

Significance
Sensing double-stranded RNA (dsRNA) is an important antiviral defense mechanism, as dsRNAs are commonly generated during viral infection and trigger different innate immune responses. The OAS3-RNase L pathway quickly detects dsRNA in virus-infected cells and degrades host and viral RNAs to block viral replication and propagation to other cells. However, how cells regulate OAS3 and RNase L during viral infection is still poorly understood. We developed a CRISPR-Cas9 knockout screening method and identified IRF2 as a critical regulator of OAS3. We showed that IRF2-deficient cells failed to properly activate RNase L in response to viral infection, leading to a significant increase in virus replication and infectivity. This underscores IRF2's crucial role in innate immunity to protect cells against viruses.
Keywords: innate immunity, RNase L, RNA decay, CRISPR screen, virus
Abstract
OAS-RNase L is a double-stranded RNA-induced antiviral pathway triggered in response to diverse viral infections. Upon activation, OAS-RNase L suppresses virus replication by promoting the decay of host and viral RNAs and inducing translational shutdown. However, whether OASs and RNase L are the only factors involved in this pathway remains unclear. Here, we develop CRISPR-Translate, a FACS-based genome-wide CRISPR-Cas9 knockout screening method that uses translation levels as a readout and identifies IRF2 as a key regulator of OAS3. Mechanistically, we demonstrate that IRF2 promotes basal expression of OAS3 in unstressed cells, allowing a rapid activation of RNase L following viral infection. Furthermore, IRF2 works in concert with the interferon response through STAT2 to further enhance OAS3 expression. We propose that IRF2-induced RNase L is critical in enabling cells to mount a rapid antiviral response immediately after viral infection, serving as the initial line of defense. This rapid response provides host cells the necessary time to activate additional antiviral signaling pathways, forming secondary defense waves.
The innate immune system is the primary line of defense against viruses after they gain entry into cells (1, 2). The first step of the innate immune response relies on the host cell’s ability to recognize conserved features of pathogens that are not present in the host (3–5). Virus-associated molecules such as genomic DNA and RNA or double-stranded RNA (dsRNA) produced in virally infected cells are recognized by pattern-recognition receptors (PRRs) expressed in the host cells (3–5). Upon detection, PRR-mediated signaling pathways are activated and trigger different types of responses to suppress viral replication and subsequent rounds of infections (6, 7).
The 2′,5′-oligoadenylate (2-5As) synthase (OAS)-RNase L is one of the antiviral signaling systems that effectively limits viral replication upon detection of cytoplasmic dsRNAs produced during viral replication and transcription (8). Viral dsRNAs are recognized by OAS enzymes, which produce 2-5As from ATP and act as a secondary messenger to trigger homodimerization and activation of RNase L, a latent cytoplasmic endonuclease (9–12). Once activated, RNase L promotes rapid and widespread degradation of viral and cellular RNAs, including tRNAs, rRNAs, and mRNAs, leading to translation arrest in host cells to inhibit viral replication (13–18). Human cells encode three catalytically active OAS genes (OAS1, OAS2, and OAS3), which are structurally homologous to cGAS, a cytoplasmic double-stranded DNA (dsDNA) sensor involved in the stimulation of interferon gene expression (19–21). Although all three OASs have the ability to synthesize 2-5As, OAS3 is the primary enzyme responsible for activating the RNase L pathway in response to viral infections (22, 23). OAS3 is a 120 kDa protein with a high affinity for dsRNA of 50 base pairs or more (24, 25). Once bound to dsRNA, OAS3 undergoes a conformational change, allowing for ATP binding and the synthesis of 2-5A chains required for RNase L dimerization in cells (12, 26). However, it is still unclear whether additional factors other than OAS3 and RNase L are involved in the regulation of this pathway.
Since its first discovery, the RNase L pathway has been studied for its role in restricting various viruses, including West Nile virus (WNV), SARS-CoV-2, dengue virus (DENV), and Sindbis virus (SINV) (23, 27, 28). However, many other viruses have developed strategies to counteract the activation of RNase L (29). Influenza A virus (IAV) and vaccinia virus (VACV) code for NS1 proteins and E3L RNA-binding proteins, respectively, which bind and sequester dsRNA to prevent OAS3 activation (23, 29–31) Poliovirus transcribes a highly structured competitive inhibitor RNA (ciRNA) within its open reading frame that functions as an RNase L inhibitor (32, 33). Moreover, MERS-CoV expresses NS4b, which degrades 2-5A (34). On the other hand, Zika virus (ZIKV) exploits the RNase L protein from the host cells independently of RNase L’s catalytic activity to enhance its replication factories and increase infectious virus production (35, 36). Therefore, characterizing the mechanism by which cells regulate the activation of RNase L is crucial to a better understanding of how viruses antagonize different steps of the pathway to propagate in cells.
In this study, we developed an unbiased approach using a FACS-based genome-wide CRISPR library screening method to identify novel regulators of RNase L. Using RNase L-driven translation arrest as a readout, we identified IRF2 as a mediator of the RNase L pathway. IRF2 promotes OAS3 basal expression in unstressed cells, allowing rapid activation of OAS3, followed by RNase L when a virus gains entry into the cells. Importantly, we demonstrated that OAS3/RNase L regulation by IRF2 in host cells directly impacts viral replication. Moreover, we found that IRF2 cooperates with the interferon response to enhance OAS3 expression at a later time during the infection, and when IRF2 is not functional, the interferon pathway acts as an alternative pathway to promote RNase L activity. We propose that IRF2 plays a critical role in enabling cells to mount a rapid response immediately following viral infection, serving as the initial line of defense to impede viral replication. This early suppression of viral replication provides host cells with the necessary time to activate other PRR-mediated responses, constituting a secondary wave of protective mechanisms.
Results
CRISPR-Translate: A CRISPR-Cas9 Library Screening Method to Identify Factors Regulating Translation.
To identify unknown essential factors regulating RNase L-driven RNA decay and global translation arrest in virally infected cells, we developed CRISPR-Translate, a FACS-based CRISPR-Cas9 screening strategy that exploits ongoing translation levels as a readout (Fig. 1A). In addition to activating RNase L, dsRNAs produced during viral replication and transcription are detected by protein kinase R (PKR), which in turn phosphorylates eIF2α to stop translation initiation. Cells depleted of both PKR and RNase L fully suppressed poly(I:C)-induced translation shutdown, indicating that these two factors are solely responsible for modulating translation in response to poly(I:C) (13, 37). Therefore, to exclusively monitor translation regulation by RNase L, we knocked out (KO) PKR from U2OS cells (SI Appendix, Fig. S1A). We transduced U2OS PKR KO cells with the genome-wide Brunello CRISPR library targeting 19,114 genes at a multiplicity of infection (MOI) of ~0.3, and uninfected cells were removed with puromycin selection (Fig. 1A, steps 1 and 2). We then transfected the in-cell library covering 97.33% of the Brunello library gRNAs with poly(I:C) (polyinosinic-polycytidylic acid), a synthetic analog of dsRNA that is used to mimic infections by RNA viruses (Fig. 1A, step 3). For the screen, we selected U2OS cells that showed a higher efficiency of poly(I:C) transfection compared to other cell lines, including A549. To measure translation levels in poly(I:C) transfected cells, we treated them with azidohomoalanine (AHA), an analog of methionine that is incorporated into newly synthesized polypeptide chains and then labeled with a 488-tagged alkyne probe using click-It reaction (Fig. 1A, step 4). Cells were subsequently FACS sorted into two populations: cells with positive 488 fluorescence signals and those with negative 488 fluorescence signals, representing cells undergoing active translation and translation arrest, respectively (Fig. 1A, step 5). Genomic DNA was next extracted from both cell populations, and gRNA sequences were amplified by PCR for deep sequencing analysis and quantification to determine which genes are essential to promote translation arrest after poly(I:C) transfection (Fig. 1A, step 6).
Fig. 1.
