

Abstract
Viral proteins that evade the host’s innate immune response play a crucial role in pathogenesis, significantly impacting viral infections and potential therapeutic strategies. Identifying these proteins through traditional methods is challenging and time-consuming due to the complexity of virus-host interactions. Leveraging advancements in computational biology, we present VirusHound-II, a novel tool that utilizes machine learning techniques to predict viral proteins evading the innate immune response with high accuracy. We evaluated a comprehensive range of machine learning models, including ensemble methods, neural networks, and support vector machines. Using a dataset of 1337 viral proteins known to evade the innate immune response (VPEINRs) and an equal number of non-VPEINRs, we employed pseudo amino acid composition as the molecular descriptor. Our methodology involved a tenfold cross-validation strategy on 80% of the data for training, followed by testing on an independent dataset comprising the remaining 20%. The random forest model demonstrated superior performance metrics, achieving 0.9290 accuracy, 0.9283 F1 score, 0.9354 precision, and 0.9213 sensitivity in the independent testing phase. These results establish VirusHound-II as an advancement in computational virology, accessible via a user-friendly web application. We anticipate that VirusHound-II will be a crucial resource for researchers, enabling the rapid and reliable prediction of viral proteins evading the innate immune response. This tool has the potential to accelerate the identification of therapeutic targets and enhance our understanding of viral evasion mechanisms, contributing to the development of more effective antiviral strategies and advancing our knowledge of virus-host interactions.
Keywords: Virus, Machine learning, Deep learning, Protein, Immune system
Introduction
Through millions of years of coevolution with their hosts, viruses have become experts in manipulating the immune system. This has driven viruses to develop an arsenal of tactics to evade immune responses [1–6]. The host innate immune response is the first line of defense against pathogens, aiming to prevent infection and combat invading microorganisms [7, 8]. In the context of viral infections, the host innate immune system is tasked with serving as the foremost protective barrier, striving to impede viral invasion or replication before the development of more targeted defenses by the adaptive immune system [9].
The innate immune system employs numerous proteins known as pattern recognition receptors (PRRs) to detect unique molecular structures specific to pathogens, termed pathogen-associated molecular patterns (PAMPs), found in viruses and bacteria. The recognition of PAMPs by PRRs initiates a sophisticated response from the innate immune system that inhibits viral protein synthesis and ultimately viral replication, in addition to releasing signaling molecules to induce an immune response in neighboring cells, preparing them to suppress the spread of the infection [10, 11]. PAMPs are conserved molecular motifs essential for the pathogen’s survival [12, 13] and are found in their glycoproteins, lipopolysaccharides, proteoglycans, and nucleic acids [14, 15]. PRRs consist of various protein families distinguished by ligand specificity, cellular localization, and the activation of specific signaling pathways that lead to distinct anti-pathogen responses [16]. One of the main roles of many PRRs is to induce the production of type I interferons in response to viral infections. The expression of type I interferons triggers signaling pathways that activate the transcription of a diverse set of proteins that establish an antiviral response in target cells and act as effector proteins with direct antiviral activity [11, 17, 18]. Human cells express PRR proteins that specifically interact with viral PAMPs (including dsRNA and proteins) and trigger the immune response [19–21].
Viruses have developed various strategies to evade the host innate immune response. These strategies include inhibiting the host signaling pathways that induce the innate antiviral immune response, such as preventing activation of host innate immune system, cleaving host innate immune proteins, inducing mitophagy to limit interferon induction, inhibiting transcription factors, and targeting antiviral proteins [22–25]. Viral proteins targeting host proteins to evade an innate immune response is a critical area of study in virology and immunology, offering insights into how viruses persist in the host and how we might develop therapeutic interventions [26, 27]. By targeting viral proteins involved in immune evasion, new therapeutics can be developed to restore the host’s innate immune response, offering a more effective treatment strategy against viral infections [28]. On the other hand, identifying host proteins targeted by viral evasion mechanisms can lead to the discovery of biomarkers for early detection of viral infections or for monitoring the efficacy of antiviral therapies [29].
Machine learning plays a crucial role in studying the host’s innate immune response. Recent studies have used machine learning classifiers to distinguish viral and non-viral acute infections based on host immune response mRNAs [30]. Additionally, machine learning of flow cytometry data has been employed to understand host immune responses to SARS-CoV-2 infection [31]. In the realm of artificial immune systems, the study of innate immune-based algorithms, such as the dendritic cell algorithm and toll-like receptor algorithm, has been explored to enhance self-adaptation and self-learning in computational intelligence [32].
Currently, there is no tool for predicting virus proteins that evade the host’s innate immune response. This work aims to develop a first-of-its-kind robust predictive model and application to identify such pathogenic virus proteins efficiently and reliably. In our previous work, we developed VirusHound-I, a tool for predicting viral proteins that evade the adaptive immune response [6]. The present study introduces VirusHound-II, which significantly distinguishes itself from its predecessor by focusing on the prediction of proteins that evade the innate immune response, a fundamentally different problem given the distinction in evasion mechanisms between these two types of immune responses. VirusHound-II represents a significant innovation in the field of computational virology, being the first tool specifically designed to predict viral proteins that evade the host’s innate immune response. This specificity is crucial, as the mechanisms for evading the innate immune response are fundamentally distinct from those affecting the adaptive immune response [3, 33].
Methods
Data sets
Amino acid sequences of pathogenic virus proteins that evade the host’s immune response were identified and downloaded from the UniProt database [34]. After a careful manual review of each of the sequences, only those that met the following two criteria were selected: (1) Amino acid sequences with revised notation (2) and scientific literature support demonstrating the functionality of these proteins. After manual curation, the sequences were separated into different groups according to their type as follows: a dataset comprised of 1337 corresponding to pathogenic virus proteins that evade the host’s innate immune response (VPEINRs) and 1337 virus proteins without this activity (Non-VPEINRs). All sequences used are available for the reproducibility of this work in the code repository https://github.com/jfbldevs/virushound-II.
