

Abstract
Single nucleotide exact amplicon sequence variants (ASV) of the human gut microbiome were used to evaluate if individuals with a depression phenotype (DEPR) could be identified from healthy reference subjects (NODEP). Microbial DNA in stool samples obtained from 40 subjects were characterized using high throughput microbiome sequence data processed via DADA2 error correction combined with PIME machine-learning de-noising and taxa binning/parsing of prevalent ASVs at the single nucleotide level of resolution. Application of ALDEx2 differential abundance analysis with assessed effect sizes and stringent PICRUSt2 predicted metabolic pathways. This multivariate machine-learning approach significantly differentiated DEPR (n = 20) vs. NODEP (n = 20) (PERMANOVA P < 0.001) based on microbiome taxa clustering and neurocircuit-relevant metabolic pathway network analysis for GABA, butyrate, glutamate, monoamines, monosaturated fatty acids, and inflammasome components. Gut microbiome dysbiosis using ASV prevalence data may offer the diagnostic potential of using human metaorganism biomarkers to identify individuals with a depression phenotype.
Introduction
Depression is the leading cause of medical disability worldwide [1]. Reliably diagnosing individuals with depression, and understanding the biomedical and neurophysiological mechanisms underlying depression with its attending medical comorbidities, has the potential to radically transform diagnosis and treatment, substantially reduce disability-adjusted-life-year impacts on the economy and quality of life, and mitigate stigma.
Depression is associated with gut dysbiosis that disrupts a microbiome/systemic pathophysiology/brain bidirectional axis, based on preclinical rodent models and human studies [2–16]. Notwithstanding this basic dogma, recent Pubmed meta-analyses of human depression-specific gut microbiome studies [6, 12] have assessed that there is no consensus identifying the particular net gut microbial ecology of taxa and attending metabolomic interactions of microbiota with their hosts’ physiology. This extends to lack of accord regarding differences in taxa relative abundances and diversity in depression, as compared with healthy reference subjects, although there is universal agreement that taxon significant differences do indeed exist. The literature is inconclusive regarding possible interplay between particular species and antidepressants use, experimentally limited to rodent models [16–20], although ketamine animal studies implicate a gut microbiome-associated anti-inflammatory aspect to the antidepressant mechanism of this drug [15]. Thus, understanding specific biological mechanisms, identification of microbiome taxa and functional pathway patterns as biomarkers, and development of microbiota-centric medical interventions have been confounded by the literature’s wide variation in microbial compositional and abundance dataset analyses. These translational lapses hold for all the mental disorders, and particularly for depression [6].
Next generation sequencing has launched an expansion of microbiome studies, but often at the cost of data relevancy due to sequencing and pipeline shortcomings. The recent bioinformatics literature has challenged status quo microbiome analyses [21–24], establishing—yet it is not yet widely appreciated—that the commonly used high throughput sequencing methods and popular software yield compositional datasets of relative operational taxonomic unit (OTU) abundances are inherently flawed due to differences in 16S rRNA gene sequencing platforms and bioinformatics methodologies, many of which do not account for covariate effects.
Metagenomic shotgun sequencing approaches using composition-based, abundance-based, or combined hybrid binning analyses, can, in many respects, improve on the 16S OTU approach to differentiating cohorts, due to of feature specificity assigned to microbial metabolic pathways and genes [25–28]. Nevertheless, the specificity of closed-database metagenomics in general is not ideal for definitive biomarker identification nor microbiota/host interactive mechanisms, due to limitations incurred by the potential of sequencing errors and to confining microbial compositions within a defined catalog, often altering orthologous groups to conform to artificial enterotypes determined by Dirichlet analysis [29].
Inappropriate use of such compositional data has subsequently resulted in conflicting conclusions in the literature due to pipeline errors and OTU clustering methods. Publications are often widely quoted as authoritative major advances leading the field regarding the role of gut microbiome in mental disorders and comorbid health issues, yet their unchallenged compositional data analyses can be flawed thereby leading to deep propagation of misinformation in the literature [30, 31].
In the present study we avoid the dangers of closed-database OTU clustering, and of the downside of shotgun sequencing metagenomics, by instead demonstrating a novel machine-learning pipeline that involves taxa binning and prevalences of exact amplicon sequence variants (ASV) [21]. Prevalence is defined as the proportion of individuals within a specific cohort who share an OTU or taxon at least once, regardless of abundance; it is a frequency of occurrence, in contrast to abundance which is the average fractional representation of a single OTU or taxon only when present. For example, using our (Prevalence Interval for Microbiome Evaluation (PIME) R package [32] a prevalence cutoff of 55% means that the taxa selected at this prevalence interval are found in 55% of the subjects.
We hypothesized that the prevalences of exact ASV within noisy gut microbiome data can readily identify pathophysiology-associated differences in gut signatures of microbial taxa and their metabolic pathway in people living with depression as compared with healthy reference subjects, at high resolution without resorting to OTU relative abundance clustering nor shotgun sequencing.
Materials and methods
Stool samples were obtained from 40 volunteer subjects, median age 34 y.o. distributed as groups of n = 20 subjects (ten females and ten males) meeting DSM-IV criteria for depression (DEPR) as outpatients seeking help at the University of Florida Department of Psychiatry, plus n = 20 healthy reference control subjects (14 females and 6 males) without a diagnosed mental illness (NODEP). Subjects refrained from probiotics and antibiotics during 30 days prior to providing stool samples, and they reported no gastrointestinal disorders. Fifteen of the 20 DEPR subjects had clinically charted antidepressants that included fluoxetine, venlafaxine, duloxetine, lamotrigine, mirtazapine, gabapentin, citalopram, sertraline, or bupropion, while five DEPR subjects had charts that did not specify a particular antidepressant medication by name. The possible effect of antidepressant medication use on metadata clustering was examined, but this resulted in no outliers of any significant differences that influenced data interpretation, as shown with statistical details in “Results” below; therefore, data from all 20 DEPR patients were pooled and included in all subsequent analyses regardless of specific antidepressant or whether or not their chart specified an antidepressant. The University of Florida Institutional Review Board approved the study (clinicaltrials.govNCT02693327) for which participating subjects provided written informed consent and were remunerated.
Stool samples were collected with OMNIgeneGUT fecal collection kits (DNAGenotek, Ottawa, Ontario, Canada) and stored at −80 °C until DNA extraction. DNA from each sample was extracted from ~200 mg of stool using the E.Z. N.A Stool Extraction Kit following the manufacturer’s protocol (Omega Bio-tek, Doraville, CA). Samples were randomized during extraction to avoid processing order bias, with the absence of processing and kit contamination verified by parallel blank negative controls. High DNA quality was determined by spectrophotometry. The region V3–V4 of 16S rRNA gene amplicons was amplified and sequenced with Illumina MiSeq (2 × 300 cycles run) as described previously [33].
