

Abstract
Neuroendocrine tumor (NET) is a rare malignant tumor, notably small cell lung cancer (SCLC), a type of lung neuroendocrine tumor, which has a survival rate of less than 7%. Although various biomarkers including CHGA (Chromogranin A), INSM1 (Insulinoma-associated protein 1), and SYP (Synaptophysin) are extensively used for the diagnostic testing of NET, their diverse specificities and sensitivities are acknowledged as limitations. Here, we demonstrate that RepID (Replication initiation determinant protein), a component of CRL4 (Cullin-RING ubiquitin E3 ligase 4), holds promise as a biomarker for identifying NET and SCLC. Analysis of the Cancer Cell Line Encyclopedia (CCLE) via the CellMinerCDB portal reveals a high correlation between RepID transcript levels and mRNA expression of NE signature genes. Additionally, RepID protein is highly expressed in SCLC patient tissues and a subset of SCLC cell lines. Viability analysis following treatment with pevonedistat and SZL-P1-41 in SCLC cell lines and human SCLC-organoid models indicates that RepID expression determines the sensitivity to CRL-targeting anti-cancer drugs. These findings suggest that RepID represents a novel biomarker for NET and SCLC, and insights from RepID research in these cancers could lead to innovative therapeutic strategies.
Keywords: Neuroendocrine tumor, Small cell lung cancer, RepID, CRL
Subject terms: Cell-cycle proteins, Proteins, Ubiquitins, Chemotherapy, Targeted therapies, Small-cell lung cancer, Tumour biomarkers
Introduction
Neuroendocrine tumor (NET) accounts for approximately 2% of all malignant tumors diagnosed in the western world and is classified as a rare tumor type1,2. NETs can originate from NE cells throughout the body, including the gastrointestinal tract, lungs, appendix, and pancreas1–5. Among these organs, 22–27% of NETs originate from the lungs, making it the second most frequent primary site3. In the three major types of lung cancer, approximately 80–85% of small cell lung cancers (SCLC) exhibit NE characteristics, whereas most non-small cell lung cancers (NSCLC) and other cancers do not show NE signatures4,5.
SCLC is one of the most lethal tumors, with fewer than 7% of diagnosed patients surviving up to 5 years3,6. The poor prognosis of SCLC is attributed to its aggressive characteristics, which include rapid proliferation, extensive metastasis, and high recurrence rates. SCLC is categorized into the following subtypes: (1) ASCL1 (Achaete-scute homolog 1), (2) NeuroD1 (Neurogenic differentiation 1), (3) POU2F3 (POU class 2 homeobox 3), and (4) YAP1 (Yes-associated protein 1)7,8. SCLC-A (ASCL1) and SCLC-N (NeuroD1) exhibit high levels of NE markers as neuroendocrine subtypes, whereas SCLC-P (POU2F3) and SCLC-Y (YAP1) are characterized as non-neuroendocrine subtypes and show low levels of NE markers8. Depending on the subtype, SCLC exhibits varied responses to representative NE markers such as chromogranin A (CHGA), synaptophysin (SYP), and insulinoma-associated protein 1 (INSM1) (1). Consequently, the lack of specific biomarkers and the varied biomarker patterns continue to pose challenges in NET diagnosis. Additionally, the stagnation of therapies for SCLC, unchanged for approximately 30 years, alongside the development of resistance to chemo/radiotherapy, present significant limitations. The identification of novel biomarkers is imperative to address the diagnostic and therapeutic challenges in NET.
Replication initiation determinant protein (RepID/DCAF14/PHIP) belongs to the DDB1/CUL4-associated factor (DCAF) family, acting as a substrate receptor for the Cullin-RING ubiquitin ligase complex 4 (CRL4)9. RepID possesses various chromatin binding domains, including bromo and cryptic tudor domains9,10, enabling it to strongly bind to chromatin and recruit the CRL4 complex to chromatin via the WD40 domain11. The recruitment of CRL4 by RepID is essential to prevent excessive replication origin licensing, known as re-replication, and to ensure proper mitotic exit through ubiquitination of chromatin licensing and DNA replication factor 1 (CDT1) or BUB3, a member of the spindle assembly checkpoint (SAC) complex9,11–16. Variations in RepID expression among different cancer cells lead to changes in drug sensitivity, affecting responses to p97/VCP segregase inhibitor, paclitaxel, and CRL-targeting anti-cancer drugs, including CRL1 inhibitor or pevonedistat, NAE1 inhibitor11,16,17. RepID also promotes cell proliferation by inhibiting apoptotic cell death and is recognized as a biomarker for aggressive metastatic melanoma18–22. Identifying cancer cells with high RepID expression levels could be crucial for developing new cancer therapies.
Here, we report that RepID is expressed at high levels in NET and SCLC, indicating that RepID could serve as a potential biomarker. We noted that both mRNA and protein levels of RepID were generally elevated in NET and SCLC. This expression pattern was significantly associated with high NE marker and RepID transcript levels. Additionally, we established that the sensitivity to CRL-targeting anti-cancer drugs correlates with RepID expression, utilizing both cancer cell lines and SCLC-organoids. These findings suggest the potential for innovative therapeutic strategies in NE tumors based on the expression of RepID.
