

Abstract
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2, SARS2) is responsible for the COVID-19 pandemic and infections that continue to affect the lives of millions of people worldwide, especially those who are older and/or immunocompromised. The SARS2 main protease enzyme, Mpro (also called 3C-like protease, 3CLpro), is a bona fide drug target as evidenced by potent inhibition with nirmatrelvir and ensitrelvir, the active components of the drugs Paxlovid and Xocova, respectively. However, the existence of nirmatrelvir and ensitrelvir-resistant isolates underscores the need to develop next-generation drugs with different resistance profiles and/or distinct mechanisms of action. Here, we report the results of a high-throughput screen of 649,568 compounds using a cellular gain-of-signal assay. In this assay, Mpro inhibits expression of a luciferase reporter, and 8,777 small molecules were considered hits by causing a gain in luciferase activity 3x SD above the sample field activity (6.8% gain-of-signal relative to 100 μM GC376). Single concentration and dose-response gain-of-signal experiments confirmed 3,522/8,762 compounds as candidate inhibitors. In parallel, all initial high-throughput screening hits were tested in a peptide cleavage assay with purified Mpro and only 39/8,762 showed inhibition. Importantly, 19/39 compounds (49%) re-tested positive in both SARS2 assays, including two previously reported Mpro inhibitors, demonstrating the efficacy of the overall screening strategy. This approach led to the rediscovery of known Mpro inhibitors such as calpain inhibitor II, as well as to the discovery of novel compounds that provide chemical information for future drug development efforts.
Keywords: antiviral drugs, cell-based ultra-high throughput screening (uHTS), protease inhibitors, SARS-CoV-2 main protease (Mpro/3CLpro)
Introduction
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2, SARS2) is estimated to have caused over 10 million deaths worldwide from 2019 to present (1–3). Although no longer considered a pandemic by the World Health Organization or the United States Center for Disease Control, SARS2 has become endemic and it continues to infect millions each year, causing cold-like symptoms in the majority, as well as long-term effects collectively termed long-COVID in a minority of patients. Long-COVID pathologies include neurological symptoms (brain fog, changes in taste or smell), respiratory symptoms (heart arrythmia, chest paint, etc.), and general symptoms such as fatigue and high fever (1,4,5). SARS2 variants also remain a serious threat to immunocompromised people, including the elderly population, and can trigger pulmonary issues including inflammatory responses that lead to death (6–9).
Vaccines have proven effective in protecting from SARS2 infection and, minimally, lessening its pathogenic effects. However, rapid virus evolution continues to lead to new variants, defined by viral spike protein alterations, that undermine vaccine efficacy and drive periodic vaccine updates from pharmaceutical companies. However, SARS2 infections can also be treated with orally available drugs such as Paxlovid, which includes nirmatrelvir to directly inhibit the activity of the viral main protease enzyme, Mpro, and ritonavir to inhibit cytochrome P450-mediated metabolism of nirmatrelvir (10–13). Nirmatrelvir blocks Mpro from cleaving viral polyprotein substrates into functional units required for viral replication and pathogenesis. Additional Mpro inhibitors are in various stages of development ranging from early-stage tool compounds to late-stage clinical trials with ensitrelvir, FB-2001, and PF-07817883 (the active components of Xocova, Bofutrelvir, and Ibuzatrelvir, respectively) (14–19).
Based on clear precedents from prior antiviral drug development campaigns for HIV-1 and hepatitis C virus (HCV) protease enzymes (20–23), it is important to continue to develop and refine Mpro inhibitors until potent, long-lasting, orally available compounds are achieved. Next-generation Mpro drugs should also be weaned away from ritonavir-dependent regimes, which inhibit cytochrome P450 function and can complicate the use of other medicines. Additional desirable properties for next-generation Mpro drugs include broader spectrum activity such that they also inhibit the replication of other coronavirus species including known pathogens such as Middle East Respiratory Syndrome Coronavirus (MERS-CoV), pathogens that do not pose a current threat such as Severe Acute Respiratory Syndrome Coronavirus-1 (SARS1), and related beta-coronaviruses found in bats and many different mammals. Broader spectrum activity against present-day viruses is likely to be a key predictor (though not perfect) of future efficacy against the next pandemic coronavirus (SARS3), which most experts predict will emerge (albeit unclear with respect to timeline) (24–26). Last, but not least, next-generation Mpro inhibitors should exhibit different resistance profiles. For instance, the covalent inhibitor nirmatrelvir has a distinct resistance profile from ensitrelvir and FB-2001 (48, 27, 34, 47, 46, 33,35, 43). Here we report the results of an ultra-high-throughput screen (uHTS) of nearly 650,000 compounds using a cell-based assay as a primary screen (33–36). Additionally, as secondary screens, an analogous HCoV-NL63 cell-based assay and a biochemical assay were used to further help delineate candidate inhibitors. The primary screen leverages our original observations that SARS2 Mpro overexpression suppresses cellular gene expression (including luciferase reporter gene expression) and that bona fide Mpro inhibitors recover gene expression in a dose-responsive manner (33–36). Two advantages of using this cellular system as a primary screen are a requirement for cell-permeable molecules and that cytotoxic compounds, which also inhibit gene expression, are unlikely to be identified as positive hits. This cell-based approach led to the rediscovery of known Mpro inhibitors, such as calpain inhibitor II, as well as to the discovery of several small molecules that provide chemical information for future drug development efforts.
Materials and Methods
Gain-of-Signal Assays for uHTS
First, large batches of 293T cells were pre-transfected (16 μg / 1×107 cells) with pcDNA5/TO-Src-SARS2 Mpro-Tat-fLuc or Src-NL63 Mpro-Tat-Luc using Electroporator, ExPERT Stx (SW Version: 4.1.11, MaxCyte, USA). SARS2 and NL63 Mpro protein sequences match Genbank accession numbers QII57165.1 and AWK59972.1, respectively. After 4 h incubation at 37°C and 5% CO2, cells were harvested and resuspended in Recovery Cell Culture Freezing media (Gibco catalog no. 12648010). Aliquoted cells were frozen slowly at −80°C, then stored in liquid nitrogen until use. The 1,536-well plate format assay begins with thawing batches of pre-transfected cells and dispensing 1250 cells (5 μl) into each well of a 1,536-well plate (Aurora EWB0–42000A). After addition of 50 nl compound or vehicle (for high reference wells, a final concentration of 100 μM GC367 was added), plates were incubated for 48 h at 37°C and 5% CO2. Plates were then removed from that environment and incubated at room temperature for 10 min to prevent condensation. Gain-of-signal readouts were initiated by adding 5 μl/well of Bright-Glo reagent (Promega catalog no. E2650), and after an additional 10 min room temperature incubation, firefly luciferase activity was measured using Pherastar instrument (BMG Labtech). The final DMSO concentration per reaction well was 0.75%.
Z’-Factor Determination
Reproducibility was assessed by calculating a Z-factor (Z’). A Z’-factor of one is considered ideal, and Z’ values measured here (0.47–0.87) are considered robust and significant statistically. Additionally, assay quality can be inferred through a signal-to-noise ratio (S/N) or signal-to-background ratio (S/B). In our efforts to calculate Z’, we used a low reference (24), transfected cells treated with DMSO, and high reference (HR), transfected cells treated with 100 μM GC376, a broad spectrum coronavirus Mpro inhibitor (27–33). The following equations were used in which ABS is the absolute value of a number, SD is the standard deviation, and AVR is the average.
Calculating Percent Inhibition in Gain-of-Signal Assay
To determine percent inhibition of Mpro at single point concentrations of tool compound GC376, and other relevant chemicals reported in this study, the raw luminescent values (RLU) for each reaction well were used to calculate % inhibition:
The median low control is derived from transfected cells treated with DMSO, which yields the lowest raw luciferase signal (0% inhibition). The median high control is derived from transfected cells treated with 100 μM GC376, which yields the highest raw luciferase signal (100% inhibition). Candidate inhibitors caused a gain in luciferase signal 3x SD above the sample field activity (6.8% gain-of-signal relative to 100 μM GC376).
Recombinant Protein Preparation
A pGEX6P-1-SARS2-Mpro-His6x expression vector, which encodes a glutathione S-transferase (GST)-SARS2 Mpro-His6x fusion protein was provided by Dr. Shaun Olsen (UT Health San Antonio) (pGEX6P-1 GenBank accession no. QLL57165.1). In this construct, the natural N-terminal cleavage site for Mpro is included to facilitate self-cleavage and purification from GST. An P132H derivative (matching Omicron Mpro) was created by site-directed mutagenesis using primers 5’- ATG-TGC-TAT-GCG-TCA-TAA-TTT-TAC-CAT-TAA-GGG-TAG-3’ and 3’-TAA-TGG-TAA-AAT-TAT-GAC-GCA-TAG-CAC-ATT-GAT-AAA-CGC-5’. After DpnI digestion (New England Biolabs catalog no. 10196884), the PCR product was transformed into chemically competent E. coli DH10B cells (Thermo Fisher Scientific catalog no. EC0113). Single colonies were picked, expanded in liquid Luria-Bertani (34) medium supplemented with 100 mg/mL carbenicillin, mini-prepped, and verified by Sanger DNA sequencing.
