

Abstract
Data independent acquisition (DIA also termed SWATH) is an emerging technology in the field of mass spectrometry based proteomics. Although the concept of DIA has been around for over a decade, the recent advancements, in particular the speed of acquisition, of mass analyzers have pushed the technique into the spotlight and allowed for high-quality DIA data to be routinely acquired by proteomics labs. In this chapter we will discuss the protocols used for DIA acquisition using the Sciex TripleTOF mass spectrometers and data analysis using the Sciex processing software.
Keywords: Data independent acquisition (DIA), SWATH, Quantitative proteomics, Mass spectrometry, Spectral ion library
1. Introduction
Data Independent Acquisition Mass Spectrometry (DIA-MS) is a long-standing technique [1, 2] that has garnered increased attention recently due to the development of new pipelines for extracting, identifying, and quantifying peptides using a targeted analysis approach [3, 4]. SWATH™ couples DIA-MS with direct searching of individual samples against an established, and often a more exhaustive, peptide MS spectral library [3, 5, 6]. SWATH™ is, therefore, a two-step process (Fig. 1), development of the MS spectral library, commonally on a pooled sample representing the breath of the experimental collection, using information dependent acquisition (IDA also termed data dependent acquistion (DDA)) (see Note 1) and then the subsequent analysis of each individual sample by DIA. Thus, a major advantage of SWATH™ is that it can maximize the peptides observed both within an individual sample and across all of the samples in an experimental set, thereby increasing proteome coverage, experimental efficiency, reducing quantitative variability, and minimizing missing data across an experimental matrix. It is important to note that SWATH™ is an emerging approach and methods for estimating peptide identification confidence and false discovery rates as well as the ideal approach for estimating peptide and protein quantity from transition extracted ion chromatograms are continuing to evolve along with the sensitivity and capabilities of the instrumentation itself. As with any large-scale quantitative screening method, care should be taken to confirm and validate the biological differences and conclusions that are derived from a SWATH™ experiment.
Fig. 1.
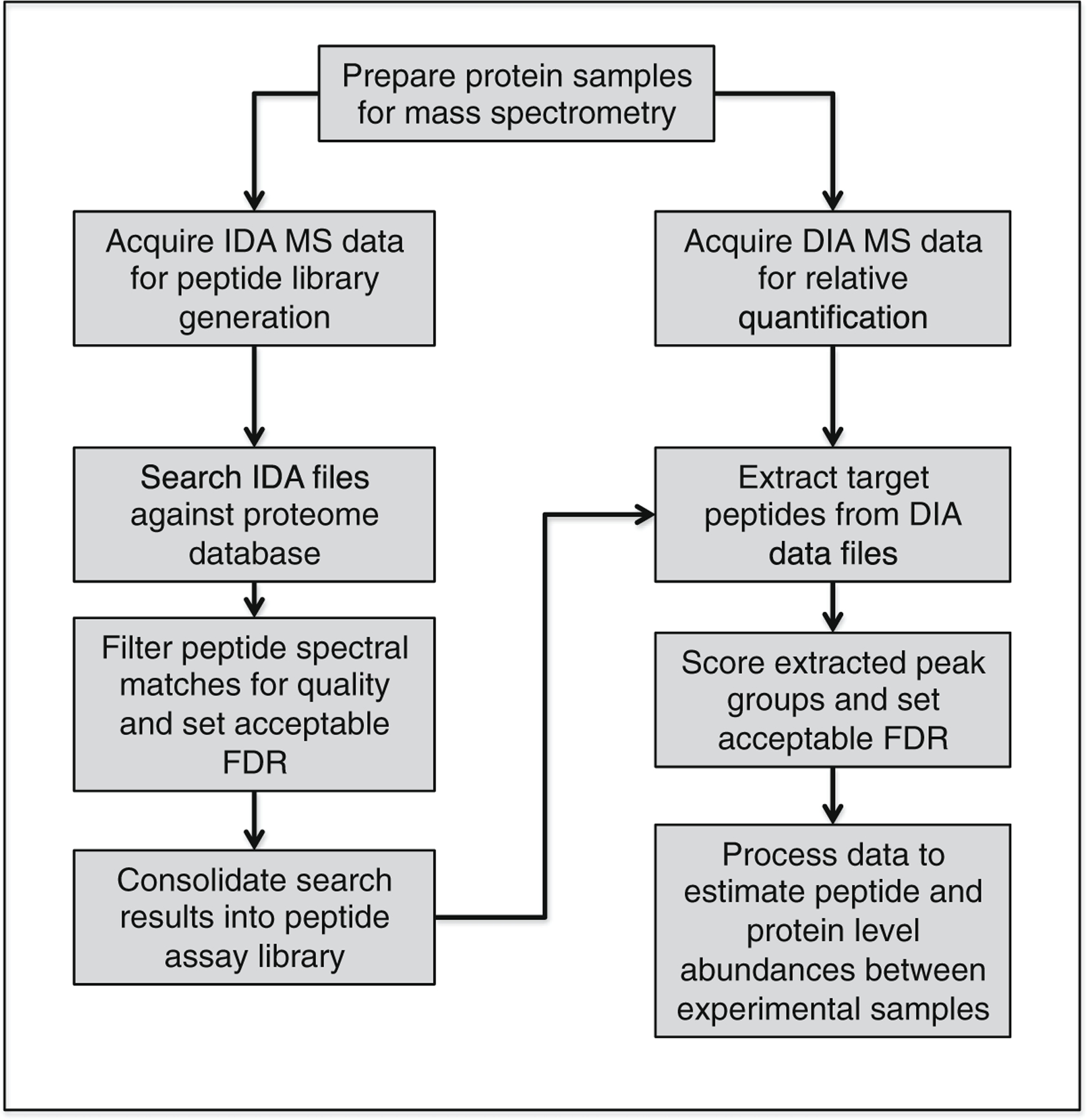
Schematic of general workflow for SWATH-MS acquisition and analysis
In a SWATH™ experiment, proteins are digested and either directly infused or, more often, separated by liquid chromatography (LC) prior to analysis on a TripleTOF mass spectrometers (5600 or 6600, Sciex), a Q-Exactive mass spectrometer (Thermo Scientific), or any instrument with sufficiently high scan speed and a quadrupole mass filter. On the Triple TOF instruments, precursor peptide ion selection is performed by filtering precursors collectively through mass-to-charge windows, typically 4–10 m/z wide, sequentially across the entire m/z range of interest rather than selectively isolating a single precursor mass/charge (m/z) per MS/MS scan as performed in IDA-MS experiments. Due to the typically wider isolation windows used in DIA experiments, two or more co-eluting precursors are often fragmented collectively to produce an MS2 spectrum containing a convoluted mixture of fragment ions from multiple precursor ions.