The Illumina demultiplexed paired-end sequenced dataset was processed by the R package DADA2 [34] to correct for amplicon errors, to identify chimeras, and to merge paired-ends reads. The end product of DADA2 yielded a total of 2724 unique ASV each trimmed to 400 nucleotides in length, cataloged, and tallied as the number of times each exact ASV was observed for each sample. A phyloseq R object was generated comprised of all 2724 ASVs, a lookup table of taxonomy assignments to each ASVs obtained by using the naive Bayesian classifier method, and the ILVA ribosomal RNA gene database [35] version v132, plus subject sample metadata.
The DADA2-derived ASV dataset was initially analyzed by permutational analysis of variance (PERMANOVA) (Adonis R package), then the ALDEx2 R package [36, 37], as described below. This dataset was then treated using our PIME R package [32], which generated ASV prevalences by machine-learning, as validated by comparison with control Monte Carlo simulations with randomized variations of sequences. This prevalence-filtered dataset was then processed by ALDEx2, and output from the DADA2/PIME/ALDEx2 workflow resulted in a denoised, filtered dataset comprised of 86 unique ASV sequences each 400 nucleotides in length (Supplementary Information Table 1-SI). PICRUSt2 [38] was employed for stringent predictions of functional metabolic pathways. As advantages over PICRUSt 1.0, the new PICRUSt2 pipeline inputs sequences as single nucleotide resolution ASVs, references ten times more genomes than PICRUSt1, and yields output as MetaCyc [39] pathway abundances referenced to shotgun metagenomics. ASV analyses included ALDEx2 differential abundances with Mann–Whitney and Bland–Altman plots, effects sizes distances, principal component analyses (PCA), and principal coordinates analyses (PCoA), network analysis, and pathway differences’ odds ratios. Software included Python and R packages, run either standalone on Mac OS X or Calypso online [40].
Results
Initial comparison of microbial communities from DEPR vs. NODEPR using the entire unfiltered dataset of 2724 unique ASVs was assessed by multivariate PERMANOVA and Bray–Curtis distances, which yielded no significant difference (p = 0.654, R2 = 0.0289). A multivariate PERMANOVA-like differential abundance analysis of the 2724 ASV DADA2 dataset was then assessed by employing the ALDEx2 R package, resulting in the Mann–Whitney plot and Bland–Altman-type plot shown in Fig. 1a, b. According to the pattern and color of ASV points in Fig. 1a, b, no red prevalent data points and no significant differences between the groups were obtained using the full unfiltered 2724 ASV DADA2 dataset; the high number of black points indicates high relative abundance of taxa with low prevalence.
Fig. 1. ASV prevalences and metadata analyses.
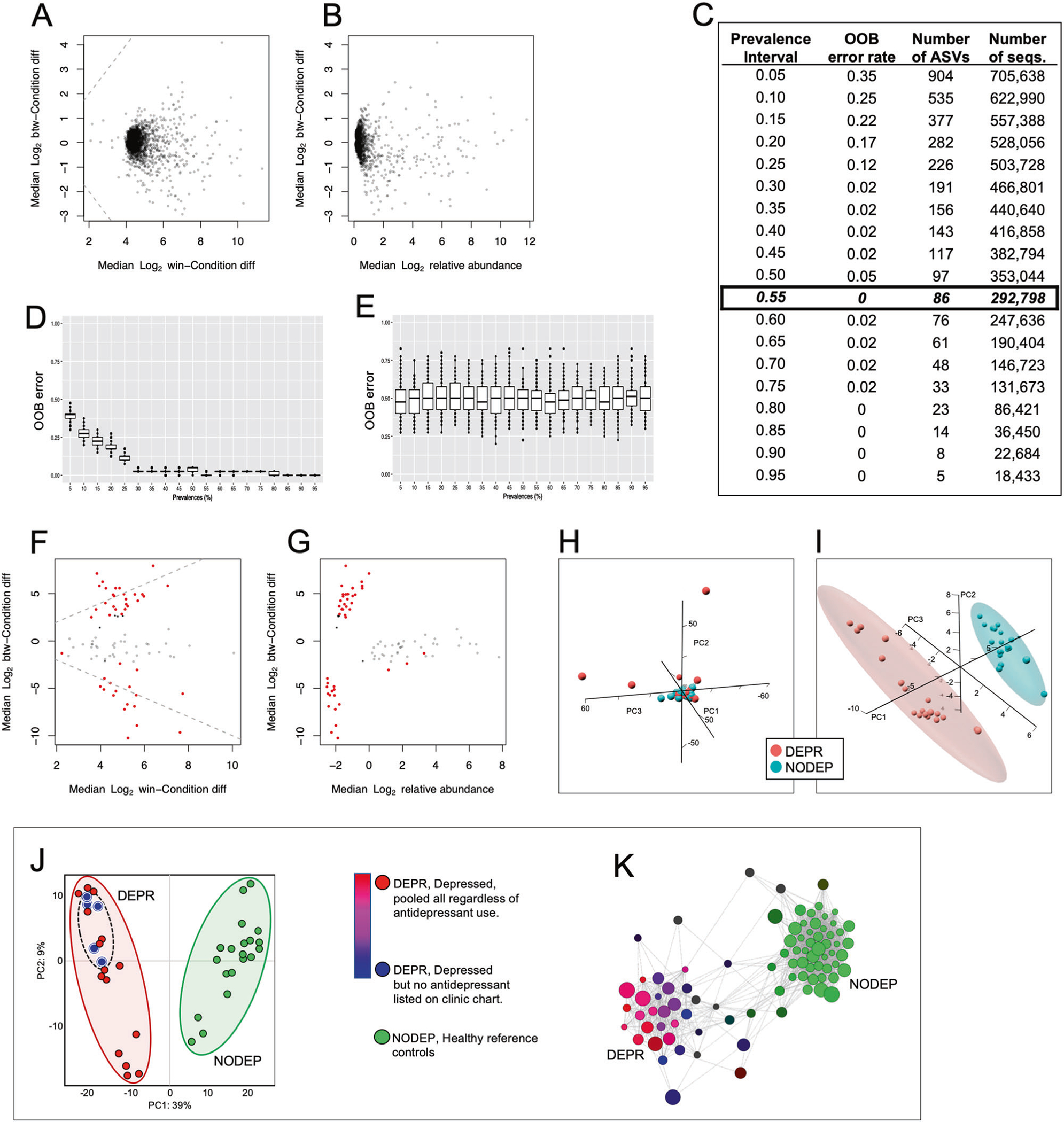
a Mann–Whitney plot of ALDEx2 output using complete DADA2 dataset of 2724 Illumina demultiplexed paired-end unique DNA sequences (Supplementary Information Table 1-SI) without PIME treatment. b Bland–Altman MA-type plot of same dataset described for a. c PIME prevalence bins and out-of-bag (OOB) errors for intervals 5–95%. Based on the criterion of greatest number of random forest sequence combinations at the minimal OBB error = zero, the 55% interval with 86 AVSs (listed in Supplementary Information Table 2-SI) was ultimately employed for all subsequent downstream analyses. d PIME OOB error predictions at each prevalence interval, showing box plots of treatments randomly assigned to the 86 ASV dataset samples. These simulated predictions match the empirical data of c. e Scrambled control validation of the PIME simulations of d, assessed by running randomized variations of OOB errors for each prevalence bin, resulting in box plots with OOB errors at all bins hovering at the predicted value of 0.55. Each plot in d and e was generated by machine-learning using the 55% prevalence dataset of 86 ASVs run through 100 bootstrap iterations of Monte Carlo simulations of random forest classifications on each prevalence interval in 5% increments. f Mann–Whitney plot of pipeline output from DADA2 that derived from the PIME 55% prevalence dataset of 86 ASV seqs, then subsequently processed with ALDEx2. g Bland–Altman MA-type plot of same dataset described for d. a, b, f, and g each point represents a unique microbial taxon exact amplicon sequence variant (ASV). Red points represent taxa assigned as differentially abundant at q < 0.1; gray points are abundant, but not nondifferentially abundant; black points are rare and not differentially abundant. Based on f and g, the 55% interval with 86 AVSs was ultimately employed for all subsequent downstream analyses. h Principal component analysis (PCA) of entire 2724 sequence dataset from the pipeline of DADA2 plus ALDEx2 but not treated using PIME, revealing no metadata clustering by PERMANOVA (Adonis) Bray–Curtis p = 0.654, R2 = 0.02893. i PCA of the 55% prevalence dataset of highly prevalent 86 ASVs from the complete DADA2/ALDEx2/PIME pipeline, revealing significant clustering of metadata by PERMANOVA (Adonis) Bray–Curtis with P < 0.001, R2 = 0.531, with group clusters shown within predicted 