Methods
Database analysis
We analyzed correlations of gene expression among 26 DCAF proteins, NE markers, and seven cancer-type signatures in the Cancer Cell Line Encyclopedia (CCLE) dataset using CellMiner cross-database (CellMinerCDB, https://discover.nci.nih.gov/cellminercdb/). The expression of RepID and NE signature was examined according to tissue origin. Furthermore, we investigated the expression of RepID in SCLC, NSCLC, and other cancer types, including four SCLC subtypes defined by the master transcription factors ASCL1, NEUROD1, POU2F3, and YAP1 (NAPY). All data are accessible online.
We analyzed correlation between the expression of RepID and MKI67 transcripts in the SCLC-CCLE-Borad-MIT using SCLC CellMiner cross-database (SclcCellMinerCDB, https://discover.nci.nih.gov/rsconnect/SclcCellMinerCDB/). Correlation of transcripts level between MKI67 and RepID, and overall survival (months) according to RepID expression in human SCLC patient were analyzed through cBioPortal for Cancer Genomics (U cologne, Nature 2015, Small Cell Lung Cancer dataset). Pearson correlation, p-value and hazard ratio (HR) were determined using Graph Pad Prism.
Cell culture and chemicals
Human SCLC (NCI-H69, H82, H146, H209, H211, H446, and DMS114) and NSCLC (NCI-H1299) cell lines were cultured in RPMI1640 medium (Invitrogen, 11,875–119). Also, human cancer cell lines, including osteosarcoma (U2OS, SAOS2), colon cancer (HCT116), myelogenous leukemia (K562), prostate cancer (DU145), immortalized lung fibroblast (VA13/WI38), and immortalized embryonic kidney (HEK293) were cultured in Dulbecco’s Modified Eagle’s Medium (Invitrogen, 11,965–092). The media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, 10,082–147) and 1 × penicillin/streptomycin (Invitrogen, 10,378–016), and maintained in a 37 °C/5% CO2 humidified incubator. All original cancer cell lines were sourced from the American Type Culture Collection (ATCC) (www.atcc.org) and tested for mycoplasmas (Lonza, LT07-418). Pevonedistat and SZL-P1-41 were sourced from Sigma-Aldrich (Cat. 505,477) and Cayman-Chemical (Cat. 26,142), respectively.
RepID depletion in the cells occurred via the CRISPR-CAS9 system. Cells were transfected with the RepID single-guide RNA (sgRNA) plasmid and linearized pCR2.1 vector containing a puromycin-resistance gene expression cassette11,23. Transfected cells were cultured for 7 days with 1 μg/mL of puromycin (Gibco, A1113803) to establish a stable cell line. An overexpressing RepID stable cell line was generated by culturing with 200 μg/mL of G418 (Roche, 108,321-42-2) for 14 days.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted following the manufacturer’s protocols from Qiagen (RNeasy Mini kit, 74,104). qRT-PCR was subsequently performed according to the protocols specified for each reagent, utilizing Takara (PrimeScript RT Master Mix, RR036A) and Invitrogen (SYBR Green PCR Master Mix, 4,309,155). The expression levels of human RepID genes in cells were determined through qRT-PCR, with GAPDH as the internal control for normalization. Primers for GAPDH were as follows: forward: 5′-GAG TCA ACG GAT TTG GTC GT-3′ and reverse: 5′-TTG ATT TTG GAG GGA TCT CG-3′. For RepID #1, the primers used were: forward: 5′-CGC ACA AAT AAA AGC TGC AA-3′ and reverse: 5′-CAG TTG GAA CAA GTC GCT CA-3′. The primers for RepID #2 included: forward: 5′-TGA GCG ACT TGT TCC AAC TG -3′ and reverse: 5′-AAC CTC CCA TCA TCT GTT GC-3′. Finally, the primers for RepID #3 were: forward: 5′- ATG AGC CTG GAA GCC CTA TT-3′ and reverse: 5′-TGA TGG TCC TTC CTT TTT CG-3′.
Soft agar assay
Two thousand H69, H82, H146, and DMS114 cells, three days post CRISPR/Cas9-mediated RepID depletion, were suspended in 1 ml of RPMI1640 medium containing 0.35% top agar and seeded onto a base layer of 0.5% agar medium (Invitrogen, 16,500,500) in 24-well plates. Following an incubation at 37 °C in a 5% CO2 humidified incubator for three weeks, colonies were stained with 0.005% crystal violet and their areas were quantified using ImageJ software.
Subcellular protein fractionation
Cytosolic, membrane-bound, soluble nuclear, and chromatin-bound protein fractions from both wild type and transfected H69 and H82 cells were prepared using a subcellular protein fractionation kit (Thermo Fisher Scientific, 78,840), augmented with PMSF, and a cocktail of protease and phosphatase inhibitors (Abcam, ab201112). Lysis of cells was achieved in accordance with the manufacturer’s recommended protocols.