Molecular descriptor computation
For all the amino acid sequences in this study, the molecular descriptors, pseudo amino acid composition (PAAC) and dipeptide composition (DPC) were calculated. This approach allows representing the sequences through a set of features that incorporate both the information on the amino acid composition and the relevant physicochemical properties, as well as the sequential distribution of these components. Calculations were carried out with the Python 3.11 programming language (https://www.python.org/) and the propy3 package (https://propy3.readthedocs.io/).
Training, cross-validation, and testing
Based on the determined molecular descriptors (PAAC and DPC), datasets were created for each, labeling the molecular descriptors with binary notation: “1” for peptides with activity (VPEINRs = 1337) and “0” for those without activity (non-VPEINRs = 1337). These datasets were divided into proportions of 80% and 20% to form the training and test (independent) sets, respectively. A tenfold stratified cross-validation was implemented on the training set. Various machine learning algorithms were evaluated, including Random Forest (RF), Extra Trees (ET), Light Gradient Boosting Machine (LGBM), Linear Discriminant Analysis (LDA), Extreme Gradient Boosting (XGBOOST), Multi-Layer Perceptron (MLP), Support Vector Machine with radial (SVM-RK) and linear (SVM-LK) kernels, Gaussian Process Classifier (GPC), Gradient Boosting Classifier (GBC), K-Nearest Neighbors (KNN), AdaBoost Classifier (ABC), Decision Tree (DT), Quadratic Discriminant Analysis (QDA), Naive Bayes (NB), Ridge Classifier (RC), Dummy Classifier (DC), and Logistic Regression (LR). The performance of these models was monitored both in the training stage with cross-validation and in the evaluation on the independent test set, using various performance metrics mentioned bellow. To carry out all the aforementioned analyses, the popular automated machine learning library called PyCaret (https://pycaret.org/) was used (Fig. 1).
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
Fig. 1.
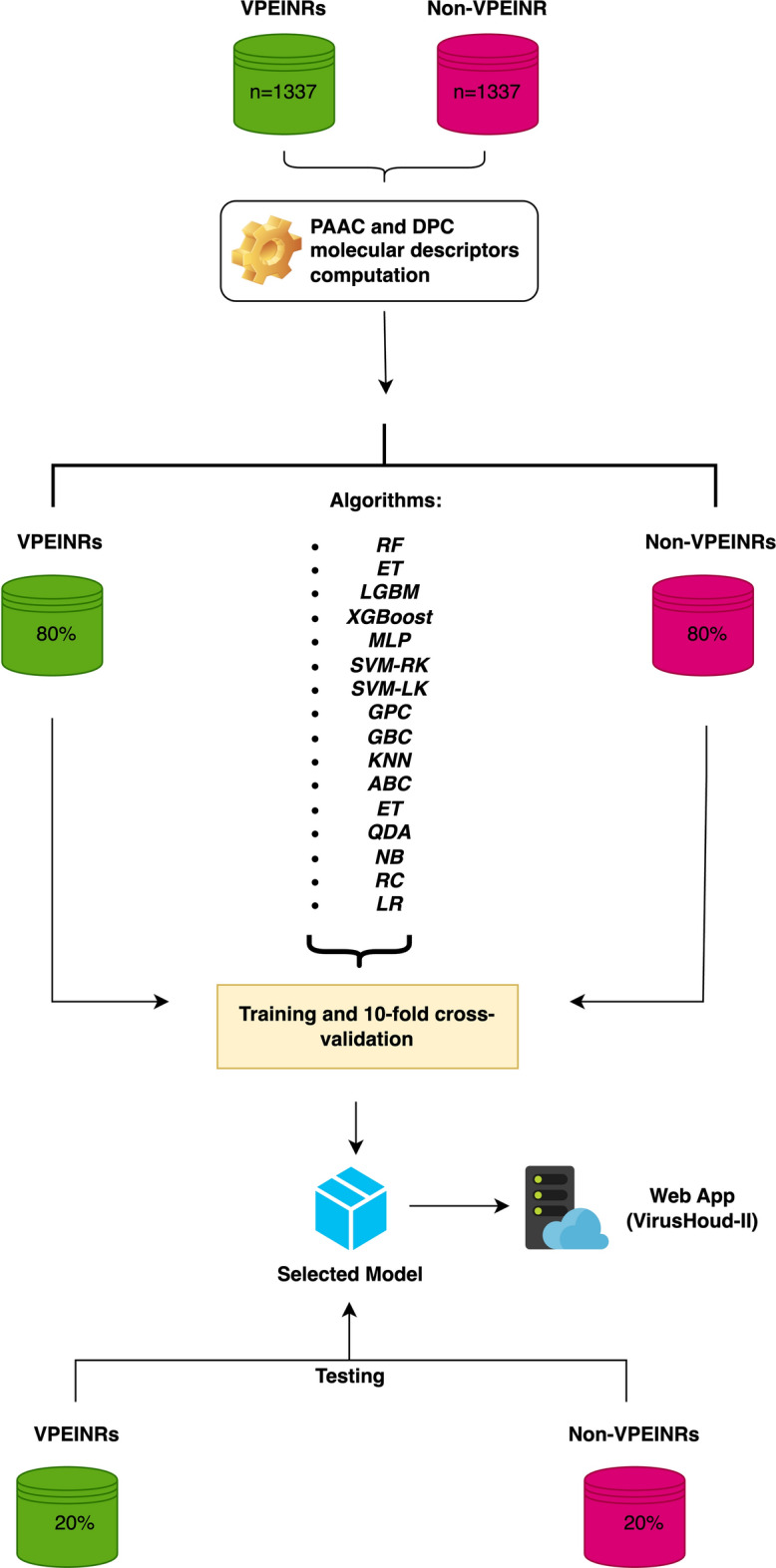
General workflow used in this study. Starting from sequences of real VPEINRs and non-VPEINRs, molecular descriptors (PAAC and DPC) were computed. The obtained descriptor values were used to create datasets with binary labels (1 for VPEINRs, 0 for non-VPEINRs). These datasets were split into 80% training and 20% testing sets. Various machine learning algorithms, including RF, ET, LGBM, XGBoost, MLP, SVM (radial and linear kernels), GPC, GBC, KNN, ABC, DTC, QDA, NB, RC, and LR, were evaluated using tenfold stratified cross-validation on the training set. The models were then assessed on the independent test set. Performance was monitored using multiple metrics including accuracy, precision, recall, F1 score, kappa, and MCC. Based on these performance measures, the best predictive model was selected for incorporation into a web application developed in Python 3.11
Finally, a web application called VirusHound-II was developed in Python 3.11 (https://www.python.org/) with a modern and intuitive interface to carry out predictions on pathogenic virus proteins. VirusHound-II not only determines whether a specific viral protein exhibits the activity in question but also assigns a probabilistic value ranging from zero to one, based on its capacity to evade the host's innate immune response. Viral proteins with a score higher than 0.5 are considered to have a high probability of evading such immune response. VirusHound-II is freely available online, with its official site located at https://www.biochemintelli.com/VirusHound-II. It can also be accessed at https://biochemintelli.streamlit.app/VirusHound-II.
