

Abstract
Background:
Although the concept of immunothrombosis has established a link between inflammation and thrombosis, the role of inflammation in the pathogenesis of deep vein thrombosis remains to be fully elucidated. Further, although various constituents of venous thrombi have been identified, their localizations and cellular and molecular interactions are yet to be combined in a single, multiplexed analysis.
Objectives:
The objective of this study was to investigate the role of the von Willebrand factor (VWF) in inflammation-associated venous thrombosis. We also performed a proof-of-concept study of imaging mass cytometry to quantitatively and simultaneously analyze the localizations and interactions of 10 venous thrombus constituents.
Methods:
We combined the murine inferior vena cava stenosis model of deep vein thrombosis with the lipopolysaccharide model of endotoxemia. We also performed a proof-of-concept study of imaging mass cytometry to assess the feasibility of this approach in analyzing the structural composition of thrombi.
Results:
We found that lipopolysaccharide-treated mice had significantly higher incidences of venous thrombosis, an effect that was mitigated when VWF was inhibited using inhibitory αVWF antibodies. Our detailed structural analysis also showed that most thrombus components are localized in the white thrombus regardless of endotoxemia. Moreover, although endotoxemia modulated the relative representation and interactions of VWF with other thrombus constituents, the scaffolding network, comprised VWF, fibrin, and neutrophil extracellular traps, remained largely unaffected.
Conclusions:
We observe a key role for VWF in the pathogenesis of inflammation-associated venous thrombosis while providing a more comprehensive insight into the molecular interactions that constitute the architecture of venous thrombi.
Keywords: animal models, image cytometry, thromboinflammation, venous thrombosis, von Willebrand factor
1 |. INTRODUCTION
Venous thromboembolism (VTE) is a global health burden that affected nearly 4 in every 1000 people in 2016 [1,2]. Evidence shows an especially high VTE incidence in critically ill patients with sepsis, a systemic inflammatory response syndrome resulting from a dysregulated immune reaction to a pathogenic infection [3,4]. Breakthrough VTE incidences, despite the use of guideline-recommended thromboprophylaxis in these patients [3,5], highlight the need for an improved understanding of the inflammation-thrombosis interplay.
“Immunothrombosis” represents a physiological, reciprocal relationship between the innate immune system and coagulation [6]. However, dysregulated immunothrombosis could contribute to the pathogenesis of thrombosis. For example, pathogens and pathogen-derived materials, such as bacterial endotoxin and lipopolysaccharide (LPS), activate platelets to initiate platelet aggregation and induce the release of neutrophil extracellular traps (NETs) [7,8]. Activated monocytes express and release tissue factors, although the accompanying cytokine storm can provoke endothelial dysfunction [9,10]. Simultaneously, impaired anticoagulant and fibrinolytic mechanisms during sepsis can further augment the thrombotic response [4].
Sepsis is also associated with elevated levels of von Willebrand factor (VWF) and a coincident reduction in the activity of its regulator, ADAMTS13 [11,12]. In addition to its hemostatic activities, reports of VWF interactions with venous thrombus constituents such as leukocytes [13], red blood cells (RBCs) [14], and NETs [15] have generated significant interest in VWF’s role in venous thrombosis. Moreover, despite previous structural studies of venous thrombi, much of this data remains as a series of independent studies, lacking a comprehensive and simultaneous analysis.
To address the role of VWF in venous thrombosis in an inflammatory context, we combined the established murine inferior vena cava (IVC) stenosis model of deep vein thrombosis (DVT) [16] with an LPS model of endotoxemia. We conducted the first imaging mass cytometry (IMC) in this model system to determine the feasibility of this approach in performing a multiplexed analysis of the structural architecture of venous thrombi. This proof-of-concept study of small replicate numbers would merit further validation with larger sample sizes. We hypothesized that endotoxemia-induced inflammation would enhance DVT development through elevated levels of plasma VWF and that an inflammatory milieu would alter the structural composition of venous thrombi.
2 |. METHODS
2.1 |. Production of recombinant human ADAMTS13 (rhADAMTS13)
rhADAMTS13 was produced by stable transfected HEK293T cells and purified using a combination of anion-exchange column chromatography and centrifugal filtration. rhADAMTS13 activity was measured using the Technozym ADAMTS-13 Activity ELISA Kit (Technoclone GmBH).
2.2 |. Mice
All mice used in this study were 8- to 10-week-old male wildtype C57BL/6 mice (Jackson Laboratory, Bar Harbour). Female mice were excluded due to anatomical incompatibility with the IVC stenosis model as described elsewhere [17]. Littermate controls were used for all experiments. All animal procedures were approved by Queen’s University Animal Care Committee.
2.3 |. Measurement of blood and plasma characteristics
Retro-orbital blood samples were collected prior to the stenosis procedure. Retro-orbital sampling and cardiac punctures were performed at the time of euthanasia. Complete blood counts were obtained using the ABC Hematology Analyzer (Scil Vet). Blood was centrifuged at 10,000 × g for 10 minutes, and plasma samples were stored at −80 °C.
Cell-free DNA (cfDNA) was measured using the Quant-iT Pico-Green dsDNA Assay Kit (P7589, Invitrogen, Thermo Fisher Scientific). Interleukin 6 (IL-6) was measured using the Mouse IL-6 ELISA Kit (ab100712, Abcam). Plasma VWF (VWF:Ag) was measured by enzyme-linked immunosorbent assay (A0082 and P0226, Agilent) as previously described [18]. VWF-collagen binding activity (VWF:CB) assay was performed as previously described (A0082 and P0226, Agilent) [19], with some modifications as follows: plates were coated with bovine collagen types I and III (193492, MP Biomedicals) for 2 hours. Washes and blocking were performed in phosphate-buffered saline containing Tween-20 (0.1%) and bovine serum albumin (5%) in phosphate-buffered saline containing Tween-20 (0.1%), respectively. Sample incubations were conducted at room temperature. The detecting antibody was diluted 1:1000. VWF-glycoprotein Ib binding assay (VWF:GpIbM) was performed using the automated INNOVANCE VWF Ac Kit (OPHL03, Siemens Healthcare Diagnostics).
2.4 |. Murine model of venous thrombosis
The IVC stenosis model of DVT was used as previously described [16,20]. Immediately after closing the abdomen, 0.5 mg/kg of E. coli O111:B4 LPS (L3024, Sigma-Aldrich) or 250 μl of saline as the vehicle control was administered intraperitoneally. Mice were euthanized after 1.5 or 24 hours, and thrombi were collected if present.
For the VWF inhibition experiments, 4 mg/kg of a polyclonal rabbit αVWF antibody (A0082, Agilent) or rabbit IgG (X0936, Agilent) as the isotype control (ISO) antibody was administered intravenously via the tail vein prior to the midline incision. After the stenosis procedure, mice received either LPS or saline, as described above.
For rhADAMTS13 infusions, 750 U/kg of rhADAMTS13 or an equivalent amount of tris-buffered saline as the vehicle control was administered to anesthetized mice intravenously via the tail vein prior to the midline incision. After the stenosis procedure, all mice received 0.5 mg/kg of LPS.
The veterinary technician performing these procedures was blinded to all treatments.
2.5 |. Tissue staining
Longitudinal sections from formalin-fixed, paraffin-embedded thrombi were stained with hematoxylin and eosin. The slides were scanned using an Olympus VS120 slide scanner at 20 × magnification. Classification and quantification of red and white thrombi were performed using the random forest classifier module in the HALO software (Indica Labs).
For the IMC tissue staining procedure, refer to the Supplementary materials. Longitudinal punches of thrombi from 3 control and 3 LPS-treated mice were assembled onto a tissue microarray block (Supplementary Table S1). The IMC antibody targets were: CD41 (platelets), CD62P (P-selectin), HMGB1, Ly6C (monocytes), Ly6G (neutrophils), Ter119 (RBCs), VWF, fibrin(ogen), and CitH3 (citrullinated histones) (Supplementary Table S2). Cell-ID Intercalator-Ir (Fluidigm) was used to stain for DNA.
2.6 |. IMC
For detailed methods, refer to the Supplementary materials. Briefly, the stained slide was loaded into a tuned Hyperion imaging module under the control of CyTOF software (Fluidigm). Laser intensity was validated in situ on excess tissue, and regions of interest (ROIs) were laser ablated at a 1-μm resolution.
2.7 |. IMC data analysis
For detailed methods, refer to the Supplementary materials. Briefly, false color images generated using the MCD Viewer software (Fluidigm) were imported into the Ilastik software (https://www.ilastik.org/). The Pixel Classification module was used to create probability maps for each red, white, or whole thrombus region against the background per ROI. The probability maps were applied as masks in quantification pipelines to measure the areas occupied by each antibody target in the CellProfiler software (www.cellprofiler.org). CellProfiler was also used to generate grid masks used in the HistoCAT software (https://github.com/BodenmillerGroup/histoCAT/releases) to conduct dimensionality reduction with T-distributed Stochastic Neighbor Embedding (tSNE) and Phenograph clustering analyses [21]. Images of thrombus scaffolds were generated in CellProfiler and overlaid with colors using the ImageJ software. Data manipulation and correlational heatmaps were generated using the ggplot2 package in RStudio software (Boston).
2.8 |. Statistical analysis
Statistical analysis was performed using Prism 8.3.0 (GraphPad) for Windows. Thrombus incidences were compared using Fisher exact test. Normality for each data set was analyzed using the Shapiro-Wilk test. Parametric data were compared using unpaired Student’s t-test or one-way analysis of variance with Tukey’s multiple-test comparisons. Non-parametric data were compared using the Mann-Whitney U test or Kruskal-Wallis test with Dunn’s multiple-test comparisons. Data are presented as mean ± standard deviation. Significance was reached when p < .05.
The correlations between thrombus targets for the IMC data were analyzed using Spearman correlation with Bonferroni multipletests correction in SPSS Statistics (IBM).
2.9 |. Data sharing statement
For original data, please contact the corresponding author D.L.
