genomic data was performed using the hierBAPS (Bayesian Analysis Population Structure) algorithm, implemented in R language v4.3.1, using the following parameters: (a) Include singleton SNPs, (b) maximum depth = 2, and (c) maximum number of populations = 1561. The results were visualized using Microreact62, associating phylogeny with MLST data, virulence and resistance genes, as well as mobile genetic elements detected in each strain.
Detection of virulence factors, antimicrobial resistance genes and mobile genetic elements
Pathogenicity islands were detected using the online tool SPIFinder v2.063, including regions with > 60% coverage alignment and > 90% identity regarding the reference. Moreover, homologous virulence genes were identified from the virulence factor database (VFDB) of pathogenic bacteria64. Coding sequence prediction was performed using Prodigal v2.6.365. The detection of virulence genes was performed using a Python script developed to run Blast Score Ratio (BSR), including genes with > 60% coverage alignment and > 90% identity regarding the reference66.
The genes associated with antimicrobial resistance were detected using Staramr67 with two databases: The first for chromosomal point mutations, PointFinder68, and the second for resistance genes acquired through horizontal gene transfer, ResFinder69. Plasmid replicons were identified using PlasmidFinder v2.141, and complete prophages regions were detected using the online tool PHASTER70. All the information obtained was associated with the previously generated phylogeny as a gene presence/absence matrix.
Pangenome and comparative analysis
Anvi’o v1.2.271 was used to analyze the pangenome of the most frequent STs circulating in Peru from 1998 to 2018. To achieve this, 10 genomes from each of the most frequent STs were randomly selected and used to create the matrix database using the anvi-gen-contigs-database script. Subsequently, the anvi-setup-ncbi-cogs script was executed to perform the annotation of those genomes, followed by the use of the anvi-pan-genome script to construct of the pangenome. Finally, the anvi-compute-genome-similarity script was run to calculate the similarity between the analyzed genomes. The results were displayed using the anvi-display-pan script, and the accessory genes from each observed group obtained from this analysis were evaluated using BLAST72.
In addition, the information obtained from PlasmidFinder was utilized to conduct a comparative analysis of the most frequently detected plasmids among the STs. This analysis was performed using BRIG v0.9573, with an identity threshold > 60% of the reference used.
Supplementary Information
Acknowledgements
We thank the Earlham Institute and the Global Challenges Research Fund for sequencing falicities, and also the entire team of the Laboratorio de Referencia Nacional de Bacteriología Clínica at the Instituto Nacional de Salud, Lima, Peru, for their assistance during the execution of this study.
Author contributions
Conceptualization: JCC, RG. Methodology: JCC, DFL, FG. Investigation: JCC. DFL, FG. Resources: RG. Formal Analysis: JCC, FG. Writing (original draft): JCC, RG. Writing (review, edit, and approval): JCC, WQ, DFL, FG, WQ, RGG, RG. Supervision: RGG, RG.
Funding
This study was developed within the framework of the project “1000 Genomes: Phylogeography and population structure of Salmonella enterica in Peru 1999–2017” supported by the Instituto Nacional de Salud, Lima, Peru (D.R. No. 101-2019-OGITT-OPE/INS).
Data availability
Sequences from NTS strain were deposited to the National Center for Biotechnology Information (NCBI) BioProject PRJNA1085984. Accession numbers for strains available in NCBI are provided in Supplementary Table 5. All data presented in this manuscript are provided in Supplementary Tables S1-S19 as well as Supplementary Figures S1-S6.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-78331-4.
References
- 1.Popa, G.L. & Papa, M.I. Salmonella spp. infection - A continuous threat worldwide. Germs11, 88–96 (2021). [DOI] [PMC free article] [PubMed]
- 2.Tassinari, E. et al. Whole-genome epidemiology links phage-mediated acquisition of a virulence gene to the clonal expansion of a pandemic Salmonella enterica serovar Typhimurium clone. Microb. Genom.6, mgen000456 (2020). [DOI] [PMC free article] [PubMed]
- 3.Seribelli, A. A. et al. Insights about the epidemiology of Salmonella Typhimurium isolates from different sources in Brazil using comparative genomics. Gut Pathog.13, 27 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuypers, W. L. et al. Fluoroquinolone resistance in Salmonella: Insights by whole-genome sequencing. Microb. Genom.4, e000195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tyson, G. H. et al. A multidrug-resistant Salmonella infantis clone is spreading and recombining in the United States. Microb. Drug Resist.27, 792–799 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.CDC - Centro Nacional de Epidemiología, Prevención y Control de Enfermedades. Análisis de Situación de las EDA. https://www.dge.gob.pe/portal/docs/tools/eda/2024/Situacion_EDA_se07.html (2024).