Results
The performance of various ML algorithms in predicting viral proteins that evade the innate immune response (VPEINRs) was evaluated using two molecular descriptors: PAAC and DPC. Models were assessed through tenfold cross-validation on the training set, followed by testing on an independent dataset.
Overall, PAAC-based models outperformed DPC-based models, with the best-performing algorithms achieving high metrics across both training and testing phases. Tables 1 and 2 present the results for PAAC-based models during cross-validation and testing, respectively, while Tables 3 and 4 show the corresponding results for DPC-based models. For PAAC-based models, the ET classifier demonstrated the best performance during cross-validation, achieving an ACC of 0.9322, SEN of 0.9206, PREC of 0.9434, and F1 of 0.9314 (Table 1). Interestingly, during testing, the RF classifier slightly outperformed ET, with an ACC of 0.9290, SEN of 0.9213, PREC of 0.9354, and F1 of 0.9283 (Table 2). The SVM-RK and LGBM also showed strong performance, consistently achieving ACC, PREC, and F1 scores above 0.92 in both phases.
Table 1.
Stratified tenfold cross-validation of the models generated with different machine learning algorithms on the training dataset using the PAAC descriptor
| Model | Accuracy | Recall | Prec | F1 | Kappa | MCC |
|---|---|---|---|---|---|---|
| ET | 0.9322 | 0.9206 | 0.9434 | 0.9314 | 0.8644 | 0.8655 |
| SVM-RK | 0.9247 | 0.9393 | 0.9135 | 0.9258 | 0.8495 | 0.8505 |
| RF | 0.9121 | 0.915 | 0.9105 | 0.9124 | 0.8242 | 0.8249 |
| MLP | 0.9079 | 0.9262 | 0.8955 | 0.9097 | 0.8158 | 0.818 |
| LGBM | 0.9074 | 0.9224 | 0.8969 | 0.9087 | 0.8149 | 0.8167 |
| XGBOOST | 0.906 | 0.9215 | 0.8952 | 0.9074 | 0.8121 | 0.8138 |
| GPC | 0.8939 | 0.9626 | 0.8475 | 0.901 | 0.7877 | 0.796 |
| GBC | 0.878 | 0.9 | 0.863 | 0.8808 | 0.756 | 0.7572 |
| KNN | 0.8574 | 0.9477 | 0.8035 | 0.8694 | 0.7148 | 0.7272 |
| ABC | 0.8364 | 0.8579 | 0.8231 | 0.8398 | 0.6727 | 0.6741 |
| DT | 0.8172 | 0.8271 | 0.8129 | 0.8189 | 0.6344 | 0.6363 |
| QDA | 0.8153 | 0.8748 | 0.7828 | 0.8257 | 0.6306 | 0.6361 |
| NB | 0.7714 | 0.8383 | 0.7412 | 0.7858 | 0.5427 | 0.5492 |
| LDA | 0.7298 | 0.7178 | 0.7365 | 0.7267 | 0.4596 | 0.4602 |
| RC | 0.7293 | 0.7178 | 0.7359 | 0.7264 | 0.4587 | 0.4593 |
| LR | 0.7256 | 0.7215 | 0.7284 | 0.7246 | 0.4512 | 0.4516 |
| SVM-LK | 0.6489 | 0.5776 | 0.7297 | 0.5554 | 0.2976 | 0.3473 |
| DC | 0.4998 | 0.9 | 0.45 | 0.6 | 0.0 | 0.0 |
Table 2.