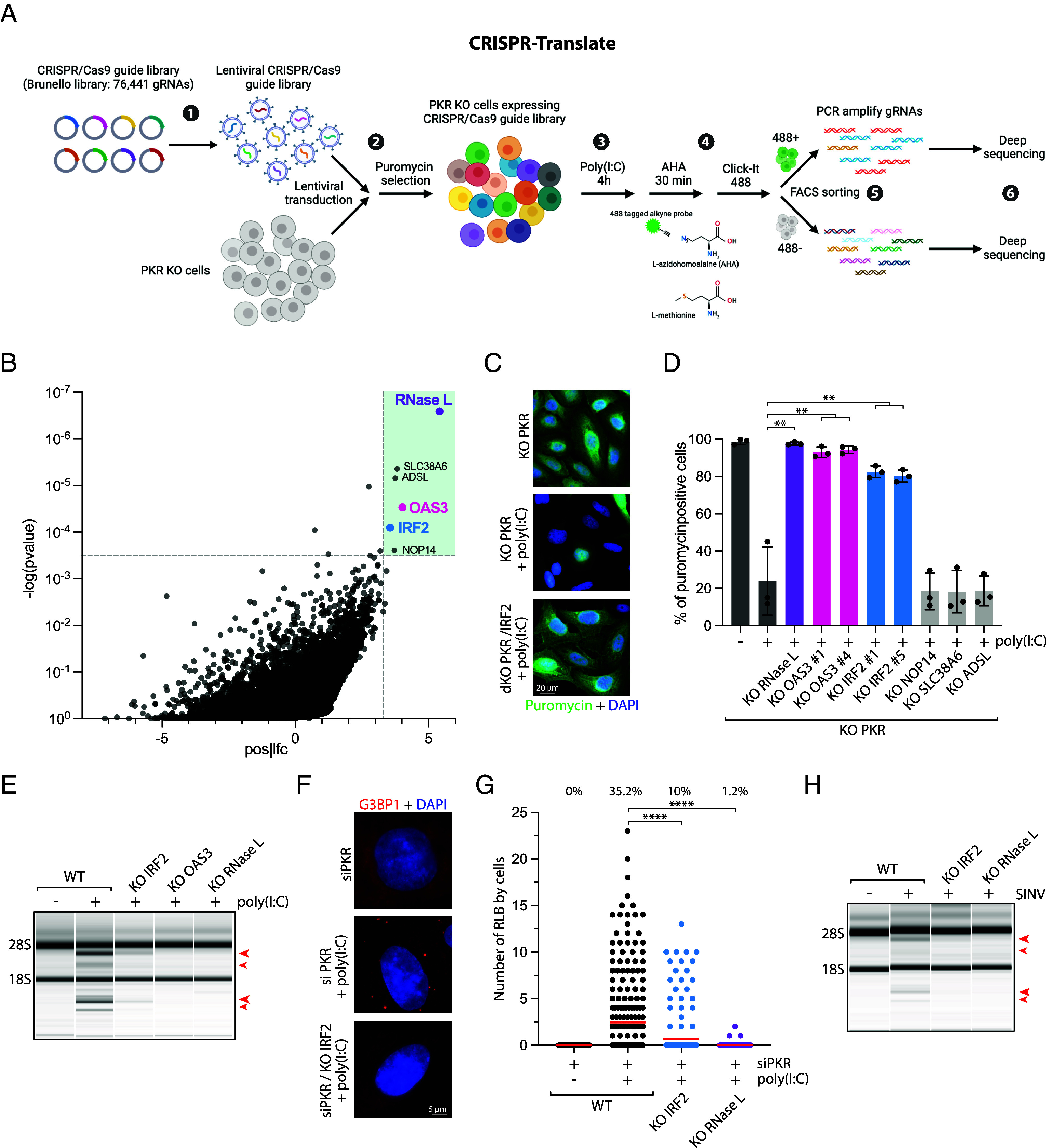
CRISPR-Translate identifies IRF2 as a regulator of the RNase L pathway. (A) Schematic of the CRISPR-Translate screening strategy. (B) Dot plots graph representative of the genes enriched in the 488-positive FACS-sorted cell population and analyzed using MAGeCK computational tool. Each dot represents a unique gene. Genes found to be highly significantly enriched in the 488-positive cell population and thus promoting translation arrest after poly(I:C) transfection in U2OS PKR KO cells are highlighted in the green box. (C) Representative immunofluorescence for puromycin in U2OS PKR KO and PKR/IRF2 dKO cells transfected with poly(I:C) (1 μg/mL, 4 h). (D) Quantification of puromycin-positive cells (%) in the indicated cell lines transfected with poly(I:C) for 4 h (1 μg/mL). (E) Total RNAs were isolated from A549 cells or indicated A549 KO cell lines after transfection with poly(I:C) (10 ng/mL; 4 h) and monitored for integrity by a bioanalyzer. The red arrows indicated ribosomal RNA cleavage products. (F) Representative immunofluorescence for G3BP1 in A549 cells knocked down with PKR siRNA and transfected with poly(I:C) (25 ng/mL, 4 h). (G) Quantification of the number of G3BP1 foci by cells in the indicated A549 cell lines knocked down with PKR siRNA followed by poly(I:C) transfection (25 ng/mL, 4 h). Top: percentage of cells with RLB foci. (H) Total RNAs were isolated from indicated U2OS cell lines infected with SINV [MOI = 1, 24 h postinfection (hpi)] and monitored for integrity by a bioanalyzer. The red arrows indicated ribosomal RNA cleavage products.
CRISPR-Translate Uncovers IRF2 as a Regulator of the RNase L Pathway.
Following gRNA sequencing, we used MAGeCK (Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout) analysis (38) to identify which gRNAs were specifically enriched in the cell population with ongoing translation after poly(I:C) transfection and thus promote translation shutdown caused by RNase L. As expected, both RNase L and OAS3 were the top two targets found in the screen based on fold enrichment (Fig. 1B and Dataset S1). This result validates CRISPR-Translate as a method for identifying factors that modulate translation. Additionally, we identified SLC38A6, ADSL, IRF2, and NOP14 as potential genes involved in promoting RNase L activity and translation shutdown post-poly(I:C) (Fig. 1B). We first confirmed these potential targets by knocking out each of these genes individually and monitoring translation level after poly(I:C) transfection, this time using puromycin, a structural analog of aminoacyl tRNAs that is incorporated into nascent peptides by translating ribosomes, as a marker of translation levels (39). We selected puromycin rather than AHA to eliminate potential off-target effects that might affect AHA incorporation itself in cells. From this secondary screen, we found that only IRF2 KO cells significantly restored translation levels post-poly(I:C), similar to RNase L and OAS3 KO cells (Fig. 1 C and D). We then asked whether IRF2 is important for promoting RNase L after poly(I:C) transfection by monitoring 18S and 28S ribosomal RNA degradation (23). IRF2 KO in both U2OS and A549 cells showed a strong decrease in RNase L-mediated ribosomal RNA cleavage (Fig. 1E and SI Appendix, Fig. S1 B–D), demonstrating that IRF2 directly regulates RNase L activity in cells. In addition to promoting RNA decay in cells, RNase L induces the formation of a unique ribonucleoprotein complex termed RNase L-dependent bodies (RLBs) that are small punctate G3BP1-positive foci present in the cell cytoplasm and are distinct from stress granules (40, 41). To monitor RLBs and not stress granules, we knocked down PKR, which mediates stress granule formation following poly(I:C) transfection (13, 40, 41). The formation of RLBs was significantly decreased posttransfection with poly(I:C) in IRF2 KO cells, reaching levels nearly similar to those observed in RNase L KO cells (Fig. 1 F and G and SI Appendix, Fig. S1E), further demonstrating that IRF2 regulates RNase L activity. Finally, we infected wild-type or IRF2 KO cells with SINV, a virus known to stimulate RNase L in host cells (23). In the absence of IRF2, consistent with the results obtained after poly(I:C) transfection, cells infected with SINV did not induce ribosomal RNA cleavage (Fig. 1H). Together, these results establish IRF2 as a factor essential for promoting RNase L during viral infection.
IRF2 Regulates OAS3 Basal Expression.
IRF2 is a transcription factor that belongs to the family of interferon regulatory transcription factors (IRFs) involved in regulating interferon-related genes. IRF2 acts as a dual-function transcription factor. IRF2 negatively regulates gene expression by competing with IRF1 for binding to the same promoter elements (42–46). On the other hand, IRF2 promotes the transcription of a few specific genes (47), including TAP1 (48), histone H4 (49), FAM111A (50), caspase 1/4 (47), and major histocompatibility class II genes (51). We showed that IRF2 was highly expressed in unstressed cells (cancer cells or noncancerous cells) and localized in the nucleus independently of the presence of dsRNA in cells (SI Appendix, Fig. S2 A and B). To investigate the mechanism by which IRF2 controls RNase L activity, we first asked whether IRF2 regulates OAS3 and/or RNase L expression in cells. We monitored OAS3 and RNase L levels in IRF2 KO cells compared to wild-type cells. Strikingly, OAS3 expression in U2OS and A549 cells was strongly suppressed at the protein and mRNA levels without IRF2, while RNase L expression levels were not significantly affected (Fig. 2 A–C). To further validate our IRF2 KO clones, we monitored several known human targets of IRF2 (including CASP1 and FAM111A) (47, 50). Consistent with previous studies, both FAM111A and CASP1 mRNA levels were down-regulated in IRF2 KO U2OS and A549 cells (SI Appendix, Fig. S2 C and D). Finally, we further confirmed IRF2 regulation of OAS3 mRNA expression by knocking down IRF2 in RPE-1, HeLa, and TOV21G cell lines (SI Appendix, Fig. S2E). Together, these results suggest that RNase L regulation by IRF2 is mediated through the regulation of OAS3 expression.
Fig. 2.