95% CI ellipses. Each dataset in h, i was log2 transformed, centered and scaled, and run with Bray–Curtis distances with the pca3d subroutine of the prcomp R package [41]. j Lack of antidepressant influence on DEPR cohort taxa clustering by PCA. Metadata were assigned as: five DEPR subjects with no specific antidepressant listed on their chart (blue), 20 DEPR subjects pooled regardless of antidepressant use (red; 15 DEPR subjects with charted use of an antidepressant plus five DEPR with no specifically listed antidepressant), or 20 NODEP subjects (green). There were no outliers from the clustered 20 subject pooled DEPR cohort (PERMANOVA P = 0.355) which were collectively isolated from NODEP (P < 0.001) relating to the 86 prevalent taxa. k Bray–Curtis dissimilarity network analysis of 86 prevalent taxa and lack of antidepressant influence on cohort distances. Pearson correlation algorithm was employed with positive taxa nodes placed with dissimilarity ordination distances connected by principal coordinates analysis (PCoA) edge placement (false discovery rate, FDR P < 0.05), with similarity cutoff at 0.25. Node sizes and colors are proportional to relative magnitude within the dataset. Note taxa clustering and purple color blend resulting from the overlay of DEPR subjects whose charts listing an antidepressant (red) on DEPR subjects whose charts did not list a specific antidepressant (blue), and of which pooled DEPR metadata were collectively segregated from clustered NODEP (green).
Subsequently, the noisy full dataset of 2724 ASVs was filtered using our PIME R package [32]. PIME removed the within-group variations and captured only biologically significant differences which have high sample prevalence levels. PIME employs a supervised machine-learning algorithm to predict random forests and estimates out-of-bag (OOB) errors, resulting in the Fig. 1c sets of ASV prevalence bins at 5% intervals. Here, high OOB errors indicate a given prevalence dataset bin is noisy representing a high relative abundance of taxa with low prevalence. Therefore in Fig. 1c the minimal OOB error = zero with the highest signal to noise ratio occurring within the 55% prevalence interval based on 292,798 sequence comparisons. This 55% prevalence dataset was comprised of 86 ASVs (Supplementary Information Table 1-SI) that were used for all subsequent downstream analyses.
Using the 55% prevalence 86 ASV dataset, OOB errors were predicted by a Monte Carlo simulation of random forest classifications by running 100 bootstrap aggregations on each prevalence interval. The simulation results shown in Fig. 1d matches Fig. 1c, thus reinforcing the appropriate choice of utilizing the 55% prevalence empirical dataset (Fig. 1c). In order to evaluate the likelihood of introducing bias while building the prevalence-filtered datasets, the data were also randomly scrambled from the two subject groups and then run through the PIME error prediction algorithm again using 100 bootstraps. The resulting control randomization OBB errors were not significantly different from predicted value of 0.55 at all prevalence bins, as shown in Fig. 1e, confirming lack of false-positive type I errors. Thus, taken together the prediction simulation (Fig. 1d) and randomization control (Fig. 1e) simulation collectively validate our PIME algorithm [32] outcome of the 86 ASVs (Fig. 1c).
The 55% prevalence dataset of 86 ASVs was then reintroduced into ALDEx2 analysis, as shown in the Mann–Whitney (Fig. 1f) and Bland–Altman-type (Fig. 1g) outcome plots. Unlike Fig. 1a, b, the data in Fig. 1f, g appear as red points representing taxa assigned as differentially abundant at q < 0.1, along with nondifferentially abundant gray points, but no black points that represent noise of high relative abundance taxa with low prevalence.
PCA [41] was executed using the unfiltered pre-PIME DADA2 dataset of 2724 ASVs and post-PIME-treated 86 ASVs. The pre-PIME 2724 sequence dataset could not be resolved into sample group clusters (p = 0.238, R2 = 0.02893) as shown in Fig. 1h. In contrast, in Fig. 1i the post-PIME-treated 86 ASV 55% prevalence dataset yielded PERMANOVA (Adonis) Bray–Curtis P < 0.001, R2 = 0.531, with PCA ordination readily resolved cluster differentiation of DEPR vs. NODEP, as shown with all points within the 95% confidence interval (CI) ellipses. These PCA results are consistent with the ADLEx2 Mann–Whitney and Bland–Altman results (Fig. 1f, g) and PERMANOVA described above.
The possible influence of antidepressant medication usage on taxa clustering of the 86 ASV dataset by the DEPR metadata phenotype was examined. This resulted in no outliers from the DEPR metadata by PCA analysis (PERMANOVA P = 0.355) (Fig. 1j), nor influence on clustering distances by Bray–Curtis dissimilarity network analyses (P > 0.05) (Fig. 1k). Therefore, data from all 20 DEPR patients were pooled and included as a single cohort in subsequent analyses regardless of specific antidepressant or whether or not their chart specified an antidepressant. Potential influences of subject sex were also assessed; however, data parsed by male/female were not significantly different from random scrambling of sex (P > 0.05, data not shown).