One approach used to increase the ability to find and confidently identify peptides from these complex mixed spectra is to associate specific peptides with defined regions within the chromatographic elution profile. Currently, in order to accomplish this, retention time (RT) determination and alignments across samples are key aspects of searching IDA data. Exogenous supplied RT standards [6] or endogenous RT standards [7] that are composed of peptides consistently observed across large number of samples must be used for RT calibration in order to properly align individual ion chromatograms across the entire sample’s elution profile.
Optimization of m/z window number and dwell time/ion accumulation time per window is performed so that the instrument cycles through the entire desired precursor m/z range (e.g., 400–1250 m/z). This is largely instrument and sample specific. For the 6600 triple TOF, you can go up to 2250 m/z but we typically analyze between 400 and 1250 m/z for tryptic digests. When analyzing middle down or any peptides larger than the average tryptic peptides, the full range can be used with the appropriate considerations to SWATH™ windows and cycle times. Ultimately, the key is to allow the instrument to cycle rapidly enough to capture multiple observations across the chromatographic elution profile for a given ion.
The data are subsequently searched against a sample-specific peptide library that allows a set number of transition ion chromatograms to be extracted for a peptide within the window of its predicted RT (determined by its observed or normalized RT from the peptide library). The peak groups are scored according to several factors intended to discriminate a “true” peptide target from nonspecific noise, and the distribution of these target scores is modeled against the distribution of scores attributed to decoy peak groups to determine a score cut off resulting in an acceptable false discovery rate. Relative peptide abundance is then inferred from the aggregate of the area under the curve for each transition extracted ion chromatograms (XICs), and various statistical approaches are used to roll transition intensity XICs into peptide intensity estimates, which can then be used to estimate the overall protein intensity. In this chapter, we present the typical workflow used currently by our group to prepare, acquire, and analyze proteomic data for a DIA-MS experiment of cell or tissue samples. For simplicity and pragmatism, we present the workflow as completed using SCIEX TripleTOF® instruments and data analysis platform exclusively, with mention of alternative approaches as appropriate.
1.1. Quality Assurance and Quality Control (QA/QC) Considerations
Robust quality assurance (QA) or quality control (QC) protocols are essential to monitor instrument performance and improve reproducibility and reliability of data. A QC standard run can be analyzed at fixed times such as the beginning and end of an experiment or day to assess variation in a variety of quality control metrics [8]. For the TripleTOF instruments, we conduct internal mass calibrations of mass accuracy and sensitivity for both MS1 and MS2 scans every 3–5 runs by monitoring at least eight peptides from 100 fmol digested beta-galactosidase standard (Sciex) and seven transition ions from the 729.3652 [M+2H]2+ ion (Table 1). What also needs to be tracked is sample processing to ensure the quality of the peptide mixture being analyzed, which is not addressed at in this manuscript but is well established in targeted multiple and selective monitoring workflows. To do this one can include an exogenously protein, such as beta-galactosidase, into the sample prior to digestion. Beta-galactosidase selected peptides can be quantified (if N15 labeled peptides are added after digestion to the sample) or assessed in each sample (for more details see Chen et al., in this book).
Table 1.
Beta-galactosidase peptides used for autocalibration and quality control
| Beta-galactosidase peptide sequence | [M+2H]2+ | Transition ions for 729.36 | Fragment |
|---|---|---|---|
| YSQQQLMETSHR | 503.2368 | ||
| RDWENPGVTQLNR | 528.9341 | ||
| GDFQFNISR | 542.2654 | ||
| IDPNAWVER | 550.2802 | ||
| DVSLLHKPTTQISDFHVATR | 567.0565 | ||
| VDEDQPFPAVPK | 671.3379 | ||
| WENPGVTQLNR | 714.8469 | ||
| APLDNDIGVSEATR | 729.3652 | ||
| 175.1190 | y1 | ||
| 347.2037 | y3 | ||
| 563.2784 | y5 | ||
| 729.3652 | b7 | ||
| 832.4523 | y8 | ||
| 1061.5222 | y10 | ||
| 1289.6332 | y12 |
Internal peptide retention time (RT) standards are currently an essential component of both peptide library generation and DIA-MS data analysis, and must be (1) detectable across all individual samples and (2) spread evenly across the chromatogram. Retention time of a given peptide from the library is used to set an extraction window for its peak group identification from the SWATH™ /DIA-MS data file, and subsequently also used in scoring the confidence of a given peak group assignment to a peptide sequence from the library. If SWATH™/DIA-MS data files and peptide library files are collected absolutely sequentially with nearly identical chromatography, one might bypass the use of RT alignment standards. Much more commonly, differences in sample matrix, chromatographic setups, timing of instrument batch acquisitions, and many other factors will contribute to imperfect chromatographic alignment necessitating RT standards to normalize peptide assay library retention time to the SWATH™ acquisition file retention time. Used alone or in combination with retention time standards that are spiked into a sample, endogenous reference peptides can also be used for the calibration of retention times across samples [7]. These can be unique to a specific library (sample); however, there are common and conserved peptides that may be present in most, if not all, mammalian cells and tissues which can be used as a complement or replacement to synthetic, externally spiked RT reference peptides [7]. Note, that new methods to analyze DIA data sets are being developed and the need for RT standards may change, however, expectations are that RT alignment will remain part of the QC for assessment of LCMS runs. As well, QC tools are available to assess quality control metrics in a shotgun or targeted proteomic workflow that allows chromatographic performance and systemic error to be monitored [9]. Tracking RT standards across sample runs can also serve to assess instrument performance.
