

Abstract
Objective
Genetic syndromes (GSs) are often linked to congenital heart disease (CHD) and cardiomyopathy (CM). The effect of GSs on survival following pediatric heart transplant (HT) has not been well described. We aimed to compare outcomes following HT between children with a GS and those without a GS.
Methods
The United Network for Organ Sharing (UNOS) transplantation database was merged with the Pediatric Health Information System (PHIS) administrative database to identify children with GS who underwent HT between 2009 and 2019. Characteristics and outcomes were compared between children with a GS (GS group) and those without a GS (no GS group).
Results
GSs were present in 225 of 2429 HT recipients (9%). The most common GSs were DiGeorge syndrome (n = 28), muscular dystrophy (n = 27), Down syndrome (n = 26), and Turner syndrome (n = 14). The incidence of CHD was higher in the GS group compared to the no GS group (54% vs 38%; P < .1); however, patient demographics, hemodynamics, renal and hepatic dysfunction, and requirements for dialysis, mechanical ventilation, extracorporeal membrane oxygenation, and mechanical circulatory support were not different between the 2 groups. Time on the waitlist was not significantly different between the GS and no GS groups (55 days vs 53 days; P = .4). There also was no between-group difference in the incidence of post-transplantation complications, including dialysis (8% vs 5%; P = .38), stroke (3% vs 4%; P = .34), primary graft dysfunction (2% vs 2%; P = .75), need for pacemaker (1% vs 1%; P = .84) and rejection (3.4% vs 3.4%; P = .96). Survival at 10 years post-HT was 75% for the no GS group and 72% for the GS group (P = .59). The survival curves also did not differ between patients with CM and those with CHD.
Conclusions
Children with certain GSs and end-stage heart failure can be expected to have similar post-transplantation outcomes to those without a GS. Although early and late post-transplantation care is individualized to each patient, the presence of a GS should not influence the decision to list for HT.
Key Words: pediatric, heart transplant, outcomes/database, genetic abnormalities, syndrome
Graphical Abstract

Survival after pediatric HT is not significantly different with selected genetic syndromes.
Central Message.
Postoperative pediatric heart transplant recovery, complications, rejection, early and late survival, and risk of retransplantation are not significantly different between children with selected genetic syndromes and those without such syndromes.
Perspective.
Genetic syndromes can complicate congenital heart surgery and affect recovery, morbidity, and survival. The effects of these syndromes on pediatric heart transplant outcomes have not been well studied. Merging the United Network for Organ Sharing transplantation database with the Pediatric Health Information System database allowed for a better understanding of the effect of genetic syndromes on heart transplant outcomes.
See Discussion on page 288.
Genetic syndrome (GSs) are frequently linked to both congenital heart disease (CHD) and cardiomyopathy (CM) in children.1, 2, 3, 4 Pediatric heart transplant (HT) is now a widely accepted therapy for patients with end-stage heart failure.5 As more children with GSs develop end-stage heart disease and are considered for pediatric HT, there is growing interest in post-transplantation outcomes for these patients. Although there are published data on patients with GSs after HT from single centers, this population remains understudied.
The purpose of the present study was to compare post-HT outcomes of pediatric patients with a GS and those without a GS. We hypothesized that the 2 groups of patients would have similar post-transplantation outcomes.
Methods
This study was approved by the University of Louisville Institutional Review Board (approval 22.0617, August 4, 2022).
Study Population
The United Network for Organ Sharing (UNOS) transplantation database was merged with the Pediatric Health Information System (PHIS) administrative database to identify children with a major GS who underwent pediatric HT between 2009 and 2019. Given the UNOS database liminations and lack of granularity, the merger was performed in order to allow identification of children with a GS. The PHIS database identified encounter diagnoses and procedures based on International Classification of Diseases (ICD) Ninth and Tenth Revision codes, providing more granularity in terms of the presence of selected GS. The merging process was as follows. The PHIS database was accessed and queried between 2008 and 2021 for patients undergoing HT and with a prior diagnosis of 1 of 10 selected and a defined GS most commonly associated with the need for HT. The UNOS thoracic transplant database also was queried for patients age <18 years between 2008 and 2021. Patients who were age ≥18 years or who underwent transplant before 2009 or after 2019 were excluded. The UNOS and PHIS databases were then merged by matching center name and transplant date. A secondary match was performed using patient age and sex. Patients with heterotaxy syndrome also were excluded from the final data set. The final study cohort was then divided into the GS and no GS groups.