For protein production, E. coli strain BL21(DE3) (New England Biolabs catalog no. C2527H) was transformed with the pGEX6P-1-SARS2-Mpro-P132H-His6x plasmid, and a single colony was grown overnight to saturation in 50 ml LB medium supplemented with 100 mg/mL carbenicillin (Thermo Fisher Scientific catalog no. J6194903). 5 ml of this primary culture was used to inoculate 1 L of LB broth supplemented with 100 mg/mL of carbenicillin and incubated at 37°C, shaking at 190 rpm, until an optical density (OD) of 0.6 was reached. At this point, Mpro expression was induced by adding 0.5 mM IPTG (Thermo Fisher Scientific catalog no. 15529019), and the incubation temperature was lowered to 18°C for an additional 20 h. The cells were collected by centrifugation at 3,000 g, resuspended in 20 mM Tris, pH 8.0, 200 mM NaCl, 5 mM β-mercaptoethanol (Thermo Fisher Scientific catalog no. O33461–100), 5 mM imidazole (Thermo Fisher Scientific catalog no. A1022122), and 5% glycerol (Thermo Fisher Scientific catalog no. A16205AP), and lysed by sonication. Mpro was captured from cleared lysate using a nickel-nitrilotriacetic acid gravity flow affinity column (Fisher Scientific catalog no. R90115), washed by a gradient of imidazole, and eluted with 300 mM imidazole. The protein was concentrated using centrifugal filter units (Millipore catalog no. UFC910008) and further purified by size exclusion chromatography (SEC) on a Superdex 200 pg column (Cytvia Life Sciences catalog no. 28989336) operating with 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM dithiothreitol (DTT) (Thermo Fisher Scientific catalog no. R0861), and 2% glycerol. The peak fractions of SEC showing single band for Mpro in SDS-PAGE were pooled and concentrated to 5 mg/mL as determined by UV absorbance (NanoDrop 8000 spectrophotometer) and, finally, flash frozen in liquid nitrogen for long-term storage at −196°C.
Biochemical Assay for uHTS
An established biochemical assay (30,35–37) was miniaturized into a 1,536-well plate format with 5 μl/well total reaction volume, which yielded a statistically significant Z’-value of 0.89. First, 2.5 μl of 300 nM SARS2 Mpro-P132H in reaction buffer [20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.05% Tween20, 0.1 mg/mL bovine serum albumin (BSA), and 1 mM DTT] was dispensed into each well of a 1,536-well plate (Greiner catalog no. 789176-F). Second, 50 nl of each test compound, GC376 (positive control), or DMSO (negative control) was added to each well and reactions were equilibrated for 30 min at RT. Third, 2.5 μl of peptide substrate (DABCYL-KTSAVLQ|SGFRKM-EDANS; UPBio catalog no. V1010–1) in assay buffer (above) was dispensed into each well, and plates were incubated an additional 90 min at RT. The final concentration of Mpro was 150 nM, compound was 10.9 μM, and substrate was 5 μM. The final DMSO concentration per reaction well was 0.75%. Fluorescence intensity was measured using a PHERAstar instrument (Ex. 360nm / Em. 460nm filter set), and calculations for inhibition are identical to those used above for cell-based uHTS.
Mpro Inhibition Gain-of-Signal Assay for Purchased Compounds
Candidate Mpro inhibitors were purchased from commercial sources for validation studies in 96 well plate format assays (compounds, sources, and catalog numbers are listed in Supplementary Table S1). Well documented compounds were used as positive references throughout these studies including GC376 (28,31–33,38–42), nirmatrelvir (10,11,13,19,25,27,30,32,34,43–49), and boceprevir (29,32,38,39,50,51). These control compounds were purchased from Selleckchem (S0475, S9866, and S3733, respectively). For luciferase-based gain-of-signal assays, 3 × 106 293T cells were seeded in a 10-cm dish and transfected 24 h later with 2 μg of the pcDNA5/TO-Src-SARS2 Mpro-Tat-fLuc or Src-NL63 Mpro-Tat-Luc plasmids (33–36) 4 h post-transfection, the cells were washed once with PBS-EDTA, trypsinized, resuspended, and counted. Cells were diluted in growth medium to yield a suspension of 4 × 105 cells/mL, and 50 μL was plated into each well of a 96-well plate containing 50 μL of growth medium with 2x the desired drug concentration yielding (2 × 104 cells/well with varying compound concentrations). After an additional 44 h incubation, 50 μL of Bright-Glo reagent (Promega catalog no. E2610) was added directly on-top of cell media for a 5-min RT incubation. All reactions were transferred into a white flat bottom 96-well plate (Thermo Fisher Scientific catalog no. 165306) and luminescence was quantified by using a Tecan Spark plate reader (Tecan Life Sciences).
Calculations to Assess Repurchased Chemicals
To determine the percent inhibition of Mpro for a single compound concentration, we used raw luminescent values that have been normalized to DMSO low luminescent control to calculate the percentage of Mpro activity as described (33–36):
The mean low control is derived from transfected cells treated with DMSO, which yields the lowest raw luciferase signal (0% inhibition). Second, the normalized percentage of Mpro inhibition is calculated by subtracting percent activity (above) from 100:
Prior to dose response re-testing, all purchased compounds were tested at 20 μM in duplicate and considered inhibitory if 10% of the gain-of-signal activity exhibited by 20 μM of GC376 was reached (e.g., equal to or above 9.7% in Figure 4).
Figure 4. Rediscovery of calpain inhibitor II.
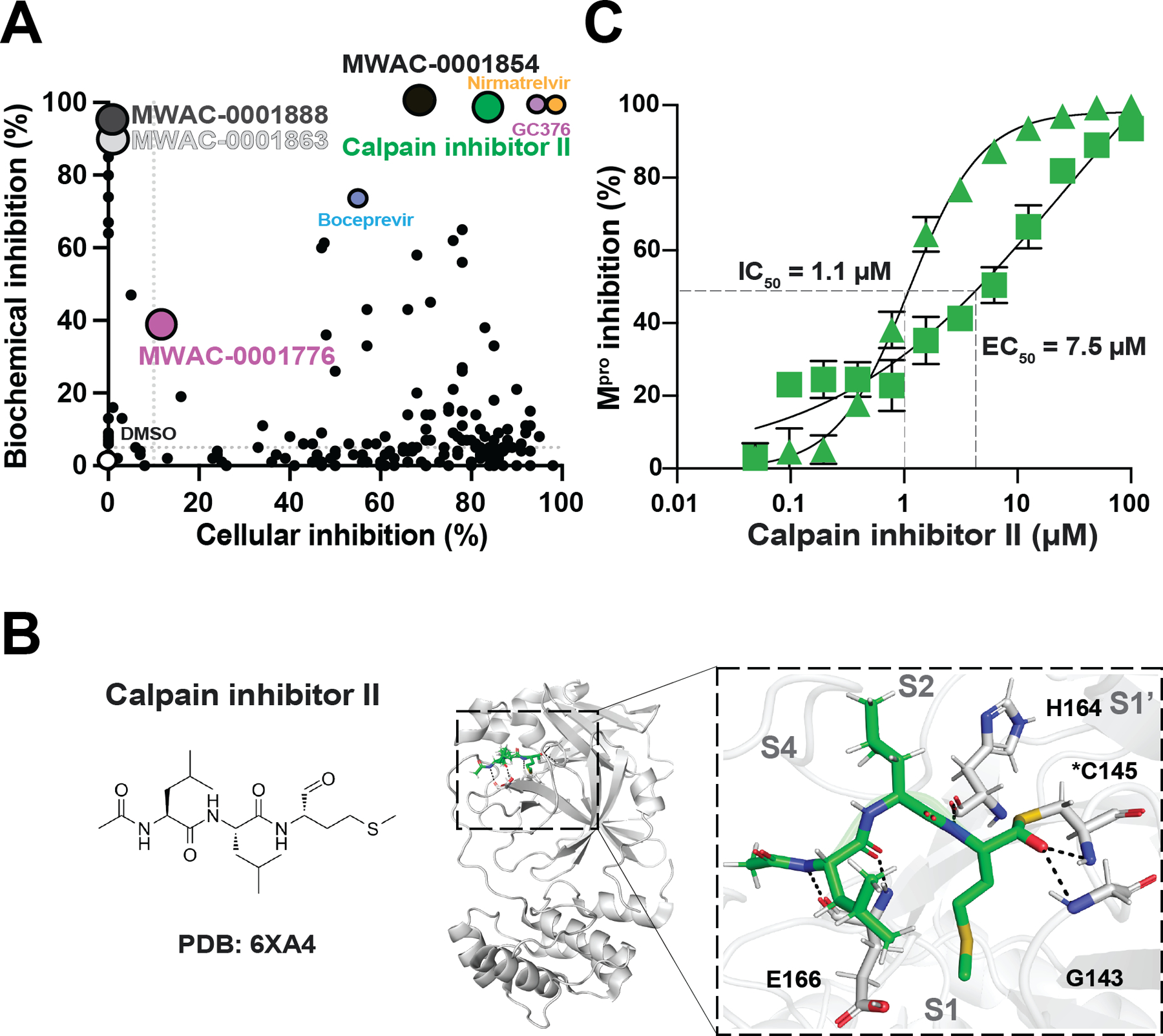
A) A dot plot comparing cell-based and biochemical SARS2 Mpro inhibition results for 20 μM of repurchased compounds. Labels are shown for compounds that were analyzed in detail. Dashed lines indicate significance cut-offs. See text for details.
(B) Ribbon schematic of the crystal structure of calpain inhibitor II in complex with Mpro (PDB 6XA4). The zoom-in (right) shows calpain inhibitor II positioned within the Mpro catalytic pocket. Black dashed lines represent hydrogen bonding.