3 |. RESULTS
3.1 |. Endotoxemia affects blood and plasma characteristics in mice
First, to confirm the inflammatory effects of LPS, we compared the blood and plasma characteristics of the LPS-treated mice with the saline-treated mice. Consistent with previous observations of thrombocytopenia in LPS-treated mice [22], an initial decrease in platelet counts at 1.5 hours post-stenosis (Supplementary Figure S1A) was exacerbated at 24 hours in the endotoxemic mice (0.34 ± 0.12–fold, p < .0001, Figure 1A). Similar trends in the control mice only reached statistical significance after 24 hours (0.66 ± 0.24–fold, p = .0002, Figure 1A, Supplementary Figure S1A). Circulating levels of cfDNA, associated with adverse clinical outcomes in sepsis [23], and IL-6, a proinflammatory cytokine commonly elevated in sepsis [24], were also significantly higher in the endotoxemic mice at 24 hours (p = .0032 and p < .0001; Figure 1B, C, respectively). Together, these changes support a proinflammatory state in the LPS-treated mice.
FIGURE 1.
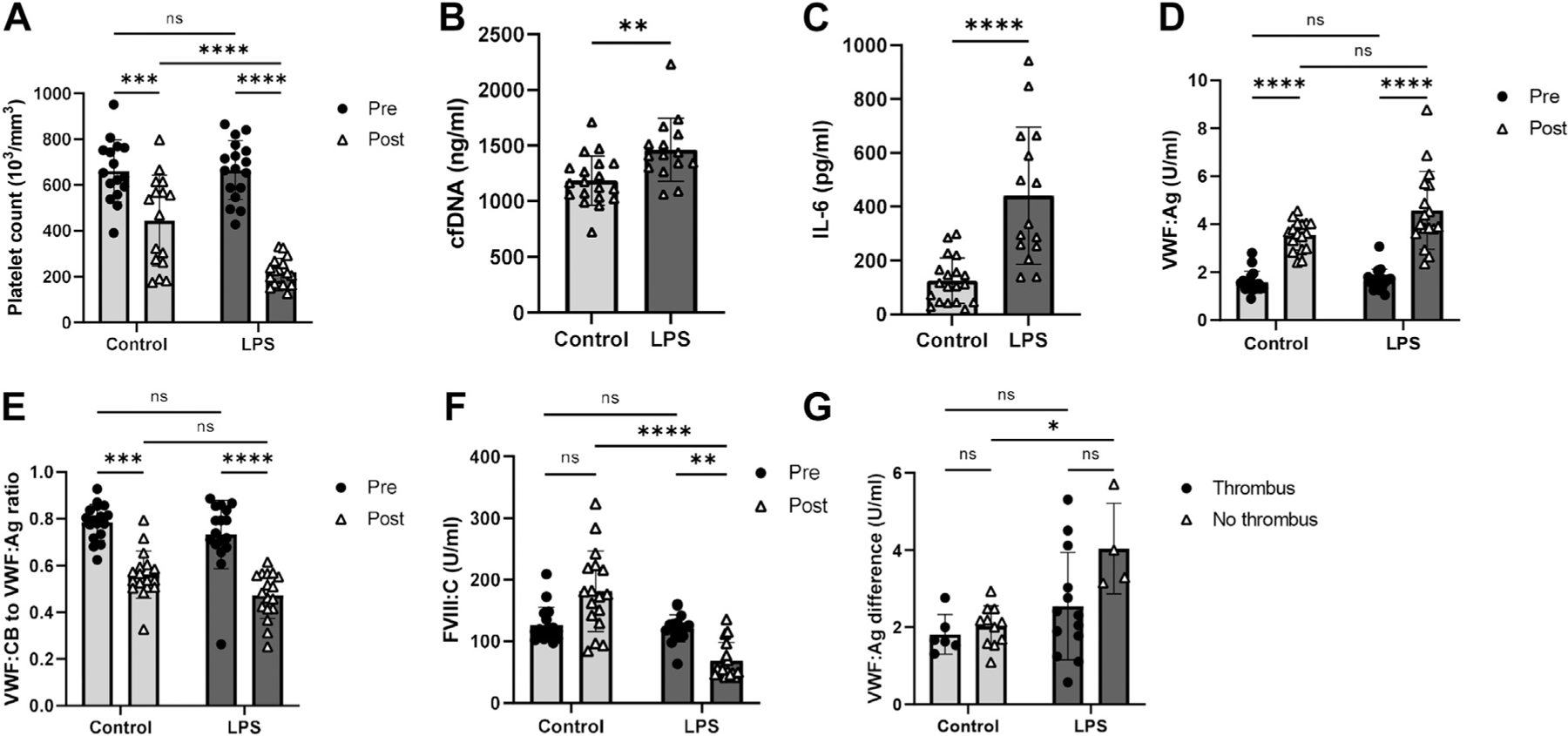
Endotoxemia affects blood and plasma characteristics in mice. (A) Circulating platelet counts in blood samples collected prior to (Pre) and 24 hours after (Post) the stenosis procedure in mice receiving vehicle control (n = 16) or LPS (n = 17). Plasma levels of (B) cfDNA and (C) IL-6 at 24 hours post-stenosis in mice receiving control (n = 19) or LPS (n = 15). Pre and Post (D) VWF:Ag and (E) ratio of VWF:CB to VWF:Ag in mice receiving control (n = 17) or LPS (n = 17). (F) Pre and post-FVIII:C in mice receiving control (n = 14) or LPS (n = 17). (G) Changes in VWF:Ag from baseline to 24 hours post-stenosis in thrombosed (control n = 6, LPS n = 13) and nonthrombosed (control n = 11, LPS n = 4) mice. All assays were performed in duplicate. Pooled plasma from normal C57Bl/6 mice was used as the reference for all VWF assays. *p < .05, **p < .01, ***p < .001, ****p < .0001. cfDNA, cell-free DNA; IL-6, Interleukin 6; LPS, lipopolysaccharide; ns, not significant; VWF:Ag, plasma von Willebrand factor; VWF:CB, von Willebrand factor–collagen binding activity.
LPS has been found to increase VWF:Ag in mice [25]. In this study, the endotoxemic mice also had significantly elevated VWF:Ag levels at 1.5 hours (Supplementary Figure S1B) and 24 hours post-stenosis (2.80 ± 0.89–fold, Figure 1D). This increase was only statistically significant in the controls at 24 hours (2.32 ± 0.46–fold, p < .0001, Figure 1D and Supplementary Figure S1B). Despite absolute increases in the VWF:CB post-stenosis (Supplementary Figures S1C and S2), the VWF:CB to VWF:Ag ratios progressively decreased in both the control and LPS cohorts (−0.22 ± 0.10 and −0.26 ± 0.14, respectively, Figure 1E and Supplementary Figure S1D).
In contrast, FVIII:C levels initially decreased in both the control and LPS cohorts at 1.5 hours post-stenosis (0.75 ± 0.16–fold and 0.73 ± 0.11–fold, respectively, Supplementary Figure S1E). However, FVIII:C levels showed an increasing trend by 24 hours post-stenosis in the controls, albeit not statistically significant, although FVIII:C continued decreasing in the LPS-treated mice (1.23 ± 0.43–fold and 0.58 ± 0.25–fold, respectively, Figure 1F).
The VWF may be partially depleted in the plasma once it is incorporated into a thrombus. To exclude this confounding effect, we compared the changes in VWF levels between the thrombosed and nonthrombosed mice from pre-stenosis to 24 hours post-stenosis. In the nonthrombosed mice, the magnitude of VWF:Ag increase was significantly greater in the endotoxemic mice than the controls (3.18 ± 0.40 and 2.42 ± 0.37–fold, respectively, p = .0108, Figure 1G). Collectively, these results suggest that endotoxemia induces a greater magnitude of increase in plasma VWF levels than control treatment at 24 hours after the IVC stenosis.
3.2 |. Endotoxemia increases thrombus incidence, an outcome that is mitigated by a reduction in VWF activity
At 24 hours post-stenosis, a significantly higher proportion of the LPS-treated mice (13/17, 76.5%) developed thrombi compared with the controls (8/20 [40%]) (p = .0453, Figure 2A). However, this was not accompanied by an increase in thrombus burden as measured by thrombus weights (p = .5131, Figure 2B).
FIGURE 2.
