

Abstract
Thrombosis remains a leading cause of death worldwide despite technological advances in prevention, diagnosis, and treatment. The traditional view of arterial thrombus formation is that it is a platelet‐dependent process, whereas that of venous thrombus formation is a coagulation‐dependent process. Current pathological and basic studies on atherothrombosis and venous thrombosis have revealed the diverse participation of platelet and coagulation activation mechanisms in both thrombus initiation and growth processes during clinical thrombotic events. Atherosclerotic plaque cell‐derived tissue factor contributes to fibrin formation and platelet aggregation. The degree of plaque disruption and a blood flow alteration promote atherothrombotic occlusion. While blood stasis/turbulent flow due to luminal stenosis itself initiates venous thrombus formation. The coagulation factor XI‐driven propagation phase of blood coagulation plays a major role in venous thrombus growth, but a minor role in hemostasis. These lines of evidence indicate that atherothrombosis onset is affected by the thrombogenic potential of atherosclerotic plaques, the plaque disruption size, and an alteration in blood flow. Upon onset of venous thrombosis, enhancement of the propagation phase of blood coagulation under blood stasis and a hypercoagulable state contribute to large thrombus formation.
Keywords: atherothrombosis, factor XI, thrombus formation, thrombus growth, venous thromboembolism
Abbreviations
- AMI
acute myocardial infarction
- COVID‐19
coronavirus disease 2019
- DVT
deep vein thrombosis
- FVIII
factor VIII
- FIX
factor IX
- FX
factor X
- FXI
factor XI
- GP
glycoprotein
- IVC
inferior vena cava
- MRI
magnetic resonance imaging
- NET
neutrophil extracellular trap
- PE
pulmonary embolism
- SMC
smooth muscle cell
- VTE
venous thromboembolism
- VWF
von Willebrand factor
INTRODUCTION
Thrombosis is a leading cause of death worldwide and includes myocardial infarction, ischemic stroke, and venous thromboembolism (VTE) that comprises deep vein thrombosis (DVT) and pulmonary embolism (PE). 1 Thrombus formation is affected by alterations of the vascular wall, blood flow, and blood content. It is considered that atherosclerosis and its plaque disruption are fundamental processes in atherothrombosis and that alterations of blood and the hypercoagulative state are crucial for VTE onset. Clinical imaging and pathological autopsies have revealed that atherothrombosis formation does not always result in thrombotic occlusion and acute symptomatic events. 2 , 3 , 4 Clinical studies have shown that a substantial number (40%) of DVT cases with or without PE were asymptomatic. 5 , 6 These findings indicate that thrombus growth after thrombus formation is also critical for the onset of atherothrombosis and VTE.
The basic mechanisms of hemostasis and thrombus formation are platelet adhesion and aggregation at the site of endothelial injury or denudation and activation of the coagulation cascade. Platelets initially adhere at a site of vascular injury via an interaction between von Willebrand factor (VWF), an adhesion molecule, and the platelet surface receptor glycoprotein (GP) Ibα‐V‐IX complex. Platelet aggregation follows a subsequent shape change and granule secretion, which is accompanied by fibrinogen binding to integrin αIIbβ3 on activated platelets. 7 Blood coagulation is a series of amplifying enzymatic reactions triggered by binding of tissue factor to coagulation factor VII, which leads to insoluble fibrin formation. In this reaction, four enzymes, thrombin, activated factor X (FX), IX (FIX), and XI (FXI), and two cofactors, factor V and VIII (FVIII), constitute an amplifying circuit of thrombin generation called the propagation phase. 8 Blood coagulation is tightly regulated by coagulation inhibitors and the fibrinolytic system (Figure 1). The in vivo function of these proteins has been established by gene knockout techniques. However, most evidence is based on mouse thrombus models using chemically/physically injured normal arteries or veins. Thus, there are substantial differences between animal thrombosis models and the human pathology of atherothrombosis and VTE, and the difference in mechanisms between hemostasis and thrombosis has received little attention.
Figure 1.

Scheme of coagulation factors and endothelium‐dependent anticoagulation and fibrinolysis. Coagulation factors include enzymes (circles) and cofactors (triangles), and are present in blood except for tissue factor (TF). Thrombin (IIa), activated factor XI (FXIa), activated factor IX (FIXa), activated FX (FXa), activated factor VIII (FVIIIa), and activated factor V (FVa) drive the propagation phase of coagulation (red arrows) for thrombin generation. Thrombin contributes to fibrin formation and further platelet aggregation. Activated protein C (aPC) and protein S inactivate FVIIIa and FVa in the presence of thrombin and the thrombomodulin (TM) complex on the endothelium. The endothelium also releases tissue type plasminogen activator (t‐PA) that activates fibrinolysis. The scheme does not show each zymogen, factor XIII, or platelets that provide a lipid membrane for the coagulation reaction.
Traditionally, it has been considered that arterial thrombus is a platelet‐rich thrombus due to rapid arterial flow, and that venous thrombus is composed of erythrocytes and fibrin due to static venous flow. The consideration may be derived from macroscopic and/or microscopic findings. In contrast to the progress in the pathology and pathophysiology of atherosclerosis, 9 the pathological evidence of atherothrombosis and DVT has been limited because of the following reasons. Almost all pathological findings have been obtained from autopsy cases of acute myocardial infarction (AMI) and VTE. 10 , 11 Technological advances in endovascular devices have made it possible to evaluate the thrombus composition immediately after onset. 12 The findings of aspirated arterial and venous thrombi have provided important information on thrombus formation without medical treatments, the time‐lag after the clinical event, and postmortem artifacts compared with pathological findings obtained from autopsies.
This review focuses on the underlying mechanisms of thrombus formation and growth in atherothrombosis and DVT, and discusses the differences between hemostasis and thrombosis.
ATHEROTHROMBOSIS
Thrombosis is a major complication of atherosclerosis. Sudden morphological changes of the plaque initiate thrombus formation. In contrast to the concept of platelet‐rich arterial thrombus, 13 our studies revealed that acute coronary thrombi in patients with AMI consist of not only platelets, but also fibrin, erythrocytes, and leukocytes (Figure 2). 14 , 15 The platelet, VWF, and fibrin contents do not differ for 24 h after onset. 14 These findings suggest that the acute change in the coronary plaque induces both platelet aggregation and coagulation activation. The presence of erythrocytes also suggests a blood flow alteration in occlusive thrombus formation.
Figure 2.

Constituents of coronary atherothrombus in AMI patients. Double immunofluorescence of an aspirated coronary thrombus. The coronary thrombus is composed of platelets (GPIIb/IIIa), fibrin, and von Willebrand factor (VWF). Adapted from Yamashita et al. 14 with permission.
INITIATION OF THROMBUS FORMATION ON DISRUPTED PLAQUES
Atherothrombosis is initiated by plaque rupture and erosion as well as calcified nodule disruption. 16 Accumulated evidence has revealed that chronic inflammation and matrix degradation destabilize the fibrous cap atheroma and the subsequent fibrous cap rupture triggers thrombus formation. The inflammation can be triggered by various stimuli such as oxidized low‐density lipoprotein, plasma‐derived endogenous molecules and smoking‐related chemicals, and cholesterol crystals. 17 These stimuli activate innate and acquired immunities that contribute to the initiation and progression of atherosclerotic lesions. The inflammation destabilizes the fibrous cap through an imbalance of matrix generation and degradation, and degenerative changes. Matrix metalloproteinases produced by macrophages and smooth muscle cells (SMCs) play major roles in plaque instability and subsequent plaque rupture. 9 Because the pathophysiology of plaque destabilization is well known, we focus on thrombus initiation at ruptured sites and the mechanisms of plaque erosion in this section. To date, the mechanisms of calcified nodule formation and its disruption remain unclear.