Evaluation of the models generated with different machine learning algorithms on the test dataset using the PAAC descriptor
| Model | ACC | Recall | Prec | F1 | k | MCC |
|---|---|---|---|---|---|---|
| MLP | 0.9065 | 0.9363 | 0.8834 | 0.9091 | 0.8131 | 0.8146 |
| LR | 0.7495 | 0.7453 | 0.7509 | 0.7481 | 0.4991 | 0.4991 |
| KNN | 0.8467 | 0.9625 | 0.7812 | 0.8624 | 0.6936 | 0.713 |
| NB | 0.8131 | 0.8727 | 0.7793 | 0.8233 | 0.6263 | 0.6308 |
| DT | 0.8262 | 0.8352 | 0.8199 | 0.8275 | 0.6523 | 0.6525 |
| SVM-LK | 0.7477 | 0.7715 | 0.7357 | 0.7532 | 0.4954 | 0.496 |
| SVM-RK | 0.9215 | 0.9401 | 0.9061 | 0.9228 | 0.843 | 0.8436 |
| GPC | 0.8897 | 0.9625 | 0.8399 | 0.897 | 0.7795 | 0.7879 |
| RC | 0.7495 | 0.7453 | 0.7509 | 0.7481 | 0.4991 | 0.4991 |
| RF | 0.929 | 0.9213 | 0.9354 | 0.9283 | 0.8579 | 0.858 |
| QDA | 0.8224 | 0.8801 | 0.7886 | 0.8319 | 0.6449 | 0.6493 |
| ABC | 0.8729 | 0.8989 | 0.8541 | 0.8759 | 0.7458 | 0.7468 |
| GBC | 0.8991 | 0.9139 | 0.8873 | 0.9004 | 0.7981 | 0.7985 |
| LDA | 0.7495 | 0.7453 | 0.7509 | 0.7481 | 0.4991 | 0.4991 |
| ET | 0.929 | 0.9139 | 0.9421 | 0.9278 | 0.8579 | 0.8583 |
| XGBOOST | 0.9196 | 0.9288 | 0.9118 | 0.9202 | 0.8393 | 0.8394 |
| LGBM | 0.9308 | 0.9326 | 0.9291 | 0.9308 | 0.8617 | 0.8617 |
| DC | 0.4991 | 1.0 | 0.4991 | 0.6658 | 0.0 | 0.0 |
Table 3.
Stratified tenfold cross-validation of the models generated with different machine learning algorithms on the training dataset using the DPC descriptor
| Model | Accuracy | AUC | Recall | Prec | F1 | Kappa | MCC |
|---|---|---|---|---|---|---|---|
| LGBM | 0.9261 | 0.9723 | 0.9252 | 0.9269 | 0.9259 | 0.8522 | 0.8526 |
| RF | 0.9238 | 0.9658 | 0.9065 | 0.939 | 0.9222 | 0.8476 | 0.8485 |
| XGBOOST | 0.9219 | 0.9732 | 0.9206 | 0.9239 | 0.9218 | 0.8438 | 0.8448 |
| ET | 0.921 | 0.956 | 0.8888 | 0.9502 | 0.9182 | 0.842 | 0.8442 |
| GBC | 0.9028 | 0.9641 | 0.9093 | 0.8979 | 0.9032 | 0.8055 | 0.8063 |
| MLP | 0.8911 | 0.9502 | 0.9449 | 0.8542 | 0.8969 | 0.7821 | 0.7873 |
| SVM-RK | 0.8654 | 0.9376 | 0.9243 | 0.8274 | 0.8729 | 0.7307 | 0.7363 |
| LR | 0.8453 | 0.8892 | 0.9121 | 0.8056 | 0.8551 | 0.6905 | 0.6976 |
| LDA | 0.8448 | 0.9164 | 0.9299 | 0.7953 | 0.8571 | 0.6896 | 0.7004 |
| ABC | 0.8434 | 0.9187 | 0.8598 | 0.8327 | 0.8456 | 0.6867 | 0.6879 |
| RC | 0.8424 | 0.9146 | 0.9271 | 0.7944 | 0.8551 | 0.6849 | 0.6958 |
| DT | 0.8237 | 0.8292 | 0.8486 | 0.8088 | 0.8279 | 0.6475 | 0.6488 |
| QDA | 0.8205 | 0.914 | 0.6888 | 0.9362 | 0.7929 | 0.641 | 0.6652 |
| SVM-LK | 0.8186 | 0.8815 | 0.8262 | 0.8144 | 0.8175 | 0.6372 | 0.6413 |
| NB | 0.7648 | 0.8196 | 0.8645 | 0.7217 | 0.7864 | 0.5296 | 0.5409 |
| GPC | 0.64 | 0.8639 | 0.9701 | 0.5846 | 0.7295 | 0.2797 | 0.3727 |
| KNN | 0.625 | 0.7453 | 0.9439 | 0.5767 | 0.7159 | 0.2499 | 0.3242 |
| DC | 0.4998 | 0.5 | 0.9 | 0.45 | 0.6 | 0.0 | 0.0 |
Table 4.
Evaluation of the models generated with different machine learning algorithms on the test dataset using the DPC descriptor
| Model | Accuracy | Recall | Prec | F1 | Kappa | MCC |
|---|---|---|---|---|---|---|
| MLP | 0.8804 | 0.8876 | 0.8745 | 0.881 | 0.7608 | 0.7608 |
| LR | 0.8393 | 0.8914 | 0.8068 | 0.847 | 0.6786 | 0.6823 |
| KNN | 0.6449 | 0.9288 | 0.5919 | 0.723 | 0.2905 | 0.3528 |
| NB | 0.7645 | 0.8652 | 0.7196 | 0.7857 | 0.5291 | 0.5403 |
| DT | 0.8318 | 0.8165 | 0.8417 | 0.8289 | 0.6635 | 0.6638 |
| SVM-LK | 0.8393 | 0.8652 | 0.8221 | 0.8431 | 0.6785 | 0.6795 |
| SVM-RK | 0.8505 | 0.8839 | 0.8281 | 0.8551 | 0.701 | 0.7026 |
| GPC | 0.6299 | 0.9476 | 0.5789 | 0.7188 | 0.2607 | 0.3374 |
| RC | 0.8449 | 0.9101 | 0.8046 | 0.8541 | 0.6898 | 0.6958 |
| RF | 0.9121 | 0.8577 | 0.9622 | 0.9069 | 0.8243 | 0.8292 |
| QDA | 0.843 | 0.7303 | 0.942 | 0.8228 | 0.6858 | 0.7038 |
| ABC | 0.8579 | 0.8502 | 0.8631 | 0.8566 | 0.7159 | 0.716 |
| GBC | 0.9065 | 0.8801 | 0.9289 | 0.9038 | 0.8131 | 0.8142 |
| LDA | 0.8411 | 0.9213 | 0.7935 | 0.8527 | 0.6823 | 0.6913 |
| ET | 0.9065 | 0.839 | 0.9697 | 0.8996 | 0.813 | 0.8205 |
| XGBOOST | 0.914 | 0.8876 | 0.9368 | 0.9115 | 0.828 | 0.8292 |
| LGBM | 0.9178 | 0.8764 | 0.9551 | 0.9141 | 0.8355 | 0.8383 |
| DC | 0.4991 | 1.0 | 0.4991 | 0.6658 | 0.0 | 0.0 |
Moderate performance was observed for algorithms such as GBC and ABC, which achieved ACC and F1 scores between 0.85 and 0.90 during testing (Table 2). While not top performers, these models still demonstrated reasonable predictive capability. The worst-performing models for PAAC-based prediction included LR, LDA, and SVM-LK, which had ACC and F1 scores below 0.75 during both cross-validation and testing (Tables 1 and 2). Notably, SVM-LK performed particularly poorly, with an ACC of only 0.6489 during cross-validation. For DPC-based models, LGBM showed the best performance during cross-validation (ACC: 0.9261, SEN: 0.9252, PREC: 0.9269, F1: 0.9259; Table 3) and testing (ACC: 0.9178, SEN: 0.8764, PREC: 0.9551, F1: 0.9141; Table 4). RF and XGB also performed well, consistently achieving ACC and F1 scores above 0.90.