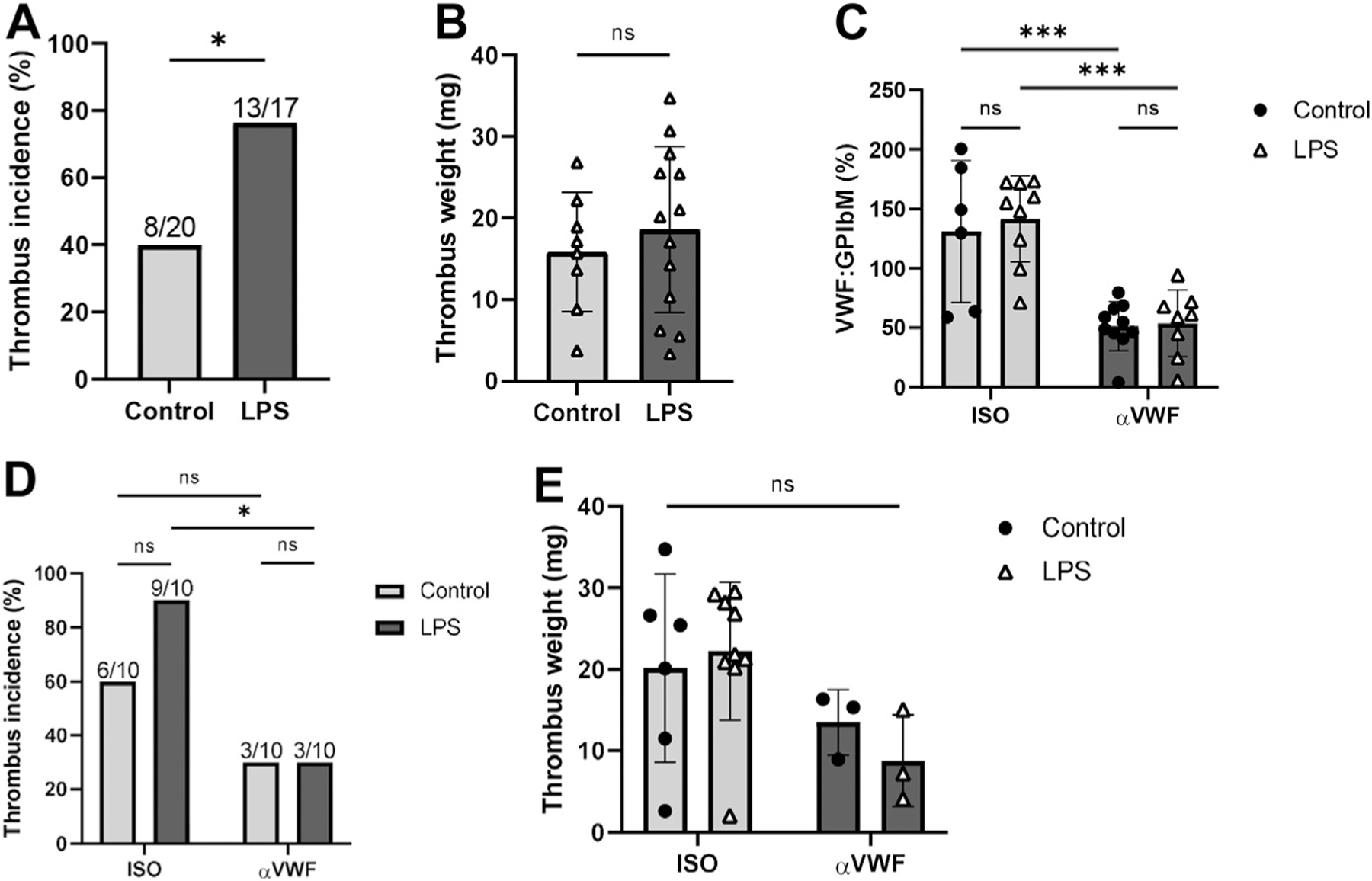
Endotoxemia increases thrombosis incidence, an outcome that is mitigated by reduced VWF activity. (A) Thrombus incidence and (B) thrombus weights (>0 mg) of vehicle control and LPS-treated mice at 24 hours post-stenosis. (C) VWF:GPIbM at 24 hours post-stenosis in vehicle control and LPS-treated mice treated with αVWF antibody (n = 10 and n = 8, respectively) or ISO (n = 6 and n = 9, respectively), expressed as a percentage of activity relative to pooled murine plasma as reference. (D) Thrombus incidence and (E) thrombus weights in control and LPS-treated mice in αVWF and ISO cohorts at 24 hours post-stenosis. ns: not significant. *p < .05, ***p < .001. ISO, isotope control antibody; LPS, Lipopolysaccharide; ns, not significant; VWF:Ag, plasma von Willebrand factor; VWF:CB, von Willebrand factor–collagen binding activity; VWF:GbIb, VWF-GPIb binding activity.
Given these results, we investigated the role of VWF in this pathological outcome using inhibitory αVWF antibodies. The significantly reduced VWF-platelet binding activity in mice receiving the αVWF versus the ISO antibodies in both the LPS (53.8 ± 28.0% vs 141.6 ± 36.1%, respectively, p = .0001, Figure 2C) and control cohorts (51.4 ± 20.6% vs 131.0 ± 59.6%, respectively, p = .0010, Figure 2C) confirmed the inhibitory activity of the αVWF antibodies. Notably, only 3 of 10 (30%) of the αVWF antibody-treated mice developed thrombi regardless of endotoxemic state, although 6 of 10 (60%) of the control and 9 of 10 (90%) of the LPS-treated mice in the ISO cohort developed thrombi (Figure 2D). The reduced thrombus incidence in the αVWF-LPS mice was statistically significant compared with the ISO-LPS mice (p = .0198), despite the residual levels of VWF activity (Figure 2C). Inhibition of VWF also reduced thrombus incidence in the control mice compared with the ISO-treated mice (30% vs 60%) but did not reach statistical significance (p = .3698). Finally, there were no significant differences in the thrombus weights of the αVWF- and the ISO-treated mice (Figure 2E). Therefore, we conclude that VWF may play an important role in initiating, rather than propagating, inflammation-associated thrombosis, manifested as thrombus incidence and weights (burden), respectively.
3.3 |. Enhanced ADAMTS13 activity does not affect endotoxemia-associated thrombosis outcome
Since VWF inhibition negatively regulated endotoxemia-associated thrombosis, we combined rhADAMTS13 (or vehicle control) infusions with LPS treatment to determine whether enhanced ADAMTS13 activity could also mitigate this pathological process. We first confirmed the effects of the rhADAMTS13 by comparing high-molecular weight (HMW) VWF multimers at 15 minutes post-treatment, which showed overall decreased HMW VWF multimers in the rhADAMTS13-treated mice and no significant change in the vehicle control-treated mice (Supplementary Figure S4). When combined with the stenosis procedure, we confirmed that although post-stenosis VWF:Ag levels were significantly increased in the mice receiving rhADAMTS13 (3.77 ± 1.79–fold, p < .0001, Figure 3A), these changes were comparable to those exhibited by mice receiving the vehicle control (3.01 ± 1.49–fold, p = .0037, Figure 3A). The VWF:CB to VWF:Ag ratios also decreased without significant differences between the control and rhADAMTS13 cohorts (−0.29 ± 0.16 and −0.34 ± 0.18, respectively, p = .5308, Figure 3B). Likewise, significant differences between the control and rhADAMTS13 cohorts were lacking in both thrombus incidences (p > .9999, Figure 3C) and weights of the resulting thrombi (p = .5925, Figure 3D). Given these results, we conclude that enhanced ADAMTS13 activity, at least by 750 U/kg, does not modulate overall VWF levels, activity, or thrombosis outcomes.
FIGURE 3.
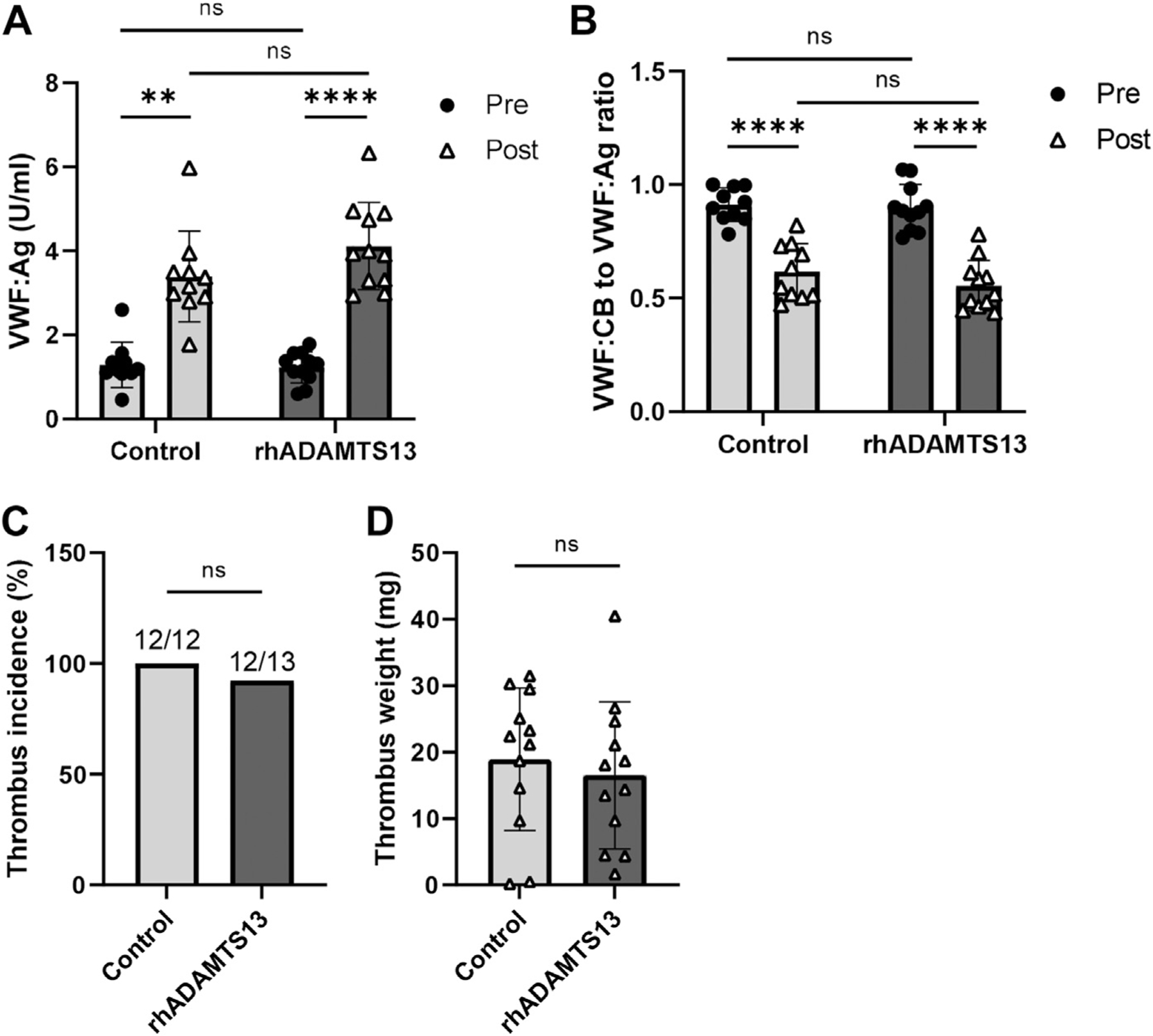
Enhanced ADAMTS13 activity does not affect endotoxemia-associated thrombosis outcomes. (A) VWF:Ag and (B) ratios of VWF:CB to VWF:Ag in blood samples collected prior to (Pre) and 24h after (Post) the stenosis procedure from mice receiving vehicle control (n = 10) or recombinant human ADAMTS13 (rhADAMTS13) (n = 11). (C) Thrombus incidence and (D) thrombus weights (>0 mg) of vehicle control and rhADAMTS13-treated mice at 24h post-stenosis. Pooled plasma from normal C57Bl/6 mice was used as the reference for all assays. **p < .01, ****p < .0001. ns, not significant; VWF:Ag, plasma von Willebrand factor; VWF:CB, von Willebrand factor–collagen binding activity.