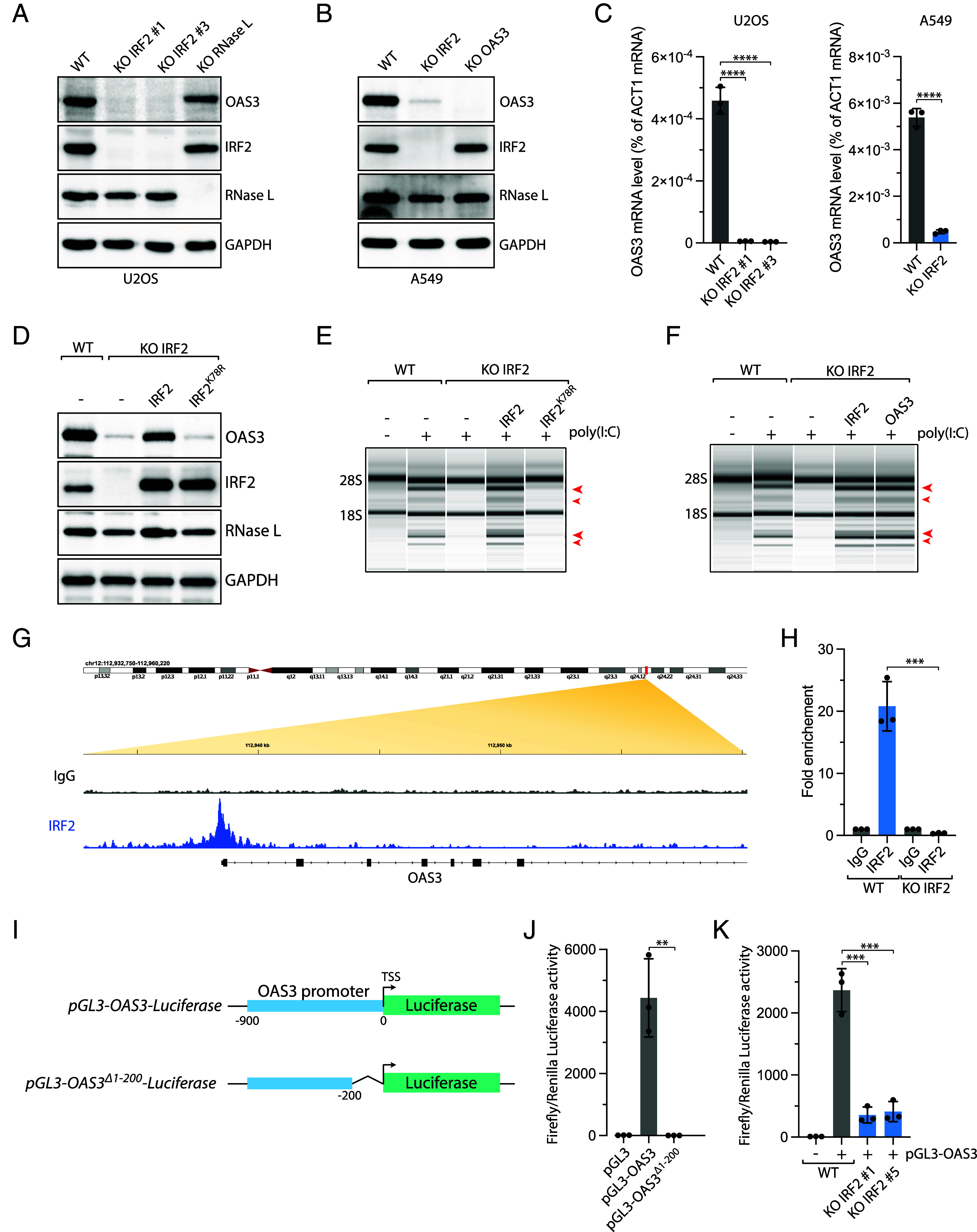
IRF2 promotes OAS3 expression in unstressed cells. (A and B) The levels of OAS3, IRF2, RNase L, and GAPDH were analyzed in the indicated U2OS (A) or A549 (B) cell lines by western blot. (C) The OAS3 mRNA levels were monitored by RT-qPCR in indicated U2OS (Left) or A549 (Right) cell lines. Mean values ± SD (n = 3). ****P < 0.0001 (two-tailed t test). (D) The levels of OAS3 were analyzed by western blot using indicated antibodies in A549 IRF2 KO cells expressing either wild-type IRF2 or DNA binding mutant IRF2K78R. (E and F) Total RNAs were isolated from A549 WT or IRF2 KO cells expressing wild-type IRF2, IRF2K78R, or OAS3 and monitored for integrity by a bioanalyzer after transfection with poly(I:C) (10 ng/mL, 4 h). The red arrows indicated ribosomal RNA cleavage products. (G) IgG and IRF2 ChIP-sequencing in A549 cells. Analysis of ChIP-sequencing data focuses on the promoter region of OAS3. (H) IRF2 ChIP was performed in A549 WT and IRF2 KO cells. IRF2 binding on the OAS3 promoter was determined by qPCR. Mean values ± SD. ***P < 0.001 (two-tailed t test). (I) Schematic of pGL3-OAS3-luciferase constructs harboring OAS3 WT promotor region (−1 to −900 bp upstream of transcription start site [TSS]) and IRF2 binding motif region deleted (Δ−1 to −200 bp). (J) The relative luciferase activity (ratio of Firefly:Renilla) was measured in U2OS cells transiently transfected with the empty pGL3 and pGL3 vector harboring either WT and Δ200 bp OAS3 promoter region. (K) The relative luciferase activity was monitored U2OS WT or IRF2 KO cells 24 h following transfection with the pGL3-OAS3 vector.
To determine the role of IRF2 in regulating OAS3-induced RNase L activation, we complemented IRF2 KO cells with IRF2 wild-type (IRF2WT) or a DNA binding mutant (IRF2K78R) and monitored OAS3 expression levels and RNA ribosomal cleavage. While IRF2WT fully restored both OAS3 protein levels and RNase L activity, IRF2K78R failed to induce either OAS3 expression and RNA decay post-poly(I:C) (Fig. 2 D and E). This result demonstrates that IRF2 DNA binding is critical for its function in regulating RNase L in cells. We then ectopically expressed OAS3 in IRF2 KO cells. We found that OAS3 expression was sufficient to restore ribosomal RNA cleavage in the absence of IRF2 (Fig. 2F and SI Appendix, Fig. S2F), suggesting that IRF2's control of the RNase L pathway is solely orchestrated via the regulation of OAS3.
Next, we examined whether IRF2 directly binds to the OAS3 promoter. We performed IRF2 chromatin immunoprecipitation sequencing (ChIP-seq) and revealed that IRF2 was constitutively present on the promoter of OAS3 in unstressed cells (Fig. 2G). We confirmed these data by performing ChIP-qPCR using primers targeting the region bound by IRF2. We showed a strong enrichment of IRF2 on the OAS3 promoter in unstimulated WT cells but not in IRF2 KO cells (Fig. 2H), further suggesting that IRF2 directly controls OAS3 expression in cells. To better characterize the transcriptional role of IRF2 in regulating OAS3, we constructed a pGL3 luciferase reporter plasmid controlled by the OAS3 promoter region (Fig. 2I). Luciferase activity was only detected when the promoter of OAS3 was present, and deletion of the IRF2 binding site on the OAS3 promoter abrogated luciferase luminescence (Fig. 2 I and J). Importantly, luciferase activity significantly decreased in IRF2 KO cells compared to wild-type cells transfected with the pGL-OAS3 luciferase reporter (Fig. 2K), confirming that IRF2 directly drives OAS3 expression. Altogether, these results reveal the critical role of IRF2 in maintaining the constitutive expression of OAS3 by directly promoting its expression in unstressed cells.
IRF2 Drives Rapid Activation of RNase L.
To further investigate the role of IRF2 in regulating OAS3 expression, we quantified mRNA and protein levels of OAS3 in wild-type A549 cells compared to IRF2 KO cells following poly(I:C) transfection. We found that both OAS3 mRNA and protein levels increased starting at 8 h posttransfection with poly(I:C) (Fig. 3 A and B). Although OAS3 was not expressed in the absence of IRF2 in unstressed cells, OAS3 expression becomes induced after poly(I:C) in IRF2 KO cells at the same time as wild-type cells but at lower levels (Fig. 3 A and B), suggesting that OAS3 expression after poly(I:C) is driven independently of IRF2. Similar results were obtained with U2OS cells and in IRF2 KO cells infected with SINV (Fig. 3C and SI Appendix, Fig. S3 A and B). Next, we monitored RNase L activation over time after poly(I:C) transfection. In A549 wild-type cells, RNase L activation showed a gradual increase from 2 to 16 h. However, in IRF2 KO cells, only very weak RNA ribosomal degradation mediated by RNase L was detected at a later time (8 to 16 h) when OAS3 was expressed back (Fig. 3D). Although OAS3 protein levels were restored to similar levels as in WT cells at 16 h, RNase L activity remained low, possibly due to the clearance of poly(I:C) from the cells preventing OAS3 activation. Alternatively, poly(I:C) may be masked by the binding of other PRRs or RNA binding proteins in the cells over time, preventing OAS3 activation. Therefore, to demonstrate that OAS3 expression after poly(I:C) was sufficient to promote RNase L activity, we treated cells with IFNα to induce OAS3 in IRF2 KO cells prior to transfection with poly(I:C) or SINV infection (Fig. 3E). IFNα-mediated OAS3 expression was enough to restore RNase L activity in IRF2 KO cells treated with poly(I:C) (Fig. 3F) or infected by SINV (SI Appendix, Fig. S3C), demonstrating that the reexpression of OAS3 during viral infection can stimulate RNase L activity.
Fig. 3.