Moderate performance in DPC-based models was seen with algorithms like GBC and ABC, achieving ACC and F1 scores between 0.85 and 0.90 during testing (Table 4). The poorest performance for DPC-based prediction was observed with KNC and GPC, both achieving ACC below 0.65 during testing (Table 4). This stark underperformance compared to other algorithms suggests these methods may be ill-suited for this specific prediction task using DPC descriptors.
A critical analysis of these results reveals two important points: (1) the consistently high performance of ensemble methods (RF, ET, LGBM, XGB) across both descriptor types suggests these algorithms are particularly well-suited for this prediction task, and (2) the discrepancy in performance between PAAC and DPC descriptors indicates that PAAC may capture more relevant features for VPEINR prediction.
The selection of the RF model using PAAC descriptors for incorporation into the VirusHound-II application is justified by several key factors. Firstly, RF demonstrated exceptional performance during the testing phase, achieving an ACC of 0.9290, PREC of 0.9354, and an F1 of 0.9283 (Table 2), the highest values among all models evaluated with PAAC descriptors. Furthermore, RF showed notable consistency between the cross-validation and testing phases, suggesting good generalization capability. However, it is important to note that the LGBM model with DPC descriptors showed comparable performance (Table 4) and could be considered as a viable alternative or complementary approach.
Discussion
The human immune system is a complex network designed to defend the body against infections and diseases. It can be broadly divided into two main categories: the innate immune response and the adaptive immune response. Each has distinct characteristics and plays a crucial role in protecting the body [35]. Throughout evolution, viruses have developed highly specialized proteins that allow them to evade the host’s immune system. These proteins mimic or target specific components of the immune system, prevent immune recognition, and modulate immune responses [36, 37]. For example, Vaccinia virus and Molluscum contagiosum virus produce proteins that bind to and neutralize immune molecules like interleukin 18 (IL-18). Similarly, viruses like Human cytomegalovirus (HCMV) have proteins like US28 vCKR that interact with chemokines [38]. The Epstein-Barr Virus protein vIL-10 mimics human interleukin-10 to suppress the activation of effector immune cells, a strategy that affects both innate and adaptive immunity [39–42]. These interactions enable viruses to evade immune detection and response, showcasing the sophisticated mechanisms viruses have developed to counteract the host’s immune system.
In recent years, machine learning has significantly advanced immunology by providing robust tools for analyzing complex data, predicting disease susceptibility, and creating personalized medicine strategies. By utilizing large datasets, these techniques reveal patterns not discernible through conventional methods, enhancing both our understanding of immune mechanisms and the development of diagnostic and therapeutic solutions [43–46]. Machine learning has been extensively applied in immunology, particularly in predicting B-cell and T-cell epitopes. These epitopes are crucial for designing epitope-based vaccines and understanding immune responses. In this context, various algorithms, including random forests, artificial neural networks, and support vector machines, have been used to predict epitopes accurately [47–49]. However, to date, no tool has been reported capable of predicting proteins that evade the host’s innate immune response. The closest in this line is our tool named VirusHound-I, which allows the prediction of viral proteins that evade the host’s adaptive immune response with high efficiency [6].
The RF has been highlighted as essential tool in the field of bioinformatics, particularly in the development of predictive models for peptides and proteins based on the calculation of molecular descriptors [50]. To date, many studies have been reported that make use of RF for the development of predictive models based on the computation of molecular descriptors. These research areas include the prediction of antimicrobial peptides [51], antiviral peptides [50], prediction of T [52] and B [53] cell epitopes, peptides with antitumor activity [54], antigens [55, 56], and toxins [57], among many others. In this study, we observed that the RF classifier outperformed the other algorithms during both the training and testing phases. The superiority of RF in this and work on this type of data has not been studied yet. However, we believe this could be because the RF is especially effective in tabular data classification due to its resistance to overfitting, attributable to multiple decision trees and random feature selection [58–60]. It can efficiently handle high-dimensional data without requiring dimension reduction and operates well with data at its original scales, eliminating the need for normalization [60]. Moreover, RF handles missing data during training, provides valuable information about the importance of features to facilitate model interpretation, and is flexible and adaptable for different types of tasks and parameter settings, optimizing its performance in various applications [58–60].