Output from PIME/ALDEx2 yielded effect sizes for the 86 unique ASV sequences (Supplementary Information Table 2-SI), along with uncovering certain multiplicities of assigned taxa names assembled at the levels of Family, Genus, and species. The bar graph of Fig. 2a shows the taxa differential effect size values over the cutoff range of ≥0.5 for NODEP and ≤−0.5 for DEPR. ASV sequences of Supplemental Table 2-SI were assigned a unique code used for downstream assessment of the 55% prevalence dataset, regardless of whether multiple sequences could be assigned the same taxonomic name, and then parsed for redundant multiple copies (n > 1) or unique (n = 1) taxonomic names assigned to the ASVs as shown in Fig. 2b, c. Note the large ASV prevalence representation from the Firmicutes phylum that occurs in both DEPR and NODEP, particularly in Lachnospiraceae, Ruminococcaceae, and Bacteroidetes Families. And also note the diversity of taxa names assigned to the prevalent ASVs is greater for DEPR than for NODEP, as corroborated by Chao1 and Shannon alpha diversity analyses (data not shown). The data of Fig. 2 are further discussed below in the Discussion.
Fig. 2. ASV and taxa prevalences for DEPR vs. NODEP.

a ALDEx2 effect sizes for taxons assigned from ASVs. Displayed cut-offs are effect size ≥0.5 (NODEP) or ≤−0.5 (DEPR). b DEPR ASVs and taxons. c NODEP ASVs and taxons. a–c The DADA2/ALDEx2/PIME pipeline output 55% prevalence dataset taxa names were assigned to each 86 unique ASV, regardless of whether multiple sequences could be assigned the same taxonomic name, based on the naive Bayesian classifier method and the SILVA ribosomal RNA gene database [35] version v132. Redundant multiple copies (N > 1) or unique (N = 1) taxonomic names for the ASVs are shown at the levels of Family, Genus, and species. The full set of all ALDEx2 effect sizes ASV sequences are listed in Supplementary Information Table 1-SI and Table-2SI.
PICRUSt2 treatment of the 86 ASVs from the 55% prevalence dataset predicted 284 MetaCyc microbiome metabolic pathways. Fig. 3a shows the network analysis revealing clustering of gut microbiome pathways common to DEPR, as segregated from clustering common to healthy control NODEP. The possible influence of antidepressant medication use on metabolic pathway clustering by the DEPR metadata phenotype was examined in Fig. 3a, resulting in no outliers within the DEPR cluster by Bray–Curtis dissimilarity network analyses (P > 0.05). Therefore, pathway data from all 20 DEPR patients were pooled and considered as a single cohort (purple circles in Fig. 3a) regardless of specific antidepressant (assigned red in Fig. 3a) or whether or not their chart specified an antidepressant (assigned blue in Fig. 3a). Based on PICRUSt2 pathway data of Fig. 3a, odds ratios and AUC were generated, with the top most salient pathway differences shown in the forest plot of Fig. 3b. Notably, these data highlight untoward pathways in subjects living with depression as contrasted with healthy pathways in reference subjects without depression.
Fig. 3. Metabolic pathway segregation of DEPR vs. NODEP.
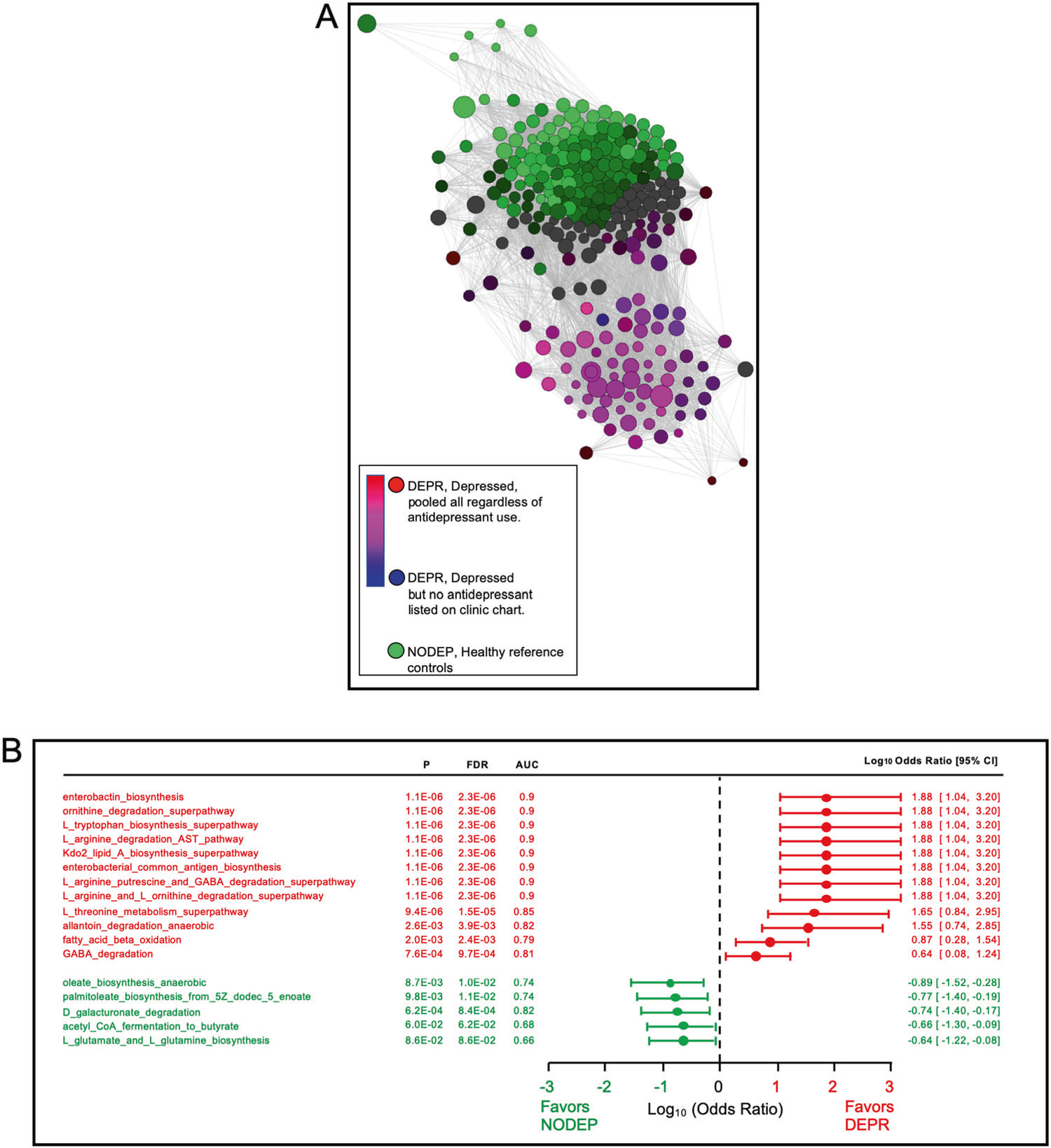
a Bray–Curtis dissimilarity network analysis of microbiome metabolic pathways, and lack of antidepressant influence on cohort distances. Data are 284MetaCyc pathways predicted by PICRUSt2 using the DADA2/ALDEx2/PIME 55% bin prevalence taxa dataset of 86 ASVs. Pearson correlation algorithm was employed with pathway nodes placed by PCoA, showing significant (FDR P < 0.05) positive associations connected by edges, with similarity cutoff at 0.25. Node sizes and colors are proportional to relative magnitude within the dataset. Note pathway clustering and purple color blend resulting from the overlay of DEPR subjects whose charts listed an antidepressant (red) on DEPR subjects whose charts did not list a specific antidepressant (blue), and of which pooled DEPR metadata were collectively segregated from clustered NODEP. b. Odds ratios in forest plot of select microbiome metabolic pathway data of Fig. 2a. Based on the PICRUSt2 MetaCyc pathway data, odds ratios and AUC were generated, with salient pathway differences shown. Note the untoward pathways associated with depression pathophysiology phenotype in DEPR, in contrast to healthy pathways in NODEP.