3.4 |. LPS-induced inflammation enhances the red thrombus area, but most thrombus constituents localize in the white thrombus
We next evaluated whether endotoxemia-induced inflammation would alter the structural architecture of venous thrombi. Human venous thrombi comprise 2 distinct regions of red and white thrombus, mainly consisting of RBCs and fibrin and platelets and fibrin, respectively [26]. We observed these morphological features in our thrombus samples (Figure 4A) and documented that the proportion of the red thrombus in the endotoxemic mice was significantly increased compared with the controls (80.5 ± 11.1% and 66.4 ± 16.2%, respectively, p = .0283, Figure 4B), with corresponding decreases in the white thrombus.
FIGURE 4.
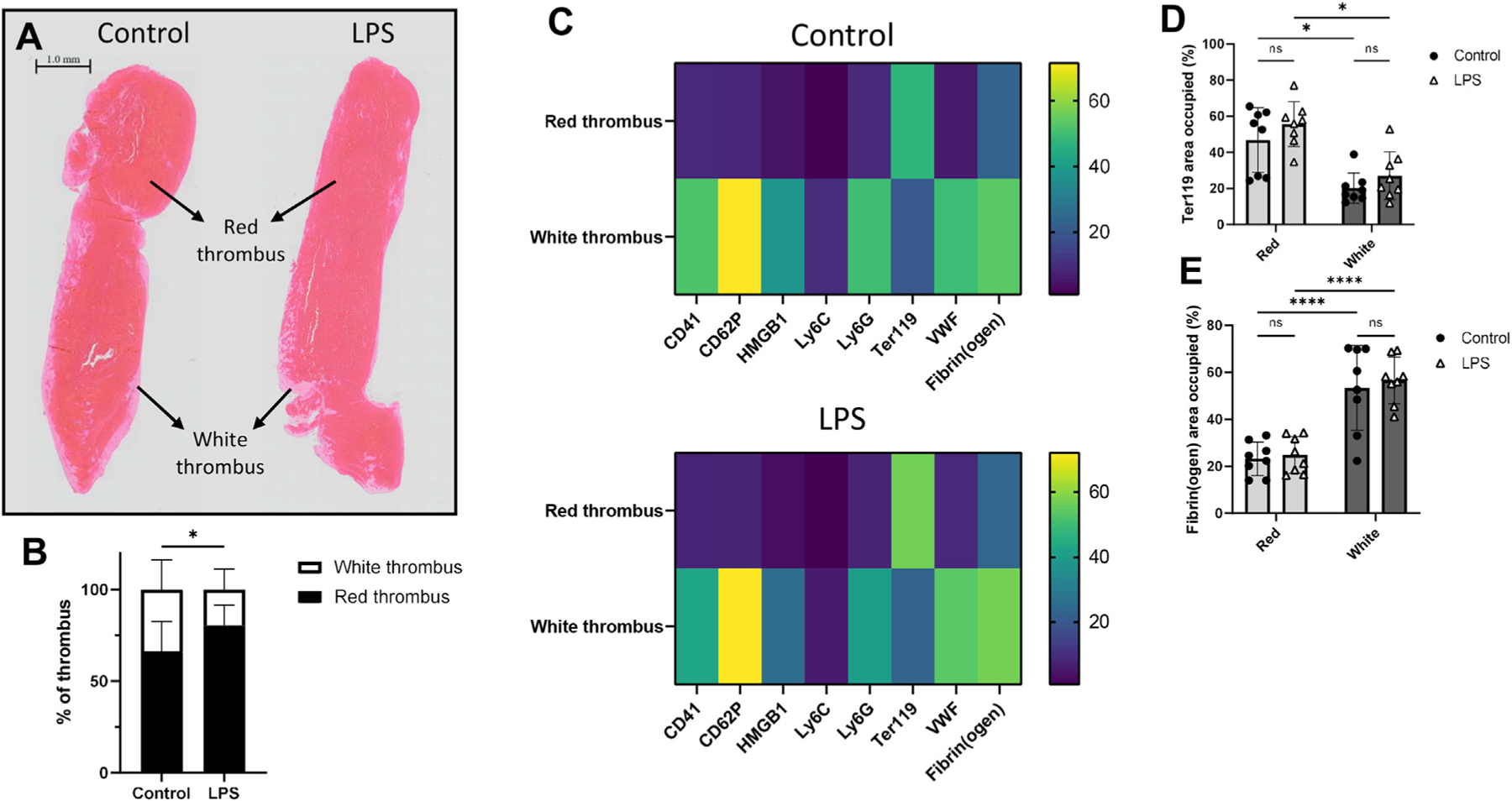
Endotoxemia significantly increases the proportion of the red thrombus, but most thrombus components largely localize in the white thrombus. (A) Hematoxylin and eosin stains of thrombi obtained from control (left) and LPS- (right) treated mice at 24 hours post-stenosis show distinct red and white thrombi. Images are representative of thrombi from n = 7 control and n = 12 LPS mice. Scale bar, 1.0 mm. (B) Mean areas of red and white thrombi as percentages of the entire thrombus in control and LPS cohorts. (C) Heatmaps representing the areas occupied by CD41 (platelets), CD62P (P-selectin), HMGB1, Ly6C (monocytes), Ly6G (neutrophils), Ter119 (RBCs), VWF, and fibrin(ogen) as percentages of the red and white thrombi in the control (top) and LPS (bottom) cohorts. The scale on the right represents percentages. Areas occupied by (D) Ter119 and (E) fibrin(ogen) in thrombi derived from control and LPS-treated mice as percentages of red and white thrombi. *p < .05, ****p < .0001. LPS, lipopolysaccharide; ns, not significant; VWF, von Willebrand factor; VWF:Ag, plasma von Willebrand factor; VWF:CB, von Willebrand factor–collagen binding activity.
Our further structural characterization of thrombi involved detailed localization and interaction analyses using an IMC approach. Since this is a proof-of-concept experiment, this preliminary analysis was performed on a small sample size, and the conclusions are presented accordingly. By quantifying the areas occupied by various thrombus components in the red and white thrombi, we determined that most targets we analyzed were localized mainly in the white thrombus (Figure 4C and Supplementary Figure S5). The exceptions were Ter119, a marker for RBCs, which localized in the red thrombus (Figure 4D). Interestingly, we also observed that fibrin(ogen), which has been characterized as a component of both thrombus regions [16,26], was concentrated in the white thrombus, albeit with a greater distribution in the red thrombus compared with the other markers (Figure 4E). These localization patterns remained constant regardless of endotoxemia.
3.5 |. Endotoxemia increases the relative representation of VWF in thrombi and affects its localization in relation to other thrombus constituents
The results presented in the following sections are derived from the workflow depicted in Figure 5. Since VWF, our target of interest, was significantly localized in the white thrombus regardless of endotoxemia (p < .0001, Figure 6A), we focused our subsequent analyses on this region. First, we generated a grid mask over the white thrombus, where each megapixel of 10 × 10 pixels represented one unit of analysis. Using this mask, we ran a Phenograph clustering analysis for each treatment as previously described [21]. This generated clusters based on the expression profiles of each antibody target across all ROIs per treatment. For every cluster, the average expression intensity for each antibody target was used to generate a correlational heatmap (Figures 6B, C). Full clustering results are shown in Supplementary Figure S6.
FIGURE 5.
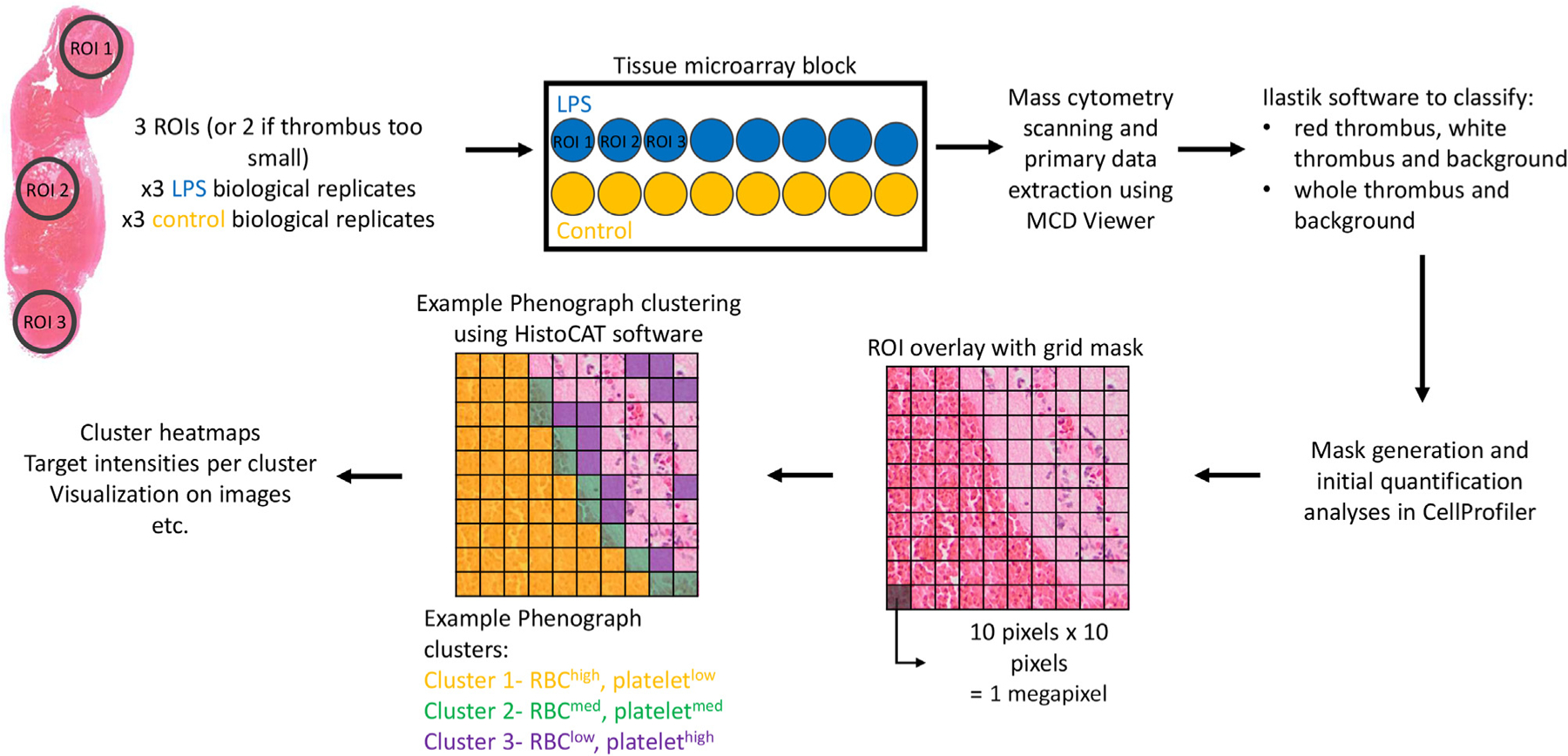
Imaging mass cytometry data analysis pipeline. Two or 3 ROIs from 3 biological replicates of thrombi from control and LPS-treated mice were stained for mass cytometry imaging. Data analysis workflow using various software is shown above. LPS, lipopolysaccharide; ROI, regions of interest.