Finally, as larger numbers of individual samples are analyzed adopting other routine QC such as randomization or blocking of sampled to minimize sample analysis bias and regular collection of quality control samples spaced evenly and strategically throughout acquisition batches will be necessary components of SWATH™ experimental design.
1.2. Spectral Library Building—Data Generation
A spectral ion library is most often used for the targeted analysis of SWATH™/DIA-MS data, although other methods (as mentioned above) are being explored and developed [10, 11], and can be primarily cell or tissue and species specific or a broader library assembled from all relevant peptide observations from a given species [5]. Spectral ion libraries are most commonly built using traditional shotgun proteomics in IDA-MS mode. In some cases spectral ion libraries previously generated have been made available to the public from various labs [5, 12, 13]. Here we will discuss the creation of new spectral ion libraries from IDA analysis of proteolytic digestions. Additional detailed information regarding the generation of spectral ion libraries, including the management of protein redundancy and isoform specificity, can be found in Schubert et al. [5]. It is important to consider differences in peptide fragmentation patterns between instruments, and ideally use IDA data acquired on the same instrument from which you will perform your SWATH™/DIA-MS acquisition [14].
Spectral ion libraries can be constructed in a number of ways. The first and most straightforward way to create an ion library is to analyze a proteolytic digestion in IDA mode of a pooled sample created from all of the individual samples that will be subsequently analyzed by DIA or of samples composing the extremes of the phenotype. This will give the most basic ion library comprising the peptides identified in a single IDA run that can then be used against the SWATH™ acquired version of itself and any other SWATH™/DIA-MS acquired sample of the same general proteome. In an attempt to expand the number of ions selected for fragmentation for library generation from a single IDA run of the pooled sample, multiple runs or technical replicates might help increase the proteome coverage provided to the sample library beyond what may be obtained from a single run and thus may help compensate for the error in sampling that is inherent to DIA methods. Alternatively, deeper and more inclusive ion libraries can be constructed post-digestion using off-line peptide fractionation and analysis of these fractions independently in IDA mode. The IDA runs are then combined to create a more complete and inclusive ion library for the given sample proteome. This should ultimately increase the power of DIA-based protein identifications by increasing the number of peptides used to quantitate highly abundant proteins while harnessing the sensitivity of MS2-based quantitation necessary for low abundance proteins and peptides. Some methods commonly used for peptide fractionation are basic-reverse phase HPLC (bRP-HPLC) [15], strong cation exchange (SCX), and strong anion exchange (SAX) [16] (see Notes 2 and 3). Our lab typically uses bRP-HPLC or a solid phase extraction SCX [17] method for peptide fractionation prior to MS analysis. For SWATH™ analysis of post-translational modifications, it is recommended to employ enrichment strategies (if applicable) either independently or in combination with the peptide fractionation techniques described and as typically performed in shotgun experiments.
The following protocol is for library generation using Sciex TripleTOF™ systems with an Eksigent® 415 nano LC and ekspert 400 autosampler, although alternative LC and autosamplers may be used with the TripleTOF systems.
2. Materials
Proteolytic peptide mixture, most often MS-grade trypsin (Promega).
5600 or 6600 TripleTOF system.
Nano-LC and autosampler (e.g., Eksigent® 415 nano LC, ekspert™ 400 autosampler) and ekspert™ cHiPLC (optional).
Trap and analytical LC columns (Eksigent® P/N 804–00006 and 804–00001).
Proteolytic peptide mixture, most often MS-grade trypsin (Promega).
5600 or 6600 TripleTOF system.
Retention time standards, either commercial peptides that are spiked in right before MS analysis (e.g., Biogynosis cat# KI-3002–2) or endogenous peptides present in all samples, can be used (Parker et al., in press) (see Note 4).
Software Needed (See Note 5)
Analyst TF 1.7.
PeakView 2.0 or higher.
Variable Window Calculator.
Protein Pilot 4.5 or higher.
SWATH™ microapp.
Microsoft Excel.
MarkerView (optional).
3. Methods
3.1. IDA Analysis of Proteolytic Digests for Spectral Ion Library Building
Create an IDA method in Analyst TF 1.7 with one survey scan and 20 candidate ion scans per cycle (see Note 6). Check the Rolling Collision Energy box.
- For TOF MS (MS1)
- Under the Include/Exclude tab put in any masses you want to monitor or exclude in your analysis.
- Under the IDA Advanced tab make sure Rolling Collision Energy is checked and make any other necessary changes that would be pertinent to your experiment.
- Default settings do not need to be changed under the Advanced MS tab.
- For Product Ion (MS2)
- Under the MS Tab set the accumulation time to 100 ms and the mass range from 100 to 1800 Da and check whether you want high resolution or high sensitivity (the high sensitivity function is most commonly selected for proteomics experiments).
- All other tabs should maintain the same parameters as for the TOF MS and do not need to be changed.
Load the sample appropriate Gradient, Loading Pump, and autosampler methods and save your Acquisition File.
Analyze your peptide samples.
Fig. 2.
