

ABSTRACT
Background
Congenital muscular dystrophies (CMDs) are diverse early‐onset conditions affecting skeletal muscle and connective tissue. This group includes collagen VI‐related dystrophies such as Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy (BM), caused by mutations in the COL6A1, COL6A2 and COL6A3 genes. We report a consanguineous Malian family with three siblings affected by UCMD due to a novel homozygous splice site variant in the COL6A1 gene.
Methods
After obtaining consent, three affected siblings and their relatives underwent physical examinations by specialists and laboratory tests where possible. DNA was extracted from peripheral blood for genetic testing, including Whole Exome Sequencing (WES). Putative variants were confirmed through Sanger Sequencing and assessed for pathogenicity using in silico tools.
Results
The three siblings and their healthy parents, from a consanguineous marriage, presented with early‐onset progressive muscle weakness, walking difficulty, proximal motor deficits, severe muscle atrophy, hypotonia, skeletal deformities, joint hyperlaxity, ankyloses at the elbows and knees, keloid scars and dental crowding. No cardiac involvement was detected and creatine kinase (CK) levels were normal. All had low serum calcium levels, treated with oral supplements. Needle myography indicated myopathic patterns. WES identified a novel splice site variant in the first intron of COL6A1 (c.98‐1G>C), which segregated with the disease within the family. This variant is predicted to cause exon 2 skipping in COL6A1, with a high CADD score of 33 and Splice AI predicting it as deleterious.
Conclusion
We identified a novel COL6A1 variant in a consanguineous family, highlighting the need for further studies in larger African cohorts to enhance genetic epidemiology and prepare for future therapeutic research.
Keywords: Africa, COL6A1, collagen VI‐related muscular dystrophies, Mali, splicing variant, Ullrich congenital muscular dystrophy
A novel homozygous splice site variant in COL6A1 causing Ullrich congenital muscular dystrophy in a Malian family, emphasizing the importance of genetic studies in African cohorts to advance epidemiological knowledge and therapeutic development.
Abbreviations
- BM
Bethlem Myopathy
- CADD
combined annotation dependent depletion
- CK
creatine kinase
- CM
congenital myopathies
- CMDs
congenital muscular dystrophies
- EMG
electromyography
- Fathmm
functional analysis through hidden Markov models
- SS
sanger sequencing
- UCMD
Ullrich congenital muscular dystrophy
- WES
whole exome sequencing.
1. Introduction
Congenital muscular dystrophies (CMDs) are a clinically and genetically diverse group of muscular diseases that are generally characterized by early‐onset global hypotonia and muscle weakness. They are divided into subgroups, including collagen VI‐related congenital myopathies that comprise Ullrich congenital myopathy (UCMD) [MIM#254090] and Bethlem myopathy (BM) [MIM#158810]. These diseases are caused by variations in the genes coding for the major α‐chains of collagen type VI, including COL6A1 (OMIM#120220), COL6A2 (OMIM#120240) and COL6A3 (OMIM#120250) (Aumailley, von der Mark, and Timpl 1985; Chu et al. 1987, 1988).
The first description of UCMD was in 1930 by Otto Ullrich, who reported it as a slowly progressive condition characterized by global hypotonia, proximal joint contractures and distal joint hyperlaxity (Ullrich 0tto. 1930). However, intelligence is usually intact in affected individuals (Bönnemann 2011).
UCMD is described as an ultrarare condition with an estimated prevalence of 0.13 per 100,000 individuals in the UK (Norwood et al. 2009). Furthermore, collagen VI‐related myopathies are now reported to be one of the most common entities under the banner of congenital muscular dystrophy (Okada et al. 2007; Peat et al. 2007). UCMD patients of African origins were described in some studies in Europe (Graziano et al. 2015), but data are scarce in Africa. In this report, we describe three affected siblings from a consanguineous marriage afflicted with UCMD and the underlying genetic defect.