Thrombus formation at a ruptured plaque
It has been traditionally considered that arterial thrombus formation is initiated by platelet adhesion to the exposed subendothelial collagenous matrix via VWF. 7 However, the ruptured plaque is characterized by paucity of SMCs and fibrous matrix. To reveal the scaffold of platelet adhesion on exposed plaque tissue, we pathologically examined plaque components and the thrombus interface using coronary aspirated thrombi with ruptured plaques obtained within 24 h of AMI onset. Necrotized tissue debris with macrophages were frequently observed at the ruptured plaque and thrombus interface. Fibrous matrix and calcification were found as small fragments. Tissue factor localized in the necrotic core, and macrophages, fibrin and VWF had deposited within the plaques and beneath the acute thrombus (Figure 3). 18 This was the first pathological evidence suggesting that platelet adhesion to fibrin and/or VWF rather than matrix proteins is a possible initial step for thrombus formation at ruptured plaques. An animal model of plaque rupture of thin‐cap fibroatheroma has not been reported to date. Further basic study is required to confirm the concept.
Figure 3.
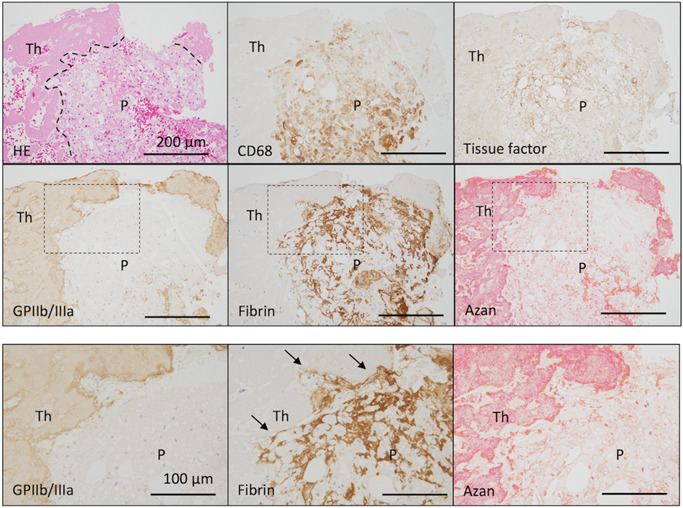
Tissue factor expression and fibrin deposition in ruptured plaques beneath platelet aggregations in patients with acute myocardial infarction. Tissue factor localizes in the necrotic core and macrophages. The thrombus on the ruptured plaque is rich in platelets, and some fibrin formation is visible in the thrombus. Fibrin formation is present around macrophages and cholesterin clefts, and in the necrotic core. In high magnification images (dashed squares), fibrin formation is present at the interface between the ruptured plaques and thrombus (arrows), and no fibrous matrix is observed (Azan staining). Interfaces are indicated by dashed lines. HE, hematoxylin and eosin; P, ruptured plaque; Th, thrombus. Adapted from Yamashita et al. 18
Mechanisms of plaque erosion
Plaque erosion is characterized by superficial erosive injury of an SMC‐rich atherosclerotic plaque, but the mechanisms of the erosive injury remain under debate. Arterial spasm is clinically suggested to be a trigger of arterial injury because vasospastic angina enhances platelet aggregation and activates coagulation in coronary circulation. 19 , 20 Experimental aortic stenosis induces endothelial damage of the nonatherosclerotic normal aorta in canines. 21 Therefore, disturbed arterial flow by stenosis or vasoconstriction may be an initiator of superficial erosive injury. To investigate whether acute luminal stenosis induces intimal injury and thrombus formation on an SMC‐rich plaque, we generated an SMC‐rich neointima in the rabbit femoral artery by a balloon catheter injury of the artery, and the artery was narrowed with a vascular occluder (Figure 4a). The SMC‐rich neointima enhanced tissue factor expression compared with the normal intima. The arterial narrowing induced disruption of blood flow in the post‐stenotic portion, superficial erosive injury with endothelial and superficial SMC detachment, endothelial and SMC apoptosis, and platelet–fibrin thrombus formation on the neointima within 30 min (Figure 4b,c). 22 Thus, disturbed blood flow by acute luminal narrowing induces superficial erosive injury and thrombus formation in an SMC‐rich plaque, and tissue factor expression in intimal SMCs plays a major role in platelet–fibrin thrombus formation on the plaque. Our data indicate that physical injury induced by disturbed blood flow contributes to the superficial erosive injury and are the first evidence on mechanisms of plaque erosion.
Figure 4.

Disturbed blow flow induces superficial erosive injury and thrombus formation in SMC‐rich plaques. The SMC‐rich neointima was induced by a balloon catheter injury of the rabbit femoral artery. (a) Rabbit femoral arteries with SMC‐rich neointima were constricted using a vascular occluder (actuating tube) to reduce the blood flow volume to 75%. (b) Immunohistochemistry of VWF, an endothelial marker, and smooth muscle actin, a smooth muscle cell marker. Images show endothelial cell and SMC detachment (arrows). N indicates the SMC‐rich neointima. (c) Longitudinal section of a rabbit femoral artery showing mural thrombus formation (arrows) at the poststenotic portion 30 min after vascular narrowing. (d) Representative histological and immunohistochemical images of occlusive thrombus formation and SMC involvement (arrows) in the thrombus. N indicates the SMC‐rich neointima. Adapted from Sumi, et al. 22 (b, d) with permission and Sawaguchi et al. 23 (a). HE, hematoxylin and eosin; SMC, smooth muscle cell; VWF, von Willebrand factor.
Neutrophils are a candidate for plaque erosion. Arterial narrowing of the mouse carotid artery with neointima induces endothelial apoptosis, neutrophil adhesion, and neutrophil extracellular trap (NET) formation. 24 , 25 Additionally, deficiency of an enzyme required for NET formation in mice protects against thrombus formation in the carotid artery. 26 Although such evidence suggests that neutrophils and NET formation play major roles in erosive injury and thrombus formation, magnified views by electron microscopy have shown neutrophil accumulation at the platelet aggregation and fibrin formation site, but not on endothelial cells. 23 Further studies are required to clarify whether NET formation is an initiator or promotor of erosive injury and thrombus formation.
MECHANISMS OF ATHEROTHROMBOTIC OCCLUSION
Plaque disruption does not always result in cardiovascular events 3 , 27 , 28 and large thrombus formation is the final step of the onset. We examined coronary thrombus area, pathological findings of the disrupted plaques, and expression of thrombogenic factors in the plaques in autopsy cases of noncardiac death and AMI. The autopsy study revealed that the coronary thrombus size correlates positively to the length of the plaque disruption and expression of tissue factor and hexokinase II, a glycolysis enzyme. 29 In this section, we focus on mechanisms of platelet–fibrin thrombus formation and thrombotic occlusion on atherosclerotic plaques.