In this study, we used PAAC to analyze viral protein sequences because it effectively captures protein composition and physicochemical properties [61–63]. PAAC offers an enriched representation by incorporating important amino acid characteristics directly impacting protein function. These characteristics include hydrophobicity, side chain volume, and amino acid composition, which are crucial for the immune evasion mechanisms of viruses [64–66]. This molecular descriptor has been extensively evaluated due to its characteristics and the excellent results it has produced, as seen in our study, where it enabled the generation of predictive models with strong performance when used alongside the RF algorithm. In this regard, several studies have reported similar findings, such as the prediction of ion channel inhibitors [67], bitter peptides [68], animal toxins [69], anti-inflammatory peptides [70], HIV-1 and HIV-2 proteins [71], cell-penetrating peptides [72], amyloidogenic regions of proteins [73], bacterial cell wall lyase [74], among many other cases.
The model based on RF was selected and incorporated into a web application called VirusHound-II (www.biochemintelli.com), which offers a minimalist interface facilitating the efficient prediction of VPEINRs. VirusHound-II complements VirusHound-I [6], differentiating both in their capacity to predict proteins that evade the two types of immune responses: the innate immune response and the adaptive immune response, respectively. VirusHound-II represents a potentially transformative tool in the field of antiviral therapy discovery and development. Its ability to accurately predict viral proteins involved in evading the innate immune response could significantly accelerate the identification of new therapeutic targets. This model not only facilitates a deeper understanding of viral evasion mechanisms but could also guide the design of more effective intervention strategies. Looking ahead, integrating VirusHound-II with broader genomic data platforms, and applying it to emerging pathogens could substantially improve our responses to epidemics and pandemics. In terms of real impact, the implementation of this tool in clinical practice and public health programs could translate into faster and more precise diagnoses, as well as more targeted and personalized treatments, underscoring its value in improving global health.
While several computational approaches have been developed to study virus-host interactions, most focus on general protein–protein interactions (PPIs) between viruses and hosts. Tools such as MP-VHPPI [75], Trans-PPI [76], and LSTM-PHV [77] have made significant contributions to predicting virus-host PPIs. However, VirusHound-II distinguishes itself by specifically targeting viral proteins involved in evading the host's innate immune response. This specialized focus offers several advantages over general PPI prediction tools. Firstly, it provides direct insights into a critical aspect of viral pathogenesis. Secondly, it offers higher interpretability, as the results are directly applicable to understanding immune evasion mechanisms. Lastly, by bypassing the need to analyze all possible virus-host protein interactions, VirusHound-II provides a more efficient approach to identifying immune evasion factors.
VirusHound-II represents an advancement in the prediction of viral proteins that evade the host innate immune response. Its high accuracy and generalization capacity demonstrate its potential as a valuable tool in virological research and the development of antiviral therapies. However, as with any predictive model, there are opportunities for future improvements and expansions. We plan to continuously update our dataset to include information on emerging viruses and new variants, maintaining the model relevance and adaptability. The incorporation of protein structural data could further enrich our predictions, providing insights into the molecular mechanisms of immune evasion. Furthermore, we are exploring the possibility of predicting not only the evasion capability but also the specific mechanisms employed by viral proteins.
Conclusions
VirusHound-II represents a significant advance in the field of computational virology, being the first tool specifically designed to predict viral proteins that evade the host’s innate immune response. Our Random Forest model, using pseudo amino acid composition descriptor, demonstrated superior performance, achieving an accuracy of 92.90% on the independent test set. The robustness of our results is supported by a rigorous methodology that includes tenfold cross-validation and testing on an independent dataset. The comprehensive comparison of various machine learning algorithms provides a solid foundation for future studies in this field. VirusHound-II, accessible through a user-friendly web application, has the potential to significantly accelerate research on virus-host interactions and the development of antiviral strategies. This study highlights the importance of computational approaches in understanding viral evasion mechanisms, providing a valuable tool for the scientific community. Overall, VirusHound-II represents an important step towards the efficient prediction of viral proteins that evade the innate immune response, with potential implications for the development of antiviral therapies and a better understanding of viral pathogenesis.
Availability and requirements
Project name: VirusHound-II.
Project home page: https://www.biochemintelli.com/VirusHound-II
Operating system(s): Platform independent.
Programming language: Python 3.10.
Other requirements: Python 3.7 or higher, scikit-learn, biopython, numpy, and pandas. License: MIT License. Any restrictions to use by non-academics: None.
Acknowledgements
We thank the FONDECYT INICACIÓN N°11240026; and Fondap-INCAR N°1523A0007.
Author contributions
J.F. wrote the main manuscript, developed the predictive models and the software. L.H. Participated in the research feedback, planned and organized the work methodology, and wrote part of the manuscript. A.J. and L.J. Participated in the research feedback.
Funding
Not applicable.