2. Materials and Methods
This study was approved by the Ethics Committee of the Faculty of Medicine and Dentistry of Bamako, Mali. Affected siblings and their parents and relatives were enrolled after giving full consent or assent for children. Family history was recorded to draw the pedigree. A physical examination was performed by specialists (neurologists, rheumatologists, medical geneticists and cardiologists) and blood chemistries, including blood cell counts, blood ions and CK and LDH levels, were checked in all three affected siblings. Needle Electromyography was also performed in the two boys. DNA was extracted from peripheral blood from all available family members for genetic testing, including whole exome sequencing (WES). Putative variants were confirmed and checked for segregation in all available family members. Variant pathogenicity was predicted using assessment tools including Splice Vault (SV), Splice AI, ACMG score and CADD Score.
3. Results
Three affected siblings (two boys and one girl) aged 10 years, 5 years and 18 months from a sibship of seven were referred to our clinic for genetic evaluation of early‐onset progressive proximal muscle weakness. They were born from asymptomatic first‐degree consanguineous parents of Arab ethnicity (Figure 1A). The first born was a girl who suddenly passed away at the age of 7 days from an unspecified disease. The inheritance pattern was suggestive of autosomal recessive. Below are the clinical findings in the three siblings.
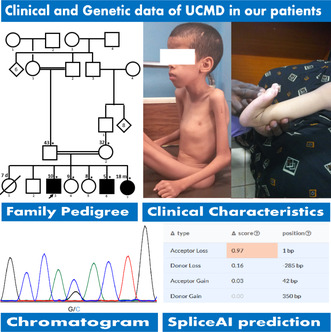
FIGURE 1.
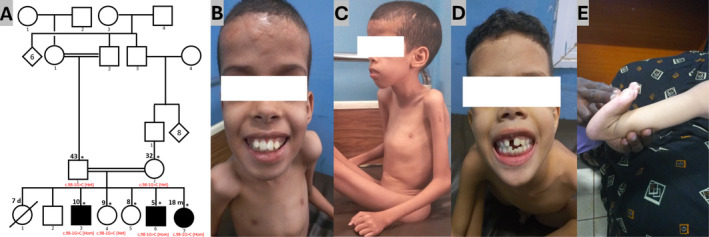
Phenotypic features of the family with COL6A1 variant. (A) Consanguineous pedigree showing three affected probands and the healthy carrier parents and siblings. (B, C) Photographs of Proband 1 showing the keloid scars on the forehead, the Pectus carinatum and severe muscle waste in the upper body. (D, E) Photographs of Probands 2 and 3 displaying dental crowding and hyperlaxity of distal joints.
3.1. Patient 1 (IV.3)
A 10‐years old male patient was seen for progressive muscle wasting. Parents report gross motor development delay. He started crawling at age 1 and was able to make only a few steps for a moment but progressively lost that ability around the age of 2 due to proximal muscle weakness and was later grounded. The symptoms worsened overtime, leading to severely reduced motor strength in four limbs, crawling became impossible. His intellectual abilities, language acquisition and social development were unremarkable. Physical examination revealed a predominantly proximal muscle weakness in both the upper and lower limbs, severe atrophy of muscles of the same area, global hypotonia and bilateral upper joint contractures and ankyloses involving the elbows and knees. Bilateral distal joint hyperlaxity was also noted in the wrists, fingers and ankles. The patient also displayed skeletal deformities, including kyphoscoliosis and pectus carinatum (Figure 1B,C). In addition, he had dental crowding and keloid scars on the forehead. Cardiovascular and pulmonary systems were uneventful. The CK levels were in the normal range (102 UI/l; range: 20–200 UI/L), but he had a low level of serum calcium (85 mg/L; range: 90–107 mg/L) and an elevated level of serum lactate (448.7 UI/l; range: 90–320 UI/L). Echocardiography was normal and needle EMG showed myogenic patterns. The patient benefited from Kinesitherapy and oral calcium supplementation.