Mechanisms of platelet–fibrin thrombus formation on atherosclerotic plaques
Most evidence on arterial thrombus formation is based on mouse thrombus models using chemically/physically injured normal arteries. 30 Because atherothrombosis occurs on disrupted atherosclerotic lesion, it is important to understand the mechanisms of thrombus formation on an atherosclerotic lesion. Rabbits are susceptible to atherosclerosis compared with mice and rats, and lipoprotein metabolism in rabbits, but not mice or rats, is similar to that in humans. 9 To examine mechanisms of atherothrombosis formation, we established a rabbit model of atherothrombosis with a repeated balloon catheter‐induced injury of iliac and femoral arteries and a conventional or 0.5% cholesterol diet. 31 , 32 , 33 , 34 The first balloon injury of arteries and the diets induced SMC proliferation, extracellular matrix production, macrophage infiltration, and expression of tissue factor in SMCs and/or macrophages. The enhanced tissue factor activity in the neointima corresponded to human atherosclerotic plaques. 35 The second balloon injury induced platelet–fibrin thrombus formation on the injured neointima. The balloon‐induced injury of the normal intima also induced thrombus, but it consisted of small aggregated platelets without fibrin formation. 32 , 34 Excess thrombin generation through activation of the coagulation cascade on the injured neointima contributed not only to fibrin formation, but also to further platelet aggregation. The source of tissue factor for arterial thrombus formation is an issue in thrombosis research because of the presence of tissue factor in circulating blood and/or leukocytes. The tissue factor associated with hematopoietic cell‐derived microvesicles contributes to laser‐induced arterial thrombus formation in mouse small arteries. 36 Conversely, normal carotid artery thrombus formation initiated by photochemical injury is driven by vascular wall cell‐derived tissue factor. 37 In our rabbit thrombosis model, the tissue factor pathway in blood coagulation played an important role in platelet–fibrin thrombus formation on the neointima. 34 , 38 Additionally, the thrombus formation was independent of blood‐derived tissue factor. 34 Our results suggest that the vascular wall tissue factor‐driven coagulation pathway plays a major role in atherothrombosis formation and proves the pathological significance of tissue factor expression in human atherosclerotic plaques.
Mechanisms of atherothrombus growth and thrombotic occlusion
Although tissue factor expression in the neointima plays a major role in platelet–fibrin thrombus formation, the thrombus remains mural and not occlusive in rabbits as observed in cases of human unstable angina. AMI results from acute total occlusion of the coronary artery, whereas unstable angina is related to mural thrombus formation in most cases. 39 Additional conditions have been considered to be required for thrombus growth and occlusive thrombus formation. A longitudinal section of a human coronary occlusive thrombus showed an irregular vortex structure, suggesting disturbed blood flow during thrombus formation. Coronary blood flow can be reduced by 80% in patients with unstable angina and microembolisms are frequently observed in the myocardium in AMI patients. 40 Therefore, it is possible that microembolisms and/or vasoconstriction reduce coronary blood flow and affect thrombus growth at disrupted plaques. Balloon injury‐induced platelet–fibrin thrombi remain mural up to a 50% reduction in arterial blood flow and become occlusive over a 75% reduction in flow. 32 During occlusive thrombus formation, serotonin secreted from activated platelets induces vascular contraction and augments platelet aggregation via the serotonin 2A receptor on SMCs and platelets. 41 Interestingly, arteries with an SMC‐rich neointima are susceptible to serotonin and show a hypercontractile response partly through Rho kinase. Additionally, SMC‐rich neointima itself has a contractile response to serotonin. 42
As mentioned above, acute luminal narrowing induces superficial erosive injury of the SMC‐rich neointima and platelet–fibrin thrombus formation in rabbits. The erosive injury longitudinally progresses over time and 60% of the mural thrombus becomes occlusive at 3 h (Figure 4d). 22 This supports the relationship between the plaque disruption length and atherothrombus size in the human coronary artery. 29
VWF in thrombotic occlusion
As a thrombus gradually becomes occlusive, it is conceivable that thrombus formation is physically inhibited by an increase in blood velocity within the narrowing lumen. VWF, a multimetric protein, is a candidate for platelet adhesion and aggregation under high shear stress within the narrowing lumen. The binding potential of VWF for GPIbα is dependent on shear stress, and high shear stress induces VWF to change from its globular form to an extended string form. 43 This enhances platelet adhesion to VWF through exposure of GPIbα‐binding sites on the VWF multimer. Additionally, the excessive shear stress induces platelet aggregation in the absence of platelet agonists. 44 In fact, our and other studies have shown that inhibition of the interaction between VWF and platelet GPIbα or ADAMTS‐13, a VWF protease, protects against occlusive thrombus formation on the neointima of rabbits and in an in vitro flow chamber system. 32 , 45
PLAQUE THROMBOGENICITY
Tissue factor is a high affinity cell surface receptor for plasma factor VII/activated factor VII, which initiates the coagulation cascade reaction by promoting proteolytic activation of coagulation FIX and FX (Figure 1). As mentioned above, pathological and animal studies indicate that tissue factor is a major determinant of thrombogenic potential in atherosclerotic plaques. A recent review described in detail tissue factor regulation in atherosclerotic plaques. 46 Tissue factor expression in macrophages and SMCs was affected by Th1 and Th2 cytokines, hypoxia, C‐reactive protein deposition, and even vascular cell metabolites in the data published by our institute. 46 , 47 , 48 , 49 , 50 , 51 Glycolysis and tryptophan kynurenine pathways are associated with regulation of tissue factor expression in macrophages. 29 , 49 , 50 Interestingly, an increase in the intracellular glutamine level downregulates procoagulant activity in coronary artery SMCs. 51
Recent evidence suggests a possible interaction between intraplaque hemorrhage and coronary thrombotic events. A pathological study found intraplaque hemorrhage in advanced atherosclerotic plaques, suggesting that accumulation of cholesterol from the erythrocyte membrane accelerates plaque progression. 52 Intraplaque hemorrhage might be a potent marker of plaque instability and thrombogenic potential. We reported that erythrocyte contents, iron deposition, and thrombus formation are higher in coronary plaques from unstable angina pectoris than stable angina pectoris. 53 High‐signal intensity coronary plaques on a T1‐weighted image are a strong predictor for future cardiovascular events. Noguchi et al. 54 reported that one‐sixth of patients with high intensity plaques develop cardiovascular events within 2 years. The signal intensity in T1‐weighted magnetic resonance imaging (MRI) correlates to erythrocytes and fibrin deposition, and expression of matrix metalloproteinase‐9 and tissue factor in plaques (Figure 5a). 55 We pathologically examined ruptured coronary plaques of AMI patients, and found that bilirubin and iron depositions indicated previous hemorrhage in up to one‐fourth of the plaques. 18 Bilirubin deposition is a novel finding of advanced atherosclerotic plaques. Carotid and coronary ruptured plaques variously express enzymes of hemo‐metabolism and endoproducts, heme oxygenase‐1, biliverdin reductase, and ferritin (Figure 5b). 18 , 56 These lines of evidence suggest that intraplaque hemorrhage precedes plaque rupture, destabilizes lipid‐rich plaques, and promotes thrombogenic potential in coronary plaques.
Figure 5.