Availability of data and materials
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol. 2000;18:861–926. [DOI] [PubMed] [Google Scholar]
- 2.Doherty PC, Tripp RA, Sixbey JW. Evasion of Host immune responses by tumours and viruses. 2007;245–70. [DOI] [PubMed]
- 3.Vossen MT, Westerhout EM, Söderberg-Nauclér C, Wiertz EJ. Viral immune evasion: a masterpiece of evolution. Immunogenetics. 2002;54:527–42. [DOI] [PubMed] [Google Scholar]
- 4.Benedict CA, Norris PS, Ware CF. To kill or be killed: viral evasion of apoptosis. Nat Immunol. 2002;3:1013–8. [DOI] [PubMed] [Google Scholar]
- 5.Lucas M, Karrer U, Lucas A, Klenerman P. Viral escape mechanisms—escapology taught by viruses. Int J Exp Pathol. 2001;82:269–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beltrán JF, Belén LH, Farias JG, Zamorano M, Lefin N, Miranda J, et al. VirusHound-I: prediction of viral proteins involved in the evasion of host adaptive immune response using the random forest algorithm and generative adversarial network for data augmentation. Brief Bioinform. 2023;25:bbad434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tosi MF. Innate immune responses to infection. J Allergy Clin Immunol. 2005;116:241–9. [DOI] [PubMed] [Google Scholar]
- 8.Koenderman L, Buurman W, Daha MR. The innate immune response. Immunol Lett. 2014;162:95–102. [DOI] [PubMed] [Google Scholar]
- 9.Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;43:336–41. [DOI] [PubMed] [Google Scholar]
- 10.Gale M, Sen GC. Viral evasion of the interferon system. J Interferon Cytokine Res. 2009;29:475–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malmgaard L. Induction and Regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–54. [DOI] [PubMed] [Google Scholar]
- 12.Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. [DOI] [PubMed] [Google Scholar]
- 13.Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86:2900–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 15.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. [DOI] [PubMed] [Google Scholar]
- 16.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. [DOI] [PubMed] [Google Scholar]
- 17.Der SD, Zhou A, Williams BRG, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci. 1998;95:15623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1979;1994(264):1918–21. [DOI] [PubMed] [Google Scholar]
- 19.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3:920–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. 2021;6:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beachboard DC, Horner SM. Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol. 2016;32:113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelemans T, Kikkert M. Viral Innate immune evasion and the pathogenesis of emerging RNA virus infections. Viruses. 2019;11:961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasuga Y, Zhu B, Jang K-J, Yoo J-S. Innate immune sensing of coronavirus and viral evasion strategies. Exp Mol Med. 2021;53:723–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minkoff JM, tenOever B. Innate immune evasion strategies of SARS-CoV-2. Nat Rev Microbiol. 2023. 10.1038/s41579-022-00839-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maarouf M, Rai K, Goraya M, Chen J-L. Immune ecosystem of virus-infected host tissues. Int J Mol Sci. 2018;19:1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rashid F, Xie Z, Suleman M, Shah A, Khan S, Luo S. Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front Immunol. 2022;13:940756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gargan S, Stevenson NJ. Unravelling the immunomodulatory effects of viral ion channels, towards the treatment of disease. Viruses. 2021;13:2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng TI, Dorr PK, Krishnan P, Cohen DE, Rhee S, Wang SX, et al. Biomarkers for the clinical development of antiviral therapies. Cytom B Clin Cytom. 2021;100:19–32. [DOI] [PubMed] [Google Scholar]
- 30.Pandya R, He YD, Sweeney TE, Hasin-Brumshtein Y, Khatri P. A machine learning classifier using 33 host immune response mRNAs accurately distinguishes viral and non-viral acute respiratory illnesses in nasal swab samples. Genome Med. 2023;15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu J, Chen T, Mao X, Fang Y, Sun H, Wei D-Q, et al. Machine learning of flow cytometry data reveals the delayed innate immune responses correlate with the severity of COVID-19. Front Immunol. 2023;14:974343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Liang Y, Dong H, Tan C, Xiao Z, Liu S. Innate immune memory and its application to artificial immune systems. J Supercomput. 2022;78:11680–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Trends Microbiol. 2000;8:410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bateman A, Martin M-J, Orchard S, Magrane M, Agivetova R, Ahmad S, et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pulendran B, Davis MM. The science and medicine of human immunology. Science. 1979;2020:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herbert JA, Panagiotou S. Immune response to viruses. In: Encyclopedia of Infection and Immunity. Elsevier; 2022. p. 429–44.
- 37.Alcami A, Ghazal P, Yewdell JW. Viruses in control of the immune system. EMBO Rep. 2002;3:927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol. 2010;10:514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012;8: e1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bejarano MT, Masucci MG. Interleukin-10 abrogates the inhibition of Epstein-Barr virus-induced B-Cell transformation by memory T-cell responses. Blood. 1998;92:4256–62. [PubMed] [Google Scholar]
- 41.de Silva JM, de Alves CEC, Pontes GS. Epstein-Barr virus: the mastermind of immune chaos. Front Immunol. 2024;15:1297994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jog NR, Chakravarty EF, Guthridge JM, James JA. Epstein Barr virus interleukin 10 suppresses anti-inflammatory phenotype in human monocytes. Front Immunol. 2018;9:2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katayama Y, Yokota R, Akiyama T, Kobayashi TJ. Machine learning approaches to TCR repertoire analysis. Front Immunol. 2022;13:858057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Culos A, Tsai AS, Stanley N, Becker M, Ghaemi MS, McIlwain DR, et al. Integration of mechanistic immunological knowledge into a machine learning pipeline improves predictions. Nat Mach Intell. 2020;2:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barone SM, Paul AG, Muehling LM, Lannigan JA, Kwok WW, Turner RB, et al. Unsupervised machine learning reveals key immune cell subsets in COVID-19, rhinovirus infection, and cancer therapy. Elife. 2021;10:e64653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shetab Boushehri S, Essig K, Chlis N-K, Herter S, Bacac M, Theis FJ, et al. Explainable machine learning for profiling the immunological synapse and functional characterization of therapeutic antibodies. Nat Commun. 2023;14:7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubinstein ND, Mayrose I, Pupko T. A machine-learning approach for predicting B-cell epitopes. Mol Immunol. 2009;46:840–7. [DOI] [PubMed] [Google Scholar]
- 48.Bukhari SNH, Jain A, Haq E, Mehbodniya A, Webber J. Machine learning techniques for the prediction of B-cell and T-cell epitopes as potential vaccine targets with a specific focus on SARS-CoV-2 pathogen: a review. Pathogens. 2022;11:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bravi B. Development and use of machine learning algorithms in vaccine target selection. NPJ Vaccines. 2024;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefin N, Herrera-Belén L, Farias JG, Beltrán JF. Review and perspective on bioinformatics tools using machine learning and deep learning for predicting antiviral peptides. Mol Divers. 2023. 10.1007/s11030-023-10718-3. [DOI] [PubMed] [Google Scholar]
- 51.Xu J, Li F, Leier A, Xiang D, Shen H-H, Marquez Lago TT, et al. Comprehensive assessment of machine learning-based methods for predicting antimicrobial peptides. Brief Bioinform. 2021;22:bbab083. [DOI] [PubMed] [Google Scholar]
- 52.Huang J-H, Xie H-L, Yan J, Lu H-M, Xu Q-S, Liang Y-Z. Using random forest to classify T-cell epitopes based on amino acid properties and molecular features. Anal Chim Acta. 2013;804:70–5. [DOI] [PubMed] [Google Scholar]
- 53.Huang J-H, Wen M, Tang L-J, Xie H-L, Fu L, Liang Y-Z, et al. Using random forest to classify linear B-cell epitopes based on amino acid properties and molecular features. Biochimie. 2014;103:1–6. [DOI] [PubMed] [Google Scholar]
- 54.Deng Y, Ma S, Li J, Zheng B, Lv Z. Using the random forest for identifying key physicochemical properties of amino acids to discriminate anticancer and non-anticancer peptides. Int J Mol Sci. 2023;24:10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beltrán Lissabet JF, Herrera Belén L, Farias JG. TTAgP 1.0: A computational tool for the specific prediction of tumor T cell antigens. Comput Biol Chem. 2019;83:107103. [DOI] [PubMed] [Google Scholar]
- 56.Herrera-Bravo J, Farías JG, Contreras FP, Herrera-Belén L, Norambuena J-A, Beltrán JF. VirVACPRED: a web server for prediction of protective viral antigens. Int J Pept Res Ther. 2022;28:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pallavi M, Valsan AS, Thoufi KU. Toxicity prediction in peptides and proteins using Random forest,Decision Tree and Logistic Regression. In: 2022 international conference on futuristic technologies (INCOFT). IEEE; 2022. p. 1–6.