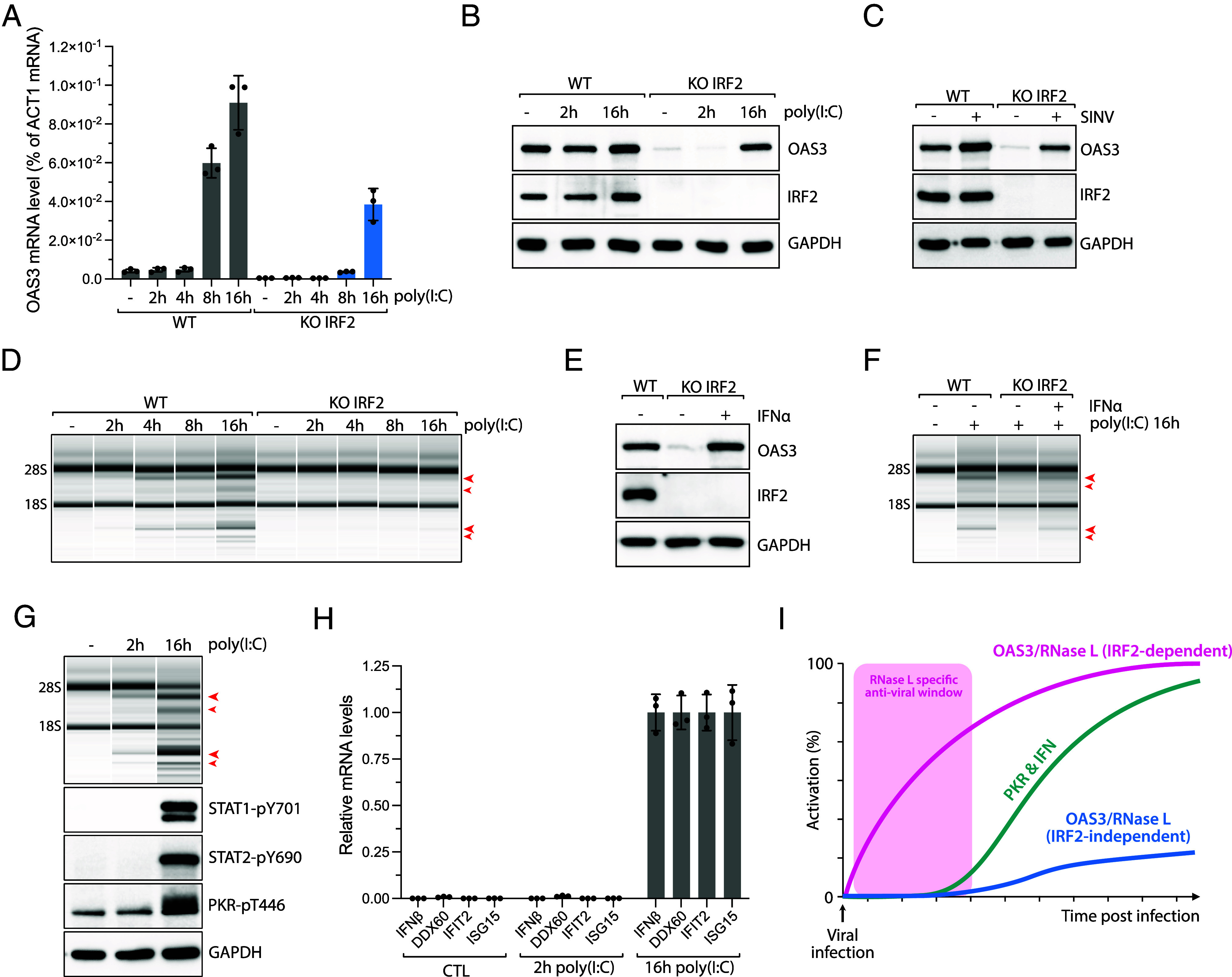
IRF2-independent expression of OAS3 in response to dsRNAs. (A) A549 WT and IRF2 KO cells were transfected with poly(I:C) (10 ng/mL). The level of OAS3 mRNA was monitored at indicated time points by RT-qPCR. Mean values ± SD. (B) A549 WT and IRF2 KO cells were transfected with poly(I:C) (10 ng/mL). The protein levels of OAS3, IRF2, and GAPDH were monitored at indicated time points by western blot. (C) A549 WT or IRF2 KO cells infected with SINV (MOI = 10) were collected at 24 hpi, and the protein levels of OAS3, IRF2, and GAPDH were analyzed by western blot. (D) A549 WT and IRF2 KO cells were transfected with poly(I:C) (10 ng/mL), and total RNAs were isolated at the indicated time points and analyzed for integrity by a bioanalyzer. The red arrows indicated ribosomal RNA cleavage products. (E) A549 IRF2 KO cells were treated with IFN-α (1,000 U/mL) for 16 h, and the protein levels of OAS3, IRF2, and GAPDH were analyzed by western blot. (F) A549 WT and IRF2 KO cells were pretreated with IFN-α (1,000 U/mL) for 8 h and transfected with poly(I:C) (10 ng/mL) for 16 h. Total RNA was isolated and analyzed for ribosomal integrity by a bioanalyzer. The red arrows indicated ribosomal RNA cleavage products. (G) A549 WT cells were transfected with poly(I:C) (10 ng/mL), and cell lysates were collected at 2 or 16 h time points. Total RNAs were analyzed for ribosomal degradation by a bioanalyzer, and the phosphorylation levels of STAT1-pY701, STAT2-pY690, and PKR-pT446 were monitored by western blot. The red arrows indicated ribosomal RNA cleavage products. (H) Quantification of mRNA levels of IFN genes (IFNβ, DDX60, IFIT2, and ISG15) at indicated time points post-poly(I:C) transfection (10 ng/mL) in A549 cells. (I) Proposed model of the role of IRF2 promoting RNase L during viral infection. IRF2 induces rapid activation of the OAS3/RNase L pathway immediately after viral infection, while IRF2 deficient cells result in a delayed response in the activation of the RNase L pathway.
Next, we compared RNase L activation kinetics to other innate immune response pathways induced by dsRNAs in cells. The interferon response was stimulated at 16 h following poly(I:C) transfection as evidenced by STAT1/2 phosphorylation and by the induction of interferon-stimulated genes (IFNβ, DDX60, IFIT2, and ISG15) (Fig. 3 G and H). Likewise, we detected PKR autophosphorylation 16 h post-poly(I:C) transfection (Fig. 3G). However, at 2 h posttransfection with poly(I:C), neither PKR nor the interferon pathway had yet been activated, despite the detection of RNase L-induced ribosomal RNA degradation (Fig. 3G). These results suggest that RNase L acts very quickly in response to foreign dsRNAs in cells and prior to other dsRNA-mediated innate immune responses. They also support previous studies reporting that RNase L activation precedes PKR activation (13, 52). Therefore, we propose that IRF2-mediated OAS3 basal expression is critical for RNase L’s quick response to viral infection by ensuring the availability of OAS3. This gives host cells the necessary time to build an interferon response and activate PKR as a secondary wave of defense against viruses (Fig. 3I).
Interplay between IRF2 and STAT2 to Promote OAS3 Expression During Viral Infections.
We next asked which factors drive OAS3 expression post-poly(I:C) or SINV infection. Transfected poly(I:C) in cells is detected by RIG-I/MDA-5/MAVS dsRNA sensors that trigger an IFN response and the expression of hundreds of ISGs, including OAS enzymes (53). We found that RIG-I knockdown or the inhibition of the IFN response using the JAK kinase inhibitor ruxolitinib suppressed OAS3 expression 16 h post-poly(I:C) (Fig. 4 A and B and SI Appendix, Fig. S4A). Consistently, IRF2 KO cells infected with SINV failed to express OAS3 at 24 h postinfection (hpi) when treated with ruxolitinib (Fig. 4C). We then knocked down STAT1 or STAT2, the main transcription factors phosphorylated by JAK kinases, which drive ISG expression during the IFN response. Knockdown of STAT2 but not STAT1 strongly decreased OAS3 levels in poly(I:C) transfected cells and SINV-infected cells (Fig. 4 D–F and SI Appendix, Fig. S4 B and C). Notably, STAT2 knockdown did not affect OAS3 expression in unstressed cells (Fig. 4D and SI Appendix, Fig. S4C), indicating that STAT2 is only required for IFN-induced OAS3 expression. Moreover, we confirmed STAT2-mediated OAS3 expression after poly(I:C) transfection in U2OS cells (SI Appendix, Fig. S4D). To further establish that STAT2 directly promotes OAS3 expression in response to viral infection, we performed STAT2 ChIP-seq and ChIP-qPCR. STAT2 was detected on the OAS3 promoter in cells treated with poly(I:C) for 16 h, whereas IRF2 was present regardless of the treatment status (Fig. 4 G–I), demonstrating that STAT2 only regulates OAS3 expression in response to viral infections. Together, these results reveal that OAS3 expression is independently controlled by both IRF2 and STAT2. IRF2 maintains a constitutive basal expression of OAS3 in unstressed cells, while STAT2 promotes OAS3 levels following viral infection-mediated IFN response.
Fig. 4.