Relationship between intraplaque hemorrhage and coronary thrombosis. (a) Representative in vitro MRI of a nonculprit lesion in an AMI patient, and histological and immunohistochemical images of a high‐signal intensity plaque on a T1‐weighted image (WI). T1 high‐intensity portions (asterisks) in the plaque show a low signal intensity on T2WI and are variably immunopositive for CD68, glycophorin A, fibrin, and matrix metalloproteinase 9 (MMP9). There are tissue factor‐immunopositive foci in the plaque (arrows). The foci are close to the lumen (L). Adapted from Kuroiwa et al. 55 with permission. (b) Bilirubin deposition and expression of ferritin and biliverdin reductase in a coronary ruptured plaque in an AMI patient. Ferritin and biliverdin reductase are predominantly expressed in yellowish (middle row, arrows) and grayish (bottom row, arrows) macrophages. Bilirubin deposition is present in the middle row (arrowhead). AMI, acute myocardial infarction, HE, hematoxylin and eosin; MRI, magnetic resonance imaging. Adapted from Yamashita et al. 18


VTE
Venous thromboembolism is the third most common cardiovascular disease worldwide. Estimates of incidence range from 79 to 269 per 100 000 for VTE and 39 to 115 per 100 000 for PE. 1 The reported incidence is lower in Asian, Asian American, and Native American populations, and higher in black populations. 1 The reversible risk factors for VTE include immobilization, such as hospitalization and recent surgery, whereas active cancer and antiphospholipid syndrome are persistent or irreversible. 57 Genetic conditions that predispose to VTE are deficiencies of protein C, protein S, antithrombin, Factor V Leiden mutation, and prothrombin gene mutation G20210A. 57 These can be divided into three prothrombotic states: blood flow alteration (immobilization), vascular/tissue injury (surgery, active cancer, and antiphospholipid syndrome), and a hypercoagulable state (active cancer, antiphospholipid syndrome, and genetic abnormalities).
In addition to the traditional risk factors, a high prevalence of VTE has been reported after earthquakes 58 , 59 and in severe cases of coronavirus disease 2019 (COVID‐19). Thrombotic complications, predominantly VTE, occur in 10%–20% of hospitalized patients with COVID‐19 despite routine thromboprophylaxis. 60 , 61 Although the mechanisms of COVID‐19‐associated thrombosis are under investigation, immunological reactions to viral infection are becoming the consensus as a mechanism of thrombogenesis. 62 In this section, we focus on the mechanisms of DVT formation under disturbed blood flow or endothelial injury.
Constituents of VTE
Venous thrombi are composed of a large amount of fibrin and erythrocytes, and relatively few platelets. 10 , 11 However, we revealed that DVT at venous wall attachment sites and PE contain platelets, fibrin, erythrocytes, and VWF, and their proportions do not differ between DVT and PE. 63 Platelets are predominantly localized with VWF‐ and fibrin‐rich areas, but not an erythrocyte‐rich area. FVIII localized in DVT is closely associated with VWF, platelets, and fibrin. 64 These pathological findings suggest that platelets are a major component in addition to fibrin and erythrocytes, and that VWF and FVIII play roles in venous thrombus formation. Aspirated thrombi obtained from the proximal (upper) portion of DVT are rich in erythrocytes and fibrin, and the immunopositive area for platelets is one‐third to one‐half of the erythrocyte and fibrin areas (Figure 6). 65 Neutrophils are localized along the border of platelet–fibrin‐ and erythrocyte‐rich areas. DVT shows various phases of degenerative or organizing reactions such as fresh components, cell lytic changes, macrophage infiltration, endothelialization, and myofibroblastic/fibroblastic cell proliferation (Figure 7a). These findings suggest that the proportion of the components differs between the attachment site and proximal portion, and we hypothesized that venous thrombus has multiple growth phases. The concept is supported by clinical and basic studies using MRI and an animal model of DVT. The median value of the DVT volume calculated by MRI was 20 mL. 66 The value appears to be much larger than that of atherothrombosis. The mean area of the coronary occlusive thrombus was 2 mm2. 29 The signal intensity of DVT on diffusion weighted MRI was a heterogenous high‐to‐iso intensity and the signal intensity of the proximal portion was higher than that of the distal portion of DVT (Figure 7b,c). The high signal intensity reflects fresh thrombus components within 1 week in a rabbit jugular vein thrombosis model. In vitro MRI confirmed that the erythrocyte content, but not blood coagulation, affects the diffusion capacity of water molecules, corresponding to the signal intensity of the diffusion weighted image (Figure 7d). 66 Our data indicate that large DVT consists of fresh and organizing components reflecting multiple growth phases and that diffusion weighted MRI detects the fresh component as a high signal intensity portion.
Figure 6.

Constituents of deep venous thrombus. Representative light and immunohistochemical microphotographs of an aspirated deep vein thrombosis. The thrombus is stained with hematoxylin and eosin (HE) and antibodies against glycophorin A, integrin α2bβ3 (a platelet protein), and fibrin. The thrombus is rich in glycophorin A and fibrin. Adapted from Furukoji et al. 65
Figure 7.

Heterogenous components of DVT: The possibility of multistep growth phases. (a) An aspirated DVT at 33 days after onset shows various phases of reactions, fresh components without a degenerative change, a cell lytic change, macrophage infiltration and an organizing reaction (hematoxylin and eosin staining). (b) Representative magnetic resonance images of DVT patients. High‐signal intensity and high‐to‐iso signal intensity lesions on T1‐weighted images (WI) occupied bilateral femoral to popliteal veins (arrows), indicating DVT. Merged diffusion weighted image (DWI) and T1WI‐localized heterogeneous high‐to‐iso signal intensity lesions in deep veins on DWI. Areas of DVT with a high signal intensity on DWI have a low apparent diffusion coefficient (ADC) and high‐to‐iso signal intensity on T2WI. ADC indicating the diffusion capacity of a water molecule and a low value reflecting restriction of its diffusion capacity. (c) DWI contrast‐to‐noise ratio in proximal and distal portions of DVT. (d) ADC of coagulated blood measured by in vitro MRI. Human venous blood containing sodium citrate was separated into WB (whole blood), PRP (platelet‐rich plasma), and ERB (erythrocyte‐rich blood) 1 and 2. Blood was coagulated using human placental tissue factor and a CaCl2 solution. ADC values are associated with the erythrocyte content in clotting blood. n = 9 in each. Adapted from Gi et al. 66 (b–d). DVT, deep vein thrombosis.