Study Purpose and Definitions
The purpose of this study was to compare post-HT outcomes in pediatric patients with a GS and a contemporaneous control of patients without a GS. Risk factors affecting survival, retransplantation, and rejection also were assessed. The GSs were defined by the following ICD-9 or -10 codes: Down syndrome, DiGeorge syndrome, Turner syndrome, Noonan syndrome, Danon disease, Barth syndrome, muscular dystrophy, Marfan syndrome, very long-chain acyl-CoA dehydrogenase deficiency, primary creatinine deficiency, and other diagnoses, including GSs not classified as specific diagnoses using ICD codes owing to limitations of the ICD codes themselves.
Demographic information, including age, sex, race, height and weight, and pretransplantation cardiac diagnosis, were collected. Data also were collected on pretransplantation hemodynamics, laboratory data, and use of dialysis, mechanical ventilation, extracorporeal membrane oxygenation (ECMO), or a ventricular assist device (VAD). Donor ischemia time and waitlist time also were determined. Post-transplantation data collected included survival, postoperative hospital length of stay, as well as diagnosis of primary graft dysfunction, stroke, rejection, and need for pacemaker, dialysis, or retransplantation.
Analysis
Nonparametric univariate statistical methods were initially used to evaluate baseline characteristics between the 2 groups. Continuous variables were analyzed using the Kruskal-Wallis test, and categorical variables were analyzed using the χ2 test. Kaplan-Meier survival curves were computed to evaluate the survival, and the log-rank test was used to detect any difference between the groups. Survival curves stratified by the etiology of heart failure and presence of genetic abnormality were calculated. A multivariable regression model was generated for the GS group to identify factors associated with mortality at 1 year post-transplantation. Data are presented as median (interquartile range [IQR]) or number (%). Statistical analyses were performed using SAS 9.4 software (SAS Institute). A 2-tailed 5% α level was considered to indicate statistical significance.
Results
Demographics and Clinical Characteristics
Baseline demographics and clinical characteristics of the GS and no GS groups at the time of transplant are presented in Table 1. A total of 2429 patients underwent HT during the study period, of whom 225 (9%) were diagnosed with a GS. The median age at transplantation was 5 years (IQR, 0-13 years) for both the GS and no GS groups. The percentage of patients with a pretransplantation diagnosis of CHD was higher in the GS group (54% vs 38%; P < .01). The most common GSs were DiGeorge syndrome (n = 28; 12%), muscular dystrophy (n = 27; 12%), Down syndrome (n = 26; 12%), and Turner/Noonan syndrome (n = 14; 6%), with other syndromes not specified (n = 107; 48%).
Table 1.
Patient demographics at HT
| Characteristic | No GS (N = 2204) | Confirmed GS (N = 225) | P value |
|---|---|---|---|
| Age, y, median (IQR) | 5 (0-13) | 5 (0-13) | .96 |
| Male sex, % | 54 | 58 | .32 |
| Race, % | .37 | ||
| White | 55 | 55 | |
| Black | 17 | 18 | |
| Other | 28 | 27 | |
| Height, cm, median (IQR) | 107 (68-154) | 100 (66-146) | .31 |
| Weight, kg, median (IQR) | 17 (8-46) | 18 (8-41) | .35 |
| Body mass index, median (IQR) | 17 (15-20) | 17 (15-19) | .66 |
| Blood group, % | .15 | ||
| A | 37 | 31 | |
| B | 14 | 18 | |
| AB | 45 | 48 | |
| O | 4 | 3 | |
| Underlying diagnosis, % | <.01 | ||
| Congenital heart disease | 38 | 54 | |
| Cardiomyopathy | 62 | 46 |
Significant P value for the difference between patients who have congenital heart disease and those who have cardiomyopathy. HT, Heart transplant; GS, genetic syndromes; IQR, interquartile range.