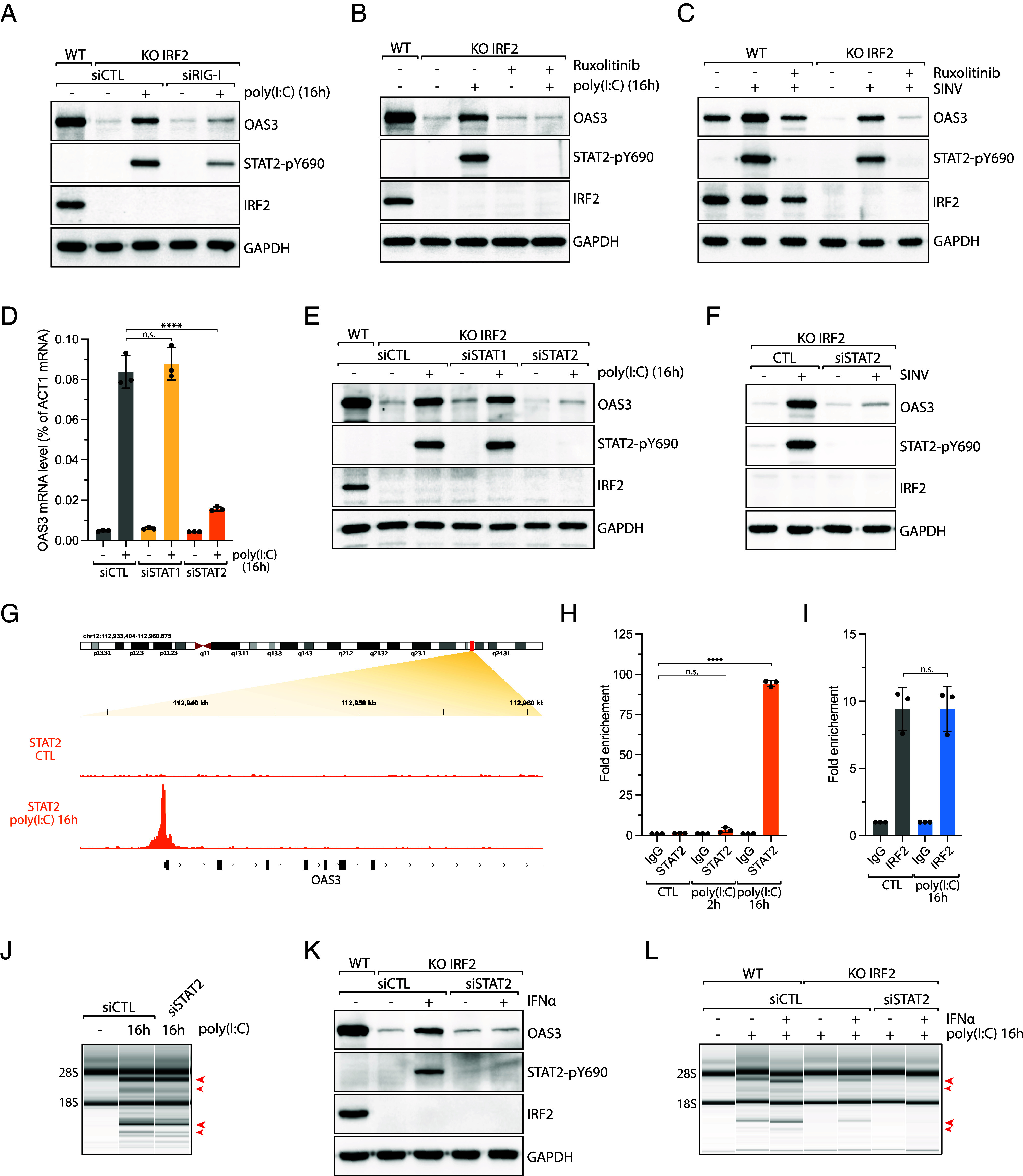
STAT2 drives IFN-induced OAS3 expression during viral infection. (A) A549 IRF2 KO cells knocked down with RIG-I siRNA for 40 h were transfected with poly(I:C) (10 ng/mL; 16 h), and the levels of the indicated proteins were analyzed by western blot. (B and C) A549 WT and IRF2 KO cells were treated with ruxolitinib (2 μM), followed by transfection with poly(I:C) (10 ng/mL, 16 h) (B) or infection with SINV (MOI =10, 24 hpi) (C). The protein levels of OAS3, STAT2-pY690, IRF2, and GAPDH were analyzed by western blot. (D) The OAS3 mRNA levels were analyzed by RT-qPCR in A549 cells knocked down with the indicated siRNAs, followed by transfection with poly(I:C) (10 ng/mL, 16 h). Mean values ± SD. ****P < 0.0001 (two-tailed t test). (E) A549 WT or IRF2 KO cells knocked down with STAT1 or STAT2 siRNAs were transfected with poly(I:C) (10 ng/mL, 16 h). The protein levels of the indicated proteins were analyzed by western blot. (F) A549 IRF2 KO cells were knocked down with siCTL or siSTAT2 and infected with SINV (MOI = 10, 24 hpi). The cell lysates were analyzed for the protein levels of OAS3, STAT2-pY690, IRF2, and GAPDH by western blot. (G) STAT2 ChIP-sequencing in A549 cells transfected with poly(I:C) (1 μg/mL, 16 h). Analysis of STAT2 ChIP-sequencing data shows the region upstream of the OAS3 TSS (transcription starting site). (H) STAT2 ChIP-qPCR was performed in A549 cells transfected with poly(I:C) for 2 or 16 h. STAT2 binding on the OAS3 promoter was determined by qPCR. Mean values ± SD. ****P < 0.0001 (two-tailed t test). (I) IRF2 ChIP-qPCR was performed in A549 cells transfected with poly(I:C) (1 μg/mL, 16 h). IRF2 binding on the OAS3 promoter was analyzed by qPCR. Mean values ± SD. (J) A549 cells knocked down with either siCTL or siSTAT2 were transfected with poly(I:C) (10 ng/mL) for 16 h and analyzed for RNA integrity by a bioanalyzer. The red arrows indicated ribosomal RNA cleavage products. (K) A549 IRF2 KO cells transfected with either siCTL or siSTAT2 were treated with IFNα (1,000 U/mL) for 16 h. Cell lysates were then analyzed for the protein level of OAS3, STAT2-pY690, IRF2, and GAPDH by western blot. (L) A549 WT or IRF2 KO cells transfected with either siCTL or siSTAT2 were pretreated with IFNα (1,000 U/mL) for 8 h and transfected with poly(I:C) (10 ng/mL) for 16 h. Total RNAs were isolated and analyzed for RNA integrity by a bioanalyzer. The red arrows indicated ribosomal RNA cleavage products.
To further investigate the role of the interplay between IRF2- and STAT2-mediated OAS3 expression, we first knocked down STAT2 in wild-type cells transfected with poly(I:C) for 16 h. In the absence of STAT2, RNase L activity was not impacted (Fig. 4J). Similar results were obtained after ruxolitinib treatment in cells transfected with poly(I:C) or infected with SINV (SI Appendix, Fig. S4 E and F), further suggesting that IRF2 is the main driver of RNase L in response to dsRNAs. We next induced the interferon response by treating IRF2 KO cells with INFα to trigger STAT2-mediated OAS3 expression prior poly(I:C) transfection or SINV infection. IFNα-mediated OAS3 expression and RNase L activity were strictly STAT2 dependent (Fig. 4 K and L and SI Appendix, Fig. S4G), demonstrating the role of STAT2 in promoting RNase L activity in the absence of IRF2 in cells during viral infection. Thus, these results suggest that interferon-induced OAS3 through STAT2 acts as a secondary pathway to promote RNase L when IRF2 is lacking.
IRF2 Suppresses SINV Replication through the Regulation of RNase L.
To assess the impact of IRF2-mediated rapid activation of RNase L on viral replication, we infected cells with SINV and monitored its replication by plaque assays. Both IRF2 KO and RNase L KO cells infected with SINV produced a significantly increased yield of infectious virus particles in the cell supernatants, which were quantified by plaque assays on culture media at 24 hpi (Fig. 5 A and B and SI Appendix, Fig. S5). The increase in viral titers was comparable to those reported in cells KO for OAS3 and RNase L in previous studies (23, 54). Complementation with wild-type IRF2, but not IRF2K78R, restored the suppression of viral replication by host cells to levels that were not statistically different (Fig. 5 C and D). We then asked whether the suppression of SINV replication by IRF2 was mediated through the regulation of RNase L. We ectopically expressed OAS3 in IRF2 KO cells, which we previously showed to be sufficient for restoring RNase L activity in the absence of IRF2 (Fig. 2F), and monitored SINV titers at 24 hpi. Strikingly, OAS3 expression in IRF2 KO cells was enough to reduce SINV infectious viral particle production (Fig. 5E), demonstrating that IRF2’s protective role against SINV infection in host cells was primarily mediated through activation of the RNase L pathway. Finally, we treated wild-type and IRF2 KO cells with ruxolitinib to suppress the IFN response and then monitored SINV replication by the plaque assay. Ruxolitinib treatment further enhanced SINV titer in both cell lines (Fig. 5F), suggesting that IRF2-induced RNase L pathway and the IFN pathway are both essential for suppressing SINV replication in host cells. Taken together, these results reveal that IRF2’s regulation of RNase L through the control of OAS3 expression levels is critical for preventing viral replication in host cells.
Fig. 5.

IRF2 suppresses SINV replication through the regulation of RNase L. (A) A549 WT, RNase L KO, or IRF2 KO cells were infected with SINV (MOI =10, 24 hpi), and the titer of infectious virus from the supernatant was determined by the plaque assay in Vero cells. Representative images of the plaque-forming assay were shown. (B) Quantification of viral titer (log10 PFU/mL) shown in A was determined by the plaque assay from three biological replicates. Mean values ± SD. *P < 0.05. (C) A549 WT or IRF2 KO cells complemented with the indicated construct were infected with SINV (MOI = 10, 24 hpi), and the titer of infectious virus from the supernatant was determined by the plaque assay in Vero cells. Representative images of the plaque-forming assay were shown. (D) Quantification of viral titer (log10 PFU/mL) shown in C was determined by the plaque assay from three biological replicates. Mean values ± SD. ***P < 0.001. (E) A549 WT or IRF2 KO cells complemented with OAS3 were infected with SINV (MOI = 10, 24 hpi), and the titer of infectious virus from the supernatant was determined by the plaque assay in Vero cells to determine the viral titer (log10 PFU/mL) from three biological replicates. Mean values ± SD. *P < 0.05. (F) A549 WT or IRF2 KO cells treated with ruxolitinib (2 μM) were infected with SINV (MOI = 10, 24 hpi), and the titer of infectious virus from the supernatant was determined by the plaque assay in Vero cells to determine the viral titer (log10 PFU/mL) from three biological replicates. Mean values ± SD. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Discussion
The activation of the OAS3/RNase L pathway in response to viral infection is highly detrimental to the replication of many viruses, including WNV, SARS-CoV-2, DENV, and SINV (23, 27, 28), by promoting RNA decay and translation arrest. This underscores the importance of understanding how cells regulate the RNase L pathway in response to viral infection. In this study, we identified IRF2 as a key factor in the rapid activation of RNase L by ensuring appropriate expression levels of OAS3 in cells. We found that cells lacking IRF2 failed to activate RNase L in response to viral infections. Moreover, we showed that the absence of rapid RNase L activation mediated by IRF2 promotes viral replication, highlighting the importance of a timely stimulation of RNase L immediately following virus infection. We propose that the quick activation of RNase L following viral infections is critical for limiting virus levels and giving cells the necessary time to mount additional antiviral immune responses.