Immunoblot analysis and quantification
Whole-cell lysates and chromatin-bound fractions underwent immunodetection following separation by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The employed primary antibodies included anti-CUL1 (Abcam, ab75851, 1:1000), anti-CUL4A (Abcam, ab92554, 1:20,000), anti-CUL4B (Sigma, C9995, 1:2000), anti-RepID (Abcam, ab86244, 1:2000), anti-CDT1 (Cell Signaling, 8064, 1:2000), anti-FLAG (Sigma, F1804, 1:1000), anti-a-tubulin (Sigma, T9026, 1:10,000), and anti-histone H3 (Millipore, 07-690, 1:25,000). Horseradish peroxidase (HRP)-linked secondary antibodies, anti-rabbit IgG (Cell Signaling, 7074, 1:5000) and anti-mouse IgG (Cell Signaling, 7076, 1:5000), were used in accordance with the recommended protocols. Quantification of protein bands was performed using ImageJ software.
Neon transfection system
Cells were electroporated for transfection using the Neon Transfection System (Invitrogen, MPK5000). The day before the experiment, cells were seeded into culture dishes. After 24 h, cells that had reached 80% confluency were harvested using Trypsin–EDTA (Biowest, L0931) and then centrifuged. The pellet was resuspended for cell counting with a hemocytometer. The cells were then transferred to a sterile microcentrifuge tube and centrifuged at room temperature. Plasmid DNA (either Lenti-CRISPR/Cas9-sgRepID or FLAG-RepID-Full Length) was added to the cell pellet, which had been depleted of the supernatant. The cells were then resuspended in resuspension buffer R from the kit (Invitrogen, MPK10096). Following gentle resuspension, the cells were electroporated and incubated in media at 37 °C in a humidified 5% CO2 incubator.
Immunofluorescence analysis
Cultured Cells (H69, H82, H146, DMS114) were collected into Eppendorf tubes, and the cell pellets were washed with ice-cold PBS. After the washing step, cell pellets were resuspended, and slides were prepared using a TXT3 Cytospin (Nasco Korea Corp., Seoul, Korea) at 1000xg for 5 min. The slides were rinsed with phosphate-buffered saline (PBS; pH 7.4), fixed in 4.0% paraformaldehyde (PFA) for 10 min, and permeabilized using PBS-T buffer [0.1% Triton X-100 in 1 × PBS, a phosphatase inhibitor cocktail (Roche, P4906845001), and a protease inhibitor cocktail (Sigma, P8340)] for 10 min at room temperature. The slides were then incubated in blocking solution (1% BSA in PBS-T buffer) for 30 min at room temperature. Primary antibody staining was carried out using anti-RepID (Abcam, ab86244, 1:100), followed by secondary antibody staining with Alexa Fluor™ Plus 488 conjugated anti-rabbit IgG (Invitrogen, A32731, 1:500). DNA was counterstained with DAPI (Thermofisher Scientific, 62,248, 1:1000). Imaging was performed using a Zeiss LSM880 with Airyscan confocal microscope.
Immunohistochemistry analysis
Analysis of RepID expression in lung cancer histological subtypes and its correlation with disease characteristics were conducted using the Biomax lung cancer tissue array (LC807b). The slide was heated at 60 °C for 2 h. Following this, deparaffinization and rehydration were performed using xylene and ethanol, while antigen retrieval was facilitated using a heat-induced epitope retrieval (HIER) buffer (10 mM Citric acid, pH 6.0). The slide, immersed in HIER buffer, was subsequently boiled in a 92 °C water bath for 10 min and then allowed to cool at room temperature. To prevent non-specific binding, the slide was incubated for 30 min with a blocking solution (2% BSA, 5% FBS, 0.1% Triton X-100 in PBS). The slide was treated overnight at 4 °C with an anti-RepID antibody (Abcam, ab86244, 1:100). Following the removal of the primary antibody solution, an Alexa Fluor™ Plus 555 conjugated anti-rabbit IgG (Invitrogen, A32732) in blocking solution was applied to the slide and incubated for 1 h at room temperature in the dark. DNA was counterstained with DAPI (Thermo Fisher Scientific, 62,248, 1:1000) and the samples were mounted using mounting medium (Sigma-Aldrich, F4680). Images were acquired using a Zeiss LSM880 confocal microscope, and the intensity of fluorescence was measured with ImageJ software.
Patient samples
Small (1–4 cm3) sections of lung cancer tissue samples were collected from resected lung specimens as part of the biobanking process for lung cancer at the Asan Bio-Resource Center of Asan Medical Center (Seoul, Korea). All tissues were used with the patients’ informed consent. The research protocol was approved by the Ethics Committee of Asan Medical Center, and the entire experimental protocol was conducted in compliance with relevant institutional guidelines. Samples were characterized as tumor or normal tissue based on their histopathological assessment. Each patient’s diagnosis was validated by pathologists at Asan Medical Center.