Patients’ clinical characteristics at the time of transplantation are shown in Table 2. There were no statistically significant differences between the no GS and GS groups at the time of transplantation in terms of mean pulmonary artery pressure (22 mm Hg in the no GS group vs 20 mm Hg in the GS group; P = .93), serum creatinine level (0.4 vs 0.4; P = .44), total bilirubin level (0.5 vs 0.6; P = .17), or use of dialysis (2.7% vs 4.0%; P = .44). There was no statistically significant difference between the 2 groups in the need for mechanical ventilation (15% in the no GS group vs 16% in the GS group; P = .77), ECMO (3.4% vs 2.2%; P = .43), LVAD (23% vs 18%; P = .26), and inotropic agents (52% vs 47%; P = .19), as well as time on the HT waitlist (53 days vs 55 days; P = .4).
Table 2.
Patient clinical characteristics at HT
| Characteristic | No GS (N = 2204) | Confirmed GS (N = 225) | P value |
|---|---|---|---|
| PAP, mm Hg, median (IQR) | 22 (17-30) | 20 (17-31) | .93 |
| Creatinine, mg/dL, median (IQR) | 0.4 (0.3-0.6) | 0.4 (0.3-0.6) | .44 |
| Dialysis, % | 2.7% | 4.0% | .29 |
| Bilirubin, mg/dL, median (IQR) | 0.5 (0.3-1.0) | 0.6 (0.3-1.0) | .17 |
| Mechanical ventilation, % | 15% | 16% | .77 |
| EMCO, % | 3.4% | 2.2% | .43 |
| VAD, % | 23% | 18% | .26 |
| Inotropes, % | 52% | 47% | .19 |
| Donor ischemia time, min, median (IQR) | 3.5 (2.9-4.1) | 3.5 (3.0-4.1) | .49 |
| Waitlist time, d, median (IQR) | 53 (20-113) | 55 (20-135) | .40 |
HT, Heart transplant; GS, genetic syndrome; PAP, pulmonary artery pressure; IQR, interquartile range; ECMO, extracorporeal membrane oxygenation; VAD, ventricular assist device.
Post-Transplantation Outcomes
The length of hospital stay after HT was longer in the GS group compared with the no GS group (median, 21 [IQR, 14-40] days vs 19 [IQR, 13-33] days; P = .03). However, the incidence of post-transplantation in-hospital complications was not significantly different between the 2 groups. This included the need for postoperative dialysis (8% in the no GS group vs 5% in the GS group; P = .38), stroke (3% vs 4%; P = .34), primary graft dysfunction (2% vs 2%; P = .75), need for pacemaker (1% vs 1%; P = .84), and graft rejection (3.4% vs 3.4%; P = .96). There remained no difference between the 2 groups when rejection was evaluated by the frequency of events prior to hospital discharge (12% vs 13%; P = .32) and within the first-year post-transplantation (15% vs 18%; P = .21) (Figure 1).
Figure 1.

Frequency of post-transplantation complications showing no statistically significant difference between the genetic syndrome (GS) and no GS groups.
Post-Transplantation Survival
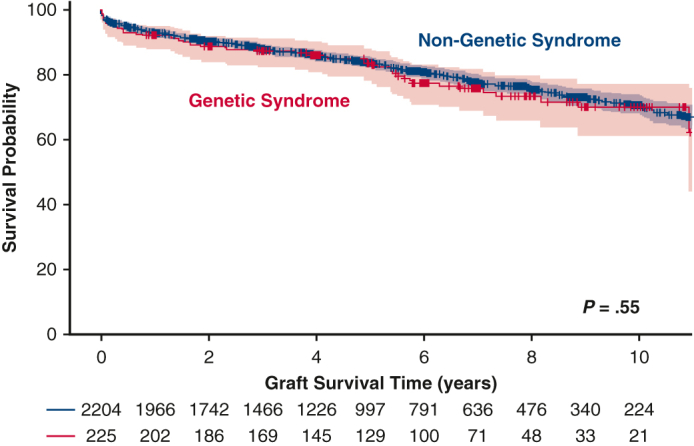
Initial 30-day mortality was not different between the 2 groups (3% for the no GS group vs 4% for the GS group; P = .36). Overall post-HT survival at 10 years also was not statistically significantly different between the 2 groups (70% vs 71%; P = .55) (Figure 2). When patients were grouped by underlying heart condition, either CHD or CM, the 10-year survival remained similar in the no GS and GS groups (Figure 3). The rate of retransplantation at 5 years post-transplantation also did not differ between the 2 groups (Figure 4).