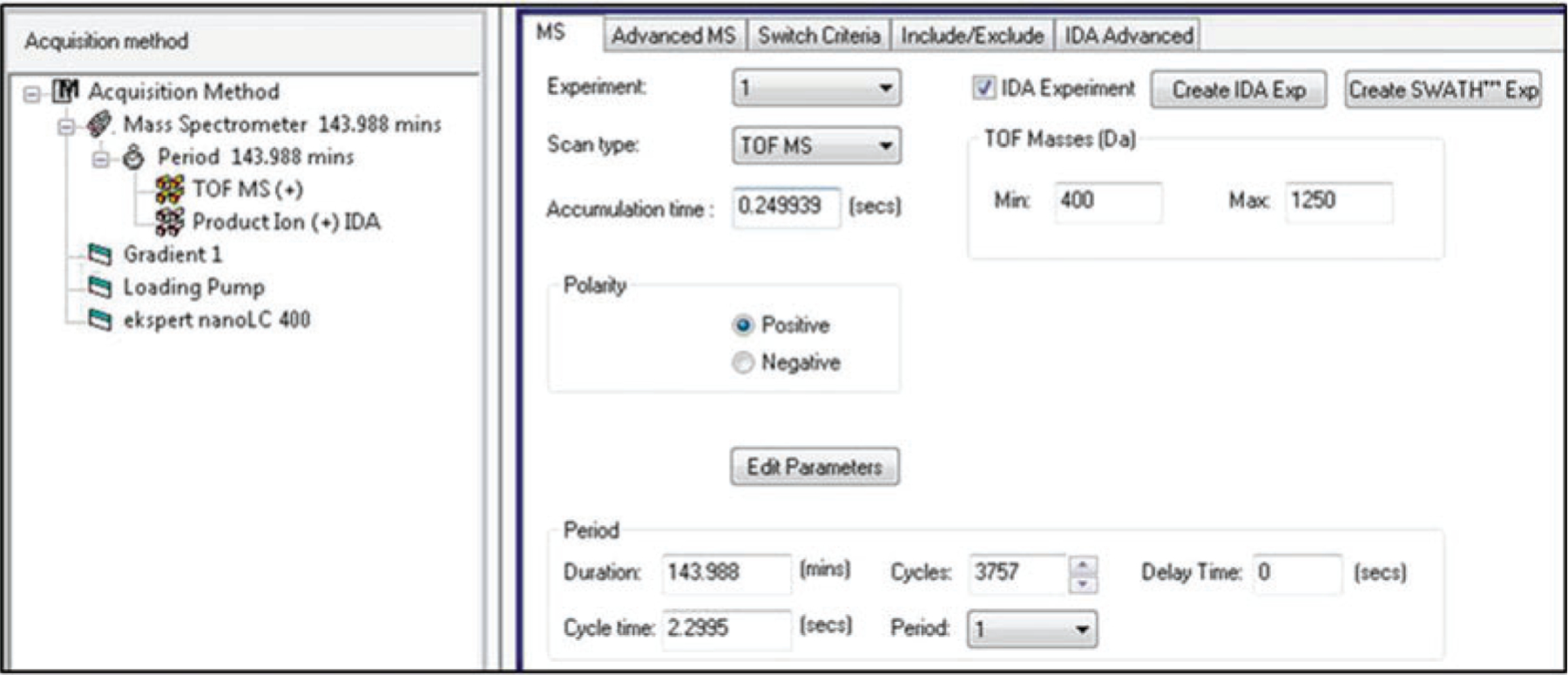
Example of TOF MS parameters for TripleTOF MS instruments
Fig. 3.
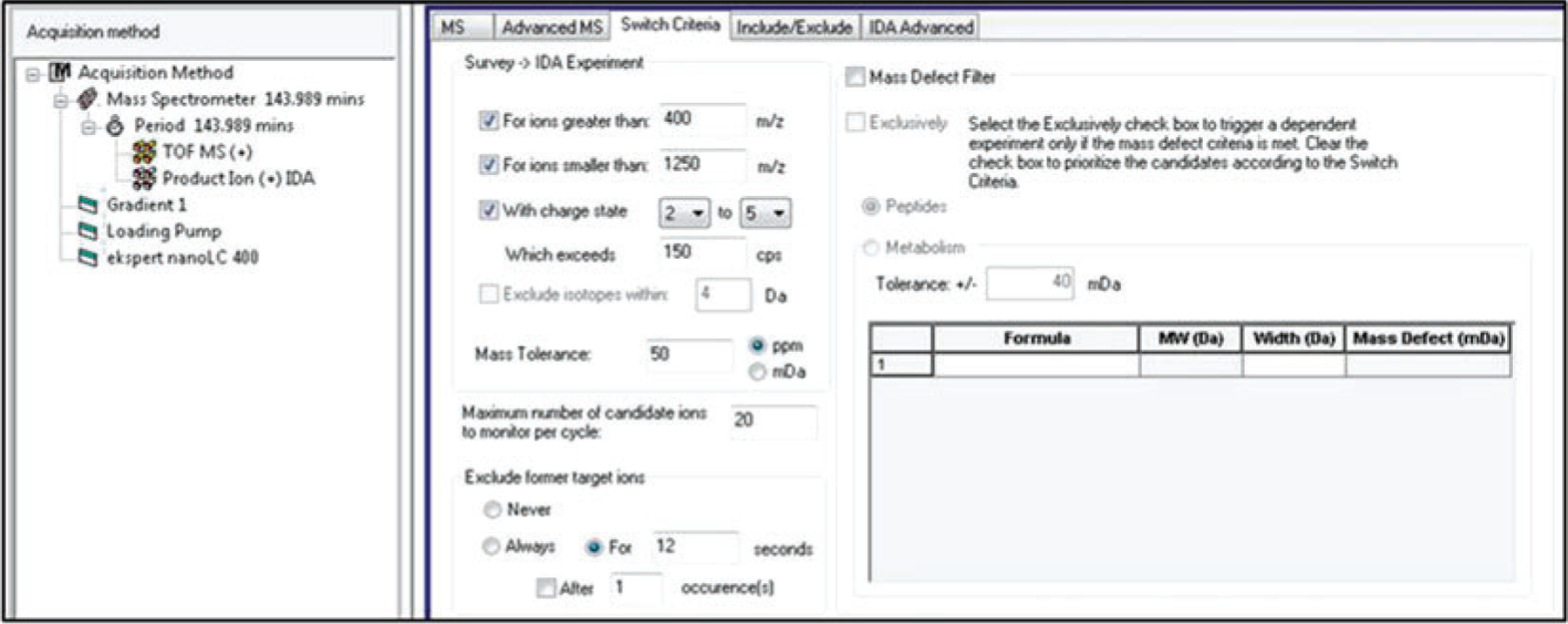
Example of Switch Criteria parameters for TripleTOF MS instruments
3.2. SWATH-MS Data Acquisition (DIA-MS acquistion)
3.2.1. Creation of Variable Window SWATH™ Methods
Optimized SWATH™ methods can be constructed for specific samples using the Sciex Variable Window Calculator application. The steps for creating the customized SWATH™ variable windows for a specific sample are listed in the Variable Window Calculator under the Instructions and Controls tab. After following these directions select the number of variable windows (see Note 9) you want to analyze in your method and the mass range of the SWATH™ analysis. For general proteomics experiments the window overlap is usually left at 1 Da and the collision energy spread (CES) is usually left at 5. The minimum window width should be set no lower than 4 due to the default parameters in the PeakView software. After the Variable Window calculator is finished creating the optimal windows for your analysis go to the OUTPUT for Analyst tab and copy columns A, B, and C into a new Excel file and save as a Text (Tab Delimited) file which can then be loaded into the SWATH™ method within Analyst TF 1.7.