INITIATION AND GROWTH OF VENOUS THROMBUS
Pathophysiological alterations involved in DVT development are called Virchow's triad, namely changes in blood flow, blood content, and the vascular wall. Although it remains the main theory of venous thrombus formation, we currently have a deeper understanding of each factor. Accumulated evidence suggests that an alteration of blood flow and endothelial/leukocyte reactions contribute to thrombus initiation and that alterations of blood flow and a hypercoagulable state contribute to thrombus growth. 67 , 68 , 69
Blood flow alteration
Blood stasis has been traditionally considered to initiate blood coagulation within blood and subsequent venous thrombus formation. However, this concept needs to be reconsidered. Blood stasis leads to neutrophil adhesion on endothelial cells and migration under the endothelium without morphological endothelial damage and thrombus formation within 1 h in the rabbit jugular vein. 70 Stasis‐induced venous thrombus can be detected by ultrasound at 1 h in the mouse inferior vena cava. 71 We reported that blood stasis induced by rabbit jugular vein ligation initiates venous thrombus formation accompanied by endothelial denudation within 4 h. 67 The thrombus consists of erythrocytes, fibrin, platelets, and leukocytes as observed in human DVT. This indicates that blood stasis itself initiates venous thrombus formation by affecting endothelial functions and denudation. Von Brühl et al. 68 assessed early cellular events that trigger DVT formation using a mouse model of DVT induced by flow restriction (<80%) in the inferior vena cava and intravital imaging system. Neutrophils and monocytes had accumulated in clusters of layers on the intact endothelium within 1 h. The leukocyte accumulation depended on an interaction of endothelial p‐selection with p‐selectin GP ligand on leukocytes. Surprisingly, tissue factor derived from myeloid cells, particularly monocytes, but not the vascular wall, was responsible for the initiation of fibrin formation. The flow restriction may induce VWF release from endothelial cells, p‐selectin expression on the endothelium, and subsequent platelet and leukocyte accumulation and fibrin formation on the endothelial surface. 68
Cancer‐associated thrombosis
Cancer cells invade and compress the venous wall at primary and metastatic sites, and can enter circulation. Cancer cells express tissue factor, podoplanin, plasminogen activator inhibitor (a fibrinolysis inhibitor), and other thrombogenic substances. 72 Basic studies have reported that cancer cells promote venous thrombus formation and its stability via cancer cell‐derived microvesicles, NET formation, thrombocytosis, or suppression of fibrinolysis. 72 Thus, cancer can physically and biologically initiate and promote venous thrombus formation. Although pathologists must recognize cancer as an initiator of venous thrombus formation, pathological evidence focusing on the thrombotic site remains limited.
FXI PARTICIPATES IN VENOUS THROMBUS GROWTH, BUT NOT HEMOSTASIS
FXI in venous thrombus growth
Increased plasma levels of factor FVIII, IX, and XI are associated with VTE. 73 , 74 , 75 , 76 These factors are involved in the propagation phase of the coagulation cascade (Figure 1). Traditional risk factors (protein C and S) are also associated with the propagation phase (Figure 1). To investigate the role of FVIII and FXI in venous thrombus formation and thrombus growth, we performed in vivo and in vitro experiments. Inhibition of FVIII activation reduces platelet aggregation and fibrin formation through thrombin generation in vitro. 64 Conversely, an elevated FVIII level by recombinant FVIII treatment markedly promotes venous thrombus growth and subsequent occlusive thrombus formation in rabbits (Figure 8a). 69 During excess thrombin generation, FXI and VWF, but not tissue factor, contribute to the thrombus growth process (Figure 8b). 69 FXI inhibition by an anti‐FXI antibody also significantly suppresses fibrin and erythrocyte‐rich venous thrombus formation in rabbits (Figure 8c). 67 FXI−/− mice also resist venous thrombus growth in stenosis‐induced and electrolytic venous thrombus models. 77 These data provide in vivo evidence of the FXI‐driven propagation phase of blood coagulation. Further pathological study is required to verify the presence or absence of FXI in human VTE.
Figure 8.

FVIII and FXI contribute to venous thrombus growth. (a) Recombinant FVIII (rFVIII) administration promotes venous thrombus formation. Fluorescence images at 15 min after rabbit jugular vein endothelial denudation and at 15 s after indocyanine green (ICG) infusion with saline (control, left) or 100 IU/kg rFVIII (right). Histological images show mural (control) or occlusive (rFVIII infusion) thrombus formation at 1 h after endothelial denudation. (b) Venous thrombus growth was monitored over time by ICG administration after endothelial denudation by rFVIII infusion. The ratio of the ICG fluorescence intensity to the background positively correlated to the thrombus size (r = 0.84). When the average fluorescence intensity of ICG exceeded three‐fold of the background, argatroban (thrombin inhibitor) or an anti‐tissue factor (TF), anti‐FXI, or anti‐VWF antibody was infused. The graph shows the difference in the fluorescence intensity of ICG before each inhibitor administration or at 1 h after endothelial denudation (n = 4 in each). (c) Venous thrombus components in rabbits and effect of the anti‐FXI antibody (XI‐5108, 3 mg/kg) on venous thrombus formation. Rabbit jugular vein thrombus is composed of erythrocytes, platelets (GPIIb/IIIa), and fibrin. Venous thrombus weight at 4 h after vessel ligation or endothelial denudation in jugular veins. Adapted from Takahashi et al. 67 and Sugita et al. 69 with permission. HE, hematoxylin and eosin.
Factor XI in hemostasis
The contributions of FVIII, FIX, and FXI to hemostasis differ significantly. Human deficiency of FVIII or FIX is well known as hemophilia A and B, respectively. However, spontaneous bleeding is rare in FXI‐deficient individuals and most bleeding manifestations are related to injury. 78 Additionally, plasma FXI activity below 50% is protective against VTE and ischemic stroke. 79 Inhibition of factor XI activation by an anti‐FXI antibody suppresses venous thrombus formation, but does not affect bleeding time in rabbits. 67 , 80 FXI−/− mice have no spontaneous bleeding and no excess bleeding in tail and abdominal wall cut models, and exhibit milder bleeding than FIX−/− mice in the saphenous vein bleeding model. 81 These lines of evidence indicate that the contribution of FXI to hemostasis is less than that of FVIII and FIX, and that FXI mainly contributes to venous thrombus growth (Figure 9).
Figure 9.

Possible difference in mechanisms between hemostasis and venous thrombosis. (a) Blood coagulation in hemostasis. Vascular wall damage induces exposure of vascular wall‐derived tissue factor (TF) and initiates the coagulation cascade. FIX is predominantly activated by the FVIIa/TF complex, and activated FIX (FIXa) and activated FVIII (FVIIIa) are required for hemostatic thrombus formation. Activated FXI (FXIa) plays a minor role in hemostasis. FXII is not essential for hemostasis. Activated protein C (aPC) and protein S inactivate FVIIIa and FVa in the presence of the thrombin (IIa) and thrombomodulin (TM) complex on the endothelium. (b) Blood coagulation in venous thrombosis. Blood flow restriction or stasis induces leukocyte accumulation on a dysfunctional endothelium and/or detachment of the endothelium. Leukocyte‐ and/or vascular cell‐derived TF initiates the coagulation reaction. FXI activated by thrombin enhances further thrombin generation via FIX and FX activation. The propagation phase and blood flow alteration promote thrombus formation. This process is augmented by diminished activity of aPC and protein S. The contribution of FXII to thrombus growth is under debate. The schemes do not show each zymogen, factor XIII, or platelets that provide a lipid membrane for the coagulation reaction.
Novel anticoagulants targeting factor XI
The properties of FXI in hemostasis and thrombosis have provided insights for the development of ideal anticoagulants that protect against VTE without bleeding. Current direct oral anticoagulants are widely used to prevent VTE and ischemic stroke. Although the bleeding risk of direct oral anticoagulants is lower than that of warfarin, they have a risk of major bleeding in 2.5% of patients per year even with lower dose usage. 82 Novel anticoagulants that target FXI/FXIa are under development. These include FXI antisense oligonucleotides, anti‐FXI antibodies, and oral direct FXIa inhibitors. 83
Factor XII in hemostasis and thrombosis
Factor XII may be a candidate novel anticoagulant because FXII‐deficient patients have no bleeding tendency and FXII promotes thrombus growth via NET‐driven FXII activation in mice. 68 However, clinical studies have shown a lack of an association between FXII plasma levels and VTE and ischemic stroke. 84 Additionally, expression of citrullinated histone 3, a NET marker, is frequently observed in organizing DVT, but not fresh DVT, suggesting that NETs are predominantly formed after thrombus formation. 85 Thus, it has been considered that FXII is not essential for hemostasis and the contribution of FXII to thrombus growth is under debate (Figure 9).