Culturing patient-derived cancer organoids
Patient samples were immersed in cold Hank’s balanced salt solution (HBSS; Lonza, Basel, Switzerland) containing antibiotics (Lonza, Basel, Switzerland) and transported to the laboratory within 1 h of excision on ice. The samples were subsequently washed three times with cold HBSS that included antibiotics and sectioned with sterile blades. Next, these sections were incubated at 37 °C for 2 h with intermittent agitation in DMEM/F12 medium (Lonza) supplemented with 1 mg/mL collagenase/dispase (Roche, IN, USA), 0.001% DNase (Sigma-Aldrich, MO, USA), 200U/mL penicillin, 200 mg/mL streptomycin, and 0.5 mg/mL amphotericin B (2%; Sigma-Aldrich, MO, USA). Following incubation, the sections were thoroughly triturated by repeated pipetting. The triturated suspensions were then filtered through 70 μm cell strainers (BD Falcon, CA, USA) and centrifuged at 112 × g for 3 min. The resultant pellet was resuspended in 100 μL MBM (a serum-free medium comprising DMEM/F12; Lonza), enriched with 20 ng/mL of bFGF (Invitrogen, CA, USA), 50 ng/mL human EGF (Invitrogen), N2 supplement (Invitrogen), B27 supplement (Invitrogen), 10 μM ROCK inhibitor (Enzo Life Sciences, NY, USA), and 1% penicillin/streptomycin (Gibco). Subsequently, 100 μL of Matrigel (Corning, NY, USA) was mixed with the remaining 50 μL of the suspension to establish organoids, and this mixture was allowed to solidify in a pre-warmed well of a 6-well culture plate (Corning) at 37 °C for 10 min. After the gel solidified, 3 mL of MBM was added to the well, with medium changes occurring every 3–4 days. Organoids were passaged every 1–3 weeks. For passaging, organoids contained in solidified Matrigel were collected in cold DPBS and centrifuged at 112 × g for 3 min at 4 °C. The pellets were washed in cold DPBS, then centrifuged again at 250 × g for 15 min at 4 °C. The washed organoids were then resuspended in 2 mL of TrypLE Express (Invitrogen) and incubated for 10 min at 37 °C to facilitate dissociation. Following dissociation, 10 mL of DMEM/F12 with 10% FBS was added, and the mixture was centrifuged at 112 × g for 3 min. After centrifugation, the pellets were washed with DPBS and again centrifuged at 112 × g for 3 min. Finally, the pellets were resuspended in a MBM + Matrigel mixture (1:3 ratio) and reseeded at ratios of either 1:3 or 1:4 to form new organoids.
Whole transcriptome sequencing and data processing
Lung cancer organoids (LCO) (> passage 3) cultured in 4–6 wells of 24-well culture plates (Corning) were utilized for RNA extraction. To collect the LCOs, solidified Matrigel drops containing the LCOs were harvested in cold DPBS, transferred to a conical tube, and centrifuged at 112 × g for 3 min at 4 °C. The resulting pellet was washed with cold DPBS and subsequently centrifuged at 250 × g for 15 min at 4 °C. Total RNA was then extracted from the pellets utilizing the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s guidance. A cDNA library was prepared using the TruSeq RNA Access Library Prep Kit (Illumina, Inc., CA, USA) with 1 mg of total RNA. All samples met the cDNA library quality assurance (minimum requirement: > 5 nM). Subsequently, 100-nt paired-end sequencing was conducted on the HiSeq 2500 (Illumina, Inc., CA, USA). Total RNA sequencing followed the protocols as previously described24.
Drug screening
LCOs, after being cultured in 24-well plates for 2 weeks, were harvested and dissociated using TrypLE Express. The dissociated LCOs were then combined with MBM + Matrigel in a 1:3 ratio and plated onto 96-well white plates (10 μl of 2 × 103 cells per well; Corning). Following gelation, 100 µl MBM was added to each well. The LCOs were cultured for an additional 10–14 days. Subsequently, 6 concentrations of pevonedistat, SZL-P1-41, and DMSO controls were administered every 3 days in triplicate. On the sixth day, the medium was replaced with 100 μl of fresh MBM per well to ascertain cell viability, and 100 μl of CellTiter-Glo (Promega) was added to each well. Plates were agitated for 30 min at RT before conducting luminescence readings. IC50 values were determined using Graph Pad Prism.
For cell counting kit-8 (CCK-8) assay, the cells were seeded onto 96-well plate (2.5 × 103 cells per well). After 24 h, pevonedistat, SZL-P1-41, and DMSO controls were administered in 96-well plate. Then, 96-well plates were incubated in a 37 °C/5% CO2 humidified incubator for 2 days or 3 days. Ten microliters of CCK-8 solution (MedChemExpress, HY-K0301) added to the 96-well plates. The plates were swirled and maintained in a 37 °C/5% CO2 humidified incubator. After 2 h, the plates were agitated at RT and absorbance of the plates were measured at 450 nm using microplate reader (Molecular Devices, SpectraMax iD3). IC50 values were determined using Graph Pad Prism.
Flow cytometry analysis
Cells were seed 6-well plate prior to treat the CRL-targeting drugs (MLN4924, SZL-P1-41). After 24 h, drugs add to cells and incubate at 37 °C in a moist atmosphere of 5% CO2 for 48 h. Cells were pulse-labeled with 10 μM EdU (5-ethynyl-2’-deoxyuridine) for 1 h. Cells were harvested with DPBS washing. EdU staining was performed according to the manufacturer’s protocol (Invitrogen, C10424). For staining of DNA, Nuclear Green DCS1 was used (Abcam. 138,905). An FACSymphony A3 cell analyzer (BD Biosciences) and FlowJo software were used for analyses.