3.2.2. Creation of a SWATH™ Method in Analyst TF 1.7
In Analyst TF 1.7 go to the Build Acquisition Method tab on the left-hand side of the window. Click on TOF MS and select Create SWATH™ Exp button then select the Manual tab within this window.
Under SWATH™ Analysis Parameters select the mass range of the analysis (typically 400–1250 Da for tryptic peptides). Under Fragmentation Conditions make sure Rolling Collision energy is checked (the CES set in the Variable Window Calculator will overwrite the CES value inputted on this screen). Under SWATH™ Detection Parameters select the mass range to monitor for the SWATH™ MS2 spectra (typically 100–1800 Da) and the accumulation time for each window (typically for 100 VW 30 ms is adequate) (see Note 10). Lastly, click the Read SWATH™ Windows from Text File box and load in your .txt file created in the Variable Window Calculator.
The accumulation time for the MS1 can be set between 50 and 150 ms to give a quick survey scan for each cycle (see Note 11). Select the appropriate loading pump, gradient, and autosampler methods for the file (see Note 12). The gradient method chosen should be the same one that was used during the IDA analysis preformed to generate the proteome-specific spectral library.
3.3. SWATH™ Data Analysis Using PeakView 2.1 and SWATH™ Microapp 2.0
3.3.1. Introduction to SWATH™ Data Analysis Procedure
As with many methodologies, there are several options for processing SWATH™ data and analyzing results. Here, we present the protocol to process data through the SCIEX proprietary software. In our lab, we also regularly utilize two alternative pipelines, Skyline [18] and OpenSWATH [4]. Skyline is a free and open-source tool built in Windows computing environments for analysis of multiple MS data types, including DIA. OpenSWATH™ is a free and open-source built within the openMS data analysis tool space, and operates optimally in a Linux computing environment. A summary of the basic information pertaining to using these two alternate data analysis pathways is provided in Table 2 located at the end of this section. In this final section, we will provide a cursory summary specific to the approach used in our lab for the general implementation of the SCIEX software tools. We recommend referring to the SCIEX software user manuals for additional guidance.
Table 2.
Selected alternative DIA-MS data analysis approaches
| Parameters | Skylinea | OpenSWATHb |
|---|---|---|
| Input DIA File format | .WIFF | .mzML/.mzXMLc |
| Peptide Ion Library | Built from DDA search result files (e.g., pep.xml,.group) or imported as a “transition list” | Built using TPP tools and custom Python scriptsd |
| SWATH workflow | Internal to Skyline | OpenSwathWorkflow.exe |
| Output File Format | .csv transition report | .tsv transition report |
| Visualization | Internal to Skyline | TAPIRe |
| Peak Picking Algorithm | mProphetf adaptation | pyProphetg |
| Multi-Run Alignment | – | Feature Alignmenth |
| Quantitative Statistics | Linked External Tool MSstatsi | External Tools (e.g., MapDIA,j MSstats) |
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010)
Röst HL et al. OpenSWATH™ enables automated, targeted analysis of data independent acquisition MS data. Nature Biotechnology 10;32(3):219–223 (2014)
Conversion to mzML or mzXML can be done using the tool msconvert, available at: (http://proteowizard.source-forge.net/tools/msconvert.html). Do not select peak picking, files may expand 10× or more from raw file size
Schubert OT et al., Building high-quality assay libraries for targeted analysis of SWATH™ MS data. Nature Protocols, 10(3):426–441 (2015). Note: Libraries generated using the pipeline described in the Schubert et al. paper can be formatted for use in the PeakView microapp, and substituted in the workflow above
Python script, available to download from https://github.com/msproteomicstools, found in folder msproteomics-tools/analysis/alignment/feature_alignment.py
3.3.2. Creation of Spectral Ion Library Using Protein Pilot Paragon Method
- Prepare the protein reference database that you will use for matching DDA spectra to peptide sequences. For instance, FASTA documents for annotated proteomes can be downloaded from the Uniprot website: (http://www.uniprot.org/proteomes). Typically, we chose to use the curated, or reference proteomes, for a given organism of interest.
- If external retention time standards were used in the experiment, such as the Biognosys iRT (see Note 13) peptides, copy their sequences and append to your FASTA file by opening it in a text editor. FASTA proteome databases should be saved in the appropriate folder within the Protein Pilot software files on your computer as per the software manual instructions.
In Protein Pilot, select the option for an LC MS search and prepare a database search method appropriate for your experiment, including all of the raw data files you would like to include to build the ion library.
Once the search is completed open the “FDR report” generated for the search and record the number of proteins identified at 1 % Global FDR to be used as input in the following section.
3.3.3. Importing Ion Libraries into the SWATH™ Microapp and Analyzing SWATH™ Data
Open PeakView and using the tabs at the top of the screen, navigate to Quantitation → SWATH™ Processing → Import Ion Library (Fig. 4).
Find the .group file produced from the Protein Pilot search and set the number of proteins to import to the 1 % Global FDR (see Note 14) recorded in the previous section from the FDR report generated by Protein Pilot. Typically peptides shared by more than one protein are not imported. Under Select sample type, chose the option appropriate for whether the samples were unlabeled (typical) or labeled with a chemical tag (i.e., iTRAQ, SILAC).