- 7.Silva, C. et al. Characterization of Salmonella enterica isolates causing bacteremia in Lima, Peru, using multiple typing methods. PLoS One12, e0189946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zamudio, M., Meza, A., Bailón, H., Martinez-Urtaza, J. & Campos, J. Experiencias en la vigilancia epidemiológica de agentes patógenos transmitidos por alimentos a través de electroforesis en campo pulsado (PFGE) en el Perú. Rev. Peru Med. Exp. Salud Publica28, 128–135 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Quino, W. et al. Phylogenetic structure of Salmonella enteritidis provides context for a foodborne outbreak in Peru. Sci. Rep.10, 22080 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quino, W. et al. Multidrogorresistencia de Salmonella infantis en Perú: Un estudio mediante secuenciamiento de nueva generación. Rev. Peru Med. Exp. Salud Publica36, 37–45 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Vallejos-Sánchez, K. et al. Whole-genome sequencing of a Salmonella enterica subsp. enterica Serovar infantis strain isolated from broiler chicken in Peru. Microbiol. Resour. Announc.8, e00826–19 (2019). [DOI] [PMC free article] [PubMed]
- 12.Carhuaricra Huaman, D. E. et al. Genomic characterization of Salmonella typhimurium isolated from guinea pigs with Salmonellosis in Lima, Peru. Microorganisms10, 1726 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurtado, R. et al. WGS-based lineage and antimicrobial resistance pattern of Salmonella typhimurium isolated during 2000–2017 in Peru. Antibiotics (Basel)11, 1170 (2022). 10.3390/antibiotics11091170. [DOI] [PMC free article] [PubMed]
- 14.Nazir, A. et al. Rising trends of foodborne illnesses in the U.S.: Short communication. Ann. Med. Surg. (Lond)85, 2280–2281 (2023). [DOI] [PMC free article] [PubMed]
- 15.Branchu, P. et al. Genome variation and molecular epidemiology of Salmonella enterica Serovar Typhimurium pathovariants. Infect. Immun.86, e00079-e118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez, D. M. et al. A review of the global emergence of multidrug-resistant Salmonella enterica subsp. enterica Serovar Infantis. Int. J. Food Microbiol.403, 110297 (2023). [DOI] [PubMed]
- 17.Benevides, V. P. et al. Genomic features and phylogenetic analysis of antimicrobial-resistant Salmonella mbandaka ST413 strains. Microorganisms12, 312 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sia, C. M. et al. Genomic diversity of antimicrobial resistance in non-typhoidal Salmonella in Victoria, Australia. Microb. Genom.7, 000725 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chattaway, M. A. et al. Evaluation of genomic typing methods in the Salmonella Reference Laboratory in Public Health, England, 2012–2020. Pathogens12, 223 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darboe, S. et al. Genomic diversity and antimicrobial resistance among non-typhoidal Salmonella associated with human disease in The Gambia. Microb. Genom.8, 000785 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu, H. et al. Characterization of Salmonella serotypes prevalent in asymptomatic people and patients. BMC Infect. Dis.21, 632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong, B. et al. Prevalence, serotype distribution and antimicrobial resistance of non-typhoidal Salmonella in hospitalized patients in Conghua District of Guangzhou, China. Front. Cell Infect. Microbiol.12, 805384 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miranda, A. L. et al. Phenotypic and genotypic characterization of Salmonella spp. isolated from foods and clinical samples in Brazil. An. Acad. Bras. Ciênc.89, 1143–1153 (2017).
- 24.Faula, L. L. et al. Phenotypic and genotypic characterization of Salmonella isolates recovered from foods linked to human Salmonellosis outbreaks in Minas Gerais State, Brazil. J. Food Prot.85, 142–154 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Retamal, P. et al. Virulence and antimicrobial resistance factors in Salmonella enterica serotypes isolated from pigs and chickens in central Chile. Front. Vet. Sci.9, 971246 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parra-Flores, J. et al. Genomic characterization of Cronobacter spp. and Salmonella spp. strains isolated from powdered infant formula in Chile. Front. Microbiol.13, 884721 (2022). [DOI] [PMC free article] [PubMed]
- 27.Webber, B. et al. Detection of virulence genes in Salmonella Heidelberg isolated from chicken carcasses. Rev. Inst. Med. Trop. Sao Paulo61, e36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Timme, R. E. et al. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol. Evol.5, 2109–2123 (2013). [DOI] [PMC free article] [PubMed]
- 29.Worley, J. et al. Salmonella enterica phylogeny based on whole-genome sequencing reveals two new clades and novel patterns of horizontally acquired genetic elements. mBio9, e02303–18 (2018). [DOI] [PMC free article] [PubMed]
- 30.Sangal, V. et al. Evolution and population structure of Salmonella enterica serovar Newport. J. Bacteriol.192, 6465–6476 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Octavia, S. & Lan, R. Frequent recombination and low level of clonality within Salmonella enterica subspecies I. Microbiology (Reading)152, 1099–1108 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Didelot, X. et al. Recombination and population structure in Salmonella enterica. PLoS Genet.7, e1002191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lozano-Villegas, K. J. et al. Molecular detection of virulence factors in Salmonella serovars isolated from poultry and human samples. Vet. Med. Int.2023, 1875253 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lou, L. et al. Salmonella pathogenicity island 1 (SPI-1) and its complex regulatory network. Front. Cell. Infect. Microbiol.9, 270 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blondel, C. J. et al. Contribution of the type VI secretion system encoded in SPI-19 to chicken colonization by Salmonella enterica serotypes Gallinarum and Enteritidis. PLoS One5, e11724 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Troxell, B. A type 6 secretion system (T6SS) encoded gene within Salmonella enterica serovar Enteritidis contributes to virulence. Virulence9, 585–587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikhimiukor, O. O. et al. Genomic characterization of