3.2. Patient 2 (IV.6)
Patient 2 is the sixth child in the sibship and the third boy. He was referred to us at the age of 5 for the same reason as patient 1. Born from a normal pregnancy, he could sit at 6 months of age and walked around the age of 2. Parents noticed a wadding gait when he started walking, along with difficulties with running and jumping, leading to frequent falls. He also had difficulties climbing stairs and raising hands above his shoulders. Symptoms progressively evolved over time to being wheelchair bound. The physical examination showed that he had features like those of patient 1 but less severe. He had reduced motor strength in both upper and lower limbs with significant muscle wasting. Hyperlaxity was noted in distal joints, including wrists, fingers and ankles. In addition, he had joint contractures in the elbows and dental crowding. No facial dysmorphism or skeletal deformities were observed at this point, but he had keloid scars located on the forehead (Figure 1D). He showed no cardiovascular or respiratory symptoms. Serum CK levels were in the normal range (90 UI/L), the lactate levels were increased (606.8 UI/L) while calcium levels were low (70.7 mg/L). EMG showed myopathic features like those in his brother. His treatment consisted of physical therapy and oral calcium supplementation.
3.3. Patient 3 (IV.7)
An 18‐month‐old girl, last born of the sibship, was seen in our clinic for the same reasons as her older brothers. Also, born from a normal pregnancy, she started sitting at the age of 6 months and stood at the age of 12 months but could not walk unassisted. As she grew up, parents reported the same symptoms as in her brothers. Intelligence, language and social abilities are age appropriate. The physical examination was challenging but revealed more proximal than distal muscle weakness, cervical hypotonia, hyperlaxity in the wrists and ankles (Figure 1D) and bilateral pes planus. She could barely walk, even with assistance. Blood chemistry showed normal serum CK levels (93 UI/l) with high serum lactate levels (614 UI/l) and low serum calcium levels (70.5 mg/L). The EMG features were consistent with muscular dystrophy. She was put on oral calcium supplements in addition to physical therapy.
The pattern of inheritance and the clinical presentation were highly suggestive of UCMD.
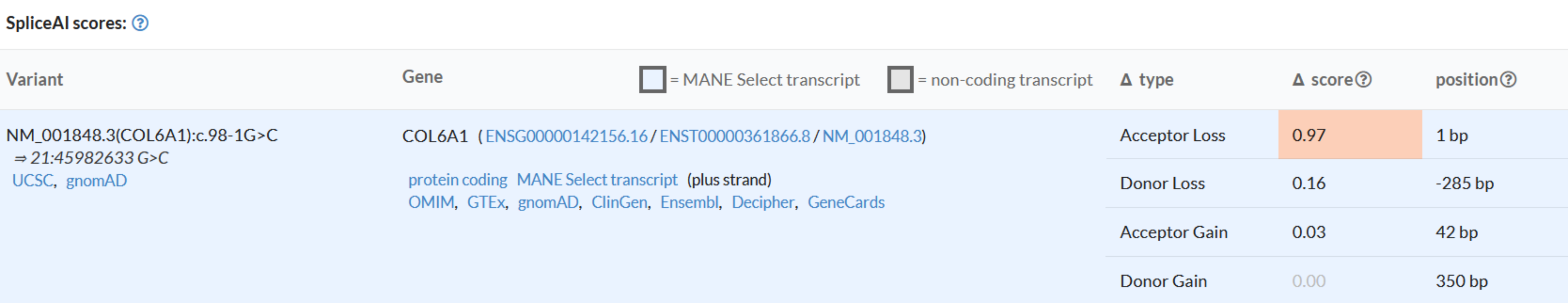
WES identified a novel segregating, homozygous, pathogenic splicing variant (c.98‐1G> C) in intron 1 of the COL6A1 gene (Figure 2A). This variant is predicted to be deleterious by CADD (score = 34) and to cause exon2 skipping in COL6A1 by SpliceVault. In addition, Splice AI prediction showed a splicing acceptor loss with a high delta score = 0.97.