Select all of the SWATH™ files to be analyzed for a given experiment.
Set your processing settings. For protein quantitation analysis, examples of typical parameter settings are given in Fig. 5 (see Note 15).
After setting your processing settings click “Process” to analyze your SWATH™ data.
Once completed you can export the data for visualization in MarkerView by clicking Quantitation→ SWATH™ Processing→ Export→ Areas or Export→ All to get a complete list of all parameters for the analysis in Excel format (Fig. 6).
Fig. 4.
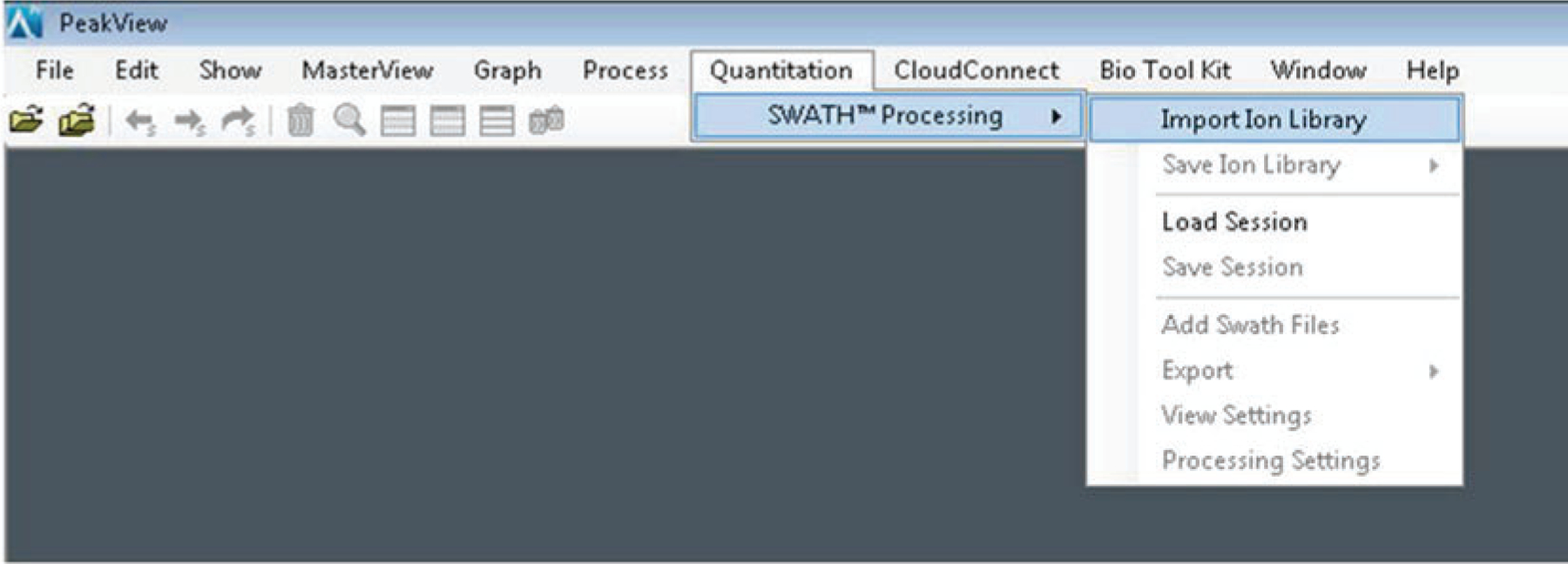
Schematic for importing ion library into PeakView software
Fig. 5.
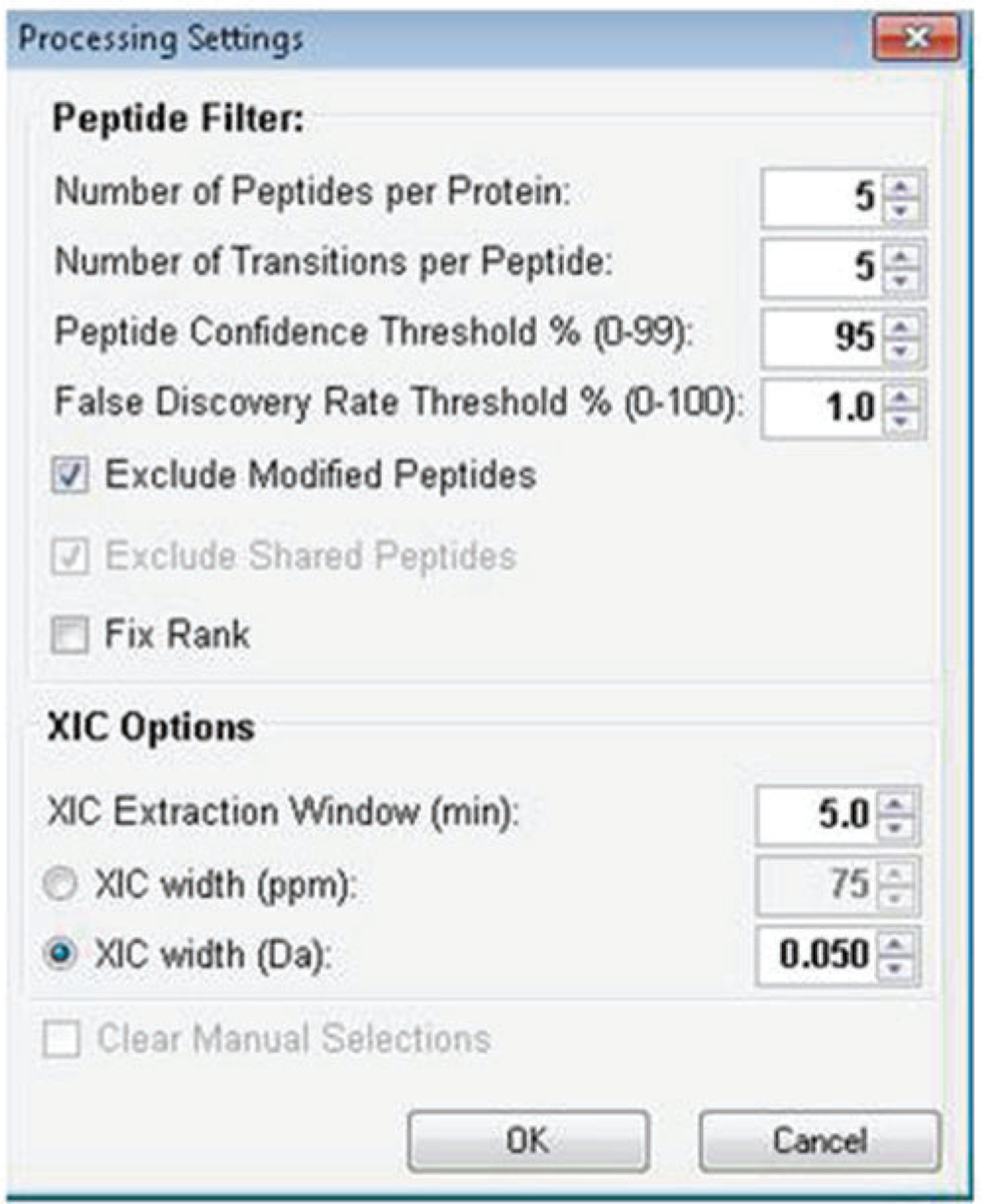
Example of typical processing settings for SWATH analysis using PeakView software
Fig. 6.