Discussion
The machine-learning pipeline of the present study unmasked a novel and useful pattern of gut microbiota taxa variants’ prevalences and functionally relevant metabolic pathways associated with depression, as compared with healthy reference subjects. This is the first implementation of advanced error suppression at the level of single nucleotide resolution in compositional gut microbiome assignment to a major mental disorder and attending functional pathways phenotype, via a unique pipeline that incorporates DADA2 error correction [34] combined with de-noising and taxa binning of exact ASV prevalences generated by the R package PIME [32], followed by differential abundance analysis with effect sizes via ALDEx2 [36, 37]. The precedent for justifying exact sequences to differentiate microbial populations without OTU clustering has been established by large scale population diversity studies [21–24].
Functional metabolic pathways
It is becoming recognized in the literature that the value of microbiome studies lies in the importance of the interactive ecology of microbial intermediary metabolism pathways over differences in taxonomy [42]. Patterns of metabolic pathways in gut microbiota taxa reflect their impact on distinguishing host physiology phenotypes, due to the interplay of microbial metabolism with host metabolome and physiology. The present ASV approach revealed the differentiation of untoward gut microbiota MetaCyc pathways in DEPR, in contrast to healthy pathways in NODEP reference subjects (Fig. 3a, b). These results are consistent with the distinguishing hallmarks of unfavorable shifts in metabolism and host inflammasome dysregulation associated with disruption of gut barrier reported for depression vs. healthy individuals, as we and others [6, 8, 11, 12, 43–47] have reported previously.
In the present results (Fig. 3) DEPR-associated pathways of butyrate degradation and GABA degradation were prominent in the gut microbiome of DEPR. In contrast, NODEP was prominently represented by multiple Lachnospiraceae NK4A136 and Lachnospiraceae UCG-001 ASVs (Fig. 2, Table 2-SI), which represent butyrate producing species associated with the physiological and behavioral health benefits of this short chain fatty acid (SCFA) [10, 13, 48, 49]. In a mouse model of depression, Lachnospiraceae UCG-001 and Lachnospiraceae NK4A136 abundances were significantly enhanced by the antidepressant fluoxetine in a subgroup of mice that behaviorally responded to fluoxetine, but these species were not enhanced in the subgroup that did not respond to antidepressant treatment [19]. Our pipeline effect size results (Fig. 2, Table 2-SI) also indicated high prevalences of Roseburia spp, Bacteroides spp, Faecalibacterium spp—in particular, Faecalibacterium prausnitzii—in both DEPR and NODEP, explainable by the notion that SCFA metabolism is highly strain-specific and diet-dependent [20, 50]. Faecalibacterium prausnitzii is the single most common human gut bacterium, with relative abundance dependent on prebiotic diet composition [50, 51].
Inflammation and dysregulation of glutamate, monoamines, and GABA neurotransmission have been associated with the pathophysiology of depression and comorbid neuropathic pain [6, 13, 52–54]. The results (Fig. 2, Table 2-SI) indicated a mixture of taxa representing species that have potential for GABA production (Parabaceroidetes merdre and certain strains of Alstipes spp, Bacteriodes spp, Eubacterium spp, and Escherichia spp) or GABA consumption (select strains of Flavonifractor plautii, Pseudomonas spp, and Acinetobacter spp) [9, 12], with somewhat greater prevalence of GABA producing taxa in DEPR compared to NODEP. Indeed, the odds ratio data of Fig. 3b favor subjects with depression possessing gut microbiota GABA degradation and reduced biosynthesis via: “L_arginine_putrescine_and_GABA_degradation_superpathway”, “L_arginine_and_ornithine_degradation_superpathway”, and “L_argi nine_degradation_AST_superpathway”. Conversely, in healthy reference subjects without depression the Fig. 3b odds ratio favored “L_glutamine_and_glutamate_biosynthesis” which promotes GABA production.
The data of Fig. 3 reflect microbiota alterations in small molecules and other amino acid pathways associated with depression, such that threonine, tryptophan, and leucine that can activate mTOR-mediated intestinal inflammation, while arginine and ornithine suppress gut inflammation [55]. Under proinflammatory gut bacteria conditions, high levels of tryptophan are converted to kynurenine at the expense of reduced serotonin synthesis, whereupon kynurenine crosses blood–brain barrier and is converted to neurotoxic kynurenic and quinolinic acids which have been correlated with depressive symptoms [56]. Microbial degradation of allantoin in DEPR, and D-galactuonate degradation NODEP in our subjects are consistent with animal models [57, 58].
Proinflammatory gut bacteria that generate Kdo2-lipid were favored in DEPR (Fig. 2). Kdo2-lipids are the primary component of LPS that activates host TLR4-MD2 signaling and myeloid differentiation [59], enterobacterial common antigen that is linked to LPS via Kdo2 [60], and iron-sequestering biofilm-enhancing enterobactin [61]. The large effect size for Enterobacteriaceae in DEPR (Fig. 2a, Table 2-S1) is consistent with LPS-related morbidity of strains that disrupt intestinal barrier and invokes inflammation from proinflammatory cytokine responses [50] in humans with depression [62]. The high ASV prevalence of Roseburia intestinalis in our NODEP subjects (Fig. 2) is consistent with prior studies showing that the flagellin of this species reduces intestinal inflammation by suppressing IL-17 in the host [63]. The pathway data (Fig. 3) favored enhancement of oleate and palmitoleate, which are inversely correlated with depression [64, 65]. Overall gut microbiota fatty acid beta oxidation was favored in DEPR (Fig. 3b).