(C) Representative dose responses with calpain inhibitor II using SARS2 Mpro cellular gain-of-signal (square points) and SARS2 Mpro biochemical proteolytic cleavage (triangle points) assays. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
Biochemical Mpro Activity Assays for Repurchased Chemicals
The proteolytic activity of SARS2 Mpro was analyzed using a quenched fluorescent peptide substrate DABCYL-KTSAVLQ|SGFRKM-EDANS (UPBio catalog no. V1010–1). Mpro cleavage between Q and S liberates fluorescence. Cleavage reactions were carried out in 50 μL reactions in Greiner 96-well chimney half-area plates (Greiner catalog no. 675076) with 5 μM substrate, 150 nM Mpro, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1mM EDTA, 0.05% Tween20, 0.1 mg/mL bovine serum albumin (BSA), 1 mM DTT. For inhibition studies, Mpro was incubated at room temperature with various concentrations of chemical (2-fold serial dilution series starting at 100 μM) for 30 m in reaction buffer containing BSA prior to addition of the substrate to initiate the reaction. Fluorescence intensity was measured once per minute using a Tecan Spark 10M plate reader (Ex. 360 nm / Em. 460 nm filter set). The final DMSO concentration per reaction well was 0.75%. Prior to dose response re-testing, all purchased compounds were tested at 20 μM in duplicate and considered inhibitory if 5% of the inhibition of the level of 20 μM of GC376 was reached (e.g., equal to or above 5% in Figure 4).
SARS2 Mpro Structures and Molecular Docking
The chemical structures of select compounds were obtained from PubChem, and ChemDraw was used for illustration (Supplementary Table S1). High-resolution x-ray structures of SARS2 Mpro with calpain inhibitor II and GC-14 were obtained from the Protein Data Bank (PDB 6XA4 and 8ACL, respectively). PDB 8ACL was also used to for molecular docking studies using Maestro (Schrödinger). The protein was prepared using the Protein Preparation Wizard using default settings with water molecules removed. MWAC-0001776 was sketched in the 2-D sketcher and loaded into the LigPrep tool using an ionization state at pH 7 ± 2 with specified chiralities retained. A docking grid was prepared using the centroid of the workspace ligand, with a hydrogen bond constraint placed at G143. Docking was performed using Glide SP, with constraints. The output of the docked ligand was displayed in the Maestro workspace and used for creating an illustration.
Results
Optimization of a Cell-Based Gain-of-Signal Assay for Mpro Inhibition
We recently reported a cell-based gain-of-signal assay based on the novel observation that wildtype SARS2 Mpro suppresses expression of a firefly luciferase (34) reporter gene in 293T cells (33–36). In this system, chemical inhibitors of Mpro proteolytic activity restore reporter gene expression and luminescent signal in a quantitative and dose-responsive manner (assay schematic in Figure 1A and a dose response of the broad-spectrum coronavirus Mpro inhibitor GC376 in Figure 1B). Owing to high sensitivity and a large signal-to-background ratio, the assay was miniaturized to 5 μl total volume and adapted to a 1536-well plate format for uHTS. Using 100 μM of the tool compound GC376 (27–33) as a positive control and DMSO as a negative control, the initial set-up signal-to-background ratio was 36 and the Z’ was 0.47 (Figure 1C).
Figure 1. Cell-based gain-of-signal assay for SARS2 Mpro inhibition.
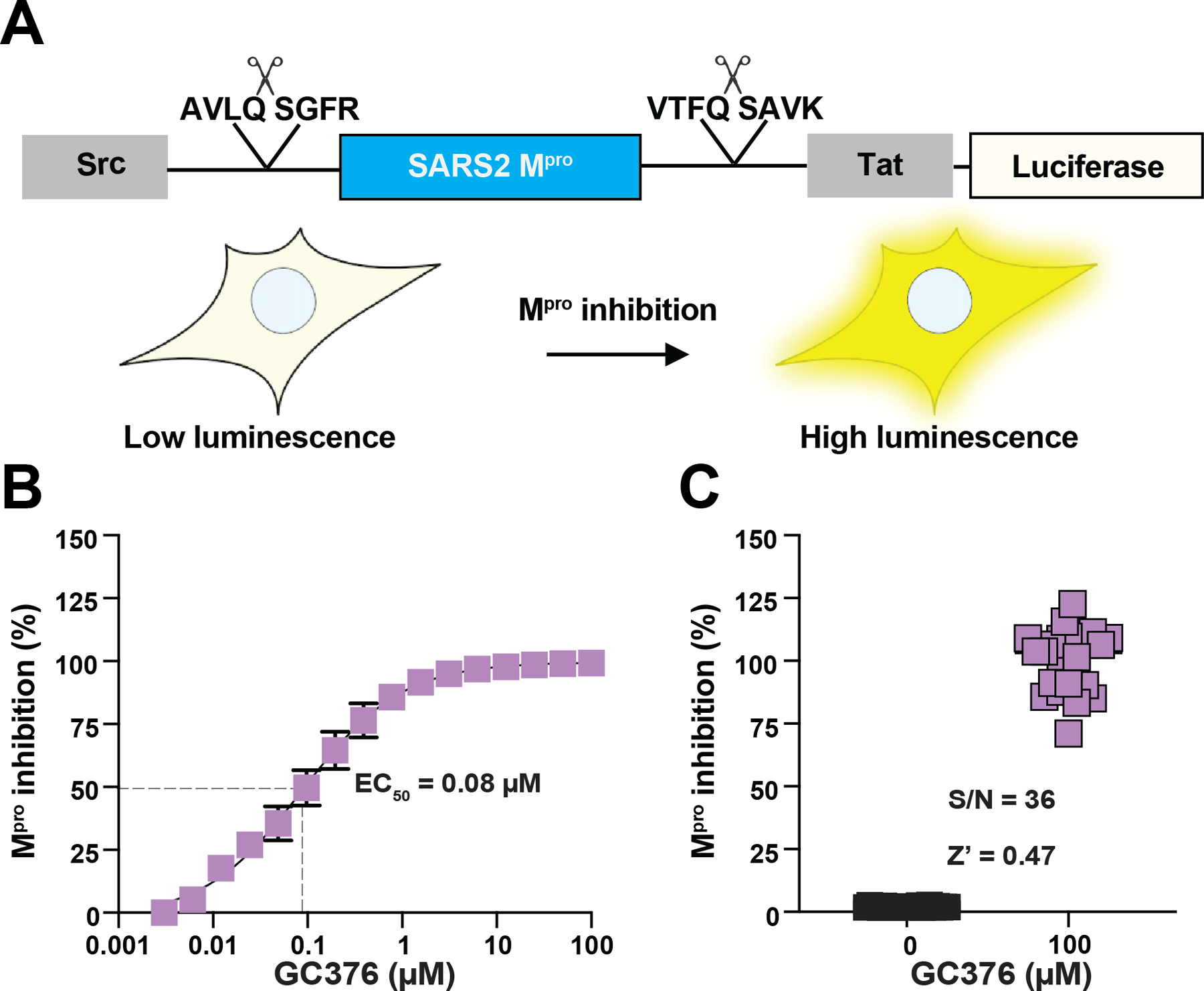
(A) Schematic of cellular gain-of-signal assay for SARS2 Mpro inhibition. See text for details.
(B) Representative dose response with GC376. Each data point is the average of two technical replicates, and the error bars report the difference between each replicate.
(C) Assay validation in 1536 well format by comparing gain-of-signal assay values for GC-376 and DMSO as a positive and negative controls, respectively (n = 24 for each condition).
Primary uHTS with Gain-of-Signal Assay for Mpro Inhibition
Primary uHTS was conducted using the 1536-well format SARS2 Mpro gain-of-signal assay and the UF-Scripps Drug Discovery Library (UF-SDDL), which is comprised of 649,568 compounds (52,53). This library is one of the largest in academia, and it is comprised of over 20 commercially sourced compound libraries, supplemented with multiple academically sourced compound series, including small molecules and sub-libraries prepared internally and, therefore, approximately 22,000 compounds in this collection are unique. In its current state, the UF-SDDL has several focused sub-libraries for screening popular drug-discovery target classes (e.g., kinases/transferases, GPCRs, ion channels, nuclear receptors, hydrolases, transporters) with diverse chemistries (e.g., click-chemistry, PAINS-free collections, Fsp3-enriched, covalent inhibitors, and natural product collections) and desirable physical properties (e.g., “rule-of-five”, “rule-of-three”, polar surface area, etc.).
Primary screening was conducted using ~10 μM of each small molecule in single point format, with 24 positive (GC376) and 24 negative (DMSO) wells on every 1536-well plate. The primary screen yielded good statistics, with an average Z’ value of 0.52 +/− 0.08 and a signal-to-background ratio of 32 +/− 7.6 over a total of 522 plates (uHTS composite dot plot in Figure 2). A hit cut-off was established as the average plus 3x SD of sample field activity (6.8% gain-of-signal relative to 100 μM GC376), resulting in a total of 8,777 hits and a final hit rate of 1.35%, in line with results from previous uHTS campaigns (54–56).
Figure 2. Primary uHTS results for SARS2 Mpro inhibition.