Results
RepID transcript is upregulated in NE and SCLC cancer
We first analyzed the correlation of the DCAF family across various human cancer cell lines using the CCLE via the CellMinerCDB database. Among the over 100 members of DCAF, we selected 26 DCAFs that are physically associated with CUL4A and CUL4B, as indicated in previous studies25. Compared to other DCAFs, the expression of RepID transcripts showed a high correlation with the NE signature (Fig. 1a). In addition, higher RepID expression was noted in cancer cells expressing NE markers, including CHGA and DPYSL5 (Fig. 1b). By analyzing the average values across all tissues, we confirmed a high correlation between RepID expression and the NE signature in liver, lung, thyroid, CNS, and bone tissues (Fig. 1c). Consistent with the predominance of SCLC in NET, RepID expression was found to be higher in SCLC compared to other types of lung cancer (Fig. 1d). We further categorized lung cancer based on NE signatures and observed high levels of RepID transcripts in all lung cancer types exhibiting NE signatures (Fig. 1d). Interestingly, non-NE SCLC cells also demonstrated significantly elevated levels of RepID expression compared to other non-NE lung cancer cells (Fig. 1d). To validate our in silico analysis, we conducted quantitative real-time PCR on a set of 13 representative cancer cell lines, which included six SCLC (NE-SCLC: H69, H146, H209, and H82; non-NE SCLC: H211 and DMS114), other non-NE cancer cell lines from different origins such as 1 NSCLC (H1299), 1 osteosarcoma (U2OS), 1 colon cancer (HCT116), 1 erythroleukemia (K562), 1 prostate cancer (DU145), and 2 immortalized human normal cell lines (lung fibroblast VA13/WI38 and human embryonic kidney HEK293). Consistent with our findings, RepID transcripts were generally higher in SCLC than in the others, with the exception of H82, known as the NEUROD1-positive SCLC (Fig. 1e). These results suggest that high levels of RepID could potentially serve as biomarkers for NET and SCLC.
Fig. 1.

RepID transcript is highly expressed in NET and SCLC. (a) RepID expression is highly correlated with the NE signature. The expression of DCAF transcripts was compared with cancer signatures using the Cancer Cell Line Encyclopedia (CCLE) database through CellMinerCDB (n = 1036 in all cancers) (NE: neuroendocrine, HRD_LOH: Homologous recombination deficiency score based on LOH regions, NtAI: Number of telomeric imbalances, LST: Large scale transition, HRD_SUM: Sum of HRD_LOH, NtAI, and LST homologous recombination deficiency signature scores, EMT: 1st principal component of non-hematopoietic cell line expression data matrix for 38 EMT genes, APM: Antigen presentation machinery transcript expression signature score). (b) Correlation analysis between selected DCAF and NE marker expression scores (Exp). (c) High correlation of RepID-NE signature in lung tissue. (d) RepID expression was analyzed in NE/non-NE cancer or SCLC/NSCLC and SCLC NAPY (*p-value < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s.; not significant). (e) RepID transcripts are higher in SCLC compared to other cell lines. The level of transcripts was confirmed by qRT-PCR using three different primer sets across 13 cell lines, including SCLC (*p-value < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s.; not significant).
RepID protein is upregulated in NE and SCLC cancer
To confirm if the observed increase in mRNA levels of RepID corresponded with an increase in protein levels, immunohistochemistry (IHC) was performed using tissue samples from lung cancer patients. These tissues were categorized based on pathological diagnoses and analyzed through the intensity measurement of fluorescence. Lung adenocarcinoma (LUAD), lung squamous carcinoma (LUSC), mixed (LUAD + LUSC), bronchioloalveolar carcinoma (BAC), and large cell carcinoma (LCC) exhibited lower levels of RepID protein compared to normal tissues (Fig. 2a, b). In contrast, the mean RepID intensity in SCLC-containing tissues was approximately three times that of normal tissue (Fig. 2a, b). Our immunoblotting analysis, conducted on various cancer cell lines, demonstrated that RepID protein expression is generally higher in SCLCs, particularly in ASCL1-type SCLCs such as H69 and H146 (Fig. 2c). The localization and intensity of RepID by immunofluorescence (IF) were further investigated in cells H69, H146, DMS114, and H82, which vary in RepID expression levels. Consistent with RepID’s reported role in recruiting CRL4 to chromatin/chromosomes, its localization was primarily nuclear, and the intensity was higher in all tested SCLC cell lines except for H82 (Fig. 2d). These findings suggest that RepID may serve as a potential biomarker for NET and SCLC.
Fig. 2.

RepID is upregulated in NET and SCLC at the protein level. (a, b) The slide containing tissues from a patient (lung cancer, normal) was analyzed by immunofluorescence with anti-RepID (red) and DAPI (blue). Representative images separated by pathology are shown (a). The scale bar represents 20 μm. The quantification of intensity was performed using ImageJ software (*p-value < 0.05, **p < 0.01, ***p < 0.001, n.s.; not significant) (b). (c, d) RepID protein is highly expressed in subsets of NET and SCLC cancer cell lines. Immunoblot analysis using antibodies against RepID was conducted on 15 cell lines (upper panel), and the quantification of RepID protein expression was represented as relative density by normalizing the RepID level in the U2OS cell line. The average RepID level is indicated (lower panel). Alpha-tubulin was used for normalization and as a loading control (*p-value < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s.; not significant) (c). The indicated SCLC cell lines were post-extracted and stained with anti-RepID (green), and DAPI (blue). The scale bar indicates 10 μm (d).