FIGURE 6.
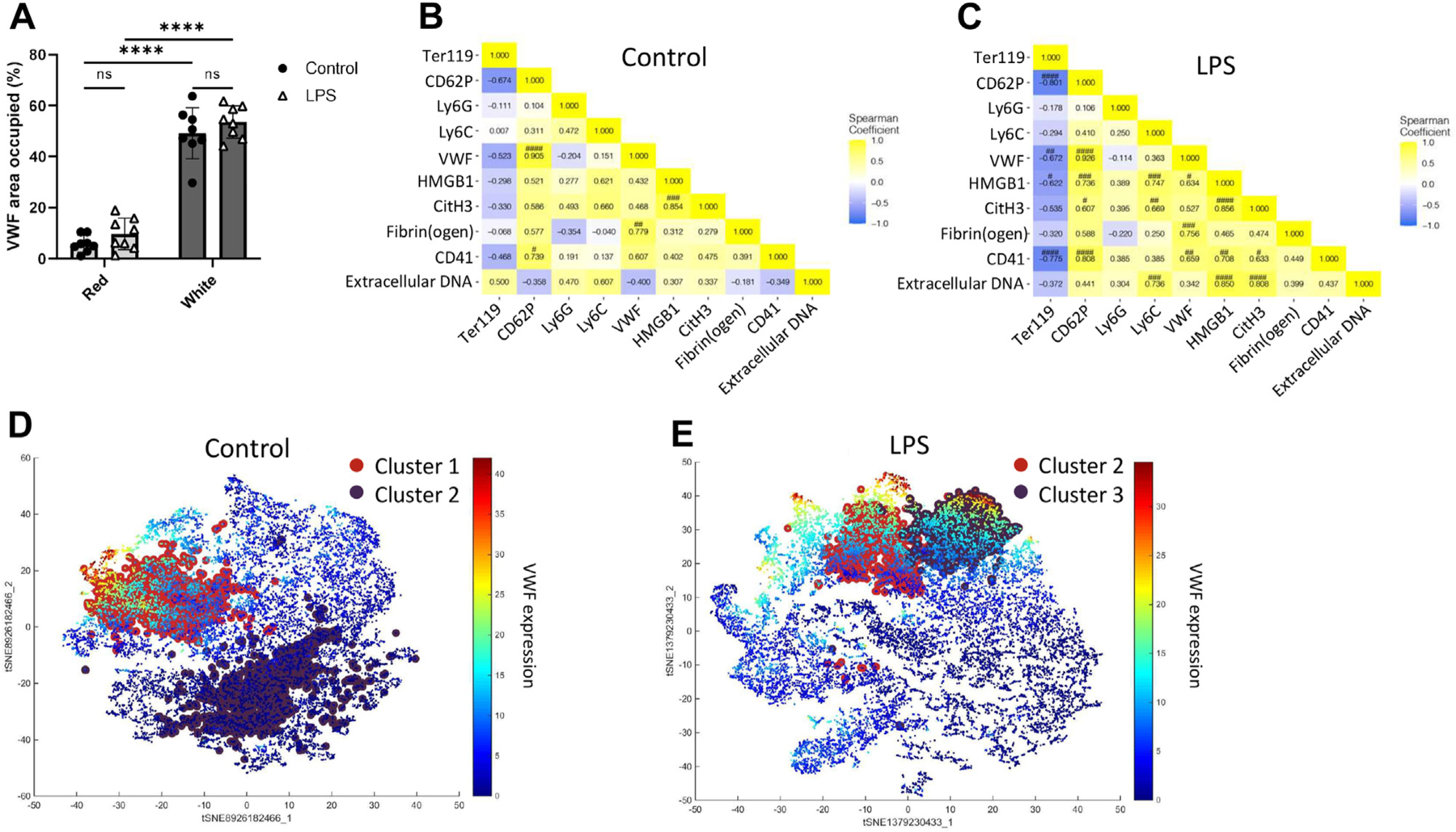
Endotoxemia increases the relative representation of VWF and affects its localization relative to other thrombus constituents. (A) Area occupied by VWF in thrombi derived from control and LPS-treated mice as percentages of red and white thrombi. Correlational heatmaps of the average intensity of expression of Ter119 (RBCs), CD62P (P-selectin), Ly6G (neutrophils), Ly6C (monocytes), VWF, HMGB1, CitH3, fibrin(ogen), CD41 (platelets), and extracellular DNA in Phenograph clusters in (B) control and (C) LPS cohorts. t-SNE representations of the 2 most common clusters in the (D) control and (E) LPS cohorts were identified on a heatmap of VWF expression intensity. ****p < .0001. #, ##, ###, #### represent significance levels after Bonferroni multiple-tests correction. LPS, lipopolysaccharide; ns, not significant; t-SNE, T-distributed Stochastic Neighbor Embedding; VWF, von Willebrand factor.
In both the control and LPS cohorts, VWF expression correlated most strongly with that of CD62P, which in turn correlated with CD41. In contrast, VWF correlations with CD41 and HMGB1 were greater in the LPS cohort. Likewise, CD41 only associated with HMGB1 in the LPS cohort, despite the significantly decreased expression of HMGB1 in this cohort (Supplementary Figure S5C). These findings suggest that VWF exhibits targeted interactions with platelets and HMGB1, and platelets with HMGB1, in inflammation-associated thrombosis. The positive correlation of VWF with CD41 in endotoxemia was also concurrent with a negative correlation with RBCs, further highlighting the red-white thrombus distinction. The relatively weak VWF associations with Ly6G and Ly6C, the markers for neutrophils and monocytes, respectively, suggest a lack of significant interactions within the growing thrombus.
To identify the most ubiquitous thrombus interactions, we also analyzed the following 2 most common Phenograph clusters in each treatment cohort: clusters 1 and 2 in the control cohort and clusters 2 and 3 in the LPS cohort. Notably, as demonstrated by the overlay of these clusters on a VWF expression heatmap (Figures 6D, E), the 2 most common clusters in the LPS cohort exhibited high VWF expressions. This contrasted with the control cohort, where only one of the 2 most common clusters had high VWF expression. Together, these results highlight the significance of VWF in the structural composition of venous thrombi, especially in inflammation-associated thrombosis. However, the distribution of these most common clusters in the white thrombus (represented by VWF and CD41 localization in Figures 7A, C) did not exhibit a discernible pattern in either treatment cohort. In these clusters, CD62P, Ly6G, VWF, fibrin(ogen), and CD41 expressed the highest intensities (Figures 7B, D).
FIGURE 7.