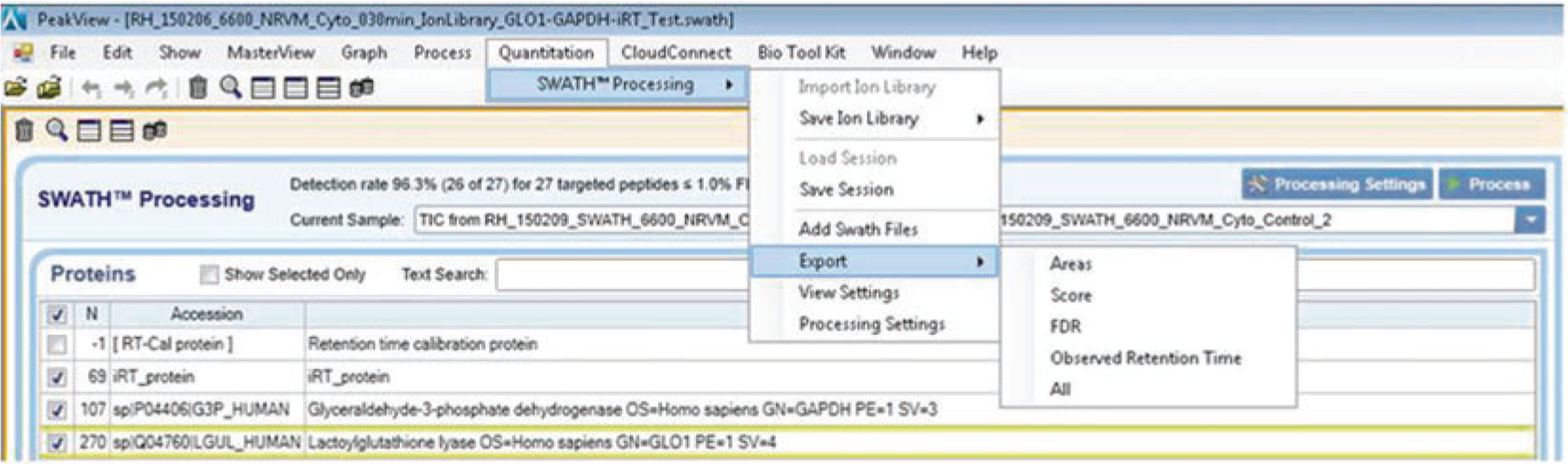
Schematic for exporting SWATH results from PeakView software
Acknowledgements
We would like to acknowledge funding to JVE from 1R01HL111362–01A, 1P01HL112730–01A1, 1U54NS091046–01, NHLBI-HV-10–05 [2], HHSN268201000032C and the Erika Glazer Endowed Chair for Women’s Heart Health and funding to SP from the National Marfan Foundation Victor E McKusick Post-Doctoral Fellowship; as well as the technical and intellectual support of Brigitt Simons and Christie Hunter at Sciex in many helpful discussions.
4 Notes
1. The Sciex terminology Information Dependent Acquisition (IDA) is the same as Data Dependent Acquisition (DDA) and this is the terminology used in the Sciex software for shotgun proteomics experiments. In this paper we will be using the IDA acronym to be consistent with the Sciex terminology and software.
2. bRP-HPLC fractionation may be preferred over SCX or SAX fractionation if downstream phosphopeptide enrichment or analysis of other negatively charged peptides is desired. This is due to a more equal distribution of phosphopeptides throughout basic RP fractions compared to SCX and SAX fractions, in which phosphopeptides are most dense in the early and late fractions, respectively.
3. The SCX method published by Dephoure and Gygi [17] was based on 10 mg of starting material and was used upstream of phosphopeptide enrichment. Our lab has used this method for both phosphoproteomic and general proteomic analysis and we have scaled back the protocol for 1 mg of starting material, in which we have cut the reagents used in the Dephoure and Gygi paper by 1/10th. If using less than 1 mg of starting material, scale back the reagents accordingly [13].
4. If large number of samples, include beta-galactosidase for sample preparation assessment and N15 labeled peptides to track (see Chen et al., this book).
5. Sciex software can be downloaded at http://www.absciex.com/downloads/software-downloads.
6. The number of survey scans desired for the analysis of concatenated or single run samples for library generation is a matter of user discretion but a typical IDA method on a TripleTOF system uses 20 candidate ions.
7. The 5600 TripleTOF system can go up to 1250 m/z and the 6600 TripleTOF can go up to 2250 m/z. However, we find that for tryptic digests there is little additional peptide data obtained above 1250 m/z. The larger mass range on the 6600 system is beneficial when doing large protein modifications such as glycoproteomics or when using alternative proteolytic methods that produce larger peptides (i.e., Lys-C, CNBr).