CONCLUSIONS
Thrombus growth and subsequent large thrombus formation are critical processes for the onset of atherothrombosis and DVT. The degree of plaque disruption is a major effector of atherothrombus size. Activated FXI is a critical enzyme in the venous thrombus growth and has a minor contribution to hemostasis. Therefore, novel anticoagulants that target FXI/activated FXI have attracted great attention as safe agents.
AUTHOR CONTRIBUTIONS
Atsushi Yamashita contributed to drafting the manuscript and figures. Yujiro Asada contributed to revision. All authors approved the final version of the manuscript.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGMENTS
We thank all laboratory members at the Division of Pathophysiology, Department of Pathology, Faculty of Medicine, University of Miyazaki, Japan, and all collaborators for their contribution to this review. This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (18H02773, 19H03445, 19K07437, 20K08085, and 21K07706) and the Setsuro Fujii Memorial, The Osaka Foundation for Promotion of Fundamental Medical Research. Atsushi Yamashita was selected as the winner of the Pathology Research Award in 2021 by the Japanese Society of Pathology. We thank Mitchell Arico from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Yamashita A, Asada Y. Underlying mechanisms of thrombus formation/growth in atherothrombosis and deep vein thrombosis. Pathol Int. 2023;73:65–80. 10.1111/pin.13305
REFERENCES
- 1. Wendelboe AM, Raskob GE. Global burden of thrombosis: epidemiologic aspects. Circ Res. 2016;118:1340–7. [DOI] [PubMed] [Google Scholar]
- 2. Asakura M, Ueda Y, Yamaguchi O, Adachi T, Hirayama A, Hori M, et al. Extensive development of vulnerable plaques as a pan‐coronary process in patients with myocardial infarction: an angioscopic study. J Am Coll Cardiol. 2001;37:1284–88. [DOI] [PubMed] [Google Scholar]
- 3. Sato Y, Hatakeyama K, Marutsuka K, Asada Y. Incidence of asymptomatic coronary thrombosis and plaque disruption: comparison of non‐cardiac and cardiac deaths among autopsy cases. Thromb Res. 2009;124:19–23. [DOI] [PubMed] [Google Scholar]
- 4. Vergallo R, Uemura S, Soeda T, Minami Y, Cho JM, Ong DS, et al. Prevalence and predictors of multiple coronary plaque ruptures: in vivo 3‐vessel optical coherence tomography imaging study. Arterioscler Thromb Vasc Biol. 2016;36:2229–38. [DOI] [PubMed] [Google Scholar]
- 5. Moser KM, et al. Frequent asymptomatic pulmonary embolism in patients with deep venous thrombosis. J Am Med Assoc. 1994;271:223–5. [PubMed] [Google Scholar]
- 6. Ota S, Matsuda A, Ogihara Y, Yamada N, Nakamura M, Mori T, et al. Incidence, characteristics and management of venous thromboembolism in Japan during 2011. Circ J. 2018;82:555–60. [DOI] [PubMed] [Google Scholar]
- 7. Mazzucato M, Cozzi MR, Pradella P, Ruggeri ZM, De Marco L. Distinct roles of ADP receptors in von Willebrand factor‐mediated platelet signaling and activation under high flow. Blood. 2004;104:3221–7. [DOI] [PubMed] [Google Scholar]
- 8. Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–12. [DOI] [PubMed] [Google Scholar]
- 9. Fan J, Watanabe T. Atherosclerosis: known and unknown. Pathol Int. 2022;72:151–60. [DOI] [PubMed] [Google Scholar]
- 10. Stein PD, Evans H. An autopsy study of leg vein thrombosis. Circulation. 1967;35:671–81. [DOI] [PubMed] [Google Scholar]
- 11. Beckering RE Jr., Crowson M. Variations in cellulose acetate membranes for lipoprotein electrophoresis. Am J Clin Path. 1971;56:765–6. [DOI] [PubMed] [Google Scholar]
- 12. Kelly RV, Cohen MG, Stouffer GA. Mechanical thrombectomy options in complex percutaneous coronary interventions. Catheter Cardiovasc Interv. 2006;68:917–28. [DOI] [PubMed] [Google Scholar]
- 13. Gurbel PA, Jeong YH, Navarese EP, Tantry US. Platelet‐mediated thrombosis. Circ Res. 2016;118:1380–91. [DOI] [PubMed] [Google Scholar]
- 14. Yamashita A, Sumi T, Goto S, Hoshiba Y, Nishihira K, Kawamoto R, et al. Detection of von Willebrand factor and tissue factor in platelets‐fibrin rich coronary thrombi in acute myocardial infarction. Am J Cardiol. 2006;97:26–8. [DOI] [PubMed] [Google Scholar]
- 15. Nishihira K, Yamashita A, Ishikawa T, Hatakeyama K, Shibata Y, Asada Y. Composition of thrombi in late drug‐eluting stent thrombosis versus de novo acute myocardial infarction. Thromb Res. 2010;126:254–7. [DOI] [PubMed] [Google Scholar]
- 16. Yahagi K, Davis HR, Arbustini E, Virmani R. Sex differences in coronary artery disease: pathological observations. Atherosclerosis. 2015;239:260–7. [DOI] [PubMed] [Google Scholar]
- 17. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20:589–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamashita A, Nishihira K, Gi T, Maekawa K, Hatakeyama K, Horiuchi S, et al. Pathological features of ruptured coronary plaque and thrombus interfaces: fibrin and von Willebrand factor as platelet scaffolds on rupture sites. Thromb Haemost. 2021;121:234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oshima S, Yasue H, Ogawa H, Okumura K, Matsuyama K. Fibrinopeptide A is released into the coronary circulation after coronary spasm. Circulation. 1990;82:2222–5. [DOI] [PubMed] [Google Scholar]
- 20. Miyamoto S, Ogawa H, Soejima H, Takazoe K, Kajiwara I, Shimomura H, et al. Enhanced platelet aggregation in the coronary circulation after coronary spasm. Thromb Res. 2001;103:377–86. [DOI] [PubMed] [Google Scholar]
- 21. Fry DL. Acute vascular endothelial changes associated with increased blood velocity gradients. Circ Res. 1968;22:165–97. [DOI] [PubMed] [Google Scholar]
- 22. Sumi T, Yamashita A, Matsuda S, Goto S, Nishihira K, Furukoji E, et al. Disturbed blood flow induces erosive injury to smooth muscle cell‐rich neointima and promotes thrombus formation in rabbit femoral arteries. J Thromb Haemost. 2010;8:1394–402. [DOI] [PubMed] [Google Scholar]
- 23. Sawaguchi A, Kamimura T, Yamashita A, Takahashi N, Ichikawa K, Aoyama F, et al. Informative three‐dimensional survey of cell/tissue architectures in thick paraffin sections by simple low‐vacuum scanning electron microscopy. Sci Rep. 2018;8:7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quillard T, Araújo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. 2015;36:1394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franck G, Mawson T, Sausen G, Salinas M, Masson GS, Cole A, et al. Flow perturbation mediates neutrophil recruitment and potentiates endothelial injury via TLR2 in mice: implications for superficial erosion. Circ Res. 2017;121:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ Res. 2018;123:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arbustini E, Grasso M, Diegoli M, Morbini P, Aguzzi A, Fasani R, et al. Coronary thrombosis in non‐cardiac death. Coron Artery Dis. 1993;4:751–60. [DOI] [PubMed] [Google Scholar]