Figure 2.

Kaplan-Meier survival curves comparing the genetic syndrome (GS) and no GS groups showing no statistically significant differences in freedom from death or retransplant at 10 years post-transplantation.
Figure 3.

Kaplan-Meier survival curves for the genetic syndrome (GS) and no GS groups for patients congenital heart disease (CHD) or cardiomyopathy (CM) prior to heart transplant (HT). The survival curves with 95% confidence intervals show no significant differences in freedom from death or retransplantation at 5 years post-HT between the CHD and CM patients in the GS and no GS groups.
Figure 4.

Kaplan-Meier survival curves for the genetic syndrome (GS) and no GS groups comparing rates of survival, death, and retransplantation after initial heart transplant (HT) showing no statistically significant differences at 5 years post-HT.
The 5-year survival following pediatric HT for selected GSs is shown in Figure 5. Survival was lower in children with DiGeorge syndrome compared to those with Down syndrome, muscular dystrophy, or Turner/Noonan syndrome; however, the difference was not statistically significant (P = .12).
Figure 5.

Kaplan-Meier survival curves for patients with the most common genetic syndromes—Turner/Noonan syndrome, Down syndrome, DiGeorge syndrome, and muscular dystrophy—showing no statistically significant differences among them.
To investigate factors related to 1-year mortality for the patients with a GS, a multivariate regression model was performed using the variables of age, sex, race, weight, body mass index, renal function (evaluated by glomerular filtration rate), total bilirubin, days on the transplantation waitlist, need for mechanical support prior to transplant, ABO blood type, and ischemic time of the transplanted graft. None of these clinical characteristics appeared to be related to mortality (Table 3).
Table 3.
Multivariable analysis
| Variable | OR | 95% CI | P value |
|---|---|---|---|
| Age (y) | 1.14 | 0.89-1.48 | .302 |
| Sex, male vs female | 2.48 | 0.76-8.13 | .133 |
| Race, black vs white | 0.69 | 0.13-3.64 | .798 |
| Race, other vs white | 0.30 | 0.05-1.65 | .257 |
| Weight (kg) | 0.91 | 0.82-1.02 | .097 |
| Body mass index | 1.29 | 0.99-1.67 | .060 |
| eGFR | 1 | 1-1.03 | .060 |
| Total bilirubin | 0.97 | 0.78-1.21 | .784 |
| Use of VAD | 0.22 | 0.02-1.91 | .168 |
| Waitlist duration (d) | 0.99 | 0.99-1.00 | .145 |
| ABO compatible vs identical | 0.48 | 0.11-2.20 | .245 |
| ABO incompatible vs identical | 2 | 0.19-21.30 | .387 |
| Ischemia time (h) | 0.69 | 0.38-1.28 | .241 |
OR, Odds ratio; CI, confidence interval; eGFR, estimated glomerular filtration rate; VAD, ventricular assist device.
Discussion
Nationwide, over a recent 10-year period, patients with a GS composed approximately 9% of the total pediatric population who underwent HT. These included patients with a heterogenous mix of underlying syndromes, including those with chromosomal anomalies, skeletal myopathies, mitochondrial disorders, and connective tissue disorders. Despite this, there were no differences in listing clinical characteristics, waitlist time, post-transplantation complications, or post-transplantation survival between patients with a GS and those without a GS. However, depending on the type of underlying syndrome, there did appear to be some differences in long-term survival between the 2 groups (Figure 6).