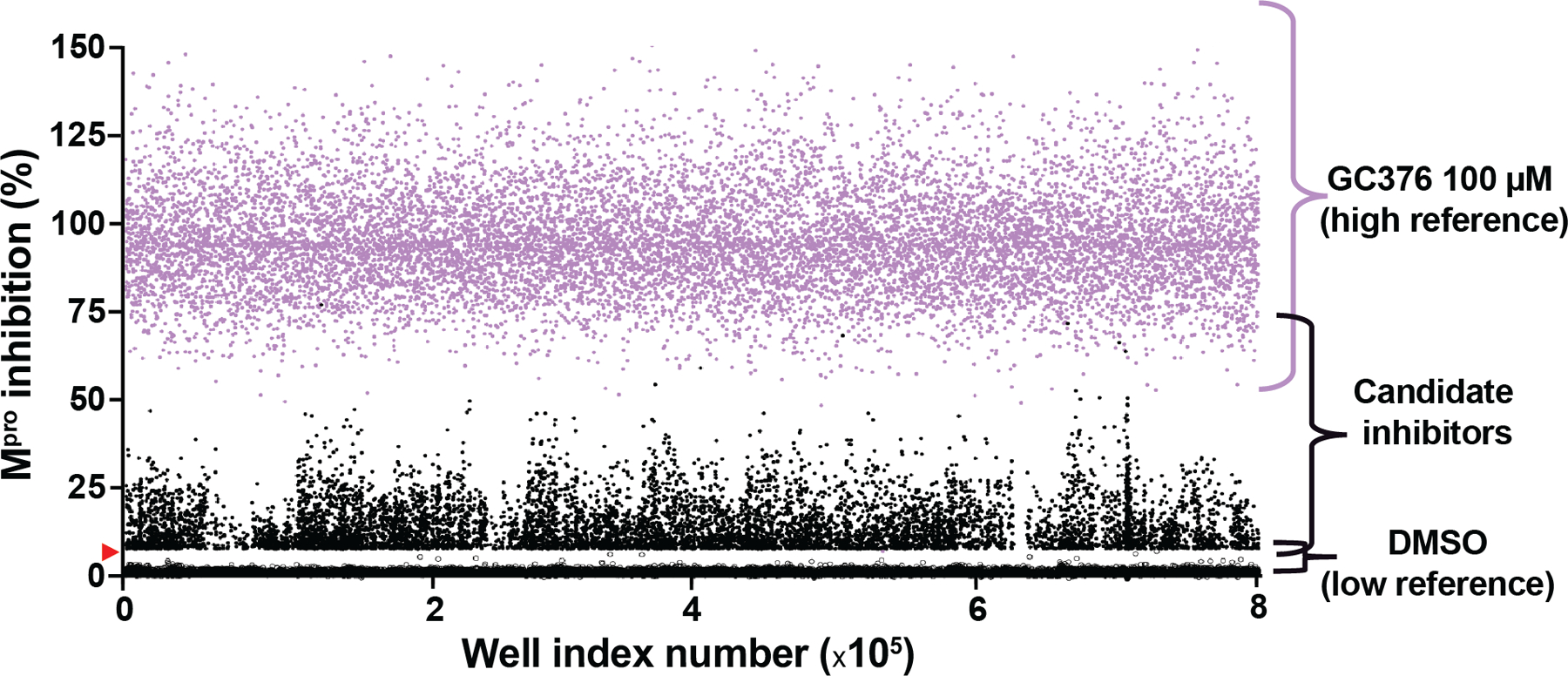
Data from each 1536 well screening plate are combined and represented as a single dot plot with DMSO values as low controls (open circles) and 100 μM GC376 values as high controls (lavender data points). Candidate inhibitors are represented by black data points with the vast majority falling below the 6.8% gain-of-signal cut-off (not shown to avoid blacking-out DMSO values).
Secondary Screens Using an Orthologous Cell-based Assay with HCoV-NL63 Mpro and a Biochemical Peptide-based Cleavage Assay with SARS2 Mpro
Primary screen hits were first re-tested in triplicate in 1536-well format with 10.9 μM of each of the 8,762 compounds (15 chemicals from the original 8,777 were unavailable), which resulted in confirmation of 40% of the initial uHTS-implicated small molecules as candidate Mpro inhibitors (n = 3,522/8,762; Figure 3A). To further increase the likelihood of discovering direct inhibitors of SARS2 Mpro activity, two secondary screens were performed. First, the available candidate SARS2 Mpro inhibitors (n = 8,762) were tested on the uHTS platform using an orthologous cell-based assay with a Human Coronavirus NL63 (HCoV-NL63) Mpro construct expressed in 293T cells (Src-NL63 Mpro-Tat-Luc assay schematic in Figure 3B). This construct is identical in amino acid sequence to the SARS2 Mpro cell-based construct, apart from the Mpro coding region (44% identity), and it was shown previously to suppress luciferase expression to a similar degree in 293T cells (33–36) (representative data with GC376 in Figure 3C). This secondary screen with the HCoV-NL63 construct tested the same 8,762 hits at 10.9 μM and yielded good statistics, with an average Z’ of 0.56 ± 0.05 over 27 plates. Interestingly, many of these compounds inhibited both SARS2 Mpro and NL63 Mpro (n = 3,328 in Figure 3A). This result was unexpected given 56% divergence between these proteins, and it suggested that these proteases may share at least one cellular target that, when engaged by compound, results in a restoration of luciferase expression. This unexpectedly large group of compounds will be considered in future studies dedicated to identifying the cellular target(s). However, 194 compounds still appeared to uniquely inhibit SARS2 Mpro through comparison of the results of these two gain-of-signal cellular assays (Figure 3A).
Figure 3. Secondary screens using an orthologous cell-based assay with HCoV-NL63 Mpro and a biochemical peptide-based cleavage assay with recombinant SARS2 Mpro.
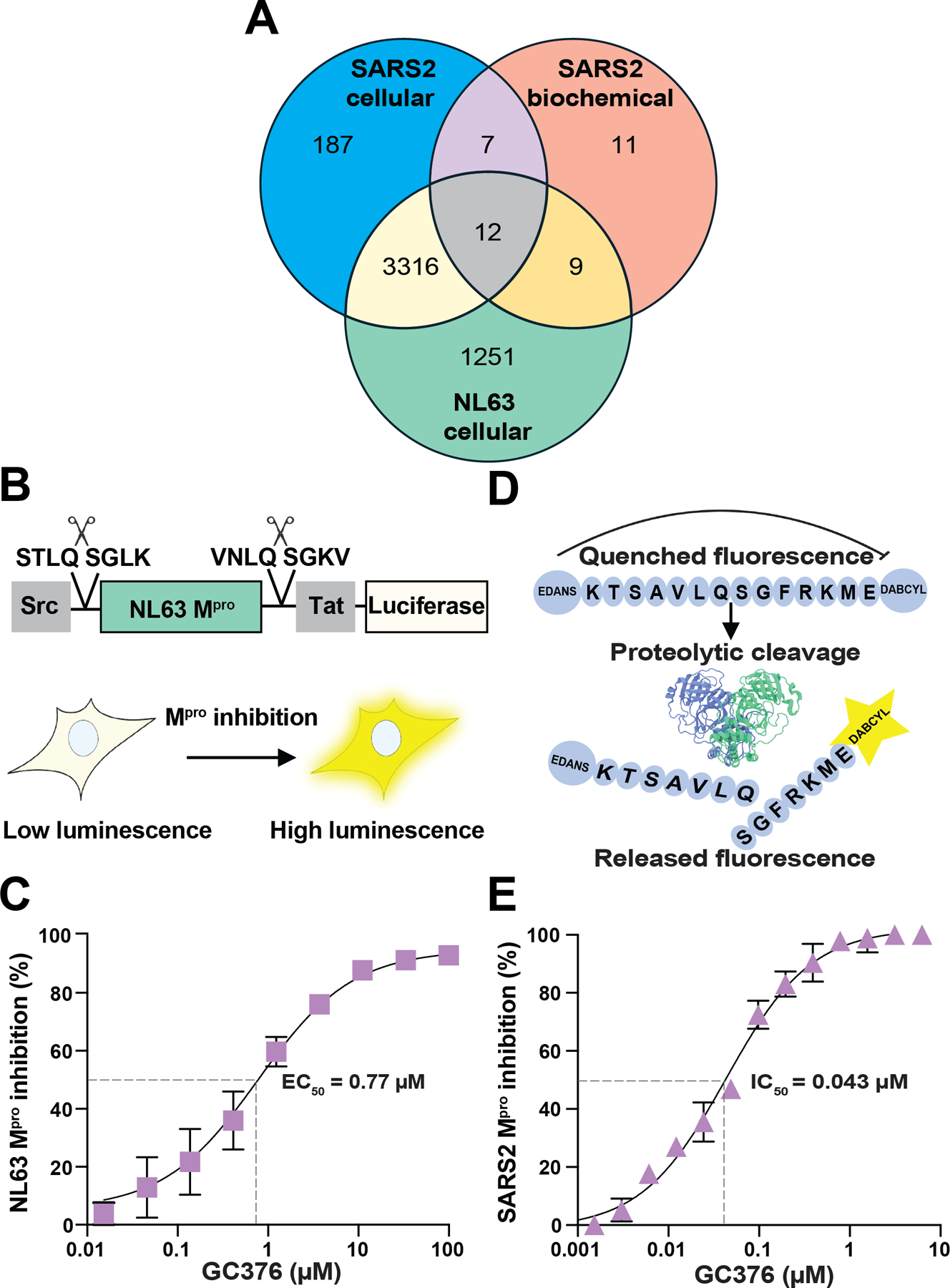
(A) Venn overlap of confirmed positive hits from SARS2 Mpro uHTS and secondary screens for inhibition in a cell-based NL63 Mpro gain-of signal assay and in a SARS2 Mpro biochemical proteolytic cleavage assay. See text for details.