Taxa
In addition to functional pathway differences, assigned taxa names and taxa linear discriminant analysis estimates of effect size differences between depressed human subjects vs. healthy control subjects have been used in previous gut microbiome compositional studies based on OTU relative abundances or shotgun sequencing metagenomics [6, 8–10, 44, 47, 48, 66–68]. Our ASV results (Figs. 1j, k and 2a–c, and Table 2-SI) are in accord with the reported general trend in increased OTU relative abundances of taxa associated with human depression and rodent models of depression with respect to Acidaminococcaceae, Enterobacteriaceae, Rikenellaceae, and Coriobacteriaceae Families, and in particular of Blautia sp, Alistipes sp, Parabacteroides spp, Phascolarctobacterium sp, Oscillibacter spp, Rosburia spp, Flavonifractor sp, and Holdemania sp [10, 11, 44, 45, 48]. Regarding trends for OTU relative abundances depleted in DEPR and increased in NODEP, select species of Faecalibacterium spp, Ruminococcus spp, Lachnospiraceae spp, and Bacteroides spp have been reported [9, 10, 44, 48], as also observed in our ASV prevalence results (Fig. 2, Table 2-SI). Previous reports have negatively correlated Faecalibacterium spp OTU abundances with magnitude of depression symptom severity [10, 69], while our results (Fig. 2, Table 2-SI) identified separate particular Faecalibacterium spp ASVs for DEPR and NODEP. Elevated Parabacteroidetes is associated with anhedonia in rat models [70], consistent with our results (Fig. 2a–c, Table 2-SI).
Fig. 2 and Table 2-SI indicated that in NODEP nearly 75% of the prevalent taxa with effect sizes >0.5 are Lachnospiraceae spp, with the balance of prevalent taxa represented by Bacteroides spp and Ruminococcaceae family. The top prevalence effect size for NODEP (Fig. 2a) was Faecalibacterium CM04-06 of the Ruminococcaceae family. Both DEPR and NODEP yielded large prevalences of ASVs tagged to Ruminococcaceae. This is not unexpected because beneficial vs untoward health effects of Ruminococcaceae are highly species- and strain-specific and diet-induced, due to variations in their fiber hydrolyzing enzyme profiles [20, 50]. Strain-dependence abundances in DEPR is reportedly associated with elevated R. flavefaciens which abrogates effects of antidepressants [18], and elevated select members of Bacteroidetes phylum, but with net decreases in Firmicutes in both human studies and in rat depression models employing relative OTU abundances alone [45] or in conjunction with LC/MS metabolomics [11]. Getachew et al. [15] reported that antidepressant ketamine reduced levels of Ruminococcus spp in rats. These OTU reports are in contrast to the high degree of representation of ASV assigned to Lachnospiraceae, Ruminococcaceae, and Bacteroidaceae in both DEPR and NODEP (Fig. 2, Table 2-SI).
Our ASV prevalence data (Fig. 2, Table 2-SI) indicated an overall greater diversity of taxa prevalences in DEPR, in concordance with a 16S closed-OTU approach of Jiang et al. [10], but in contrast to other reports of alpha diversity or richness with no difference in humans [44] or reduction with a rat depression model [45]. It has been posited that the many dimensions of “diversity” of a given ecosystem composition are not per se an index of “better” vs. “worse” [71].
Physiological anthropology of depression
Mood disorders exhibit familial transmission, but the exact genetics remain unresolved despite ongoing studies analyzing nearly 200 candidate human maker genes [43]. People are essentially metaorganisms comprised of ~1014 prokaryotic cells plus roughly the same number of eukaryotic cells—host physiology is the co-evolutionary consequence of the interplay among human plus bacterial genomes and metabolomics. The present study emphasizes the importance of gut microbiome prevalences on host depression phenotype behavior and metabolic pathways; it is the prevalence—in contrast to relative abundance—of certain bacterial metabolic functions steered by microbiome genes that ultimately shapes host-microbiota relationships. Specific human genetic loci shape heritable patterns of gut microbiome taxa prevalences in the host [72]. The Christensenellaceae family is the single most heritable gut microbial taxon, typically correlated with various healthy phenotypes [72]. Notably, our results (Table 2-SI) indicated ASV prevalence of Christensenellaceae R-7 group in the NODEP cohort (effect size 0.47), in contrast to DEPR. These data taken together with PCA discriminatory taxa clustering of NODEP subjects vs. DEPR (Fig. 1i–k) collectively imply the heritability of resistance to depression. Thus, anthropological group cohesion cultural factors such as food and dietary habits, mating preferences that sustain closed groups, and environmental communal exposure to a common set of commensal bacteria may propagate bacterial species of depression. Or conversely, perhaps certain gut microbiota may have usurped human hosts as unwitting prokaryotic propagation vessels by shaping mood and sickness behavior as an evolutionary survival advantage by withdrawing their hosts from environmental harms or from competing infectious pathogenic bacteria.
Conclusion
In conclusion, the present study describes a novel gut microbiome machine-learning approach to potentially differentiate people with depression from healthy reference controls. The process employs DADA2, ALDEx2, PIME, and PICRUSt2 R packages to evaluate prevalent ASVs from human gut microbiome 16S rRNA amplicon sequences at the level of single nucleotide resolution. This machine-learning technique approach may reduce pitfalls of OTU relative abundance clustering and shotgun metagenomics. By employing prevalent ASVs, this study led to an ability to distinguish 20 individuals with depression from 20 healthy reference subjects. Results are supported by multivariate analyses’ PERMANOVA P < 0.001, effect sizes ≥0.5, PCA ordination, network analyses, and odds ratios, which collectively conformed to current dysbiosis and pathophysiologic hypotheses of depression associated with neurocircuit-relevant neurotransmitter pathways, inflammation, and gut–brain dysregulation. Furthermore, the differential patterns of unique ASVs assigned to taxonomy and metabolic pathways associated in individuals with depression and healthy controls were generally consistent with prior OTU relative abundance and metagenomics studies, with disparities attributable to the taxonomic MetaCyc assignment of species- and strain-specific microbiota metabolomics. This is the first published report using this gut microbiome machine-learning approach and its utility as a high throughput sequencing technique of the gut microbiome to identify individuals with depressive symptoms different from healthy reference subjects. Its application in the clinical setting may lend to personalizing treatments based on ASV in patients with depression by decreasing neurobiological heterogeneity, as based on the current DSM-5 diagnostic framework. Larger studies are needed to delineate the extent to which different symptoms of depression and influences of antidepressants may correspond with functional metaorganisms tethered to underlying neurobiological dysfunction.