Figure 6.

Outcomes of pediatric heart transplantation in children with selected genetic syndromes are comparable to those without genetic syndromes.
Among children with CHD, those with an underlying GS have a higher risk of postoperative morbidity and mortality after surgical repair.6, 7, 8 However, there is limited information regarding pediatric HT waitlist times and HT outcomes for patients with an underlying GS. Various case reports and small single-center studies of patients with skeletal myopathies and connective tissues disorders undergoing HT or VAD implantation9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22 and 1 large-scale database study23 have shown similar post-HT outcomes for patients with muscular dystrophy compared to controls.23 Single-center and multicenter studies for patients with chromosomal anomalies24, 25, 26 have shown no difference in post-transplantation survival, although some data have suggested a potential increase in postoperative morbidities. Our study expands on these findings by linking 2 large-scale databases, UNOS and PHIS, to provide granular and diagnostic data to better elucidate waitlist and HT outcomes for the population of patients with a variety of syndromes. This has allowed us to compare findings both between groups and within groups.
In this study, we found that while postoperative complications were not significantly different between the no GS and GS groups, patients with a GS did have a longer hospital length of stay (median, 21 [IQR, 14-40] days vs 19 [IQR, 13-33] days; P = .03). However, although significant, this difference was small and might not be clinically relevant. Nonetheless, children with an underlying GS are known to have a higher burden of other congenital defects and health conditions that could lead to longer hospital stays.27 We also found a higher rate of CHD in the GS group compared to the no GS group (54% vs 38%; P < .01).
Interestingly, comparing survival outcomes for patients with different underlying GSs revealed differences in long-term survival. For example, patients with DiGeorge syndrome had worse 5-year survival compared to patients with other chromosomal abnormalities and those with muscular dystrophy. Previous studies have shown that although patients with DiGeorge syndrome may have more postoperative complications after initial CHD surgical repair, they do not have a higher mortality rate after surgery28 or a higher rate of complications and/or death after HT compared to patients without DiGeorge syndrome.29 However, both of those reports were from single-center studies, and further analysis of this finding is warranted.
This study is limited by the inherent factors in an observational retrospective database design. We intentionally selected patients with a broad range of GSs to reflect the HT listing practices nationwide. It is important to acknowledge that all GSs are unique and may have differing comorbidities that can affect post-HT outcomes. We were able to evaluate differences in survival outcomes by subgroup; however, given the study's retrospective observational database design, our ability to detect further differences between the groups was limited. Owing to the study's observational nature, it might not be adequately powered to detect differences between the groups; however, this premise is unmodifiable and recognized as a limitation. Further studies to analyze these differences are warranted.
Nevertheless, these data represent a multi-institutional perspective with less bias compared to single-center studies. Both the PHIS and UNOS are large administrative databases that rely on accurate coding and data entry by participating institutions. Inaccurate or incomplete ICD-9/10 coding, for example, may lead to misreporting and potentially could affect study analyses. This also could be a reason why the “other” syndrome diagnoses represent a significant proportion of our study population. This study included only patients with a confirmed diagnosis of GS recorded by an ICD-9/10 code in their medical record and also may include a more generic diagnosis. Reliance on this coding might not truly reflect a patient's phenotype, however. Of note, this study does not include patients who were evaluated for HT but not listed; thus, the results are generalizable only to patients with a GS deemed appropriate for HT listing.
In conclusion, children with underlying GSs account for 9% of the pediatric population undergoing HT. Children who have an underlying GS with end-stage heart failure and require listing for HT have similar post-transplantation complication rates and survival as children without a GS. Although all patients need individualized care post-HT, the presence of an underlying GS should not influence the decision to list for pediatric HT in patients deemed candidates for listing.
Webcast
You can watch a Webcast of this AATS meeting presentation by going to: https://www.aats.org/resources/outcomes-of-pediatric-heart-transplantation-in-children-with-major-genetic-syndromes-are-comparable-to-those-with-no-genetic-syndromes.