(B) Schematic of cellular gain-of-signal assay for NL63 Mpro inhibition.
(C) Representative dose response with GC376 in cellular gain-of-signal assay for NL63 Mpro inhibition. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
(D) Schematic of the biochemical SARS2 Mpro peptide cleavage assay.
(E) Representative biochemical dose response with GC376. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
In parallel, a 1536-well format secondary screen was done with the 8,762 available candidate inhibitors using recombinant SARS2 Mpro in an established biochemical assay (30,35–37) (schematic in Figure 3D; see Methods for details). In this assay, limiting amounts of SARS2 Mpro (150 nM) are pre-incubated for 30 min with varying concentrations of candidate inhibitor, and then an excess concentration of peptide substrate (5 μM) is added to start the reaction with single hit cleavage kinetics (representative data with GC376 in Figure 3E). Proteolytic cleavage reactions were allowed to proceed at room temperature for 90 min and then data were collected using a plate reader, resulting in similarly high Z’ scores of 0.87 +/− 0.04 over 27 plates.
Interestingly, despite an excellent Z’ scores and a signal-to-background ratio of 6.4 +/− 0.14, only 39 candidate small molecules from the primary cell-based SARS2 uHTS tested positive in this secondary biochemical screen (Venn schematic in Figure 3A). This secondary biochemical screen was stringent in helping to identify direct-binding compounds, as only 19 out of 39 small molecules tested positive both in vitro using this assay and in living cells using the SARS2 Mpro gain-of-signal assay. Interestingly, 7 of these small molecules appeared specific to SARS2 Mpro and the other 12 also showed cross-inhibition of HCoV-NL63 Mpro in cells. Both specific and broader-spectrum inhibitors are of interest. Therefore, as an additional test for specificity, these candidate SARS2 Mpro inhibitors were tested against purified Zika virus NS2B-NS3 protease in a similar substrate cleavage assay (57). However, 18/19 compounds had no effect on NS2B-NS3 activity, and the outlier (MWAC-0001204) is likely a false positive hit that interferes with the fluorescent readout (Supplementary Table S1).
Dose Response Studies with Repurchased Compounds
The studies described above were all done with UF Scripps library compounds. To verify these results, all the SARS2 Mpro biochemical candidate inhibitors (n = 39), regardless of overlap with the two cell-based assays, together with all the SARS2 Mpro gain-of-signal candidate inhibitors (n = 187) were ordered from commercial vendors as powders and solubilized in 100% DMSO for testing. Unfortunately, several of these compounds were unavailable, but a total of 176 small molecules were obtained and tested against SARS2 Mpro in our biochemical and cellular gain-of-signal assays (see Methods for details; Supplementary Table S1).
First, these 176 compounds were tested at a single 20 μM concentration in duplicate and in parallel to various positive controls (Figure 4A and Supplementary Table S1). These experiments yielded a two-dimensional distribution of compound inhibitory activities with the majority showing strong inhibition in the cell-based assay (as identified originally) (Figure 4A). Importantly, half of the compounds tested positive in both assays (87/176; see Methods), alongside positive controls including the strong covalent inhibitor GC376 and the weak covalent inhibitor boceprevir (Figure 4A and Supplementary Table S1). Of note, we re-discovered calpain inhibitor II as a Mpro inhibitor with intermediate potency (29, 59, 61). Calpain inhibitor II is a covalent peptidomimetic compound bearing an aldehyde warhead that inhibits calpains and cathepsins, and it was shown to inhibit SARS2 Mpro with an IC50 of 970 nM in a biochemical assay (29,32,39,58,59). In addition, a SARS2 Mpro-calpain inhibitor II co-crystal structure revealed that the methionine side chain in the P1 position occupies the S1 subsite (29,32,39,58,59) (Figure 4B). Consistent with these results, our studies with re-purchased compound indicated that calpain inhibitor II has an IC50 of 1.1 μM in our biochemical assay and a dose-response EC50 of 7.5 μM in our cellular gain-of-signal assay (Figure 4C). This compound also showed no toxicity in 293T cells up to the highest tested concentration (100 μM). These results confirmed that calpain inhibitor II in indeed capable of SARS2 Mpro inhibition and further demonstrated the robustness and feasibility of our overall screening approach.
A New Mpro Inhibitor with Similarity to a Reported Small Molecule
An additional hit from our screening efforts was a non-covalent, disubstituted piperazine, MWAC-0001776, which shares chemical features with a reported compound called GC-14 (60–63) (MWAC-0001776 and GC-14 in Figure 5A–B, respectively). MWAC-0001776 inhibits Mpro with an IC50 of 17 μM in our biochemical assay and an EC50 of 6.8 μM in our gain-of-signal assay (Figure 5C–D). By comparison, GC-14 was reported to exhibit greater potency in a similar biochemical assay (IC50 = 0.40 μM) and show activity against SARS2 replication (EC50 = 1.1 μM) (60–61). Although GC-14 was not obtained for testing, an obvious difference between MWAC-0001776 and GC-14 is the addition of an amide-linked, 2-aminomethylthiophene on the piperazine of the former compound, which is predicted to occupy the S3/S4 subsite and make polar interactions between the carbonyl of the additional amide bond with the backbone amine of E166 of Mpro (Figure 5A). This additional ligand also helps explain why the reported biochemical potency of GC-14 is greater than our observed value for MWAC-0001776. The nicotinyl group that occupies the S1 subsite and the dichlorophenyl that rests in the hydrophobic S2 pocket are identical in the two compounds; this core chemotype may serve as a start point for additional modifications to improve potency.
Figure 5. A new Mpro inhibitor with similarity to a reported small molecule.
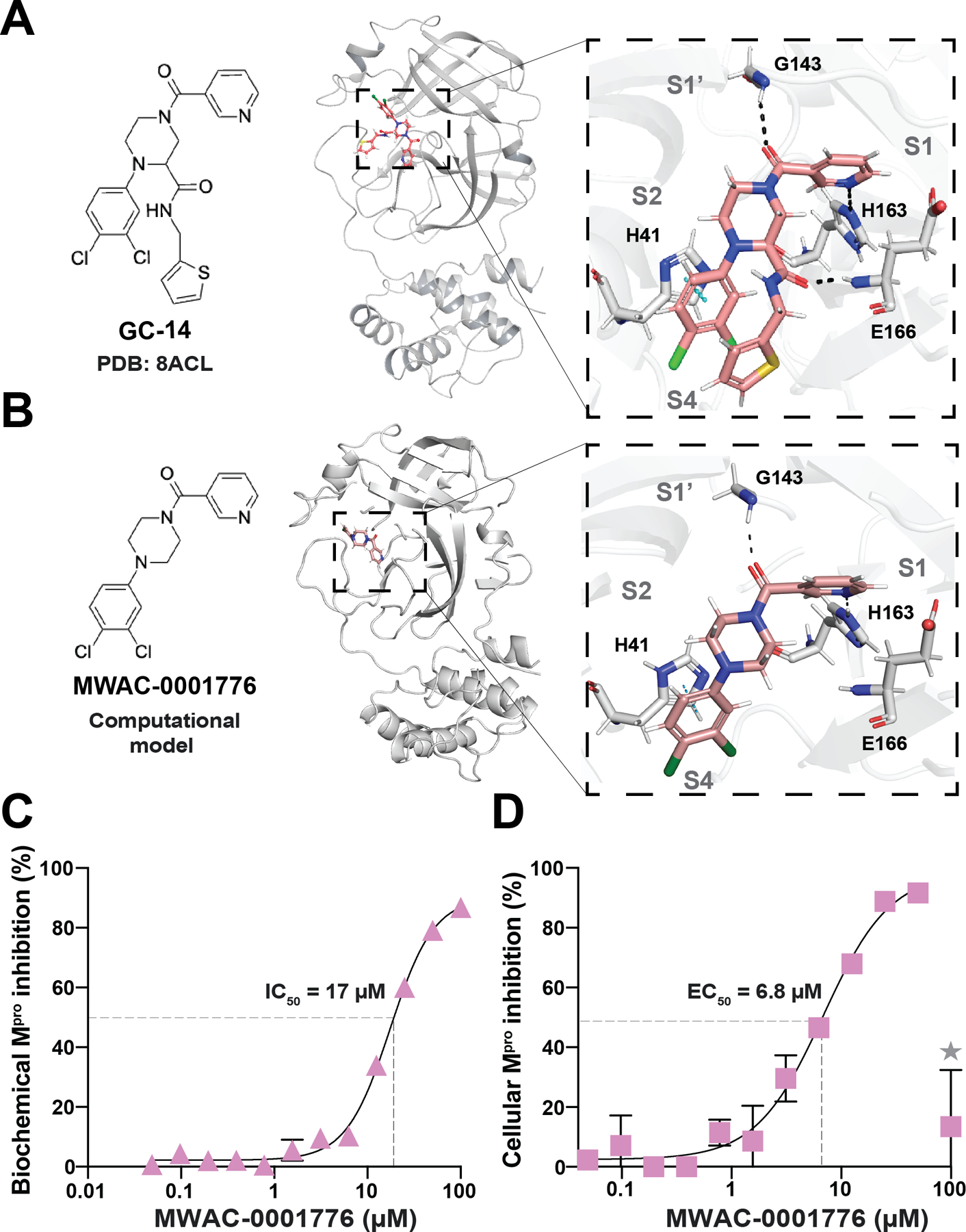
(A) Ribbon schematic of the crystal structure of GC-14 in complex with Mpro (PDB 8ACL). The zoom-in (right) shows GC-14 positioned within the Mpro catalytic pocket. Black dashed lines represent hydrogen bonding. Blue dashes represent pi-stacking.