FIGURE 2.
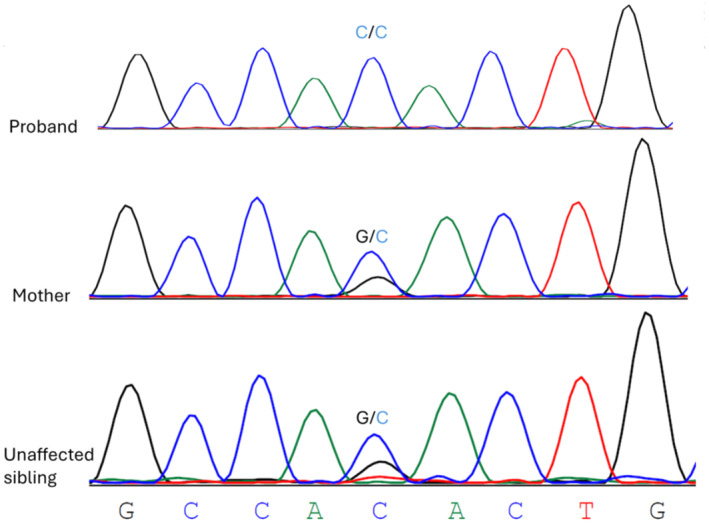
Partial chromatograms of the novel COL6A1 c.98‐1G> C variant in the proband, the mother and one unaffected sibling.
Details of clinical and laboratory findings are summarized in Table 1.
TABLE 1.
A summary of the main clinical and laboratory findings.
| Patients | Clinical features | Laboratory and genetic findings | Gene/variant/zygosity | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | Gender | Age of onset (years) | Frist symptom | Hypotonia | Proximal muscle weakness | Muscle atrophy | Joints contractures | Joints hyperlaxity | Skeletal deformities | Serum CK level | Serum lactate level | Serum calcium level | EMG findings | ||
| Proband 1 | 10 | M | 2 | Walking difficulty | Present | Present | Present | Present | Present | Present | Normal | Elevated | Low | Myopathic pattern | COL6A1/c.98‐1G>C/Hom. |
| Proband 2 | 5 | M | 2 | Walking difficulty | Present | Present | Present | Present | Present | Present | Normal | Elevated | Low | Myopathic pattern | COL6A1/c.98‐1G>C/Hom. |
| Proband 3 | 1.5 | F | 1 | Walking difficulty | Present | Present | None | Present | Present | Present | Normal | Elevated | Low | ND | COL6A1/c.98‐1G>C/Hom. |
Abbreviations: CK, creatine‐kinase; EMG, electromyography; F, female; Hom., homozygous; M, male; ND, not done.
4. Discussion and Conclusions
Here, we describe the phenotypic and genetic characteristics of three siblings from Mali with early‐onset muscular dystrophy. The clinical manifestations coupled with the family history are consistent with UCMD, the most severe form of collagen VI‐related myopathies characterized by early‐onset muscle weakness, generalized hypotonia, proximal joint contracture, distal hyperlaxity and respiratory failure (Briñas et al. 2010; Bushby, Collins, and Hicks 2014; Lamandé and Bateman 2018; Nadeau et al. 2009; Yonekawa and Nishino 2015). All probands had pronounced muscle atrophy and hypotonia, leading to delayed motor milestones and early loss of ambulation. Keloid scars were noted in patients IV.3 and IV.6, as frequently reported in UCMD patients (Nadeau et al. 2009). Patient IV.3 had kyphoscoliosis and pectus carinatum, the former being the most common type of spinal deformity reported in UCMD (Bushby, Collins, and Hicks 2014; Kim et al. 2018; Lee et al. 2017; Zanoteli et al. 2020; Zhang et al. 2022). Joint abnormalities with variable severity, including proximal joint contractures and distal joint hypermobility were also noted in the patients we describe here (Kim et al. 2018; Lee et al. 2017; Nadeau et al. 2009). Moreover, patients IV.3 and IV.6 had dental crowding, which is not typically known to be associated with UCMD. The presence of this feature does not argue against the diagnosis of UCMD, as this may be due to a totally different cause or just coincidental or stochastic. As stated in previous reports, the serum CK levels in our patients were normal (Martoni et al. 2009; Park et al. 2014). However, LDH levels were moderately high in all of them, but this is a common finding in muscular dystrophies patients (Lott and Landesman 1984; Zhu et al. 2015). Serum calcium levels were low in our patients, suggesting that the dysregulation of calcium metabolism may be involved as one of the mechanisms underlying UCMD progression (Law et al. 2020; Mareedu et al. 2021). As in previously reported cases, needle EMG showed a myopathic pattern (Bardakov et al. 2021; Elisabeth and Hh 2021; Mercuri et al. 2002).