Conflict of Interest Statement
The authors reported no conflicts of interest.
The Journal policy requires editors and reviewers to disclose conflicts of interest and to decline handling or reviewing manuscripts for which they may have a conflict of interest. The editors and reviewers of this article have no conflicts of interest.
References
- 1.Hartman R.J., Rasmussen S.A., Botto L.D., et al. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol. 2011;32(8):1147–1157. doi: 10.1007/s00246-011-0034-5. [DOI] [PubMed] [Google Scholar]
- 2.Lara D.A., Ethen M.K., Canfield M.A., Nembhard W.N., Morris S.A. A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit Heart Dis. 2017;12(1):105–112. doi: 10.1111/chd.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scaglia F., Towbin J.A., Craigen W.J., et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–931. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- 4.Hermans M.C., Pinto Y.M., Merkies I.S., de Die-Smulders C.E., Crijns H.J., Faber C.G. Hereditary muscular dystrophies and the heart. Neuromuscul Disord. 2010;20(8):479–492. doi: 10.1016/j.nmd.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Singh T.P., Cherikh W.S., Hsich E., et al. The International thoracic organ transplant registry of the international society for heart and lung transplantation: twenty-fifth pediatric heart transplantation report-2022; focus on infant heart transplantation. J Heart Lung Transplant. 2022;41(10):1357–1365. doi: 10.1016/j.healun.2022.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simsic J.M., Coleman K., Maher K.O., Cuadrado A., Kirshbom P.M. Do neonates with genetic abnormalities have an increased morbidity and mortality following cardiac surgery? Congenit Heart Dis. 2009;4(3):160–165. doi: 10.1111/j.1747-0803.2009.00281.x. [DOI] [PubMed] [Google Scholar]
- 7.Michielon G., Marino B., Formigari R., et al. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann Thorac Surg. 2006;81(3):968–975. doi: 10.1016/j.athoracsur.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 8.Blue G.M., Mah J.M., Cole A.D., et al. The negative impact of Alagille syndrome on survival of infants with pulmonary atresia. J Thorac Cardiovasc Surg. 2007;133(4):1094–1096. doi: 10.1016/j.jtcvs.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Blagova O., Nedostup A., Shumakov D., Poptsov V., Shestak A., Zaklyasminskaya E. Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy: from ablation to heart transplantation. J Atr Fibrillation. 2016;9(4):1468. doi: 10.4022/jafib.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casazza F., Brambilla G., Salvato A., Morandi L., Gronda E., Bonacina E. Cardiac transplantation in Becker muscular dystrophy. J Neurol. 1988;235(8):496–498. doi: 10.1007/BF00314256. [DOI] [PubMed] [Google Scholar]
- 11.Dell'Amore A., Botta L., Martin Suarez S., et al. Heart transplantation in patients with Emery-Dreifuss muscular dystrophy: case reports. Transplant Proc. 2007;39(10):3538–3540. doi: 10.1016/j.transproceed.2007.06.076. [DOI] [PubMed] [Google Scholar]
- 12.Kichuk Chrisant M.R., Drummond-Webb J., Hallowell S., Friedman N.R. Cardiac transplantation in twins with autosomal dominant Emery-Dreifuss muscular dystrophy. J Heart Lung Transplant. 2004;23(4):496–498. doi: 10.1016/s1053-2498(03)00204-3. [DOI] [PubMed] [Google Scholar]
- 13.Oren D., Chau P., Manning M., et al. Heart transplantation in two adolescents with Danon disease. Pediatr Transplant. 2019;23 doi: 10.1111/petr.13335. [DOI] [PubMed] [Google Scholar]