Supplementary Material
Acknowledgements
We thank Seungbum Kim and Elaine Richards for useful discussions. This study was supported by University of Florida Clinical and Translational Science Institute grant (BRS, CJP) from the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR001427; National Institute of Health (NIH) grants HL33610, HL56921 (MKR, CJP); UM1 HL087366 (CJP); Gatorade Trust through funds distributed by the University of Florida, Department of Medicine (CJP); PCORI-OneFlorida Clinical Research Consortium CDRN-1501-26692 (CJP); and internal funds from the University of Florida Department of Physiology and Functional Genomics (BRS, MKR).
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Code availability
PIME is available at https://rdrr.io/github/microEcology/pime/; DADA2 is available at https://www.bioconductor.org/packages/release/bioc/html/dada2.html; ALDEx2 is at https://bioconductor.org/packages/release/bioc/html/ALDEx2.html; and picrust2 is at https://github.com/picrust/picrust2.
Supplementary information The online version of this article (https://doi.org/10.1038/s41380-020-0652-5) contains supplementary material, which is available to authorized users.
References
- 1.WHO. World Health Organization Fact Sheet, Depression. 2018. https://www.who.int/news-room/fact-sheets/detail/depression. [Google Scholar]
- 2.Heiss CN, Olofsson LE. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. J Neuroendocrinol. 2019;31:e12684. [DOI] [PubMed] [Google Scholar]
- 3.Warner BB. The contribution of the gut microbiome to neurodevelopment and neuropsychiatric disorders. Pediatr Res. 2019;85:216–24. [DOI] [PubMed] [Google Scholar]
- 4.Stefano GB, Pilonis N, Ptacek R, Raboch J, Vnukova M, Kream RM. Gut, microbiome, and brain regulatory axis: relevance to neurodegenerative and psychiatric disorders. Cell Mol Neurobiol. 2018;38:1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skonieczna-Zydecka K, Marlicz W, Misera A, Koulaouzidis A, Loniewski I. Microbiome-the missing link in the gut-brain axis: focus on its role in gastrointestinal and mental health. J Clin Med. 2018;7:E521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter G, Hart RA, Charlesworth RPG, Sharpley CF. Gut microbiome and depression: what we know and what we need to know. Rev Neurosci. 2018;29:629–43. [DOI] [PubMed] [Google Scholar]
- 7.Kelly JR, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbiota axis: challenges for translation in psychiatry. Ann Epidemiol. 2016;26:366–72. [DOI] [PubMed] [Google Scholar]
- 8.Zalar B, Haslberger A, Peterlin B. The role of microbiota in depression—a brief review. Psychiatr Danub. 2018;30:136–41. [DOI] [PubMed] [Google Scholar]
- 9.Strandwitz P, Kim KH, Terekhova D, Liu JK, Sharma A, Levering J, et al. GABA-modulating bacteria of the human gut microbiota. Nat Microbiol. 2018;4:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186–94. [DOI] [PubMed] [Google Scholar]
- 11.Yu M, Jia H, Zhou C, Yang Y, Zhao Y, Yang M, et al. Variations in gut microbiota and fecal metabolic phenotype associated with depression by 16S rRNA gene sequencing and LC/MS-based metabolomics. J Pharm Biomed Anal. 2017;138:231–9. [DOI] [PubMed] [Google Scholar]
- 12.Cheung SG, Goldenthal AR, Uhlemann AC, Mann JJ, Miller JM, Sublette ME. Systematic review of gut microbiota and major depression. Front Psychiatr. 2019;10:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vuong HE, Yano JM, Fung TC, Hsiao EY. The Microbiome and Host Behavior. Annu Rev Neurosci. 2017;40:21–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearson-Leary J, Zhao C, Bittinger K, Eacret D, Luz S, Vigderman AS, et al. The gut microbiome regulates the increases in depressive-type behaviors and in inflammatory processes in the ventral hippocampus of stress vulnerable rats. Mol Psychiatry. 2019. 10.1038/s41380-019-0380-x. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 15.Getachew B, Aubee JI, Schottenfeld RS, Csoka AB, Thompson KM, Tizabi Y. Ketamine interactions with gut-microbiota in rats: relevance to its antidepressant and anti-inflammatory properties. BMC Microbiol. 2018;18:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun L, Zhang H, Cao Y, Wang C, Zhao C, Wang H, et al. Fluoxetine ameliorates dysbiosis in a depression model induced by chronic unpredicted mild stress in mice. Int J Med Sci. 2019;16:1260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romijn AR, Rucklidge JJ, Kuijer RG, Frampton C. A double-blind, randomized, placebo-controlled trial of Lactobacillus helveticus and Bifidobacterium longum for the symptoms of depression. Aust N. Z J Psychiatr. 2017;51:810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukic I, Getselter D, Ziv O, Oron O, Reuveni E, Koren O, et al. Antidepressants affect gut microbiota and Ruminococcus flavefaciens is able to abolish their effects on depressive-like behavior. Transl Psychiatr. 2019;9:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyte M, Daniels KM, Schmitz-Esser S. Fluoxetine-induced alteration of murine gut microbial community structure: evidence for a microbial endocrinology-based mechanism of action responsible for fluoxetine-induced side effects. PeerJ 2019;7:e6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hassan AM, Mancano G, Kashofer K, Frohlich EE, Matak A, Mayerhofer R, et al. High-fat diet induces depression-like behaviour in mice associated with changes in microbiome, neuropeptide Y, and brain metabolome. Nutr Neurosci. 2019;22:877–93. [DOI] [PubMed] [Google Scholar]
- 21.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lovell D, Pawlowsky-Glahn V, Egozcue JJ, Marguerat S, Bahler J. Proportionality: a valid alternative to correlation for relative data. PLoS Comput Biol. 2015;11:e1004075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glassman SI, Martiny JBH. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. Msphere. 2018;3:e00148–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551:457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garrido-Cardenas JA, Manzano-Agugliaro F. The metagenomics worldwide research. Curr Genet. 2017;63:819–29. [DOI] [PubMed] [Google Scholar]
- 26.Cao HT, Gibson TE, Bashan A, Liu YY. Inferring human microbial dynamics from temporal metagenomics data: Pitfalls and lessons. Bioessays. 2016;39. 10.1002/bies.201600188. Epub online. [DOI] [PubMed] [Google Scholar]
- 27.Sedlar K, Kupkova K, Provaznik I. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput Struct Biotechnol J. 2017;15:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJ, et al. Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci USA. 2015;112:E2930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang ZZ, Chen G. Zero-inflated generalized Dirichlet multinomial regression model for microbiome compositional data analysis. Biostatistics. 2018;20:698–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gloor GB, Reid G. Compositional analysis: a valid approach to analyze microbiome high-throughput sequencing data. Can J Microbiol. 2016;62:692–703. [DOI] [PubMed] [Google Scholar]
- 32.Roesch LFW, Dobbler PT, Pylro VS, Kolaczkowski B, Drew JC, Triplett EW. PIME: A package for discovery of novel differences among microbial communities. Mol Ecol Resour. 2019. 10.1111/1755-0998.13116. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 33.Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol. 2014;5:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandes AD, Reid JN, Macklaim JM, McMurrough TA, Edgell DR, Gloor GB. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome. 2014;2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gloor GB. ALDEx2: ANOVA-like differential expression tool for compositional data. 2018. https://bioconductor.org/packages/release/bioc/vignettes/ALDEx2/inst/doc/ALDEx2_vignette.pdf.