(B) Chemical structure and computational model of the crystal structure of MWAC-0001776 in complex with Mpro model created using Maestro (Schrödinger). The zoom-in (right) shows MWAC-0001776 positioned within the Mpro catalytic pocket. Black dashed lines represent hydrogen bonding. Blue dashed represent pi-stacking.
(C) Representative dose response with MWAC-0001776 using SARS2 Mpro biochemical proteolytic cleavage assay. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
(D) Representative dose response with MWAC-0001776 using SARS2 Mpro cell-based gain-of-signal assay. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
Novel Hits Obtained Through uHTS
The largest group of chemically similar compounds among our uHTS hit candidates contained an electrophilic alpha-chloroketone warhead (e.g., Figure 6A). These compounds and others with the same electrophilic warhead showed a range of Mpro inhibition activity in both our biochemical and cell-based assays (compound information and single concentration results in Supplementary Table S2). Dose response testing was not done for all compounds in this series but select compounds, such as MWAC-0001888 and MWAC-0001863, showed reproducible biochemical IC50 and cellular EC50 values (Figure 6B).
Figure 6. Additional novel hits obtained through uHTS.
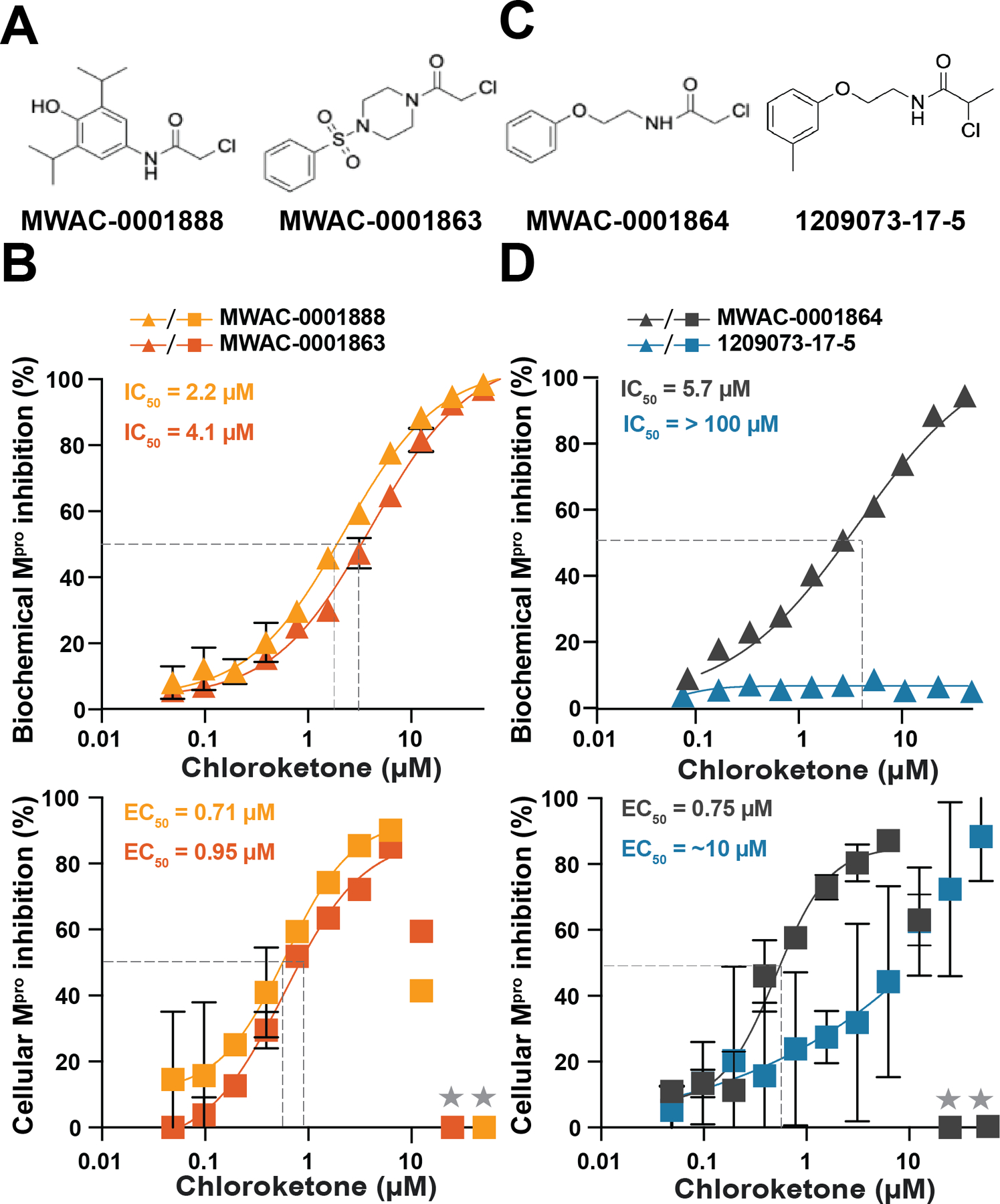
(A) Representative electrophilic α-chloroketone warhead compounds that tested positive in both SARS2 cell-based gain-of-signal and biochemical proteolytic cleavage assays.
(B) Representative dose response with MWAC-0001863, MWAC-0001854, and MWAC-0001888 using the SARS2 Mpro biochemical proteolytic cleavage assay and SARS2 Mpro cell-based gain-of-signal assay. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
(C) Representative electrophilic primary (left) and secondary (right) α-chloroketone compounds that were obtained for further characterization.
(D) Representative dose response with MWAC-000164 and 1209073-17-5 compounds using the SARS2 Mpro biochemical proteolytic cleavage assay and SARS2 cell-based gain-of-signal assay. Each data point is the average of two technical replicates, and the error bars show the difference between each replicate.
The enrichment of hits with a shared alpha-chloroketone electrophile suggested that covalent modification in the binding site, specifically with catalytic cysteine C145, likely plays a critical role in Mpro inhibition. To test this idea, we obtained a set of commercially available analogs with the alpha-chlorine removed, and all activity in the biochemical assay was abrogated (Supplementary Table S2). These did not warrant testing in cell-based studies.
We next tested a series of 15 commercially available compounds that shared an alpha-chloroketone moiety, and we found that the vast majority of these compounds retained Mpro inhibition activity (Supplementary Table S2). A key exception was a modified analog that replaced the primary chloride with a secondary chloride and consequently lost all activity (compare MWAC-0001864 and 1209073-17-5 in Figure 6C–D). To explain this, one would predict a difference in activity due to steric and electronic effects attributable to the addition of the methyl group, which makes the alpha-chloroketone a secondary alkylhalide.
Although the vast majority of the primary alpha-chloroketone-containing compound series showed Mpro inhibition, they also caused cytotoxicity at higher concentrations, which might be due to non-specific modification of host proteins and/or affecting cellular redox processes. This is evidenced in dose response curves by overt cell death and extinguished luminescence at higher compound concentrations (e.g., higher concentration data points to right of dotted line in Figure 6B). These results may be used in future studies to add specificity through further chemical modifications and/or endow a non-covalent scaffold with irreversible covalent adduction properties of the alpha-chloroketone group.
Discussion
Coronaviruses have caused three pandemics/endemics in the past 20 years, including SARS1, MERS, and SARS2/COVID-19. However, unlike SARS1 and MERS coronaviruses, which have dissipated naturally or remained restricted geographically, SARS2 has disseminated globally and is likely to continue circulating in humans with the continual emergence of new variants that may render current antiviral medicines less effective. Therefore, it is important to continue to develop and refine Mpro inhibitors until potent, long-lasting, orally available compounds are achieved. Here, we report the results of a cell-based ultra-high throughput screen and secondary screens that combined to rediscover known inhibitors and yield new chemical information. Notable small molecules include calpain inhibitor II, as reported (29,32,39,58,59) and MWAC-0001776, which shares core features with a compound called GC-14 (60–63). These two chemotypes are candidates for further development as coronavirus Mpro inhibitors.
The largest group of candidate SARS2 Mpro inhibitors shared an alpha-chloroketone motif. It is likely that the alpha-chloroketone electrophile inhibits Mpro by reacting covalently with the catalytic pocket cysteine, C145. Consistent with this predication, commercially obtained analogs that lacked the alpha-chloroketone group were no longer capable of Mpro inhibition.
We recognize that single point, IC50 measurements of covalent inhibitors are not generally accepted as rigorous measurements for covalent enzyme inhibition given the time- and concentration-dependent kinetics associated with covalent adduction. Measurements of kinact/Ki are generally required during ligand optimization studies. However, given that these hits are still early stage, assays to measure these kinetic parameters will be part of future studies with more potent analogs.