8. These values are meant to be used as a general guide in setting up an IDA method. Optimization for individual systems and sample types may be required for optimal results. For PTM and low abundant peptide analysis the accumulation times may be adjusted to allow for increased signal in both the MS1 and MS2 scans.
9. The number of variable windows chosen should be considered carefully as the more windows selected the shorter the dwell time will have to be for each window. For general purposes 100 VW and a 30 ms dwell time should be sufficient to yield good quantitation of peptides.
10. If accumulation times less than 30 ms are desired, it is recommended that they be tested prior to large-scale sample analysis to ensure the accumulation time chosen will give adequate signal for quantitation.
11. If using the 5600 TripleTOF system, the minimum accumulation time for the MS1 should be set to 150 ms to ensure the MS1 quality is sufficient to perform the background calibrations during the run. The 6600 TripleTOF system does not use this background calibration so a shorter MS1 accumulation time (50 ms) may be used to get a quick survey scan.
12. The LC and autosampler methods will vary between labs and the gradient lengths will vary depending on the complexity of the samples. Typically, for complex mixtures a gradient of 5–35 % B over 90–120 min is suitable and for less complex samples (i.e., immunoprecipitations, purified proteins) shorter gradients between 30 and 60 min may be sufficient.
13. iRT FASTA sequence is available at www.biognosys.com, or type the following into your FASTA file:
i. >Biognosys iRT Kit Fusion
A G G S S E P V T G L A D K V E A T F G V D E SANKYILAGVESNKDAVTPADFSEWSKFLLQFGAQGSPLFKLGGNETQVRTPVISGPYYERTPVITGAPYYERGDLDAASYYAPVRTGFIIDPGGVIRGTFIIDPAAIVR
14. FDR threshold can be set higher or lower depending on the user preference; the higher the FDR is set the more proteins will be incorporated into the library but the confidence of these proteins will not be as high as if a lower FDR threshold is used.
15. These parameters are meant as a guideline and can be adjusted based on user preferences. Refer to the Sciex PeakView software documentation and the literature regarding optimizing these settings for your particular experiment. Importantly, for PTM analysis, un-check the Exclude Modified Peptides box and increase the number of peptides per protein to a larger value (i.e., 100) to import all peptides identified at the confidence level selected or create a PTM enriched peptide library.
References
- 1.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods 1(1):39–45. doi: 10.1038/nmeth705 [DOI] [PubMed] [Google Scholar]
- 2.Dong MQ, Venable JD, Au N, Xu T, Park SK, Cociorva D, Johnson JR, Dillin A, Yates JR 3rd (2007) Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 317(5838):660–663. doi: 10.1126/science.1139952 [DOI] [PubMed] [Google Scholar]
- 3.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11(6):O111.016717. doi: 10.1074/mcp.O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, Aebersold R (2014) OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol 32(3):219–223. doi: 10.1038/nbt.2841 [DOI] [PubMed] [Google Scholar]
- 5.Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, MacLean B, Aebersold R (2015) Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc 10(3):426–441. doi: 10.1038/nprot.2015.015 [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Perez-Santiago J, Katz JE, Mallick P, Bandeira N (2010) Peptide identification from mixture tandem mass spectra. Mol Cell Proteomics 9(7):1476–1485. doi: 10.1074/mcp.M000136-MCP201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker SJ, Rost H, Rosenberger G, Collins BC, Malmström L, Amodei D, Venkatraman V, Raedschelders K, Van Eyk JE, Aebersold R. Mol Cell Proteomics. 2015. Oct;14(10):2800–13. doi: 10.1074/mcp.O114.042267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bereman MS (2015) Tools for monitoring system suitability in LC MS/MS centric proteomic experiments. Proteomics 15(5–6):891–902. doi: 10.1002/pmic.201400373 [DOI] [PubMed] [Google Scholar]
- 9.Bereman MS, Johnson R, Bollinger J, Boss Y, Shulman N, MacLean B, Hoofnagle AN, MacCoss MJ (2014) Implementation of statistical process control for proteomic experiments via LC MS/MS. J Am Soc Mass Spectrom 25(4):581–587. doi: 10.1007/s13361-013-0824-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI (2015) DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods 12(3):258–264. doi: 10.1038/nmeth.3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ting S, Egertson J, MacLean B, Kim S, Payne S, Noble W, MacCoss MJ (2014) Pecan: Peptide Identification Directly from Data-Independent Acquisition (DIA) MS/MS Data. American Society for Mass Spectrometry, Baltimore, MD [Google Scholar]
- 12.Toprak UH, Gillet LC, Maiolica A, Navarro P, Leitner A, Aebersold R (2014) Conserved peptide fragmentation as a benchmarking tool for mass spectrometers and a discriminating feature for targeted proteomics. Mol Cell Proteomics 13(8):2056–2071. doi: 10.1074/mcp.O113.036475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirk JA, Holewinski RJ, Kooij V, Agnetti G, Tunin RS, Witayavanitkul N, de Tombe PP, Gao WD, Van Eyk J, Kass DA (2014) Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3beta. J Clin Invest 124(1):129–138. doi: 10.1172/JCI69253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12(8):1111–1121. doi: 10.1002/pmic.201100463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, Moore RJ, Klemke RL, Camp DG 2nd, Smith RD (2011) Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11(10):2019–2026. doi: 10.1002/pmic.201000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han G, Ye M, Zhou H, Jiang X, Feng S, Jiang X, Tian R, Wan D, Zou H, Gu J (2008) Large-scale phosphoproteome analysis of human liver tissue by enrichment and fractionation of phosphopeptides with strong anion exchange chromatography. Proteomics 8(7):1346–1361. doi: 10.1002/pmic.200700884 [DOI] [PubMed] [Google Scholar]
- 17.Dephoure N, Gygi SP (2011) A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods 54(4):379–386. doi: 10.1016/j.ymeth.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26(7):966–968. doi: 10.1093/bioinformatics/btq054 [DOI] [PMC free article] [PubMed] [Google Scholar]