The functional read-out of RNase L activation relied on monitoring ribosomal cleavage, formation of RLBs, and translation arrest. Herein, we exploited RNase L-mediated translation shutdown and developed the method CRISPR-Translate to identify previously uncharacterized factors regulating the RNase L pathway. CRISPR-Translate is a FACS-based genome-wide CRISPR-Cas9 knockout screening technique that uses translation levels as read-out through labeling of cells by incorporation of AHA in nascent peptides of actively translating cells and detection in cells using Click-It chemistry (Fig. 1A). CRISPR-Translate not only confirmed the critical role of OAS3 and RNase L in shutting down translation in cells exposed to foreign dsRNA but also uncovered IRF2 as a third essential player of the pathway. Therefore, CRISPR-Translate is a powerful method to identify new factors regulating translation. However, several top target genes identified in our CRISPR screen were not confirmed as regulating translation arrest after poly(I:C) transfection. Thus, it is essential to perform secondary validation experiments to eliminate potential false positive gene candidates. Furthermore, the use of CRISPR-Translate could be extended far beyond the identification of new players involved in the regulation of RNase L. Indeed, this method can be easily adapted to study any stressors affecting translation levels in cells. The integrated stress response (ISR) is a signaling pathway stimulated in response to various stimuli and coordinated by four kinases (PKR, HRI, GCN2, and PERK) promoting the phosphorylation of eIF2α to block translation initiation (55). Hence, applying CRISPR-Translate to investigate ISR regulation would yield deeper insights into the mechanisms by which cells modulate these kinases in response to specific stressors.
Mechanistically, we revealed that IRF2 controls RNase L through the regulation of OAS3 expression. In unstressed cells, OAS3 expression is highly dependent on the presence of IRF2 binding to its promoter. This ensures basal OAS3 expression in cells, priming them to respond to any viral infection rapidly. Previous studies reported that RNase L-mediated RNA decay occurs very early in response to foreign dsRNA in cells (13, 52). This is in contrast to other innate immune pathways, such as the IFN response or the PKR pathway, which are also triggered by dsRNAs but at a later time following infection (Fig. 3 G–I) (13). Thus, we propose that IRF2-driven rapid RNase L activation is essential to delay virus-induced cell toxicity and provide the cells with the opportunity to mount additional defense mechanisms to eliminate viruses. Without early RNase L activation, our data revealed a strong increase in viral titer, demonstrating that other innate immune defense systems were not sufficient to compensate for the loss of rapid RNase L activity. This suggests that it is critical for cells to limit viral replication levels while building additional innate immune defense mechanisms. In the absence of rapid activation of RNase L, the higher levels of viral RNAs and viral proteins in cells may overwhelm the cell’s defense mechanisms, allowing the viruses to escape immune surveillance and clearing. Furthermore, it is tempting to speculate that IRF2-driven rapid activation is particularly important to protect cells infected with fast-replicating viruses. Indeed, other defense mechanisms may be activated too late to efficiently protect the cells after the virus has altered the cell biology to its advantage, including innate immunity downregulation, or has already replicated and been released from the cells.
In addition to IRF2, we showed that the IFN pathway further induces OAS3 expression through the phosphorylation of the transcription factor STAT2 in response to foreign dsRNAs. This result is consistent with previous studies showing that OAS3 is an interferon-stimulated gene (ISG) (56). Based on ChIP-seq data, both STAT2 and IRF2 bind to the same region of the OAS3 promoter. However, it remains unclear whether IRF2 and STAT2 occupy the promoter simultaneously. It is possible that IRF2 and STAT2 either cooperate to enhance OAS3 expression in response to viral infection or act independently, being mutually exclusive on the promoter. Since STAT2- and IRF2-mediated OAS3 expression occurs independently of each other, this suggests that cooperation between these transcription factors is not necessary. OAS3 expression was also reported to be regulated by STAT1 in response to enterovirus-A71 infection (57). It is conceivable that akin to STAT2, STAT1 may promote OAS3 in certain viral infection contexts. Alternatively, siRNA targeting STAT1 used in this study could potentially target STAT2 as well, given their shared homologous sequences (58). Nevertheless, our findings obtained from CRISPR-Translate along with cell treatment with JAK inhibitor showed minimal or no effect of the interferon response on RNase L activity in response to poly(I:C) or SINV infection when IRF2 is present in cells (Figs. 1B and 4J and SI Appendix, Fig. S4 E and F). These results strongly support that IRF2 is the main driver of OAS3-mediated RNase L activation during viral infection and that STAT2-promoting OAS3 expression acts as an alternative pathway for the cells. Many viruses have developed resistance mechanisms to suppress RNase L activation. It is possible that cells have evolved two distinct pathways promoting OAS3 expression to counteract potential viral factors blocking RNase L through the downregulation of OAS3. However, future studies will be required to characterize possible resistance mechanisms employed by viruses blocking OAS3 expression.
IRF2 is a transcription factor constitutively expressed in cells with dual functions. On the one hand, IRF2 negatively regulates IFN signaling by antagonizing IRF1 through competitive binding to the same promoter elements of IFNs and IFN-inducible genes (59). On the other hand, IRF2 drives the expression of a few specific genes in cells, including RNase L, TAP1 (48), histone H4 (49), FAM111A (50), caspase 1/4 (47), and MHC-II (51). IRF2 induces FAM111A, which has been shown to be important in inhibiting ZIKV replication (50), suggesting that IRF2 antiviral functions go beyond regulating RNase L for certain types of viral infections. IRF2 protects mice from lethal viral neuroinvasion caused by SINV (60). While mice deficient for IRF2 showed a clear increase in viral titer and pathologies associated with SINV infections, the dysregulation of type I IFN signaling did not cause accelerated disease and death in Irf2−/− mice infected with SINV (60). Our data revealed that ectopic expression of OAS3 was sufficient to rescue IRF2-deficient cells infected with SINV (Fig. 5E), further suggesting that IRF2’s protective role against SINV is mainly mediated through the regulation of RNase L. However, it is still unclear how IRF2 determines its pro- versus anti-transcriptional activity in cells. It is possible that different types of IRF2 DNA binding motifs or the presence of other specific transcription factors in close proximity govern IRF2’s ability to promote or suppress gene expression. However, future studies will be necessary to establish the mechanisms behind IRF2 pro- and anti-transcriptional activity.
Methods
Plasmids.
IRF2 and OAS3 cDNAs were synthesized by Gene Universal with either HA (IRF2) or Flag (OAS3) tag in the C terminus. The plasmids expressing IRF2-HA, IRF2K78R-HA, and OAS3-Flag were generated by inserting the cDNAs into a modified pBABE vector using the Gateway Cloning System (Thermo Fisher Scientific). IRF2-K78R mutant was constructed by site-directed mutagenesis. The OAS3 luciferase reporter plasmid was generated by cloning the OAS3 promoter region (TSS −1 to −900) into pGL3 basic luciferase reporter (Addgene #128046). IRF2 binding site deletion mutant (pGL3-OAS3Δ1–200) was constructed by site-directed mutagenesis.
Cell Culture.
U2OS, HEK-293FT, and Vero cells were cultured in DMEM supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. A549 cells were maintained in DMEM/F12 GlutaMAX™-I supplemented with 10 % FBS and 1 % penicillin/streptomycin. Cell lines were purchased from either ATCC or Sigma-Aldrich. A549-derived cell lines were generated by infecting A549 cells with retrovirus expressing IRF2 or OAS3 (pBABE-puro retroviral vector) and selected with puromycin (0.5 µg/mL) for 72 h.
Viruses.
SINV Ar-339 strain was purchased from ATCC (#VR-1585). Viral titer was determined by the plaque assay using Vero cells. Cells were plated into six wells, and the adsorption of the virus was performed for 1 h at 37 °C in 200 µL of virus diluted serum free DMEM. Then, Vero cells were overlaid with agarose (2 % in culture media), stained with crystal violet (0.2% crystal violet/25% EtOH), and counted to determine viral titer.
Viral Infection.
U2OS or A549 cells were infected with SINV in serum-free medium at indicated MOI at 37 °C for 1 h for adsorption. Culture medium supplemented with 10% FBS and 1% penicillin/streptomycin was added postadsorption.
Cell Treatment.
Poly(I:C)-LMW was purchased from InvivoGen (#tlrl-picw) and was transfected by forward transfection with Lipofectamine 2000 (Thermo Fisher Scientific, #11668019) at the indicated concentration and time according to the manufacturer’s instructions. For Surface sensing of translation (SUnSET) Assay (39), U2OS or A549 cells were treated with puromycin (10 μg/mL, MP Biomedicals #ICN10055210) for 10 min before fixation for analysis by immunofluorescence with an antibody against puromycin. Ruxolitinib (MedChemExpress # HY-50856) and purified human Interferon-αA/D (Sigma-Aldrich #I4401) were added directly to the cells at the indicated concentration and time.