- 14.Piccolo G., Azan G., Tonin P., et al. Dilated cardiomyopathy requiring cardiac transplantation as initial manifestation of Xp21 Becker type muscular dystrophy. Neuromuscul Disord. 1994;4(2):143–146. doi: 10.1016/0960-8966(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 15.Pick J.M., Ellis Z.D., Alejos J.C., Chang A.C. Rapidly progressive heart failure requiring transplantation in muscular dystrophy: a need for frequent screening. Cardiol Young. 2017;27(9):1836–1840. doi: 10.1017/s1047951117001251. [DOI] [PubMed] [Google Scholar]
- 16.Quinlivan R.M., Dubowitz V. Cardiac transplantation in Becker muscular dystrophy. Neuromuscul Disord. 1992;2(3):165–167. doi: 10.1016/0960-8966(92)90002-n. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann F.E.M., Schramm R., Hagl C., Eifert S., Pichlmaier M. Aortic root replacement and Berlin Heart EXCOR implantation in Marfan syndrome. Ann Thorac Surg. 2019;107:e187–e189. doi: 10.1016/j.athoracsur.2018.07.066. [DOI] [PubMed] [Google Scholar]
- 18.Knosalla C., Weng Y.G., Hammerschmidt R., et al. Orthotopic heart transplantation in patients with Marfan syndrome. Ann Thorac Surg. 2007;83(5):1691–1695. doi: 10.1016/j.athoracsur.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 19.Rao P., Keenan J.B., Rajab T.K., et al. Total artificial heart implantation in a young Marfan syndrome patient. Int J Artif Organs. 2018;41(3):175–177. doi: 10.1177/0391398817752297. [DOI] [PubMed] [Google Scholar]
- 20.Hollander S.A., Rizzuto S., Hollander A.M., et al. Obesity and premature loss of mobility in two adolescents with Becker muscular dystrophy after HeartMate II implantation. ASAIO J. 2016;62(1):e5–e7. doi: 10.1097/MAT.0000000000000292. [DOI] [PubMed] [Google Scholar]
- 21.Di Nora C., Miani D., D'Elia A.V., et al. Heart transplantation in Danon disease: long term single centre experience and review of the literature. Eur J Med Genet. 2020;63(2) doi: 10.1016/j.ejmg.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Visrodia P., Patel N.J., Burford M., et al. Heart transplantation in muscular dystrophy: single-center analysis. Clin Transplant. 2022;36(6) doi: 10.1111/ctr.14645. [DOI] [PubMed] [Google Scholar]
- 23.Wells D., Rizwan R., Jefferies J.L., et al. Heart transplantation in muscular dystrophy patients: is it a viable option? Circ Heart Fail. 2020;13(4) doi: 10.1161/CIRCHEARTFAILURE.118.005447. [DOI] [PubMed] [Google Scholar]
- 24.Carvajal H.G., Gooch C., Merritt T.C., et al. Midterm outcomes of heart transplantation in children with genetic disorders. Ann Thorac Surg. 2022;114(2):519–525. doi: 10.1016/j.athoracsur.2021.12.019. [DOI] [PubMed] [Google Scholar]
- 25.Broda C.R., Cabrera A.G., Rossano J.W., et al. Cardiac transplantation in children with Down syndrome, Turner syndrome, and other chromosomal anomalies: a multi-institutional outcomes analysis. J Heart Lung Transplant. 2018;37(6):749–754. doi: 10.1016/j.healun.2018.01.1296. [DOI] [PubMed] [Google Scholar]
- 26.Godown J., Fountain D., Bansal N., et al. Heart transplantation in children with Down syndrome. J Am Heart Assoc. 2022;11(10) doi: 10.1161/jaha.121.024883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCandless S.E., Brunger J.W., Cassidy S.B. The burden of genetic disease on inpatient care in a children’s hospital. Am J Hum Genet. 2004;74(1):121–127. doi: 10.1086/381053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald R., Dodgen A., Goyal S., et al. Impact of 22q11.2 deletion on the postoperative course of children after cardiac surgery. Pediatr Cardiol. 2013;34:341–347. doi: 10.1007/s00246-012-0454-x. [DOI] [PubMed] [Google Scholar]
- 29.Woolman P., Bearl D.W., Soslow J.H., et al. Characteristics and outcomes of heart transplantation in DiGeorge syndrome. Pediatr Cardiol. 2019;40:768–775. doi: 10.1007/s00246-019-02063-w. [DOI] [PMC free article] [PubMed] [Google Scholar]