Most Mpro inhibitor screens to-date have leveraged biochemical or computational approaches as a first step. The uHTS campaign reported here is the first to our knowledge to use a cell-based gain-of-signal assay for primary HTS. Two advantages of this approach are the immediate identification of candidate small molecules that exert activity in cells and, importantly, are not cytotoxic (at the concentration screened). However, an unexpected drawback of this approach is evidenced by the relatively small number of primary screen hits that were shown to inhibit purified Mpro in a subsequent secondary screen (n = 39). Thus, the vast majority of primary screen hits appeared to be causing a gain-of-signal luminescent read-out without directly inhibiting SARS2 Mpro inside of cells. The fact that nearly all of these compounds (n = 3328) also caused a gain-of-signal in an orthologous NL-63 Mpro cellular assay strongly suggests shared cellular targets. This phenotype may be relevant to the biology of the coronavirus main protease enzyme, and it will be the subject of future mechanistic studies.
Supplementary Material
Acknowledgements
This work was supported by National Institute of Allergy and Infectious Disease grant U19-AI171954. MT is an inaugural scholar of the South Texas Undergraduate Research Opportunities Program (STUROP) at UT Health San Antonio. RSH is an Investigator of the Howard Hughes Medical Institute and the Ewing Halsell President’s Council Distinguished Chair at University of Texas Health San Antonio.
Footnotes
Competing Interests
The Mpro gain-of-signal system is the subject of U.S. Provisional Application Serial No. 63/108,611, filed on November 2, 2020, with RSH and SAM as inventors. The other authors declare that there are no additional competing interests.
References
- 1.Du P, Li D, Wang A, Shen S, Ma Z, and Li X (2021) A Systematic Review and Meta-Analysis of Risk Factors Associated with Severity and Death in COVID-19 Patients. Can J Infect Dis Med Microbiol 2021, 6660930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ioannidis JPA (2021) Over- and under-estimation of COVID-19 deaths. Eur J Epidemiol 36, 581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor L (2022) Covid-19: True global death toll from pandemic is almost 15 million, says WHO. BMJ 377, o1144. [DOI] [PubMed] [Google Scholar]
- 4.Kavanagh KT, Cormier LE, Pontus C, Bergman A, and Webley W (2024) Long COVID’s impact on patients, workers, & society: a review. Medicine (Baltimore) 103, e37502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koch CA (2024) Long covid: hormone imbalances and/or rather complex immune dysregulations? J Endocr Soc 8, bvae043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang SW, Kim JW, Kim JY, Lim SY, Jang CY, Chang E, Yang JS, Kim KC, Jang HC, Kim D, Shin Y, Lee JY, and Kim SH (2023) Characteristics and risk factors of prolonged viable virus shedding in immunocompromised patients with COVID-19: a prospective cohort study. J Infect 86, 412–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fung M, and Babik JM (2021) COVID-19 in immunocompromised hosts: what we know so far. Clin Infect Dis 72, 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng G, Zhou Q, Meng Y, Sun H, Du S, Liu Y, and Zeng F (2023) Risk and outcomes of breakthrough COVID-19 infections in vaccinated immunocompromised patients: A meta-analysis. MedComm (2020) 4, e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antinori A, and Bausch-Jurken M (2023) The burden of COVID-19 in the immunocompromised patient: implications for vaccination and needs for the future. J Infect Dis 228, S4–S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun CK, Lee WH, Yang MH, and Tsai TH (2024) Pharmacokinetic analysis of placental transfer of ritonavir as a component of paxlovid using microdialysis in pregnant rats. Heliyon 10, e24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spiliopoulou V, Ntanasis-Stathopoulos I, Malandrakis P, Gavriatopoulou M, Theodorakakou F, Fotiou D, Migkou M, Roussou M, Eleutherakis-Papaiakovou E, Kastritis E, Dimopoulos MA, and Terpos E (2023) Use of oral antivirals ritonavir-nirmatrelvir and molnupiravir in patients with multiple myeloma is associated with low rates of severe COVID-19: a single-Center, prospective study. Viruses 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li P, Huang L, Han R, Tang M, Fei G, Zeng D, and Wang R (2024) Safety and efficacy of Paxlovid in the treatment of adults with mild to moderate COVID-19 during the omicron epidemic: a multicentre study from China. Expert Rev Anti Infect Ther, 1–9 [DOI] [PubMed] [Google Scholar]
- 13.Bege M, and Borbas A (2024) The design, synthesis and mechanism of action of paxlovid, a protease inhibitor drug combination for the treatment of COVID-19. Pharmaceutics 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yotsuyanagi H, Ohmagari N, Doi Y, Yamato M, Bac NH, Cha BK, Imamura T, Sonoyama T, Ichihashi G, Sanaki T, Tsuge Y, Uehara T, and Mukae H (2024) Efficacy and safety of 5-day oral ensitrelvir for patients with mild to moderate COVID-19: the SCORPIO-SR randomized clinical trial. JAMA Netw Open 7, e2354991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, and Wang Z (2023) Bench-to-bedside: Innovation of small molecule anti-SARS-CoV-2 drugs in China. Eur J Med Chem 257, 115503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukae H, Yotsuyanagi H, Ohmagari N, Doi Y, Imamura T, Sonoyama T, Fukuhara T, Ichihashi G, Sanaki T, Baba K, Takeda Y, Tsuge Y, and Uehara T (2022) A Randomized Phase 2/3 Study of ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in japanese patients with mild-to-moderate COVID-19 or asymptomatic SARS-CoV-2 infection. Antimicrob Agents Chemother 66, e0069722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lan Q, Yan Y, Zhang G, Xia S, Zhou J, Lu L, and Jiang S (2024) Clinical development of antivirals against SARS-CoV-2 and its variants. Curr Res Microb Sci 6, 100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferraro S, Convertino I, Cappello E, Valdiserra G, Bonaso M, and Tuccori M (2024) Lessons learnt from the preclinical discovery and development of ensitrelvir as a COVID-19 therapeutic option. Expert Opin Drug Discov 19, 9–20 [DOI] [PubMed] [Google Scholar]
- 19.Bouzidi HS, Driouich JS, Klitting R, Bernadin O, Piorkowski G, Amaral R, Fraisse L, Mowbray CE, Scandale I, Escudie F, Chatelain E, de Lamballerie X, Nougairede A, and Touret F (2024) Generation and evaluation of protease inhibitor-resistant SARS-CoV-2 strains. Antiviral Res 222, 105814. [DOI] [PubMed] [Google Scholar]
- 20.Weber IT, Wang YF, and Harrison RW (2021) HIV protease: historical perspective and current research. Viruses 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthew AN, Leidner F, Lockbaum GJ, Henes M, Zephyr J, Hou S, Rao DN, Timm J, Rusere LN, Ragland DA, Paulsen JL, Prachanronarong K, Soumana DI, Nalivaika EA, Kurt Yilmaz N, Ali A, and Schiffer CA (2021) Drug design strategies to avoid resistance in direct-acting antivirals and beyond. Chem Rev 121, 3238–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Weber IT, and Mitsuya H (2022) Beyond darunavir: recent development of next generation HIV-1 protease inhibitors to combat drug resistance. Chem Commun (Camb) 58, 11762–11782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MNL, Nalivaika EA, Ozen A, Prabu-Jeyabalan MM, Thayer K, and Schiffer CA (2010) Molecular basis for drug resistance in HIV-1 protease. Viruses 2, 2509–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore KA, Leighton T, Ostrowsky JT, Anderson CJ, Danila RN, Ulrich AK, Lackritz EM, Mehr AJ, Baric RS, Baylor NW, Gellin BG, Gordon JL, Krammer F, Perlman S, Rees HV, Saville M, Weller CL, Osterholm MT, Coronavirus Vaccines R, and Taskforce DR (2023) A research and development (R&D) roadmap for broadly protective coronavirus vaccines: A pandemic preparedness strategy. Vaccine 41, 2101–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li G, Hilgenfeld R, Whitley R, and De Clercq E (2023) Therapeutic strategies for COVID-19: progress and lessons learned. Nat Rev Drug Discov 22, 449–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cankat S, Demael MU, and Swadling L (2024) In search of a pan-coronavirus vaccine: next-generation vaccine design and immune mechanisms. Cell Mol Immunol 21, 103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daniel J, Wing-Ho Chu A, Chan WM, Cheuk-Ying Leung R, Umer Abdullah SM, Sun Y, and Kai-Wang To K (2023) Global prevalence of SARS-CoV-2 3CL protease mutations associated with nirmatrelvir or ensitrelvir resistance. EBioMedicine 91, 104559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung HC, Ke YY, Huang SY, Huang PN, Kung YA, Chang TY, Yen KJ, Peng TT, Chang SE, Huang CT, Tsai YR, Wu SH, Lee SJ, Lin JH, Liu BS, Sung WC, Shih SR, Chen CT, and Hsu JT (2020) Discovery of M protease inhibitors encoded by SARS-CoV-2. Antimicrob Agents Chemother 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, and Wang J (2020) Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 30, 678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moghadasi SA, Esler MA, Otsuka Y, Becker JT, Moraes SN, Anderson CB, Chamakuri S, Belica C, Wick C, Harki DA, Young DW, Scampavia L, Spicer TP, Shi K, Aihara H, Brown WL, and Harris RS (2022) Gain-of-Signal Assays for Probing Inhibition of SARS-CoV-2 M(pro)/3CL(pro) in Living Cells. mBio 13, e0078422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y, Shuai L, Wen Z, Wang C, Yan Y, Jiao Z, Guo F, Fu ZF, Chen H, Bu Z, and Peng G (2021) The preclinical inhibitor GS441524 in combination with GC376 efficaciously inhibited the proliferation of SARS-CoV-2 in the mouse respiratory tract. Emerg Microbes Infect 10, 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan B, Joyce R, Tan H, Hu Y, and Wang J (2023) SARS-CoV-2 main protease drug design, assay development, and drug resistance studies. Acc Chem Res 56, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vuong W, Fischer C, Khan MB, van Belkum MJ, Lamer T, Willoughby KD, Lu J, Arutyunova E, Joyce MA, Saffran HA, Shields JA, Young HS, Nieman JA, Tyrrell DL, Lemieux MJ, and Vederas JC (2021) Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: Structural enhancements, increased solubility, and micellar studies. Eur J Med Chem 222, 113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan Y, Zhou H, Liu X, Iketani S, Lin M, Zhang X, Bian Q, Wang H, Sun H, Hong SJ, Culbertson B, Mohri H, Luck MI, Zhu Y, Liu X, Lu Y, Yang X, Yang K, Sabo Y, Chavez A, Goff SP, Rao Z, Ho DD, and Yang H (2023) Molecular mechanisms of SARS-CoV-2 resistance to nirmatrelvir. Nature 622, 376–382 [DOI] [PubMed] [Google Scholar]