- 28. Nakamura E, Sato Y, Iwakiri T, Yamashita A, Moriguchi‐Goto S, Maekawa K, et al. Asymptomatic plaques of lower peripheral arteries and their association with cardiovascular disease: an autopsy study. J Atheroscler Thromb. 2017;24:921–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okuyama N, Matsuda S, Yamashita A, Moriguchi‐Goto S, Sameshima N, Iwakiri T, et al. Human coronary thrombus formation is associated with degree of plaque disruption and expression of tissue factor and hexokinase II. Circ J. 2015;79:2430–8. [DOI] [PubMed] [Google Scholar]
- 30. Sachs UJH, Nieswandt B. In vivo thrombus formation in murine models. Circ Res. 2007;100:979–91. [DOI] [PubMed] [Google Scholar]
- 31. Yamashita A, Asada Y, Sugimura H, Yamamoto H, Marutsuka K, Hatakeyama K, et al. Contribution of von Willebrand factor to thrombus formation on neointima of rabbit stenotic iliac artery under high blood‐flow velocity. Arterioscler Thromb Vasc Biol. 2003;23:1105–10. [DOI] [PubMed] [Google Scholar]
- 32. Yamashita A, Furukoji E, Marutsuka K, Hatakeyama K, Yamamoto H, Tamura S, et al. Increased vascular wall thrombogenicity combined with reduced blood flow promotes occlusive thrombus formation in rabbit femoral artery. Arterioscler Thromb Vasc Biol. 2004;24:2420–4. [DOI] [PubMed] [Google Scholar]
- 33. Moriguchi‐Goto S, Yamashita A, Tamura N, Soejima K, Takahashi M, Nakagaki T, et al. ADAMTS‐13 attenuates thrombus formation on type I collagen surface and disrupted plaques under flow conditions. Atherosclerosis. 2009;203:409–16. [DOI] [PubMed] [Google Scholar]
- 34. Yamashita A, Matsuda S, Matsumoto T, Moriguchi‐Goto S, Takahashi M, Sugita C, et al. Thrombin generation by intimal tissue factor contributes to thrombus formation on macrophage‐rich neointima but not normal intima of hyperlipidemic rabbits. Atherosclerosis. 2009;206:418–26. [DOI] [PubMed] [Google Scholar]
- 35. Hatakeyama K, Asada Y, Marutsuka K, Sato Y, Kamikubo Y, Sumiyoshi A. Localization and activity of tissue factor in human aortic atherosclerotic lesions. Atherosclerosis. 1997;133:213–9. [DOI] [PubMed] [Google Scholar]
- 36. Chou J, Mackman N, Merrill‐Skoloff G, Pedersen B, Furie BC, Furie B. Hematopoietic cell‐derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood. 2004;104:3190–7. [DOI] [PubMed] [Google Scholar]
- 37. Day SM, Reeve JL, Pedersen B, Farris DM, Myers DD, Im M, et al. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105:192–8. [DOI] [PubMed] [Google Scholar]
- 38. Funatsu T, Yamashita A, Kaku S, Iwatsuki Y, Asada Y. Plasma factor Xa inhibition can predict antithrombotic effects of oral direct factor Xa inhibitors in rabbit atherothrombosis models. Thromb Haemost. 2012;108:896–902. [DOI] [PubMed] [Google Scholar]
- 39. Ambrose JA. Thrombosis in ischemic heart disease. Arch Intern Med. 1996;156:1382–94. [PubMed] [Google Scholar]
- 40. Marzilli M, Sambuceti G, Fedele S, L'Abbate A. Coronary microcirculatory vasoconstriction during ischemia in patients with unstable angina. J Am Coll Cardiol. 2000;35:327–34. [DOI] [PubMed] [Google Scholar]
- 41. Nishihira K, Yamashita A, Tanaka N, Kawamoto R, Imamura T, Yamamoto R, et al. Inhibition of 5‐hydroxytryptamine2A receptor prevents occlusive thrombus formation on neointima of the rabbit femoral artery. J Thromb Haemost. 2006;4:247–55. [DOI] [PubMed] [Google Scholar]
- 42. Nishihira K, Yamashita A, Tanaka N, Moriguchi‐Goto S, Imamura T, Ishida T, et al. Serotonin induces vasoconstriction of smooth muscle cell‐rich neointima through 5‐hydroxytryptamine2A receptor in rabbit femoral arteries. J Thromb Haemost. 2008;6:1207–14. [DOI] [PubMed] [Google Scholar]
- 43. Siediecki C, Lestini B, Kottke‐Marchant K, Eppell S, Wilson D, Marchant R. Shear‐dependent changes in the three‐dimensional structure of human von Willebrand factor. Blood. 1996;88:2939–50. [PubMed] [Google Scholar]
- 44. Ikeda Y, Murata M, Goto S. von Willebrand factor‐dependent shear‐induced platelet aggregation: basic mechanisms and clinical implications. Ann NY Acad Sci. 1997;811:325–36. [DOI] [PubMed] [Google Scholar]
- 45. Shida Y, Nishio K, Sugimoto M, Mizuno T, Hamada M, Kato S, et al. Functional imaging of shear‐dependent activity of ADAMTS13 in regulating mural thrombus growth under whole blood flow conditions. Blood. 2008;111:1295–8. [DOI] [PubMed] [Google Scholar]
- 46. Asada Y, Yamashita A, Sato Y, Hatakeyama K. Pathophysiology of atherothrombosis: mechanisms of thrombus formation on disrupted atherosclerotic plaques. Pathol Int. 2020;70:309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matsuura Y, Yamashita A, Iwakiri T, Sugita C, Okuyama N, Kitamura K, et al. Vascular wall hypoxia promotes arterial thrombus formation via augmentation of vascular thrombogenicity. Thromb Haemost. 2015;114:158–72. [DOI] [PubMed] [Google Scholar]
- 48. Matsuda S, Yamashita A, Sato Y, Kitajima S, Koike T, Sugita C, et al. Human C‐reactive protein enhances thrombus formation after neointimal balloon injury in transgenic rabbits. J Thromb Haemost. 2011;9:201–8. [DOI] [PubMed] [Google Scholar]
- 49. Yamashita A, Zhao Y, Zhao S, Matsuura Y, Sugita C, Iwakiri T, et al. Arterial 18F‐fluorodeoxyglucose uptake reflects balloon catheter‐induced thrombus formation and tissue factor expression via nuclear factor‐κB in rabbit atherosclerotic lesions. Circ J. 2013;77:2626–35. [DOI] [PubMed] [Google Scholar]
- 50. Watanabe Y, Koyama S, Yamashita A, Matsuura Y, Nishihira K, Kitamura K, et al. Indoleamine 2,3‐dioxygenase 1 in coronary atherosclerotic plaque enhances tissue factor expression in activated macrophages. Res Pract Thromb Haemost. 2018;2:726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koyama S, Yamashita A, Matsuura Y, Saito Y, Maekawa K, Gi T, et al. Intracellular glutamine level determines vascular smooth muscle cell‐derived thrombogenicity. Atherosclerosis. 2021;328:62–73. [DOI] [PubMed] [Google Scholar]
- 52. Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. [DOI] [PubMed] [Google Scholar]
- 53. Nishihira K, Yamashita A, Imamura T, Hatakeyama K, Sato Y, Nakamura H, et al. Thioredoxin in coronary culprit lesions: possible relationship to oxidative stress and intraplaque hemorrhage. Atherosclerosis. 2008;201:360–7. [DOI] [PubMed] [Google Scholar]