RepID expression modulates drug sensitivity targeting CRL complex
RepID facilitates cell proliferation by inhibiting apoptosis and over-replication or by aiding the transition from metaphase to anaphase during mitosis11,16. Anticipating that lower RepID levels would lead to reduced proliferation, we developed RepID knockout cells using the CRISPR/Cas9 system and conducted colony formation assays. In SCLC cells H69, H146, and DMS114, which express high levels of RepID, growth rates were slightly reduced following RepID depletion, whereas the proliferation of H82 cells, which have low RepID expression, remained unchanged (Fig. 3a). Consistent with above result, we observed moderate correlation between the expression of RepID and Ki67 in SCLC cell line as well as in patient dataset (Supplementary Fig. 2a–c). We also examined whether the chromatin-bound CRL4 content varied based on RepID expression levels. H69 cells exhibited higher amounts of chromatin-bound CUL4A and CUL4B compared to H82 cells (Fig. 3b, Mock lane). RepID-depleted H69 cells showed a significant decrease in chromatin loading of CUL4A and CUL4B, alongside an increase in chromatin-bound CUL1, a compensatory mechanism in lieu of the CRL4 complex (Fig. 3b, H69 panel). Similarly, RepID-overexpressing H82 cells showed higher levels of CRL4 complex recruitment to chromatin, but chromatin-bound CRL1 also increased unexpectedly (Fig. 3b, H82 panel). These results underscore the critical role of RepID as a structural receptor influencing the chromatin loading of the CRL complex in SCLC.
Fig. 3.

RepID expression modulates drug sensitivity targeting CRL complex. (a) Knockout of RepID slightly decreases proliferation. CRISPR-CAS9 for RepID depletion was introduced into 4 SCLC cell lines (H69, H82, H146, and DMS114), and cells were seeded in soft agar and stained with crystal violet (left panel). The proliferation rate was quantified by the covered area (right-lower panel) (*p-value < 0.05, **p < 0.01, n.s.; not significant). Decreased RepID protein was confirmed by immunoblotting (right-upper panel). (b) RepID expression correlates with the amounts of chromatin-bound CRL4 complex. Whole cell lysate (WCL) and chromatin-bound (CB) fractions were separated from wild-type or RepID knockout/overexpressed cells. Alpha-tubulin and Histone H3 were used as loading controls. The numbers under the immunoblot represent the relative density ratio of each protein normalized with H3 density. (c–f) Changes in RepID protein levels correlate with sensitivity to CRL-targeting drugs, including pevonedistat and SZL-P1-41. Cells were incubated with or without pevonedistat for 2 days (c) or SZL-P1-41 for 3 days (e) and had their cell viability measured using the CCK8 assay. Relative viability was based on controls from three independent experiments. The sensitivity of the SCLC-organoid model to pevonedistat (d) or SZL-P1-41 (f) was assessed by luminescence-based measurements of viability.
We subsequently assessed the responsiveness of both H69 and H82 cells to anti-cancer drugs that target the CRL complex. RepID-depleted H69 cells exhibited diminished CDT1 accumulation upon pevonedistat treatment, a selective NEDD8-activating enzyme inhibitor, compared to those with intact RepID (Supplementary Fig. 1A). Conversely, RepID-overexpressing H82 cells showed significant CDT1 accumulation following pevonedistat treatment (Supplementary Fig. 1B). Corroborating these findings, subsequent CCK8 cell proliferation and luminescence-based analyses utilizing cells and SCLC-organoids (high RepID: 15LT-002 vs low RepID: 18LT-016) indicated a positive correlation between high levels of RepID expression and increased sensitivity to pevonedistat (Fig. 3c, d, Supplementary Figs. 1C, 2D, E, H and I).
Given the reduced chromatin-bound CRL4 caused by the loss of RepID, which primarily compensates through CRL1 function, we hypothesized an alteration in sensitivity to SZL-P1-41, a SKP2 inhibitor acting as a substrate receptor in CRL1. While H69 cells with depleted RepID showed no notable change in CDT1 accumulation, wild-type H82 cells exhibited a slight increase in CDT1 compared to RepID-overexpressing H82 cells (Supplementary Fig. 1D and E). SZL-P1-41 sensitivity was markedly demonstrated by CCK8 cell proliferation and luminescence-based analysis using cells and SCLC-organoids, with higher sensitivity observed in cells and SCLC-organoids expressing lower RepID levels (Fig. 3e, f, Supplementary Figs. 2F, G, H and I). Collectively, these results propose RepID expression as a potential biomarker for NETs and SCLC, suggesting that the level of RepID expression could be a critical standard for therapeutic strategies targeting NETs and SCLC with cullin-targeting drugs.