This splicing variant has not been reported before and is classified as likely pathogenic to pathogenic according to most in silico prediction tools (CADD, SpliceAI) and the ACMG criteria, respectively. Interestingly, this mutation is located near a variant previously reported in Clinvar (c.98‐2_103del; rs1556423703) known to cause Bethlem Myopathy (National Center for Biotechnology Information. ClinVar; [VCV000497044.9], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000497044.9 [accessed September 26, 2024].), suggesting that this region of the COL6A1 gene might be a hot spot for mutations. Notably, collagens play a central role in the formation of fibrillar and microfibrillar networks within the extracellular matrix, including basement membranes, as well as in other extracellular matrix structures. The VWA domains present in these collagens facilitate protein–protein interactions (Gelse, Pöschl, and Aigner 2003). The microfibrillar type VI collagen is characterized by robust disulfide cross‐linking, contributing to a network of beaded filaments that intertwine with other collagen fibrils (Von Der Mark et al. 1984).
Although RNA samples were unavailable to verify the COL6A1 c.98‐1G> C variant's impact on splicing, in silico tools predict that this variant likely leads to exon 2 skipping or the activation of a cryptic splice site. Exon skipping could result in an in‐frame deletion, potentially producing a shorter, dysfunctional protein (Anna and Monika 2018), while activation of a cryptic splice site might introduce a frameshift, causing premature termination and nonsense‐mediated decay (NMD) (Karousis, Nasif, and Mühlemann 2016). These outcomes would likely impair collagen VI function, consistent with the observed UCMD phenotype.
This report expands the clinical and genetic epidemiology of collagen VI‐related myopathies and opens the way to a better understanding of the phenotype–genotype correlations in UCMD.
In addition to UCMD, we considered potential differential diagnoses such as Bethlem Myopathy (BM) and Laminin α2‐related Congenital Muscular Dystrophy (MDC1A), given their overlapping clinical features of muscle weakness and contractures (Caria et al. 2019; Kwong et al. 2023; Sarkozy et al. 2020). However, genetic testing revealed no pathogenic variants in COL6A2, COL6A3, or LAMA2, effectively excluding these conditions. The identification of the novel COL6A1 c.98‐1G> C variant, alongside the clinical presentation, supports this variant as the most likely cause of the siblings' phenotype. Our probands displayed unreported clinical and laboratory features, such as dental crowding and low serum calcium, suggesting that these disorders might have a unique profile in this region of Africa caused by other genetic or environmental factors or just be an expansion of the clinical presentation.
As new sequencing technologies become widely available, screening larger cohorts in these understudied populations may uncover novel findings that could shed light into the mechanism of these diseases and trigger future therapeutic research.