- 35.Kuo CJ, Chi YH, Hsu JT, and Liang PH (2004) Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem Biophys Res Commun 318, 862–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen TT, Woo HJ, Kang HK, Nguyen VD, Kim YM, Kim DW, Ahn SA, Xia Y, and Kim D (2012) Flavonoid-mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnol Lett 34, 831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Lin D, Kusov Y, Nian Y, Ma Q, Wang J, von Brunn A, Leyssen P, Lanko K, Neyts J, de Wilde A, Snijder EJ, Liu H, and Hilgenfeld R (2020) α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J Med Chem 63, 4562–4578 [DOI] [PubMed] [Google Scholar]
- 38.Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, Song H, Shi Y, Li X, Tan W, Qi J, and Gao GF (2020) Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nature Communications 11, 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura N, Sacco MD, Ma C, Hu Y, Townsend JA, Meng X, Zhang F, Zhang X, Ba M, Szeto T, Kukuljac A, Marty MT, Schultz D, Cherry S, Xiang Y, Chen Y, and Wang J (2022) Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J Med Chem 65, 2848–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J, Chen SA, Khan MB, Brassard R, Arutyunova E, Lamer T, Vuong W, Fischer C, Young HS, Vederas JC, and Lemieux MJ (2022) Crystallization of feline coronavirus Mpro With GC376 reveals mechanism of inhibition. Front Chem 10, 852210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y, Lovell S, Tiew KC, Mandadapu SR, Alliston KR, Battaile KP, Groutas WC, and Chang KO (2012) Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol 86, 11754–11762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi D, Kim Y, Lovell S, Prakash O, Groutas WC, and Chang KO (2013) Structural and inhibitor studies of norovirus 3C-like proteases. Virus Res 178, 437–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noske GD, de Souza Silva E, de Godoy MO, Dolci I, Fernandes RS, Guido RVC, Sjo P, Oliva G, and Godoy AS (2023) Structural basis of nirmatrelvir and ensitrelvir activity against naturally occurring polymorphisms of the SARS-CoV-2 main protease. J Biol Chem 299, 103004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moghadasi SA, Heilmann E, Khalil AM, Nnabuife C, Kearns FL, Ye C, Moraes SN, Costacurta F, Esler MA, Aihara H, von Laer D, Martinez-Sobrido L, Palzkill T, Amaro RE, and Harris RS (2023) Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. Sci Adv 9, eade8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moghadasi SA, Biswas RG, Harki DA, and Harris RS (2023) Rapid resistance profiling of SARS-CoV-2 protease inhibitors. npj Antimicrobials and Resistance 1, 9 [Google Scholar]
- 46.Hu Y, Lewandowski EM, Tan H, Zhang X, Morgan RT, Zhang X, Jacobs LMC, Butler SG, Gongora MV, Choy J, Deng X, Chen Y, and Wang J (2023) Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Cent Sci 9, 1658–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Havranek B, Demissie R, Lee H, Lan S, Zhang H, Sarafianos S, Ayitou AJ, and Islam SM (2023) Discovery of nirmatrelvir resistance mutations in SARS-CoV-2 3CLpro: a computational-experimental approach. J Chem Inf Model 63, 7180–7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colson P, Delerce J, Pontarotti P, Devaux C, La Scola B, Fantini J, and Raoult D (2024) Resistance-associated mutations to the anti-SARS-CoV-2 agent nirmatrelvir: Selection not induction. J Med Virol 96, e29462. [DOI] [PubMed] [Google Scholar]
- 49.Amani B, and Amani B (2023) Efficacy and safety of nirmatrelvir/ritonavir (Paxlovid) for COVID-19: A rapid review and meta-analysis. Journal of Medical Virology 95, e28441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kneller DW, Li H, Phillips G, Weiss KL, Zhang Q, Arnould MA, Jonsson CB, Surendranathan S, Parvathareddy J, Blakeley MP, Coates L, Louis JM, Bonnesen PV, and Kovalevsky A (2022) Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nature Communications 13, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia Z, Sacco M, Hu Y, Ma C, Meng X, Zhang F, Szeto T, Xiang Y, Chen Y, and Wang J (2021) Rational Design of Hybrid SARS-CoV-2 Main Protease Inhibitors Guided by the Superimposed Cocrystal Structures with the Peptidomimetic Inhibitors GC-376, Telaprevir, and Boceprevir. ACS Pharmacology & Translational Science 4, 1408–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chulkov EG, Smith E, Rohr CM, Yahya NA, Park SK, Scampavia L, Spicer TP, and Marchant JS (2021) Identification of novel modulators of a schistosome transient receptor potential channel targeted by praziquantel. PLoS Negl Trop Dis 15, e0009898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baillargeon P, Fernandez-Vega V, Sridharan BP, Brown S, Griffin PR, Rosen H, Cravatt B, Scampavia L, and Spicer TP (2019) The Scripps Molecular Screening Center and Translational Research Institute. SLAS Discov 24, 386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith E, Davis-Gardner ME, Garcia-Ordonez RD, Nguyen TT, Hull M, Chen E, Yu X, Bannister TD, Baillargeon P, Scampavia L, Griffin P, Farzan M, and Spicer TP (2023) High throughput screening for drugs that inhibit 3C-like protease in SARS-CoV-2. SLAS Discov 28, 95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith E, Davis-Gardner ME, Garcia-Ordonez RD, Nguyen TT, Hull M, Chen E, Baillargeon P, Scampavia L, Strutzenberg T, Griffin PR, Farzan M, and Spicer TP (2020) High-Throughput Screening for Drugs That Inhibit Papain-Like Protease in SARS-CoV-2. SLAS Discov 25, 1152–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Otsuka Y, Airola MV, Choi YM, Coant N, Snider J, Cariello C, Saied EM, Arenz C, Bannister T, Rahaim R Jr., Hannun YA, Shumate J, Scampavia L, Haley JD, and Spicer TP (2021) Identification of Small-Molecule Inhibitors of Neutral Ceramidase (nCDase) via Target-Based High-Throughput Screening. SLAS Discov 26, 113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anindita PD, Otsuka Y, Lattmann S, Ngo KH, Liew CW, Kang SW, Harris RS, Scampavia L, Spicer TP, and Luo D (2024) A high throughput cell-based screening method for Zika virus protease inhibitors discovery. SLAS Discov [DOI] [PubMed] [Google Scholar]
- 58.Sacco MD, Ma C, Lagarias P, Gao A, Townsend JA, Meng X, Dube P, Zhang X, Hu Y, Kitamura N, Hurst B, Tarbet B, Marty MT, Kolocouris A, Xiang Y, Chen Y, and Wang J (2020) Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci Adv 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abhithaj J, Francis D, C SS, K GA, C S, and Variyar EJ (2022) Repurposing simeprevir, calpain inhibitor IV and a cathepsin F inhibitor against SARS-CoV-2 and insights into their interactions with Mpro. J Biomol Struct Dyn 40, 325–336 [DOI] [PubMed] [Google Scholar]
- 60.Gao S, Song L, Sylvester K, Mercorelli B, Loregian A, Toth K, Weisse RH, Useini A, Strater N, Yang M, Ye B, Tollefson AE, Muller CE, Liu X, and Zhan P (2023) Design, synthesis, and biological evaluation of trisubstituted piperazine derivatives as noncovalent severe acute respiratory syndrome coronavirus 2 main protease inhibitors with improved antiviral activity and favorable druggability. J Med Chem 66, 16426–16440 [DOI] [PubMed] [Google Scholar]
- 61.Gao S, Sylvester K, Song L, Claff T, Jing L, Woodson M, Weiße RH, Cheng Y, Schäkel L, Petry M, Gütschow M, Schiedel AC, Sträter N, Kang D, Xu S, Toth K, Tavis J, Tollefson AE, Müller CE, Liu X, and Zhan P (2022) Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high harget specificity and low toxicity. J Med Chem 65, 13343–13364 [DOI] [PubMed] [Google Scholar]
- 62.Kronenberger T, Laufer SA, and Pillaiyar T (2023) COVID-19 therapeutics: Small-molecule drug development targeting SARS-CoV-2 main protease. Drug Discov Today 28, 103579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou K, and Chen D (2023) Conventional understanding of SARS-CoV-2 Mpro and common strategies for developing its inhibitors. Chembiochem 24, e202300301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.