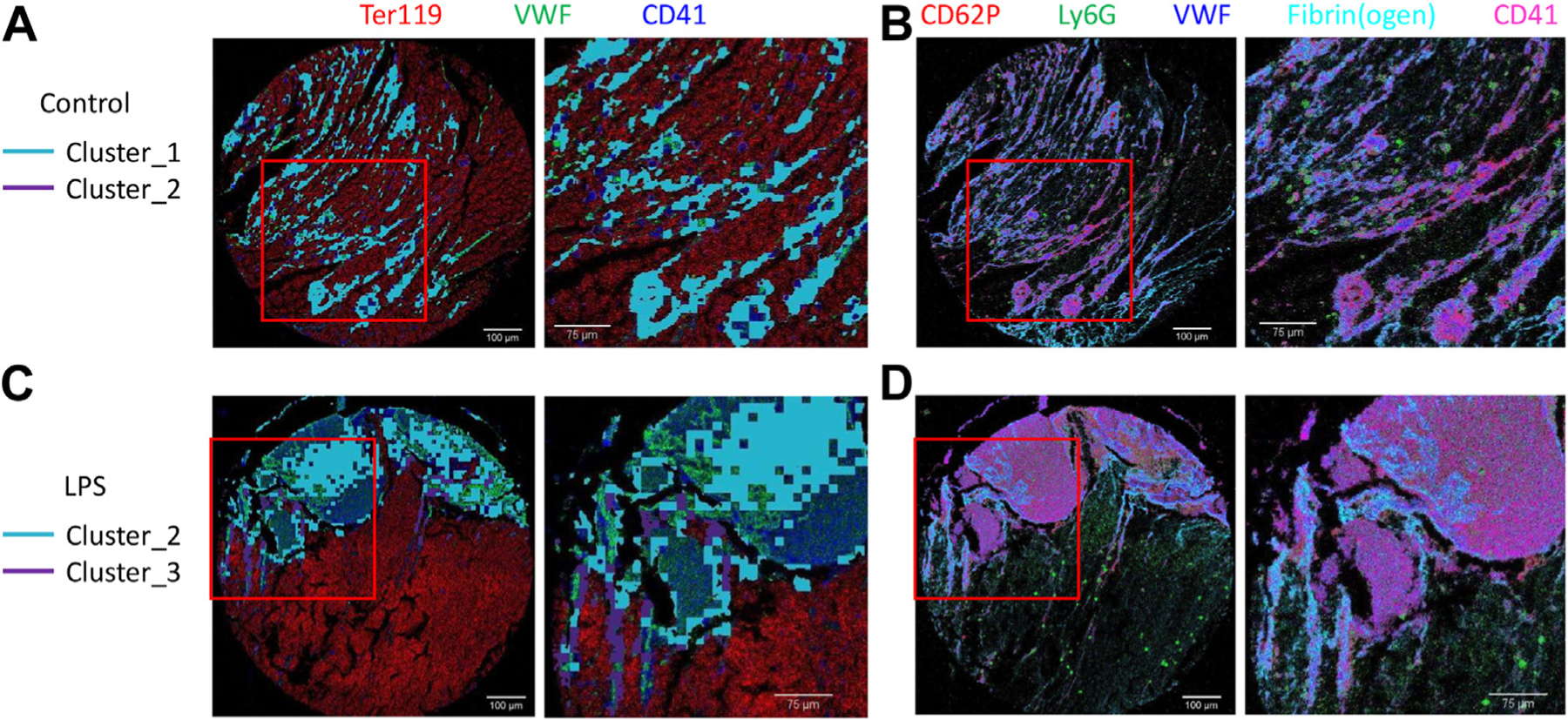
The 2 most common clusters do not exhibit a discernible pattern. The 2 most common clusters highlighted on false color overlay images of Ter119 (red), VWF (green), and CD41 (blue) in the (A) control and (B) LPS cohorts with the zoomed areas indicated by the red square shown on the right. False color overlay images of the top 5 targets with the highest relative expressions, CD62P (red), Ly6G (green), VWF (blue), fibrin(ogen) (cyan), and CD41 (magenta), in thrombi derived from (C) control and (D) LPS cohorts with the zoomed areas indicated by the red square shown on the right. LPS, lipopolysaccharide; VWF, von Willebrand factor.
3.6 |. VWF associates with fibrin(ogen), but neither correlates with extracellular DNA and citrullinated histones regardless of endotoxemia
Fibrin, VWF, and extracellular DNA, such as NETs, have been identified as the thrombus scaffolds upon which the other thrombus constituents congregate [27–29]. Given the critical role of this scaffold in the structure of thrombi, we further focused on the localization and quantification of VWF in relation to this scaffold (Figures 6B, C). We present the following results in the context of small sample size due to the preliminary nature of the IMC approach.
VWF-fibrin(ogen) correlations represented the second and the third strongest VWF correlations in the control and LPS cohorts, respectively. However, although VWF and fibrin(ogen) correlated positively with CitH3, a common marker for NETs regardless of endotoxemia, their correlations with extracellular DNA were negative in the control cohort but positive in the LPS cohort. This suggests that although the VWF-fibrin(ogen) network remains constant regardless of an inflammatory milieu, VWF and fibrin(ogen) associations with NET components, such as extracellular DNA and CitH3, may be enhanced by endotoxemia-induced inflammation.
Interestingly, CitH3 only correlated with extracellular DNA in the LPS cohort, suggesting the increased presence of NETs rather than cfDNA. As with VWF, both CitH3 and extracellular DNA correlated more strongly with CD62P, CD41, and HMGB1 in endotoxemia. The spatial localization of these scaffolding components is shown in Figures 8A and B, where extracellular DNA can be seen diffused throughout the tissue section, in contrast to the discrete colocalizations of VWF and fibrin(ogen).
FIGURE 8.
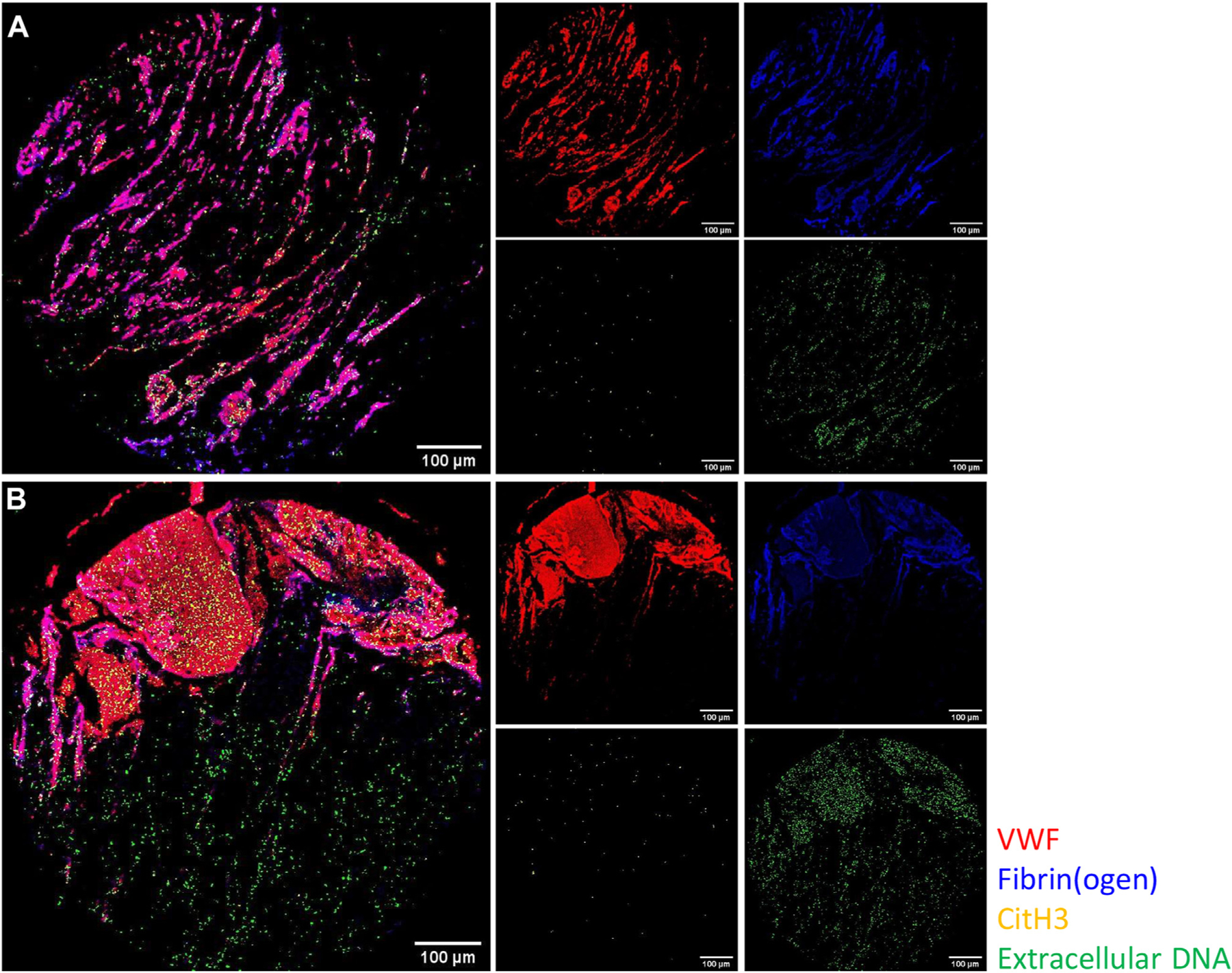
VWF associates with fibrin(ogen), but neither correlates with CitH3 and extracellular DNA, regardless of endotoxemia. False color overlay images of scaffolding components within thrombi derived from mice treated with (A) control and (B) LPS. VWF (red), fibrin(ogen) (blue), CitH3 (yellow), and extracellular DNA (green). LPS, lipopolysaccharide; VWF, von Willebrand factor.
4 |. DISCUSSION
Many studies and clinical observations have alluded to a correlation between inflammation and thrombosis, finally consolidated by the concept of immunothrombosis [6]. Experimental endotoxemia has been shown to increase venous thrombus burden in mice, and the increased risk of VTE in septic patients is well-established [3–5,30,31]. Recently, this interaction has gained further attention with reports of COVID-19-associated coagulopathies [32–35].
The VWF is an acute-phase protein rapidly released by activated endothelial cells during inflammation [36]. While the stenosis procedure is known to induce such endothelial activation [17,37], we confirmed a significantly accelerated VWF:Ag increase in the endotoxemic mice (Figure 1D). A faster rate of VWF:Ag increase than VWF activity in both the control and LPS cohorts also implied a greater presence of low-molecular weight multimers (LMWM) (Figure 1E). The continual decline of FVIII:C to 24 hours in the LPS-treated mice, likely the effects of ongoing consumption in an enhanced prothrombotic environment, contrasted with a rebound in the controls (Figure 1F and Supplementary Figure S1E). This is consistent with previous reports that mouse FVIII does not significantly respond to inflammation and aligns with previous findings that VWF, independent of FVIII, is involved in venous thrombosis [16,38].
Although inflammation is known to impair ADAMTS13 activity [11], Mimuro et al. [39] reported that low doses of LPS (5–15 μg/g) were unable to alter ADAMTS13 levels in mice. Thus, the dose used in our study may have been insufficient to modify VWF activity via reduced ADAMTS13 activity. Similarly, the low LPS dose may not have induced a significant release of ultra-large VWF multimers, overall indicating that LMWM were the predominant contributors to the rising VWF levels.