invasive typhoidal and non-typhoidal Salmonella in southwestern Nigeria. PLoS Negl. Trop. Dis.16, e0010716 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suez, J. et al. Virulence gene profiling and pathogenicity characterization of non-typhoidal Salmonella accounted for invasive disease in humans. PLoS One8, e58449 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buddhasiri, S. et al. Clinical characteristics, antimicrobial resistance, virulence genes and multi-locus sequence typing of non-typhoidal Salmonella Serovar Typhimurium and Enteritidis strains isolated from patients in Chiang Mai, Thailand. Microorganisms11, 2425 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Switt, A. I. et al. Salmonella phages and prophages: Genomics, taxonomy, and applied aspects. Methods Mol. Biol.1225, 237–287 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Carattoli, A. et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother.58, 3895–3903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aljahdali, N. H. et al. Genotypic and phenotypic characterization of incompatibility group FIB positive Salmonella enterica Serovar Typhimurium isolates from food animal sources. Genes (Basel)11, 1307 (2020). [DOI] [PMC free article] [PubMed]
- 43.Casaux, M. L., D’Alessandro, B., Vignoli, R. & Fraga, M. Phenotypic and genotypic survey of antibiotic resistance in Salmonella enterica isolates from dairy farms in Uruguay. Front. Vet. Sci.10, 1055432 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng, S. et al. Molecular characterization of IncFII plasmid carrying blaNDM-5 in a Salmonella enterica serovar Typhimurium ST34 clinical isolate in China. mSphere8, e0048023 (2023). [DOI] [PMC free article] [PubMed]
- 45.Aviv, G., Rahav, G. & Gal-Mor, O. Horizontal transfer of the Salmonella enterica Serovar infantis resistance and virulence plasmid pESI to the gut microbiota of warm-blooded hosts. mBio7, e01395–16 (2016). [DOI] [PMC free article] [PubMed]
- 46.Tate, H. et al. Comparative analysis of extended-spectrum-β-lactamase CTX-M-65-producing Salmonella enterica Serovar Infantis isolates from humans, food animals, and retail chickens in the United States. Antimicrob. Agents Chemother.61, e00488-e517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee, W. W. Y. et al. Characterization of a pESI-like plasmid and analysis of multidrug-resistant Salmonella enterica Infantis isolates in England and Wales. Microb. Genom.7, 000658 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montoro-Dasi, L. et al. Holistic strategies to control Salmonella infantis: An emerging challenge in the european broiler sector. Microorganisms11, 1765 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marston, H. D. et al. Antimicrobial resistance. JAMA.316, 1193–1204 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Guillén, A. L. Retos y problemas en el diagnóstico microbiológico en diarrea. Rev. Peru Med. Exp. Salud Publica28, 116–120 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Andrews, S. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2024).
- 52.Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coil, D., Jospin, G. & Darling, A. E. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics31, 587–589 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics29, 1072–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol.15, R46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshida, C. E. et al. The Salmonella In Silico Typing Resource (SISTR): An open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS One11, e0147101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Achtman, M. et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog.8, e1002776 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Treangen, T. J., Ondov, B. D., Koren, S. & Phillippy, A. M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol.15, 524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Didelot, X. & Wilson, D. J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol.11, e1004041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tonkin-Hill, G. et al. RhierBAPS: An R implementation of the population clustering algorithm hierBAPS. Wellcome Open Res.3, 93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Argimón, S. et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom.2, e000093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roer, L. et al. Is the evolution of Salmonella enterica subsp. enterica linked to restriction-modification systems? mSystems1, e00009–16 (2016). [DOI] [PMC free article] [PubMed]
- 64.Liu, B., Zheng, D., Zhou, S., Chen, L. & Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res.50, D912–D917 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hyatt, D. et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform.11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mestanza, O. Like Blast Score Ratio (BSR). Github Repository. https://github.com/OrsonMM/Blast-score-ratiofor-genomics (2024).
- 67.Bharat, A. et al. Correlation between phenotypic and in silico detection of antimicrobial resistance in Salmonella enterica in Canada using Staramr. Microorganisms10, 292 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zankari, E. et al. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother.72, 2764–2768 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Florensa, A. F. et al. ResFinder - An open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom.8, 000748 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arndt, D. et al. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res.44, W16–W21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eren, A. M. et al. Anvi’o: an Advanced analysis and visualization platform for ’omics data. PeerJ3, e1319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol.215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 73.Alikhan, N. F. et al. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genomics12, 402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences from NTS strain were deposited to the National Center for Biotechnology Information (NCBI) BioProject PRJNA1085984. Accession numbers for strains available in NCBI are provided in Supplementary Table 5. All data presented in this manuscript are provided in Supplementary Tables S1-S19 as well as Supplementary Figures S1-S6.