- 58.Iranzad R, Liu X. A review of random forest-based feature selection methods for data science education and applications. Int J Data Sci Anal. 2024. 10.1007/s41060-024-00509-w. [Google Scholar]
- 59.Gomes HM, Bifet A, Read J, Barddal JP, Enembreck F, Pfharinger B, et al. Adaptive random forests for evolving data stream classification. Mach Learn. 2017;106:1469–95. [Google Scholar]
- 60.Wang Q, Nguyen T-T, Huang JZ, Nguyen TT. An efficient random forests algorithm for high dimensional data classification. Adv Data Anal Classif. 2018;12:953–72. [Google Scholar]
- 61.Li C, Li X, Lin Y-X. Numerical characterization of protein sequences based on the generalized Chou’s pseudo amino acid composition. Appl Sci. 2016;6:406. [Google Scholar]
- 62.Du P, Gu S, Jiao Y. PseAAC-General: fast building various modes of general form of Chou’s pseudo-amino acid composition for large-scale protein datasets. Int J Mol Sci. 2014;15:3495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raj SS, Chandra SSV. Significance of sequence features in classification of protein-protein interactions using machine learning. Protein J. 2024;43:72–83. [DOI] [PubMed] [Google Scholar]
- 64.Esmaeili M, Mohabatkar H, Mohsenzadeh S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J Theor Biol. 2010;263:203–9. [DOI] [PubMed] [Google Scholar]
- 65.Matyášek R, Řehůřková K, Berta Marošiová K, Kovařík A. Mutational asymmetries in the SARS-CoV-2 genome may lead to increased hydrophobicity of virus proteins. Genes (Basel). 2021;12:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vormittag P, Klamp T, Hubbuch J. Ensembles of hydrophobicity scales as potent classifiers for chimeric virus-like particle solubility—an amino acid sequence-based machine learning approach. Front Bioeng Biotechnol. 2020;8:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mei J, Fu Y, Zhao J. Analysis and prediction of ion channel inhibitors by using feature selection and Chou’s general pseudo amino acid composition. J Theor Biol. 2018;456:41–8. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y-F, Wang Y-H, Gu Z-F, Pan X-R, Li J, Ding H, et al. Bitter-RF: A random forest machine model for recognizing bitter peptides. Front Med (Lausanne). 2023;10:1052923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pan Y, Wang S, Zhang Q, Lu Q, Su D, Zuo Y, et al. Analysis and prediction of animal toxins by various Chou’s pseudo components and reduced amino acid compositions. J Theor Biol. 2019;462:221–9. [DOI] [PubMed] [Google Scholar]
- 70.Zhao D, Teng Z, Li Y, Chen D. iAIPs: identifying anti-inflammatory peptides using Random Forest. Front Genet. 2021;12:773202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mei J, Zhao J. Prediction of HIV-1 and HIV-2 proteins by using Chou’s pseudo amino acid compositions and different classifiers. Sci Rep. 2018;8:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L, Chu C, Huang T, Kong X, Cai Y-D. Prediction and analysis of cell-penetrating peptides using pseudo-amino acid composition and random forest models. Amino Acids. 2015;47:1485–93. [DOI] [PubMed] [Google Scholar]
- 73.Teng Z, Zhang Z, Tian Z, Li Y, Wang G. ReRF-Pred: predicting amyloidogenic regions of proteins based on their pseudo amino acid composition and tripeptide composition. BMC Bioinform. 2021;22:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen X-X, Tang H, Li W-C, Wu H, Chen W, Ding H, et al. Identification of bacterial cell wall lyases via pseudo amino acid composition. Biomed Res Int. 2016;2016:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Asim MN, Fazeel A, Ibrahim MA, Dengel A, Ahmed S. MP-VHPPI: meta predictor for viral host protein-protein interaction prediction in multiple hosts and viruses. Front Med (Lausanne). 2022;9:1025887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang X, Yang S, Lian X, Wuchty S, Zhang Z. Transfer learning via multi-scale convolutional neural layers for human–virus protein–protein interaction prediction. Bioinformatics. 2021;37:4771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsukiyama S, Hasan MM, Fujii S, Kurata H. LSTM-PHV: prediction of human-virus protein–protein interactions by LSTM with word2vec. Brief Bioinform. 2021;22:bbab228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.