Author Contributions
A.B.M., I.B., L.C., A.Y., S.T., S.D., S.D., G.L., I.A.C. designed the study, obtained the clinical information, collected the literature data and wrote the manuscript. I.P., L.C., S.H.D., C.O.G., G.L. and I.A.C. coordinated the evaluation, critically reviewed and edited the manuscript. S.D. performed the genetic testing of the family. S.D., S.B. and F.K. analyzed the sequencing data and drafted the diagnostic report. I.P., S.T., S.D. and I.A.C. were the treating rheumatologists. A.Y. performed the protein modeling, analysis and interpretation. G.L. critically revised the final manuscript for important intellectual content and approved it. All authors read and approved the final version of the manuscript.
Ethics Statement
This study was approved by the Ethics Committee (2020/129/CE/FMOS/FAPH) of the Faculty of Medicine and Dentistry of the University of Sciences, Techniques and Technologies of Bamako. Patients and their families were enrolled in this study after giving full consent for clinical examination, genetic exploration and image taking.
Consent
Full consent was obtained from all the participants, or from legal representatives in the case of underage patients. The signed consent forms are available upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. SpliceAI prediction showing a splice acceptor loss with high delta scores (SpliceAI Δ score = 0.97).
{kind=link}
Acknowledgements
We are grateful to our patients and their family members for consenting to participate in this study.
Funding: This research study was supported by the grant #U01HG007044 funded by National Institute of Neurological Disorders and Stroke (NINDS) through the Human Hereditary and Health in Africa (H3Africa) Consortium Initiative, administered by the National Human Genome Research Institute as part of the NIH Common Fund, the Faculté de Médecine et d'Odontostomatologie, USTTB, Bamako, Mali and the Centre Hospitalier Universitaire “Point G,” Bamako, Mali.
Data Availability Statement
The datasets supporting the findings of this study are available from the corresponding author upon reasonable request.
References
- Anna, A. , and Monika G.. 2018. “Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation.” Journal of Applied Genetics 59, no. 3: 253–268. 10.1007/s13353-018-0444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley, M. , von der Mark H., and Timpl R.. 1985. “Size and Domain Structure of Collagen VI Produced by Cultured Fibroblasts.” FEBS Letters 182, no. 2: 499–502. 10.1016/0014-5793(85)80362-8. [DOI] [PubMed] [Google Scholar]
- Bardakov, S. N. , Deev R. V., Magomedova R. M., et al. 2021. “Intrafamilial Phenotypic Variability of Collagen VI‐Related Myopathy Due to a New Mutation in the COL6A1 Gene.” Journal of Neuromuscular Diseases 8, no. 2: 273–285. 10.3233/JND-200476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönnemann, C. G. 2011. “The Collagen VI‐Related Myopathies. Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy.” Handbook of Clinical Neurology 101: 81–96. 10.1016/B978-0-08-045031-5.00005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briñas, L. , Richard P., Quijano‐Roy S., et al. 2010. “Early Onset Collagen VI Myopathies: Genetic and Clinical Correlations.” Annals of Neurology 68, no. 4: 511–520. 10.1002/ana.22087. [DOI] [PubMed] [Google Scholar]
- Bushby, K. M. D. , Collins J., and Hicks D.. 2014. “Collagen Type VI Myopathies.” Advances in Experimental Medicine and Biology 802: 185–199. 10.1007/978-94-7-7893-1_12. [DOI] [PubMed] [Google Scholar]