- 54. Noguchi T, Kawasaki T, Tanaka A, Yasuda S, Goto Y, Ishihara M, et al. High‐intensity signals in coronary plaques on noncontrast T1‐weighted magnetic resonance imaging as a novel determinant of coronary events. J Am Coll Cardiol. 2014;63:989–99. [DOI] [PubMed] [Google Scholar]
- 55. Kuroiwa Y, Uchida A, Yamashita A, Miyati T, Maekawa K, Gi T, et al. Coronary high‐signal‐intensity plaques on T 1‐weighted magnetic resonance imaging reflect intraplaque hemorrhage. Cardiovasc Pathol. 2019;40:24–31. [DOI] [PubMed] [Google Scholar]
- 56. Cheng C, Noordeloos AM, Jeney V, Soares MP, Moll F, Pasterkamp G, et al. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation. 2009;119:3017–27. [DOI] [PubMed] [Google Scholar]
- 57. Zhu T, Martinez I, Emmerich J. Venous thromboembolism: risk factors for recurrence. Arterioscler Thromb Vasc Biol. 2009;29:298–310. [DOI] [PubMed] [Google Scholar]
- 58. Sakuma M, Nakamura M, Hanzawa K, Kobayashi T, Kuroiwa M, Nakanishi N, et al. Acute pulmonary embolism after an earthquake in Japan. Semin Thromb Hemost. 2006;32:856–60. [DOI] [PubMed] [Google Scholar]
- 59. Sato K, Sakamoto K, Hashimoto Y, Hanzawa K, Sueta D, Kojima S, et al. Risk factors and prevalence of deep vein thrombosis after the 2016 Kumamoto earthquakes. Circ J. 2019;83:1342–48. [DOI] [PubMed] [Google Scholar]
- 60. Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19. Thromb Res. 2020;191:145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Middeldorp S, Coppens M, Haaps TF, Foppen M, Vlaar AP, Müller MCA, et al. Incidence of venous thromboembolism in hospitalized patients with COVID‐19. J Thromb Haemost. 2020;18:1995–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loo J, Spittle DA, Newnham M. COVID‐19, immunothrombosis and venous thromboembolism: biological mechanisms. Thorax. 2021;76:412–20. [DOI] [PubMed] [Google Scholar]
- 63. Takahashi M, Yamashita A, Moriguchi‐Goto S, Marutsuka K, Sato Y, Yamamoto H, et al. Critical role of von Willebrand factor and platelet interaction in venous thromboembolism. Histol Histopathol. 2009;24:1391–8. [DOI] [PubMed] [Google Scholar]
- 64. Sugita C, Yamashita A, Moriguchi‐Goto S, Furukoji E, Takahashi M, Harada A, et al. Factor VIII contributes to platelet‐fibrin thrombus formation via thrombin generation under low shear conditions. Thromb Res. 2009;124:601–7. [DOI] [PubMed] [Google Scholar]
- 65. Furukoji E, Gi T, Yamashita A, Moriguchi‐Goto S, Kojima M, Sugita C, et al. CD163 macrophage and erythrocyte contents in aspirated deep vein thrombus are associated with the time after onset: a pilot study. Thromb J. 2016;14:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gi T, Kuroiwa Y, Yamashita A, Mizutani Y, Asanuma T, Miyati T, et al. High signal intensity on diffusion‐weighted images reflects acute phase of deep vein thrombus. Thromb Haemost. 2020;120:1463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takahashi M, Yamashita A, Moriguchi‐Goto S, Sugita C, Matsumoto T, Matsuda S, et al. Inhibition of factor XI reduces thrombus formation in rabbit jugular vein under endothelial denudation and/or blood stasis. Thromb Res. 2010;125:464–70. [DOI] [PubMed] [Google Scholar]
- 68. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sugita C, Yamashita A, Matsuura Y, Iwakiri T, Okuyama N, Matsuda S, et al. Elevated plasma factor VIII enhances venous thrombus formation in rabbits: contribution of factor XI, von Willebrand factor and tissue factor. Thromb Haemost. 2013;110:62–75. [DOI] [PubMed] [Google Scholar]
- 70. Thomas DP, Merton RE, Hockley DJ. The effect of stasis on the venous endothelium: an ultrastructural study. Br J Haematol. 1983;55:113–22. [DOI] [PubMed] [Google Scholar]
- 71. Aghourian MN, Lemarié CA, Blostein MD. In vivo monitoring of venous thrombosis in mice. J Thromb Haemost. 2012;10:447–52. [DOI] [PubMed] [Google Scholar]
- 72. Hisada Y, Mackman N. Cancer‐associated pathways and biomarkers of venous thrombosis. Blood. 2017;130:1499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Koster T, Vandenbroucke JP, Rosendaal FR, Briët E, Rosendaal FR, Blann AD. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep‐vein thrombosis. Lancet. 1995;345:152–5. [DOI] [PubMed] [Google Scholar]
- 74. Bombeli T, Jutzi M, De Conno E, Seifert B, Fehr J. In patients with deep‐vein thrombosis elevated levels of factor VIII correlate only with von Willebrand factor but not other endothelial cell‐derived coagulation and fibrinolysis proteins. Blood Coagul Fibrinolysis. 2002;13:577–81. [DOI] [PubMed] [Google Scholar]
- 75. Vlieg AH, van der Linden IK, Bertina RM, Rosendaal FR. High levels of factor IX increase the risk of venous thrombosis. Blood. 2000;95:3678–82. [PubMed] [Google Scholar]
- 76. Meijers JCM, Tekelenburg WLH, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. [DOI] [PubMed] [Google Scholar]
- 77. Grover SP, Olson TM, Cooley BC, Mackman N. Model‐dependent contributions of FXII and FXI to venous thrombosis in mice. J Thromb Haemost. 2020;18:2899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013;39:621–31. [DOI] [PubMed] [Google Scholar]
- 79. Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129:1210–15. [DOI] [PubMed] [Google Scholar]
- 80. Yamashita A, Nishihira K, Kitazawa T, Yoshihashi K, Soeda T, Esaki K, et al. Factor XI contributes to thrombus propagation on injured neointima of the rabbit iliac artery. J Thromb Haemost. 2006;4:1496–501. [DOI] [PubMed] [Google Scholar]
- 81. Ay C, Hisada Y, Cooley BC, Mackman N. Factor XI‐deficient mice exhibit increased bleeding after injury to the saphenous vein. J Thromb Haemost. 2017;15:1829–33. [DOI] [PubMed] [Google Scholar]
- 82. Carnicelli AP, Hong H, Connolly SJ, Eikelboom J, Giugliano RP, Morrow DA, et al. Direct oral anticoagulants versus warfarin in patients with atrial fibrillation: patient‐level network meta‐analyses of randomized clinical trials with interaction testing by age and sex. Circulation. 2022;145:242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gailani D. Factor XI as a target for preventing venous thromboembolism. J Thromb Haemost. 2022;20:550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Key NS. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematology. 2014;2014:66–70. [DOI] [PubMed] [Google Scholar]
- 85. Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost. 2014;12:860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]