Discussion
SCLC is a type of lethal cancer characterized by low overall survival rates in patients, with most cases classified as neuroendocrine tumors. The diagnosis of NET is challenged by varying responsiveness of markers such as CHGA, SYP, and INSM1, which depend on the molecular characteristics of NET, including NAPY. The evidence provided here demonstrates that RepID could serve as an crucial biomarker for NET and SCLC. In silico analysis via the CellMinerCDB portal elucidated the relationship between NE signatures and RepID transcripts. Moreover, high expression levels of RepID protein and mRNA in patient tissues and/or cell lines, particularly in ASCL1-type SCLC, were observed. Viability analyses indicated that sensitivity to CRL-targeting anti-cancer drugs (pevonedistat, SZL-P1-41) varied with RepID expression levels. Our findings support the potential role of RepID in diagnosing NET and SCLC.
In this study, we demonstrated that RepID transcripts were upregulated in most SCLC cell lines compared to other cell types. It has been reported that the ASCL1 and NeuroD1 types of SCLC show neuroendocrine features and high expression of neuroendocrine markers, such as INSM126,27. However, our findings revealed that the H82 cell line, which is classified under the NeuroD1 type, exhibited low expression of both RepID mRNA and protein. Previous studies have scored lung cancer specific NE signature genes to differentiate NE/non-NE subclasses of SCLC28,29. The H82 cell line was found to have a low NE signature score, while the H446 cell line of the same NeuroD1 type showed a high NE signature score28,29. This is in alignment with our observations that H446, which has a high NE signature, demonstrates greater RepID protein expression compared to H82.
We proposed RepID as a neuroendocrine biomarker due to its high expression, but its relevance in other cancer types remains to be confirmed. For instance, previous studies have recognized RepID as a predictive marker, a mediator, and a classifier of metastasis in melanoma22. Our database analysis supports these findings, indicating a relatively high correlation between RepID expression and the EMT signature, albeit lower than with the NE signature. Consequently, it may be necessary to incorporate other markers alongside RepID. Our earlier work identified CRL3 as a biomarker; additionally, BTB, a component of CRL3, and transcription factors that regulate BTB genes were found to be upregulated in NET and SCLC30. We propose utilizing a combination of RepID with either CRL3 or BTB to enhance the specificity of NET and SCLC diagnostics. A notable commonality between this and previous studies is the involvement of CRLs, which regulate approximately 20% of proteins involved in cellular functions31,32. Thus, research supports the mostly upregulated status of CRLs through deregulation across various cancer types31. Altogether, we suggest that CRL and its component receptors represent promising neuroendocrine markers and could lead to new therapeutic strategies through further research.
We propose the following model for the role of upregulated RepID as a biomarker in NET and SCLC (Fig. 4). NET or SCLC characterized by upregulated RepID predominantly contribute to an increase chromatin-bound CRL4 and show sensitivity to pevonedistat, unlike SZL-P1-41. Conversely, compensatory activities of CRL1 are crucial in non-NET or NSCLC with downregulated RepID, which increases sensitivity to the CRL1 inhibitor SZL-P1-41. In conclusion, our study (1) confirms the utility of RepID as a neuroendocrine marker, and (2) lays the groundwork for developing novel therapeutic strategies that induce cancer cell death by targeting RepID directly or by combining pevonedistat in phase III clinical trials with conventional anti-cancer drugs.
Fig. 4.
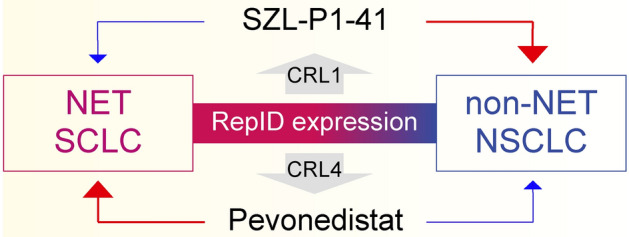
Schematic model. (a) The schematic model depicts RepID as a putative NET and SCLC biomarker. High expression of RepID is positively correlated with pevonedistat sensitivity by recruiting the CRL4 complex to chromatin, whereas NSCLC or non-NET expressing low levels of RepID sensitizes to SZL-P1-41 by increasing compensation of CRL1 function instead of CRL4.
Supplementary Information
Acknowledgements
This project received support from the “Regional Innovation Strategy (RIS)” through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (MOE) under the Grant Number 2021RIS-001. Additionally, it was supported by a grant from the National Research Foundation of Korea (NRF), funded by the Korean government (MSIT) under the grant number NRF-2021R1C1C1004165, and in part by the intramural programs of the Center for Cancer Research, National Cancer Institute (1ZIABC010411 to MIA).
Author contributions
S.M.J., S.K. and J.U.P. designed the study. J.U.P., J.H.J. and Y.S. performed experiments. S.M.J., J.U.P., C.E.R., M.I.A., Y.S. and S.J. processed the experimental data, performed the analysis, generated figures, and wrote the manuscript with support from all authors. All authors read and approved the final manuscript.
Data availability
All data are contained within the article and supporting information. Whole transcriptome sequencing has been deposited on GEO (https://www.ncbi.nlm.nih.gov/geo; accession no. GSE171682).
Declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-79104-9.