CRISPR-Translate.
The CRISPR Knockout screening using the Brunello library (61) was performed following protocols provided by Addgene (Catalog #73179) and Dr. Feng Zhang lab (62) with adaptations. For Brunello library lentiviral pool production, 293FT cells (Thermo Fisher Scientific, # R70007) were seeded in 150-cm2 culture dishes to have 70% confluency on the day of the transfection. The day after cell seeding, 15 µg of Brunello library plasmid (Addgene, #73179) and lentivirus packaging vectors (8 µg of pRSV-Rev, 8 µg of pMDL/pRRE, and 3 µg of pMD2.6) were transfected using calcium phosphate transfection method (63). 48 h following transfection, cell supernatants were collected, filtered with 0.45 µm syringe filters (Genesee Scientific, #25-246), and frozen in 1.5 mL aliquots at −80 °C.
The genome-wide CRISPR libraries were generated by transducing 140 million U2OS PKR KO cells with Brunello lentiviral pool at MOI of 0.3 to maintain at least 500× library coverage after puromycin selection. The cells were split once after reaching confluency during puromycin selection. Following 7 d of puromycin selection (0.75 µg/mL), 200 million cells were plated in 15 cm tissue culture dishes. The next day, the Brunello library harboring cells were transfected with poly(I:C) at 5 µg/mL using lipofectamine 2000 for 4 h. The cells were shifted to methionine-free media for 30 min followed by treatment with L-azidohomoalanine (AHA) (Vector Laboratory, #CCT-1066) at 25 µM for 30 min. The cells were collected and fixed with ice-cold 70% ethanol overnight. Next, the cells were labeled with a 488-tagged alkyne probe (Vector Laboratories, #CCT-1277-1) using click-It reaction (Vector Laboratories, #CCT-1263) according to the manufacturer’s protocol. The cells were then sorted on a FACS Aria Fusion into two populations: 488 positive cells and 30% of bottom 488 negative cells. 488 positive cells were subjected to a second round of sorting to eliminate any false 488-positive cells. Genomic DNA was isolated from both sorted cell populations using phenol–chloroform extraction method. Next sgRNA sequences were amplified using P5 primers with different numbers of stagger regions pooled together (for sequencing diversity) and P7 primers with unique barcode sequences using Q5 High-Fidelity DNA Polymerase (New England Biolabs, #M0491) under the following PCR condition: an initial denaturation at 98°C for 30 s, followed by 10 s at 98 °C, 30 s at 65 °C, 30 s at 72 °C for 32 cycles, and a final extension at 72 °C for 10 min. PCR products were gel extracted (Qiagen, #28706) and sequenced on a NovaSeq 6000 platform [UCI Genomics High-Throughput Facility (GHTF)]. MAGeCK analysis was performed to find enriched gRNAs in the 488-positive population relative to the 488-negative population (38). The sequences of the PCR primers used in this study are listed in SI Appendix, Table S1.
RNA Interference.
siRNA transfections were performed by reverse transfection with Lipofectamine RNAiMax (Thermo Fisher Scientific, #13778150). siRNAs were purchased from Thermo Fisher Scientific (Silencer Select siRNA). Cells were transfected with poly(I:C) or infected with viruses 40 h after siRNA transfection (4 to 8 nM). For efficient knockdown, two siRNA targeting IRF2, STAT1, or STAT2 were transfected together. The sequences of the siRNAs used in this study are listed in SI Appendix, Table S2.
Antibodies.
The antibodies used in this study are listed in SI Appendix, Table S3.
CRISPR-Cas9 Knockout Cells.
PKR and RNase L and CRISPR-Cas9 knockout cell lines were performed by transfection with Lipofectamine CRISPRMAX of TrueGuide Synthetic CRISPR gRNA (Thermo Fisher Scientific, #CMAX00003) and TrueCut Cas9 Protein v2 (Thermo Fisher Scientific, #A36498) according to the manufacturer’s instructions. IRF2 KO cells, OAS3 KO cells, and PKR KO cell lines were generated by transfecting cells with the pSpCas9(BB)-2A-Puro (PX458) plasmid containing gRNAs targeting each gene with FuGENE 6 Transfection Reagent (E2691; Promega). 24 h after transfection, GFP+ cells were sorted and selected. IRF2, PKR, RNase L, and OAS3 KO cells were validated by western blot. gRNA sequences used in this study are listed in SI Appendix, Table S4.
Immunofluorescence.
Cells were fixed with paraformaldehyde (3% paraformaldehyde and 2% sucrose in 1×PBS) for 20 min, washed twice with 1×PBS, and permeabilized with a permeabilization buffer (1×PBS and 0.2% Triton X-100) for 5 min. Subsequently, cells were washed twice with 1×PBS and blocked in PBS-T (1×PBS and 0.05% Tween-20) containing 2% BSA and 10% milk for 1 h. Cells were then incubated with the primary antibody diluted in 1×PBS containing 2% BSA and 10% milk at room temperature for 2 h. Coverslips were washed three times with PBS-T before incubation (1 h) with the appropriate secondary antibodies conjugated to fluorophores (Alexa-488 or Cy3). After three washes with PBS-T, cells were stained with DAPI (5 µg/mL, MilliporeSigma #D9542), and the coverslips were mounted using slow-fade mounting media (Thermo Fisher Scientific, # S36936). Images were captured using a Leica DMi8 THUNDER microscope.
Plaque Assay.
A549 cells were infected with SINV in a serum-free medium at the indicated MOI. At 24 hpi, the supernatants were collected, diluted with serum-free medium, and used to infect Vero cells. The virus was adsorbed for 1 h at 37 °C, and cells were overlaid with agarose (2% in full media). After 2 d, cells were fixed with trichloroacetic acid (10 %) for 20 min, stained with crystal violet (0.2% crystal violet/25% EtOH), and the number of plaques formed was counted to determine viral titer.
Bioanalyzer.
Total RNA was extracted from cells using the Quick-RNA MiniPrep Kit (Zymo Research, # R1055) according to the manufacturer’s instructions. The amount of total RNA was measured by NanoDrop (Thermo Fisher Scientific) and then diluted to 5 ng/μL. RNA samples were run on Bioanalyzer 2100 (Agilent) using the Eukaryote Total RNA Pico kit (Agilent, # 5067-1513).
RT-qPCR.
Total RNA was extracted from cells using the Quick-RNA MiniPrep Kit (Zymo Research, #R1055) according to the manufacturer’s instructions. Following extraction, total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, #4368813). RT products were analyzed by RT-qPCR using SYBR Green (PowerUp SYBR Green Master Mix, Thermo Fisher Scientific, #A25743 in a QuantStudio 3 Real-Time PCR detection system (Thermo Fisher Scientific). For each sample tested, the levels of indicated mRNA were normalized to the levels of Actin mRNA. The qPCR primers used in this study are listed in SI Appendix, Table S5.
Chromatin Immunoprecipitation.
ChIP experiments were performed using the SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling, #9003), following the Manufacturer’s protocol as previously described (64). In brief, A549 cells were fixed with 1% formaldehyde for 10 min at room temperature, followed by quenching with glycine for 5 min. The cells were then lysed, and the chromatin was fragmented by enzymatic digestion using Micrococcal Nuclease (20 min at 37 °C). IgG, IRF2, or STAT2 antibodies were incubated with 5 μg of digested and cross-linked chromatin for 16 h at 4 °C. Protein G magnetic beads were added for an additional 2 h. After immunoprecipitation, chromatin–protein complexes were eluted from protein G magnetic beads and reverse cross-linked. Eluted DNA was purified and used for qPCR and sequencing. Library generation and sequencing were performed by the UCI Genomics High-Throughput Facility (GHTF) on an Illumina NovaSeq platform. For ChiP-seq analysis of IRF2 and STAT2, raw reads were processed using STAR aligner (65) and aligned to the human genome GRCh38/hg19. Visualization was performed using Integrative Genomics Viewer.
The ChiP sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive, using the Bioproject Accession: PRJNA1125024. The sequences of ChiP-qPCR primers used in this study are listed in SI Appendix, Table S5.
Luciferase Assay.
U2OS cells were transfected with pGAL backbone, pGL3-OAS3, or pGL3-OAS3Δ1-200 plasmids using FuGENE 6 Transfection Reagent (Promega, # E2691). 24 h after transfection, the cells were collected and lysed to measure the activity of Firefly and Renilla luciferase using the Dual-Luciferase® Reporter Assay System (Promega, # E1910).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
We thank Casey Johnson and Dr. Melanie Oakes for their technical assistance. pGL3 basic luciferase reporter was a gift from Oskar Laur (Addgene, #128046). S.O. is a Dr. Lorna Calin Scholar and was supported by the Faculty Mentor Program and a Graduate Dean’s Dissertation Fellowship from the University of California, Irvine. G.S. is supported by a NIH Research Supplements to Promote Diversity in Health-Related Research (R37-CA252081-S1). L.M. is supported by a Center for Virus Research Graduate Fellowship funded by the UCI Division of Graduate Studies. This work was funded by support from the NIH NCI R37-CA252081 (R.B.) and NIAID R01-AI155962 (B.L.S). R.B. was supported by a Research Scholar Grant (RSG-24-1249960-01-DMC) from the American Cancer Society. This work was also made possible, in part, through access to the Genomics High Throughput Facility Shared Resource of the Chao Family Comprehensive Cancer Center Support Grant (P30-CA062203). Fig. 1A was generated with BioRender.com.