The critical role of VWF in DVT and obesity-associated DVT was previously demonstrated in the murine stenosis model [16,20]. In our study, we extend these findings to report that although endotoxemia-induced inflammation enhances DVT incidence, VWF plays a crucial role in this process, where even partial inhibition of its activity can be thrombo-protective (Figure 2A, D). Although the small sample size in the αVWF experiments may have underpowered the experiment, we note that even with 10 mice, thrombus incidence in the endotoxemic mice was nonetheless reduced. However, enhanced ADAMTS13 activity had negligible effects on mitigating thrombus incidence (Figure 3C), suggesting that LMWM are largely responsible for mediating inflammation-associated DVT. This finding is biologically consistent, as the activity of HMW VWF multimers is largely dependent on high shear that is characteristic of the arterial but not the venous circulation [40]. Mechanistically, our data also confirms the involvement of VWF-platelet interactions as previously reported [16], given that reduced thrombus incidence in endotoxemia was concurrent with decreased VWF-GpIb binding activity in the αVWF antibody-treated mice (Figure 2C).
Contrary to previous findings [30,31], thrombus burden as measured by thrombus weight was not altered in our study (Figure 2B), which could be attributed to differences in the thrombosis models. The ferric chloride and stasis models used in previous studies have been documented to induce endothelial damage, a characteristic largely absent in clinical DVT and reflected in the IVC stenosis model [17,41,42]. However, in contrast with the findings of Brill et al. [16], reduced VWF activity also did not affect the thrombus burden in our study. This could be due to the lack of P-selectin in the VWF knockout mice used in the previous study [43], as the role of P-selectin in leukocyte recruitment and initial VWF tethering to endothelial cells during thrombosis has previously been identified [37,44]. However, the small number of thrombosed mice in the αVWF antibody-treated cohort may also have underpowered this analysis. We were also unable to measure plasma VWF levels to confirm whether the effects of the antibodies were due to physical inhibition or accelerated clearance because the inhibitory antibodies we used bound to multiple epitopes on the VWF protein [45]. Together, this led us to conclude that the role of VWF in inflammation-associated DVT is more critical in the initiation rather than the propagation of venous thrombosis. As more VWF initially present at the nidus of thrombus formation tethers a larger quantity of prothrombotic mediators to the endothelium, the potential for thrombus development is enhanced. Indeed, Brill et al. [16] showed that initial platelet and leukocyte recruitments were only detectable on the endothelial surface where VWF had been released from Weibel-Palade bodies (WPBs), compared with areas with intact WPBs.
We also assessed the composition of venous thrombi. Consistent with previous results from our lab using a chronic inflammation model of obesity-associated DVT [20], we noted a significant increase in the proportion of the red thrombus of the endotoxemic mice (Figure 4B). Simultaneously, these mice exhibited thrombocytopenia, and their thrombi had an increased, albeit non-significant, RBC content (Figures 1A and 4D). Inflammation can promote a prothrombotic state in RBCs, increasing their adhesion to endothelial cells and exposing phosphatidylserine on their membrane surface [46]. The combined effects of decreased local availability of platelets and increased prothrombotic activity of RBCs could account for the predominance of the red thrombus in inflammation-associated thrombosis. This is also in line with the findings of Obi et al. [31] for decreased platelet content in the thrombi of LPS-treated mice.
A series of structural studies of venous thrombi have identified RBCs, fibrin, leukocytes, VWF, platelets, P-selectin, NETs, and HMGB1 as some of the main players in thrombosis [16,28,37,47]. However, no study thus far has investigated their interactions simultaneously, largely due to the technical limitations of traditional histology methods. The IMC technique provides multiplexing capabilities that overcome such limitations. Using heavy metal-tagged antibodies coupled with mass cytometry, this technique: 1) enables the multiplexing of more antibody targets than traditional methods allow; 2) capitalizes on the virtual lack of intrinsic cellular heavy metal expression to minimize background signals; and 3) allows high-resolution analysis of tissue samples [48]. For the first time, we employed this technology in our study to assess the feasibility of an IMC approach to simultaneously explore the cellular and molecular interactions of previously identified constituents of venous thrombi. Although the low biological replicate numbers of this preliminary study may reduce the power of the statistical analyses, the demonstrated robust reproducibility of this technique nonetheless provides reliable data [48]. Future studies should build upon these results with a larger number of markers and biological replicates.
Although VWF consistently exhibited a strong correlation with CD62P (Figures 6B and C), a marker of platelet activation [49], the lack of VWF association with CD41 in the control cohort indicated an alternative source of CD62P, possibly the endothelial cells [50]. Along with VWF, P-selectin is also contained in endothelial WPBs and has been proposed to anchor VWF to the endothelial surface once released [44,50]. Thus, it is possible that endothelial-derived P-selectin could be incorporated into thrombi with VWF. The VWF-CD41 correlation that was greater in the LPS cohort could be the result of an increased concentration of platelets in the white thrombus in endotoxemia, evidenced by its negative correlation with Ter119, a marker of RBCs that are the main components of the red thrombus (Figure 6C).
The HMGB1-VWF and HMGB1-CD41 correlations in the thrombi derived from endotoxemic mice supported previous reports of the crucial role of platelet-derived HMGB1 in DVT [47]. However, whether the HMGB1-VWF correlation is merely a byproduct of VWF and platelet’s proximity, or a result of their direct interactions, remains to be elucidated. Furthermore, the HMGB1-Ly6C and HMGB1-extracellular DNA correlations in the LPS cohort (Figure 6C) suggest alternative (possibly leukocyte-derived) cellular origins. Although platelet-derived, rather than cell-derived, HMGB1 has been identified as the driving factor in DVT [47], the indirect effects of leukocyte-derived HMGB1 on DVT via the inflammatory microenvironment merit further research given the immunomodulatory functions of extracellular HMGB1 [51].
VWF, fibrin, and NETs comprise the scaffolding network of a thrombus [27–29]. The VWF has been shown to interact directly with fibrin and NETs [29,52]. In our preliminary study using a novel IMC approach to provide a proof-of-concept for this method, we found that the VWF-fibrin(ogen) network largely remained stable regardless of endotoxemia, providing a constant basis of thrombosis (Figure 6B, C). In contrast, the correlation of extracellular DNA with CitH3 was greater in the thrombi derived from endotoxemic mice, indicating increased NET release in the inflammatory milieu that was absent in the control mice. Further, the variable correlations of VWF and fibrin(ogen) with extracellular DNA and CitH3 among the treatments demonstrated a more targeted interaction of these components (possibly NETs in the endotoxemic mice) with the scaffold network that may be dependent on an inflammatory microenvironment. However, we emphasize that the preliminary nature of this study warrants further analyses to confirm the findings in a larger sample size. The increased association of CD41 and HMGB1—known stimulators of NETs [49,53]—with VWF in the LPS cohort may have induced NETosis in closer proximity to VWF. However, it is possible that other yet unknown factors or mechanisms may mediate a closer association of NETs with the other scaffolding components in an inflammatory milieu.
This study suggests significant pathogenic implications for the clinical management of inflammation-associated thrombosis. This is of exceptionally high relevance in the current landscape of the COVID-19 pandemic with its associated endothelial inflammation, hypercoagulability, and thrombotic coagulopathies [32,54–56]. As in the endotoxemia model of sepsis, SARS-CoV-2 infection promptly engages a host immune response whose dysregulation could lead to collateral tissue damage. Similar to the pathogenesis of endotoxemia-associated thrombosis documented in our study, the presence of significantly increased VWF levels in severe cases of COVID-19 [55,56] may play a critical role in mediating the enhanced incidence of thrombotic complications in these patients [33,36,57].
In conclusion, we have shown that an endotoxin-induced state of inflammation increases thrombus incidence, but not thrombus burden, in the murine stenosis model of DVT. We further demonstrated that elevated VWF levels in inflammation play a critical role in mediating this pathological process. We also assessed the feasibility of using an IMC approach to conduct a structural analysis of thrombi, which shows that endotoxemia also affects the overall composition of venous thrombi, including the relative representation and cellular and molecular interactions of VWF. The integrity of the thrombus scaffold comprised VWF and fibrin(ogen) remains constant regardless of an inflammatory milieu, although variable interactions with NETs in inflammation remain to be further investigated. Together, our findings further characterize the mechanism of inflammation-associated venous thrombosis, highlighting the key functional and structural role of VWF in this pathologic process.
Supplementary Material
Essentials.
Inflammation has previously been associated with thrombosis.
Endotoxemia enhances the development of deep vein thrombosis in mice.
von Willebrand factor plays a critical role in mediating endotoxemia-associated thrombosis.
Endotoxemia modulates von Willebrand factor interactions in thrombi, but the thrombus scaffold is largely stable.
ACKNOWLEDGMENTS
The authors thank L. Boudreau, C. Notley, C. Brown, S. Virk, and A. Mo for technical laboratory assistance. D.L. is the recipient of a Canada Research Chair in Molecular Hemostasis. This work was supported by a grant-in-aid from the Heart and Stroke Foundation of Canada and a Canadian Institutes of Health Research Foundation Grant (FDN154285).
Funding information
Heart and Stroke Foundation of Canada and the Canadian Institutes of Health Research Foundation; Grant/Award Number: FDN154285.
Footnotes
DECLARATION OF COMPETING INTERESTS
There are no competing interests to disclose.
SUPPLEMENTARY MATERIAL
The online version contains supplementary material available at https://doi.org/10.1016/j.jtha.2022.11.022.