- Caria, F. , Cescon M., Gualandi F., et al. 2019. “Autosomal Recessive Bethlem Myopathy: A Clinical, Genetic and Functional Study.” Neuromuscular Disorders 29, no. 9: 657–663. 10.1016/j.nmd.2019.07.007. [DOI] [PubMed] [Google Scholar]
- Chu, M. L. , Conway D., Pan T., et al. 1988. “Amino Acid Sequence of the Triple‐Helical Domain of Human Collagen Type VI.” Journal of Biological Chemistry 263, no. 35: 18601–18606. 10.1016/s0021-9258(18)37327-7. [DOI] [PubMed] [Google Scholar]
- Chu, M.‐L. , Mann K., Deutzmann R., et al. 1987. “Characterization of Three Constituent Chains of Collagen Type VI by Peptide Sequences and cDNA Clones.” European Journal of Biochemistry 168, no. 2: 309–317. 10.1111/j.1432-1033.1987.tb13422.x. [DOI] [PubMed] [Google Scholar]
- Elisabeth, J. , and Hh J.. 2021. “Collagen VI‐Related Myopathy Caused by Compound Heterozygous Mutations of COL6A3 in a Consanguineous Kurdish Family Collagen VI‐Related Myopathy Caused by Compound Heterozygous Mutations of COL6A3 in a Consanguineous Kurdish Family Department of.” Neurology 22: 173–179. [DOI] [PubMed] [Google Scholar]
- Gelse, K. , Pöschl E., and Aigner T.. 2003. “Collagens ‐ Structure, Function, and Biosynthesis.” Advanced Drug Delivery Reviews 55, no. 12: 1531–1546. 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Graziano, A. , Bianco F., Moroni I., et al. 2015. “Prevalence of Congenital Muscular Dystrophy in Italy.” Neurology 84: 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis, E. D. , Nasif S., and Mühlemann O.. 2016. “Nonsense‐Mediated mRNA Decay: Novel Mechanistic Insights and Biological Impact.” Wiley Interdisciplinary Reviews: RNA 7, no. 5: 661–682. 10.1002/wrna.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. Y. , Kim W. J., Kim H., et al. 2018. “Collagen VI‐Related Myopathy: Expanding the Clinical and Genetic Spectrum.” Muscle and Nerve 58, no. 3: 381–388. 10.1002/mus.26093. [DOI] [PubMed] [Google Scholar]
- Kwong, A. K. , Zhang Y., Ho R. S., et al. 2023. “Collagen VI‐Related Myopathies: Clinical Variability, Phenotype‐Genotype Correlation and Exploratory Transcriptome Study.” Neuromuscular Disorders 33, no. 5: 371–381. 10.1016/j.nmd.2023.03.003. [DOI] [PubMed] [Google Scholar]
- Lamandé, S. R. , and Bateman J. F.. 2018. “Collagen VI Disorders: Insights on Form and Function in the Extracellular Matrix and Beyond.” Matrix Biology 71–72: 348–367. 10.1016/j.matbio.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Law, M. L. , Cohen H., Martin A. A., Angulski A. B. B., and Metzger J. M.. 2020. “Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy‐Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies.” Journal of Clinical Medicine 9, no. 2: 520. 10.3390/jcm9020520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. H. , Shin H. Y., Park H. J., Kim S. H., Kim S. M., and Choi Y. C.. 2017. “Clinical, Pathologic, and Genetic Features of Collagen VI‐Related Myopathy in Korea.” Journal of Clinical Neurology (Korea) 13, no. 4: 331–339. 10.3988/jcn.2017.13.4.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott, J. A. , and Landesman P. W.. 1984. “The Enzymology of Skeletal Muscle Disorders.” Critical Reviews in Clinical Laboratory Sciences 20, no. 2: 153–190. 10.3109/10408368409165773. [DOI] [PubMed] [Google Scholar]
- Mareedu, S. , Million E. D., Duan D., and Babu G. J.. 2021. “Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies.” Frontiers in Physiology 12, no. April: 1–19. 10.3389/fphys.2021.647010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martoni, E. , Urciuolo A., Sabatelli P., et al. 2009. “Identification and Characterization of Novel Collagen VI Non‐canonical Splicing Mutations Causing Ullrich Congenital Muscular Dystrophy.” Human Mutation 30, no. 5: E662–E672. 10.1002/humu.21022. [DOI] [PubMed] [Google Scholar]