Author contributions
S.O. and R.B. designed research; S.O., G.S., L.M., and J.L. performed research; S.O., A.B., and B.L.S. contributed new reagents/analytic tools; S.O. analyzed data; and S.O. and R.B. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission S.P. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
Sequencing data have been deposited in NCBI (PRJNA1125024) (66).
Supporting Information
References
- 1.Wu J., Chen Z. J., Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–88 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Brubaker S. W., Bonham K. S., Zanoni I., Kagan J. C., Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 33, 257–290 (2015), 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O., Akira S., Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Amarante-Mendes G. P., et al. , Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 9, 2379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li D., Wu M., Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 6, 291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowie A. G., Unterholzner L., Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson M. R., Kaminski J. J., Kurt-Jones E. A., Fitzgerald K. A., Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarti A., Jha B. K., Silverman R. H., New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 31, 49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naik S., Paranjape J. M., Silverman R. H., RNase L dimerization in a mammalian two-hybrid system in response to 2’,5’-oligoadenylates. Nucleic Acids Res. 26, 1522 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H., et al. , Dimeric structure of pseudokinase RNase L bound to 2–5A reveals a basis for interferon-induced antiviral activity. Mol. Cell 53, 221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Floyd-Smith G., Slattery E., Lengyel P., Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate–dependent endonuclease. Science 212, 1030–1032 (1981). [DOI] [PubMed] [Google Scholar]
- 12.Han Y., Whitney G., Donovan J., Korennykh A., Innate immune messenger 2–5A tethers human RNase L into active high-order complexes. Cell Rep. 2, 902–913 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Burke J. M., Moon S. L., Matheny T., Parker R., RNase L reprograms translation by widespread mRNA turnover escaped by antiviral mRNAs. Mol. Cell 75, 1203–1217.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rath S., et al. , Concerted 2–5A-mediated mRNA decay and transcription reprogram protein synthesis in the dsRNA response. Mol. Cell 75, 1218–1228.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen J. B., Mazan-Mamczarz K., Zhan M., Gorospe M., Hassel B. A., Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol. 6, 305–315 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennan-Laun S. E., Ezelle H. J., Li X. L., Hassel B. A., RNase-L control of cellular mRNAs: Roles in biologic functions and mechanisms of substrate targeting. J. Interferon Cytokine Res. 34, 275–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou A., et al. , Interferon action and apoptosis are defective in mice devoid of 2’,5’-oligoadenylate-dependent RNase L. EMBO J. 16, 6355–6363 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wreschner D. H., James T. C., Silverman R. H., Kerr I. M., Ribosomal RNA cleavage, nuclease activation and 2–5A(ppp(A2’p)nA) in interferon-treated cells. Nucleic Acids Res. 9, 1571–1581 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hornung V., Hartmann R., Ablasser A., Hopfner K. P., OAS proteins and cGAS: Unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 14, 521–528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chebath J., Benech P., Revel M., Vigneron M., Constitutive expression of (2’-5’) oligo A synthetase confers resistance to picornavirus infection. Nature 330, 587–588 (1987). [DOI] [PubMed] [Google Scholar]
- 21.Hartmann R., Justesen J., Sarkar S. N., Sen G. C., Yee V. C., Crystal structure of the 2’-specific and double-stranded RNA-activated interferon-induced antiviral protein 2’-5’-oligoadenylate synthetase. Mol. Cell 12, 1173–1185 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Ibsen M. S., et al. , The 2′-5′-oligoadenylate synthetase 3 enzyme potently synthesizes the 2′-5′-oligoadenylates required for RNase L activation. J. Virol. 88, 14222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., et al. , Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. U.S.A. 113, 2241–2246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donovan J., Whitney G., Rath S., Korennykh A., Structural mechanism of sensing long dsRNA via a noncatalytic domain in human oligoadenylate synthetase 3. Proc. Natl. Acad. Sci. U.S.A. 112, 3949–3954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Holleufer A., Gad H. H., Hartmann R., Length dependent activation of OAS proteins by dsRNA. Cytokine 126, 154867 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Koul A., et al. , Structural and hydrodynamic characterization of dimeric human oligoadenylate synthetase 2. Biophys. J. 118, 2726–2740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burke J. M., Gilchrist A. R., Sawyer S. L., Parker R., RNase L limits host and viral protein synthesis via inhibition of mRNA export. Sci. Adv. 7, eabh2479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., et al. , SARS-COV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 118, e2022643118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gusho E., Baskar D., Banerjee S., New advances in our understanding of the “unique” RNase L in host pathogen interaction and immune signaling. Cytokine 133, 153847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper D. A., et al. , RNase L targets distinct sites in influenza A virus RNAs. J. Virol. 89, 2764–2776 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min J. Y., Krug R. M., The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 7100–7105 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keel A. Y., Jha B. K., Kieft J. S., Structural architecture of an RNA that competitively inhibits RNase L. RNA 18, 88–99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend H. L., Jha B. K., Silverman R. H., Barton D. J., A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol. 5, 263–272 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thornbrough J. M., et al. , Middle east respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. mBio 7, e00258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whelan J. N., et al. , Zika virus employs the host antiviral RNase L protein to support replication factory assembly. Proc. Natl. Acad. Sci. U.S.A. 118, e2101713118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whelan J. N., Li Y., Silverman R. H., Weiss S. R., Zika virus production is resistant to RNase L antiviral activity. J. Virol. 93, e00313-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith J. A., Schmechel S. C., Williams B. R. G., Silverman R. H., Schiff L. A., Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J. Virol. 79, 2240–2250 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W., et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt E. K., Clavarino G., Ceppi M., Pierre P., SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Burke J. M., Lester E. T., Tauber D., Parker R., RNase L promotes the formation of unique ribonucleoprotein granules distinct from stress granules. J. Biol. Chem. 295, 1426 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manjunath L., et al. , APOBEC3B drives PKR-mediated translation shutdown and protects stress granules in response to viral infection. Nat. Commun. 14, 820 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuyama T., et al. , Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97 (1993). [PubMed] [Google Scholar]
- 43.Tanaka N., Kawakami T., Taniguchi T., Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol. Cell Biol. 13, 4531–4538 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harada H., et al. , Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 58, 729–739 (1989). [DOI] [PubMed] [Google Scholar]
- 45.Wang Y., et al. , Involvement of IFN regulatory factor (IRF)-1 and IRF-2 in the formation and progression of human esophageal cancers. Cancer Res. 67, 2535–2543 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Hida S., et al. , CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity 13, 643–655 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Benaoudia S., et al. , A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep. 20, e48235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouyez M.-C., et al. , IFN regulatory factor-2 cooperates with STAT1 to regulate transporter associated with antigen processing-1 promoter activity. J. Immunol. 174, 3948–3958 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Vaughan P. S., et al. , Activation of a cell-cycle-regulated histone gene by the oncogenic transcription factor IRF-2. Nature 377, 362–365 (1995). [DOI] [PubMed] [Google Scholar]
- 50.Ren K., et al. , IRF2 inhibits ZIKV replication by promoting FAM111A expression to enhance the host restriction effect of RFC3. Virol. J. 18, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xi H., et al. , Co-occupancy of the interferon regulatory element of the class II transactivator (CIITA) type IV promoter by interferon regulatory factors 1 and 2. Oncogene 18, 5889–5903 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Chitrakar A., et al. , Real-time 2–5A kinetics suggest that interferons β and λ evade global arrest of translation by RNase L. Proc. Natl. Acad. Sci. U.S.A. 116, 2103–2111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rehwinkel J., Gack M. U., RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551 (2020), 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y., Dong B., Wei Z., Silverman R. H., Weiss S. R., Activation of RNase L in Egyptian Rousette bat-derived RoNi/7 cells is dependent primarily on OAS3 and independent of MAVS signaling. mBio 10, e02414-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wek R. C., Role of eIF2α kinases in translational control and adaptation to cellular stress. Cold Spring Harb. Perspect. Biol. 10, a032870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samarajiwa S. A., Forster S., Auchettl K., Hertzog P. J., INTERFEROME: The database of interferon regulated genes. Nucleic Acids Res. 37, D852 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng B., Zhou X., Tian L., Wang J., Zhang W., IFN-β1b induces OAS3 to inhibit EV71 via IFN-β1b/JAK/STAT1 pathway. Virol. Sin. 37, 676–684 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oda K., et al. , Structural analysis of the STAT1:STAT2 heterodimer revealed the mechanism of Sendai virus C protein-mediated blockade of type 1 interferon signaling. J. Biol. Chem. 292, 19752–19766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Antonczyk A., et al. , Direct inhibition of IRF-dependent transcriptional regulatory mechanisms associated with disease. Front. Immunol. 10, 437612 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li M. M. H., et al. , Interferon regulatory factor 2 protects mice from lethal viral neuroinvasion. J. Exp. Med. 213, 2931–2947 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doench J. G., et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joung J., et al. , Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon M., Firestein B. L., DNA transfection: Calcium phosphate method. Methods Mol. Biol. 1018, 107–110 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Oh S., et al. , Genotoxic stress and viral infection induce transient expression of APOBEC3A and pro-inflammatory genes through two distinct pathways. Nat. Commun. 12, 4917 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A., et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sunwoo O., et al. , Data from “CRISPR-Cas9 knockout screening identifies IRF2 as a key driver of OAS3/RNase L-mediated RNA decay during viral infection.” Raw sequence reads. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1125024. Deposited 17 June 2024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
Sequencing data have been deposited in NCBI (PRJNA1125024) (66).