REFERENCES
- [1].Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, Ozaki Y, Wendelboe A, Weitz JI, ISTH Steering Committee for World Thrombosis Day. Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol 2014;34: 2363–71. [DOI] [PubMed] [Google Scholar]
- [2].Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J. Heart disease and stroke statistics–2021 update: a report from the American Heart Association. Circulation. 2021;143:e254–743. [DOI] [PubMed] [Google Scholar]
- [3].Kaplan D, Casper TC, Elliott CG, Men S, Pendleton RC, Kraiss LW, Weyrich AS, Grissom CK, Zimmerman GA, Rondina MT. VTE incidence and risk factors in patients with severe sepsis and septic shock. Chest. 2015;148:1224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Levi M, van der Poll T. Coagulation and sepsis. Thromb Res 2017;149:38–44. [DOI] [PubMed] [Google Scholar]
- [5].Hanify JM, Dupree LH, Johnson DW, Ferreira JA. Failure of chemical thromboprophylaxis in critically ill medical and surgical patients with sepsis. J Crit Care. 2017;37:206–10. [DOI] [PubMed] [Google Scholar]
- [6].Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 2013;13:34–45. [DOI] [PubMed] [Google Scholar]
- [7].Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol 2009;182(12): 7997–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, Tsung A, Zuckerbraun BS, Simmons RL, Neal MD. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep 2018;8:2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mészáros K, Aberle S, Dedrick R, Machovich R, Horwitz A, Birr C, Theofan G, Parent JB. Monocyte tissue factor induction by lipopolysaccharide (LPS): dependence on LPS-binding protein and CD14, and inhibition by a recombinant fragment of bactericidal/permeability-increasing protein. Blood. 1994;83(9):2516–25. [PubMed] [Google Scholar]
- [10].Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007;7:803–15. [DOI] [PubMed] [Google Scholar]
- [11].Kremer Hovinga JA, Zeerleder S, Kessler P, de Wit TR, van Mourik JA, Hack CE, ten Cate H, Reitsma PH, Wuillemin WA, Lämmle B. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost 2007;5: 2284–90. [DOI] [PubMed] [Google Scholar]
- [12].Peetermans M, Meyers S, Lisesenborghs L, Vanhoorelbeke K, De Meyer SF, Vandenbriele C, Lox M, Hoylaerts MF, Martinod K, Jacquemin M, Vanassche T, Verhamme P. Von Willebrand factor and ADAMTS13 impact on the outcome of Staphylococcus aureus sepsis. J Thromb Haemost 2020;18:722–31. [DOI] [PubMed] [Google Scholar]
- [13].Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, Denis CV. P-selectin glycoprotein ligand 1 and β2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108:3746–52. [DOI] [PubMed] [Google Scholar]
- [14].Smeets MWJ, Mourik MJ, Niessen HWM, Hordijk PL. Stasis promotes erythrocyte adhesion to von Willebrand factor. Arterioscler Thromb Vasc Biol 2017;37:1618–27. [DOI] [PubMed] [Google Scholar]
- [15].Grässle S, Huck V, Pappelbaum KI, Gorzelanny C, Aponte-Santamaría C, Baldauf C, Gräter F, Schneppenheim R, Obser T, Schneider SW. Von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler Thromb Vasc Biol 2014;34:1382–9. [DOI] [PubMed] [Google Scholar]
- [16].Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Köllnberger M, Wakefield TW, Lämmle B, Massberg S, Wagner DD. Von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Diaz JA, Saha P, Cooley B, Palmer OR, Grover SP, Mackman N, Wakefield TW, Henke PK, Smith A, Lal BK. Choosing a mouse model of venous thrombosis: a consensus assessment of utility and application. J Thromb Haemost 2019;17:699–707. [DOI] [PubMed] [Google Scholar]
- [18].Pruss CM, Golder M, Bryant A, Hegadorn C, Haberichter S, Lillicrap D. Use of a mouse model to elucidate the phenotypic effects of the von Willebrand factor cleavage mutants, Y1605A/M1606A and R1597W. J Thromb Haemost 2012;10:940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shida Y, Rydz N, Stegner D, Brown C, Mewburn J, Sponagle K, Danisment O, Crawford B, Vidal B, Hegadorn CA, Pruss CM, Nieswandt B, Lillicrap D. Analysis of the role of von Willebrand factor, platelet glycoprotein VI-, and α2β1-mediated collagen binding in thrombus formation. Blood. 2014;124:1799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Michels A, Dwyer CN, Mewburn J, Nesbitt K, Kawecki C, Lenting P, Swystun LL, Lillicrap D. Von Willebrand factor is a critical mediator of deep vein thrombosis in a mouse model of diet-induced obesity. Arterioscler Thromb Vasc Biol 2020;40:2860–74. [DOI] [PubMed] [Google Scholar]
- [21].Levine JH, Simonds EF, Bendall SC, Davis KL, Amir E-aD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, Finck R, Gedman AL, Radtke I, Downing JR, Pe’er D, Nolan GP. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Car BD, Eng VM, Schnyder B, Ozmen L, Huang S, Gallay P, Heumann D, Aguet M, Ryffel B. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J Exp Med 1994;179: 1437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rhodes A, Wort SJ, Thomas H, Collinson P, Bennett ED. Plasma DNA concentration as a predictor of mortality and sepsis in critically ill patients. Crit Care. 2006;10:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Matsumoto H, Ogura H, Shimizu K, Ikeda M, Hirose T, Matsuura H, Kang S, Takahashi K, Tanaka T, Shimazu T. The clinical importance of a cytokine network in the acute phase of sepsis. Sci Rep 2018;8: 13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Patel KN, Soubra SH, Bellera RV, Dong J-F, McMullen CA, Burns AR, Rumbaut RE. Differential role of von Willebrand factor and P-selectin on microvascular thrombosis in endotoxemia. Arterioscler Thromb Vasc Biol 2008;28:2225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sevitt S The structure and growth of valve-pocket thrombi in femoral veins. J Clin Path 1974;27:517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lord ST. Fibrinogen and fibrin: scaffold proteins in hemostasis. Curr Opin Hematol 2007;14:236–41. [DOI] [PubMed] [Google Scholar]
- [28].Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res 2007;120:S5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 2010;107:15880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang X Lipopolysaccharide augments venous and arterial thrombosis in the mouse. Thromb Res 2008;123:355–60. [DOI] [PubMed] [Google Scholar]
- [31].Obi AT, Andraska E, Kanthi Y, Kessinger CW, Elfline M, Luke C, Siahaan TJ, Jaffer FA, Wakefield TW, Henke PK. Endotoxaemia-augmented murine venous thrombosis is dependent on TLR-4 and ICAM-1, and potentiated by neutropenia. Thromb Haemost 2017;117:339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Malas MB, Naazie IN, Elsayed N, Mathlouthi A, Marmor R, Clary B. Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: a systematic review and meta-analysis. EClinicalMedicine. 2020;29:100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Gandet FF, Fafi-Kremer S, Castelain V, Schneider F, Grunebaum L, Anglés-Cano E, Sattler L, Mertes P-M, Meziani F, CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis). High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 2020;46:1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fogarty H, Townsend L, Cheallaigh CN, Bergin C, Martin-Loeches I, Browne P, Bacon CL, Gaule R, Gillett A, Byrne M, Ryan K, O’Connell N, O’Sullivan JM, Conlon N, O’Donnell JS. COVID19 coagulopathy in Caucasian patients. Br J Haematol 2020;189:1044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thrombo-embolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost 2020;18:1421–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost 2017;15:1285–94. [DOI] [PubMed] [Google Scholar]
- [37].von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 2012;209:819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Begbie M, Notley C, Tinlin S, Sawyer L, Lillicrap D. The factor VIII acute phase response requires the participation of NFκB and C/EBP. Thromb Haemost 2000;84:216–22. [PubMed] [Google Scholar]
- [39].Mimuro J, Niimura M, Kashiwakura Y, Ishiwata A, Ono T, Ohmori T, Madoiwa S, Okada K, Matsuo O, Sakata Y. Unbalanced expression of ADAMTS13 and von Willebrand factor in mouse endotoxinemia. Thromb Res 2008;122:91–7. [DOI] [PubMed] [Google Scholar]
- [40].De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013;121:270–7. [DOI] [PubMed] [Google Scholar]
- [41].Li W, Nieman M, Sen Gupta A. Ferric chloride-induced murine thrombosis models. J Vis Exp 2016;155:e54479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mackman N New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012;122:2331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Denis CV, André P, Saffaripour S, Wagner DD. Defect in regulated secretion of P-selectin affects leukocyte recruitment in von Willebrand factor-deficient mice. Proc Natl Acad Sci U S A 2001;98:4072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, Lóez JA, Dong J-F. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. 2004;103:2150–6. [DOI] [PubMed] [Google Scholar]
- [45].Tan FL, Ginsburg D. What a polyclonal antibody sees in von Willebrand factor. Thromb Res 2008;121:519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bateman RM, Sharpe MD, Singer M, Ellis CG. The effect of sepsis on the erythrocyte. Int J Mol Sci 2017;18:1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Brühl M-L, Gaertner F, Lorenz M, Agresti A, Coletti R, Antoine DJ, Heermann R, Jung K, Reese S, Laitinen I, Schwaiger M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128: 2435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, Varga Z, Wild PJ, Günther D, Bodenmiller B. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–22. [DOI] [PubMed] [Google Scholar]
- [49].Assinger A, Schrottmaier WC, Salzmmann M, Rayes J. Platelets in sepsis: an update on experimental models and clinical data. Front Immunol 2019;10:1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].McCormack JJ, da Silva ML, Ferraro F, Patella F, Cutler DF. Weibel-Palade bodies at a glance. J Cell Sci 2017;130:3611–7. [DOI] [PubMed] [Google Scholar]
- [51].Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol 2020;11:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mitszta A, Pelkmans L, Lindhout T, Krishnamoorthy G, de Groot PG, Hemker CH, Heemskerk JWM, Kelchtermans H, de Laat B. Thrombin-dependent incorporation of von Willebrand factor into a fibrin network. J Biol Chem 2014;289:35979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tadie JM, Bae HB, Jiang S. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol 2013;304:L342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Varga Z, Flammer AJ, Steiger P, Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Goshua G, Pine AB, Meizlish ML, Chang C-H, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, Dela Cruz CS, Dumont A, Halene S, Hwa J, Koff J, Menninger H, Neparidze N, Price C, Siner JM, Tormey C, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 2020;7:E575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mancini I, Baronciani L, Artoni A, Colpani P, Biganzoli M, Cozzi G, Novembrino C, Anzoletti MB, De Zan V, Pagliari MT, Gualtierotti R, Aliberti S, Panigada M, Grasselli G, Blasi F, Peyvandi F. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J Thromb Haemost 2021;19:513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wichmann D Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med 2020;173:1030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.