- Mercuri, E. , Yuva Y., Brown S. C., et al. 2002. “Collagen VI Involvement in Ullrich Syndrome: A Clinical, Genetic, and Immunohistochemical Study.” Neurology 58, no. 9: 1354–1359. 10.1212/WNL.58.9.1354. [DOI] [PubMed] [Google Scholar]
- Nadeau, A. , Kinali M., Main M., et al. 2009. “Natural History of Ullrich Congenital Muscular Dystrophy.” Neurology 73, no. 1: 25–31. 10.1212/WNL.0b013e3181aae851. [DOI] [PubMed] [Google Scholar]
- Norwood, F. L. M. , Harling C., Chinnery P. F., Eagle M., Bushby K., and Straub V.. 2009. “Prevalence of Genetic Muscle Disease in Northern England: In‐Depth Analysis of a Muscle Clinic Population.” Brain 132, no. 11: 3175–3186. 10.1093/brain/awp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M. , Kawahara G., Noguchi S., et al. 2007. “Primary Collagen VI Deficiency Is the Second Most Common Congenital Muscular Dystrophy in Japan.” Neurology 69, no. 10: 1035–1042. 10.1212/01.wnl.0000271387.10404.4e. [DOI] [PubMed] [Google Scholar]
- Park, Y. , Park M. S., Sung D. H., Sohn J. Y., Ki C. S., and Kim D. H.. 2014. “Ullrich Congenital Muscular Dystrophy Possibly Related With COL6A1 p.Gly302Arg Variant.” Annals of Rehabilitation Medicine 38, no. 2: 292–296. 10.5535/arm.2014.38.2.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat, R. A. , Baker N. L., Jones K. J., North K. N., and Lamandé S. R.. 2007. “Variable Penetrance of COL6A1 Null Mutations: Implications for Prenatal Diagnosis and Genetic Counselling in Ullrich Congenital Muscular Dystrophy Families.” Neuromuscular Disorders 17, no. 7: 547–557. 10.1016/j.nmd.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Sarkozy, A. , Foley A. R., Zambon A. A., Bönnemann C. G., and Muntoni F.. 2020. “LAMA2‐Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness.” Frontiers in Molecular Neuroscience 13, no. August: 1–11. 10.3389/fnmol.2020.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich 0tto . 1930. “Kongenitale, atonisch‐skleroti sc he Muskeldystrophie.” Mschr Kinderheilk 47: 502–510. [Google Scholar]
- Von Der Mark, H. , Aumailley M., Wick G., Fleischmajer R., and Timpl R.. 1984. “Immunochemistry, Genuine Size and Tissue Localization of Collagen VI.” European Journal of Biochemistry 142, no. 3: 493–502. 10.1111/J.1432-1033.1984.TB08313.X. [DOI] [PubMed] [Google Scholar]
- Yonekawa, T. , and Nishino I.. 2015. “Ullrich Congenital Muscular Dystrophy: Clinicopathological Features, Natural History and Pathomechanism(s).” Journal of Neurology, Neurosurgery and Psychiatry 86, no. 3: 280–287. 10.1136/jnnp-2013-307052. [DOI] [PubMed] [Google Scholar]
- Zanoteli, E. , Soares P. S., da Silva A. M. S., et al. 2020. “Clinical Features of Collagen VI‐Related Dystrophies: A Large Brazilian Cohort.” Clinical Neurology and Neurosurgery 192, no. September 2019: 105734. 10.1016/j.clineuro.2020.105734. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Yan H., Liu J., et al. 2022. “Clinical and Genetic Features of Infancy‐Onset Congenital Myopathies From a Chinese Paediatric Centre.” BMC Pediatrics 22, no. 1: 65. 10.1186/s12887-021-03024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Zhang H., Sun Y., et al. 2015. “Serum Enzyme Profiles Differentiate Five Types of Muscular Dystrophy.” Disease Markers 2015: 1–7. 10.1155/2015/543282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SpliceAI prediction showing a splice acceptor loss with high delta scores (SpliceAI Δ score = 0.97).
Data Availability Statement
The datasets supporting the findings of this study are available from the corresponding author upon reasonable request.