References
- 1.Rindi, G. et al. Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocr. Pathol.33(1), 115–154 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Basu, B., Sirohi, B. & Corrie, P. Systemic therapy for neuroendocrine tumours of gastroenteropancreatic origin. Endocr. Relat. Cancer17(1), R75-90 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Oronsky, B., Ma, P. C., Morgensztern, D. & Carter, C. A. Nothing but NET: A review of neuroendocrine tumors and carcinomas. Neoplasia19(12), 991–1002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisseler-Eckhoff, A. & Demes, M. Neuroendocrine tumors of the lung. Cancers (Basel)4(3), 777–798 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rekhtman, N. Neuroendocrine tumors of the lung: An update. Arch. Pathol. Lab. Med.134(11), 1628–1638 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Byers, L. A. & Rudin, C. M. Small cell lung cancer: Where do we go from here?. Cancer121(5), 664–672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rekhtman, N. Lung neuroendocrine neoplasms: Recent progress and persistent challenges. Mod. Pathol.35(Suppl 1), 36–50 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim, K. B., Dunn, C. T. & Park, K. S. Recent progress in mapping the emerging landscape of the small-cell lung cancer genome. Exp. Mol. Med.51(12), 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin, J., Arias, E. E., Chen, J., Harper, J. W. & Walter, J. C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell.23(5), 709–721 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Morgan, M. A. J. et al. A cryptic tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev.31(19), 2003–2014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang, S. M. et al. The replication initiation determinant protein (RepID) modulates replication by recruiting CUL4 to chromatin. Nat. Commun.9(1), 2782 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman, K. E. et al. Sequential replication-coupled destruction at G1/S ensures genome stability. Genes Dev.29(16), 1734–1746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senga, T. et al. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem.281(10), 6246–6252 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Sansam, C. L. et al. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev.20(22), 3117–3129 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pozo, P. N. & Cook, J. G. Regulation and function of Cdt1; a key factor in cell proliferation and genome stability. Genes (Basel)8(1), 2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang, S. M. et al. The RepID-CRL4 ubiquitin ligase complex regulates metaphase to anaphase transition via BUB3 degradation. Nat. Commun.11(1), 24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang, S. M., Redon, C. E., Fu, H. Q., Indig, F. E. & Aladjem, M. I. RepID-deficient cancer cells are sensitized to a drug targeting p97/VCP segregase. Mol Cell Toxicol.17(2), 141–149 (2021). [Google Scholar]
- 18.Farhang-Fallah, J. et al. The pleckstrin homology (PH) domain-interacting protein couples the insulin receptor substrate 1 PH domain to insulin signaling pathways leading to mitogenesis and GLUT4 translocation. Mol. Cell. Biol.22(20), 7325–7336 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li, S. et al. The full-length isoform of the mouse pleckstrin homology domain-interacting protein (PHIP) is required for postnatal growth. FEBS Lett.584(18), 4121–4127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Podcheko, A. et al. Identification of a WD40 repeat-containing isoform of PHIP as a novel regulator of beta-cell growth and survival. Mol. Cell. Biol.27(18), 6484–6496 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tokuda, E. et al. Casein kinase 2-interacting protein-1, a novel Akt pleckstrin homology domain-interacting protein, down-regulates PI3K/Akt signaling and suppresses tumor growth in vivo. Cancer Res.67(20), 9666–9676 (2007). [DOI] [PubMed] [Google Scholar]
- 22.De Semir, D. et al. Pleckstrin homology domain-interacting protein (PHIP) as a marker and mediator of melanoma metastasis. Proc. Natl. Acad. Sci. USA109(18), 7067–7072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang, Y. et al. A replicator-specific binding protein essential for site-specific initiation of DNA replication in mammalian cells. Nat. Commun.7, 11748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho, E. J. et al. Immuno-genomic classification of colorectal cancer organoids reveals cancer cells with intrinsic immunogenic properties associated with patient survival. J. Exp. Clin. Cancer Res.40(1), 230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reichermeier, K. M. et al. PIKES analysis reveals response to degraders and key regulatory mechanisms of the CRL4 network. Mol. Cell.77(5), 1092–1106 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Rudin, C. M. et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer19(5), 289–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baine, M. K. et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: A comprehensive immunohistochemical and histopathologic characterization. J. Thorac. Oncol.15(12), 1823–1835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, W. et al. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl. Lung Cancer Res.7(1), 32–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pongor, L. S. et al. Integrative epigenomic analyses of small cell lung cancer cells demonstrates the clinical translational relevance of gene body methylation. iScience.25(11), 105338 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park, J. U. et al. The differentially expressed gene signatures of the Cullin 3-RING ubiquitin ligases in neuroendocrine cancer. Biochem. Biophys. Res. Commun.636(Pt 2), 71–78 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang, S. M., Redon, C. E. & Aladjem, M. I. Chromatin-bound cullin-ring ligases: Regulatory roles in DNA replication and potential targeting for cancer therapy. Front. Mol. Biosci.5, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang, S. M., Redon, C. E., Thakur, B. L., Bahta, M. K. & Aladjem, M. I. Regulation of cell cycle drivers by Cullin-RING ubiquitin ligases. Exp. Mol. Med.52(10), 1637–1651 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and supporting information. Whole transcriptome sequencing has been deposited on GEO (https://www.ncbi.nlm.nih.gov/geo; accession no. GSE171682).