- 38.Douglas GM, Beiko RG, Langille MGI. Predicting the functional potential of the microbiome from marker genes using PICRUSt. Methods Mol Biol. 2018;1849:169–77. [DOI] [PubMed] [Google Scholar]
- 39.Caspi R, Billington R, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46:D633–D39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion MJ, Berger B, et al. Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics. 2017;33:782–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2018. https://www.R-project.org/. [Google Scholar]
- 42.Visconti A, Le Roy CI, Rosa F, Rossi N, Martin TC, Mohney RP, et al. Interplay between the human gut microbiome and host metabolism. Nat Commun. 2019;10:4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shadrina M, Bondarenko EA, Slominsky PA. Genetics factors in major depression disease. Front Psychiatr. 2018;9:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naseribafrouei A, Hestad K, Avershina E, Sekelja M, Linlokken A, Wilson R, et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil. 2014;26: 1155–62. [DOI] [PubMed] [Google Scholar]
- 45.Kelly JR, Borre Y, Patterson COB, El Aidy E, Deane JS, et al. Transferring the blues: depression-associated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res. 2016;82:109–18. [DOI] [PubMed] [Google Scholar]
- 46.Johansson R, Carlbring P, Heedman A, Paxling B, Andersson G. Depression, anxiety and their comorbidity in the Swedish general population: point prevalence and the effect on health-related quality of life. PeerJ. 2013;1:e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Editoriaol. Links between gut microbes and depression strengthened. Nature. 2019;566:7. [DOI] [PubMed] [Google Scholar]
- 48.Valles-Colomer M, Falony G, Darzi Y, Tigchelaar EF, Wang J, Tito RY, et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat Microbiol. 2019;4:623–32. [DOI] [PubMed] [Google Scholar]
- 49.Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatr. 2016;21:786–96. [DOI] [PubMed] [Google Scholar]
- 50.Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, et al. The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients. 2018;10:E988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miquel S, Martin R, Rossi O, Bermudez-Humaran LG, Chatel JM, Sokol H, et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. 2013;16:255–61. [DOI] [PubMed] [Google Scholar]
- 52.Peirce JM, Alvina K. The role of inflammation and the gut microbiome in depression and anxiety. J Neurosci Res. 2019;97:1223–41. [DOI] [PubMed] [Google Scholar]
- 53.Liu CS, Adibfar A, Herrmann N, Gallagher D, Lanctot KL. Evidence for Inflammation-Associated Depression. Curr Top Behav Neurosci. 2017;31:3–30. [DOI] [PubMed] [Google Scholar]
- 54.Halaris A. Inflammation and depression but where does the inflammation come from? Curr Opin Psychiatr. 2019;32:422–8. [DOI] [PubMed] [Google Scholar]
- 55.He F, Wu C, Li P, Li N, Zhang D, Zhu Q, et al. Functions and signaling pathways of amino acids in intestinal inflammation. Biomed Res Int. 2018;2018:9171905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waclawikova B, El Aidy S. Role of microbiota and tryptophan metabolites in the remote effect of intestinal inflammation on brain and depression. Pharmaceuticals. 2018;11:E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahn YJ, Park SJ, Woo H, Lee HE, Kim HJ, Kwon G, et al. Effects of allantoin on cognitive function and hippocampal neurogenesis. Food Chem Toxicol. 2014;64:210–6. [DOI] [PubMed] [Google Scholar]
- 58.Chung WSF, Meijerink M, Zeuner B, Holck J, Louis P, Meyer AS, et al. Prebiotic potential of pectin and pectic oligosaccharides to promote anti-inflammatory commensal bacteria in the human colon. FEMS Microbiol Ecol. 2017;93. 10.1093/femsec/fix127. [DOI] [PubMed] [Google Scholar]
- 59.Wang XY, Quinn PJ, Yan AX. Kdo(2)-lipid a: structural diversity and impact on immunopharmacology. Biol Rev 2015;90:408–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitchell AM, Srikumar T, Silhavy TJ. Cyclic enterobacterial common antigen maintains the outer membrane permeability barrier of Escherichia coli in a manner controlled by YhdP. MBio 2018;9:e01321–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H, Zeng X, Mo Y, He B, Lin H, Lin J. Enterobactin-specific antibodies induced by a novel enterobactin conjugate vaccine. Appl Environ Microbiol. 2019;85:e00358–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stevens BR, Goel R, Seungbum K, Richards EM, Holbert RC, Pepine CJ, et al. Increased human intestinal barrier permeability plasma biomarkers zonulin and FABP2 correlated with plasma LPS and altered gut microbiome in anxiety or depression. Gut. 2018;67:1555–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu C, Song K, Shen Z, Quan Y, Tan B, Luo W, et al. Roseburia intestinalis inhibits interleukin17 excretion and promotes regulatory T cells differentiation in colitis. Mol Med Rep. 2018;17:7567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolfe AR, Ogbonna EM, Lim S, Li Y, Zhang J. Dietary linoleic and oleic fatty acids in relation to severe depressed mood: 10 years follow-up of a national cohort. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:972–7. [DOI] [PubMed] [Google Scholar]
- 65.Fernandes MF, Mutch DM, Leri F. The relationship between fatty acids and different depression-related brain regions, and their potential role as biomarkers of response to antidepressants. Nutrients. 2017;9:E298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goll J, Thiagarajan M, Abubucker S, Huttenhower C, Yooseph S, Methe BA. A case study for large-scale human microbiome analysis using JCVI’s metagenomics reports (METAREP). PLoS One. 2012;7:e29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evans SJ, Bassis CM, Hein R, Assari S, Flowers SA, Kelly MB, et al. The gut microbiome composition associates with bipolar disorder and illness severity. J Psychiatr Res. 2017;87:23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang C, Fang X, Zhan G, Huang N, Li S, Bi J, et al. Key role of gut microbiota in anhedonia-like phenotype in rodents with neuropathic pain. Transl Psychiatr. 2019;9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shade A Diversity is the question, not the answer. ISME J. 2017;11:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.