

Abstract
Drosophila oogenesis is a powerful and tractable model for studies of cell and developmental biology due to the multitude of well-characterized events in both germline and somatic cells, the ease of genetic manipulation in fruit flies, and the large number of egg chambers produced by each fly. Recent improvements in live imaging and ex vivo culturing protocols have enabled researchers to conduct more detailed, longer-term studies of egg chamber development, enabling insights into fundamental biological processes. Here, we present a protocol for dissection, culturing, and imaging of late-stage egg chambers to study intercellular and directional cytoplasmic flow during ‘nurse cell dumping’. This critical developmental process towards the latter stages of oogenesis (stages 10b/11) results in rapid growth of the oocyte and shrinkage of the nurse cells and is accompanied by dynamic changes in cell shape. We also describe a procedure to record high time-resolution movies of flow of unlabeled cytoplasmic contents within nurse cells and through cytoplasmic bridges in the nurse cell cluster using reflection microscopy, and we describe two ways to analyze data from nurse cell dumping.
Keywords: Drosophila egg chamber, Oogenesis, Intercellular flow, Live Imaging, Nurse cell dumping, Reflection microscopy
1. Introduction
Oogenesis in the fruit fly Drosophila melanogaster is a process that begins in the germarium, at the anterior tip of the ovary. Germline and somatic stem cells give rise to cystoblasts and follicle cells, respectively, after which each cystoblast undergoes four rounds of incomplete cell division to produce the interconnected germline cyst. The 16-cell germline cyst is then encapsulated by somatic cells before pinching off from the germarium; at this point, the collection of germline and somatic cells is known as an ‘egg chamber’. Drosophila oocytes develop and increase dramatically in volume while remaining connected to the fifteen supportive ‘nurse cells’ in a stereotyped manner through cytoplasmic bridges called ring canals. During the roughly 96-hour course of oogenesis following encapsulation, an egg chamber progresses through 14 stages of development while moving towards the posterior of the ovary (representative images of a germarium and egg chambers in stages 2–14 are shown in Fig. 1), finally becoming a mature egg and entering the oviduct [4,5]. Oogenesis in the fruit fly is highly parallelized: egg chambers develop in strings known as ovarioles, with germaria at their anterior tip and mature eggs at the posterior, and 15–20 ovarioles are bound together to form one ovary.
Figure 1: The 14 stages of Drosophila melanogaster oogenesis.
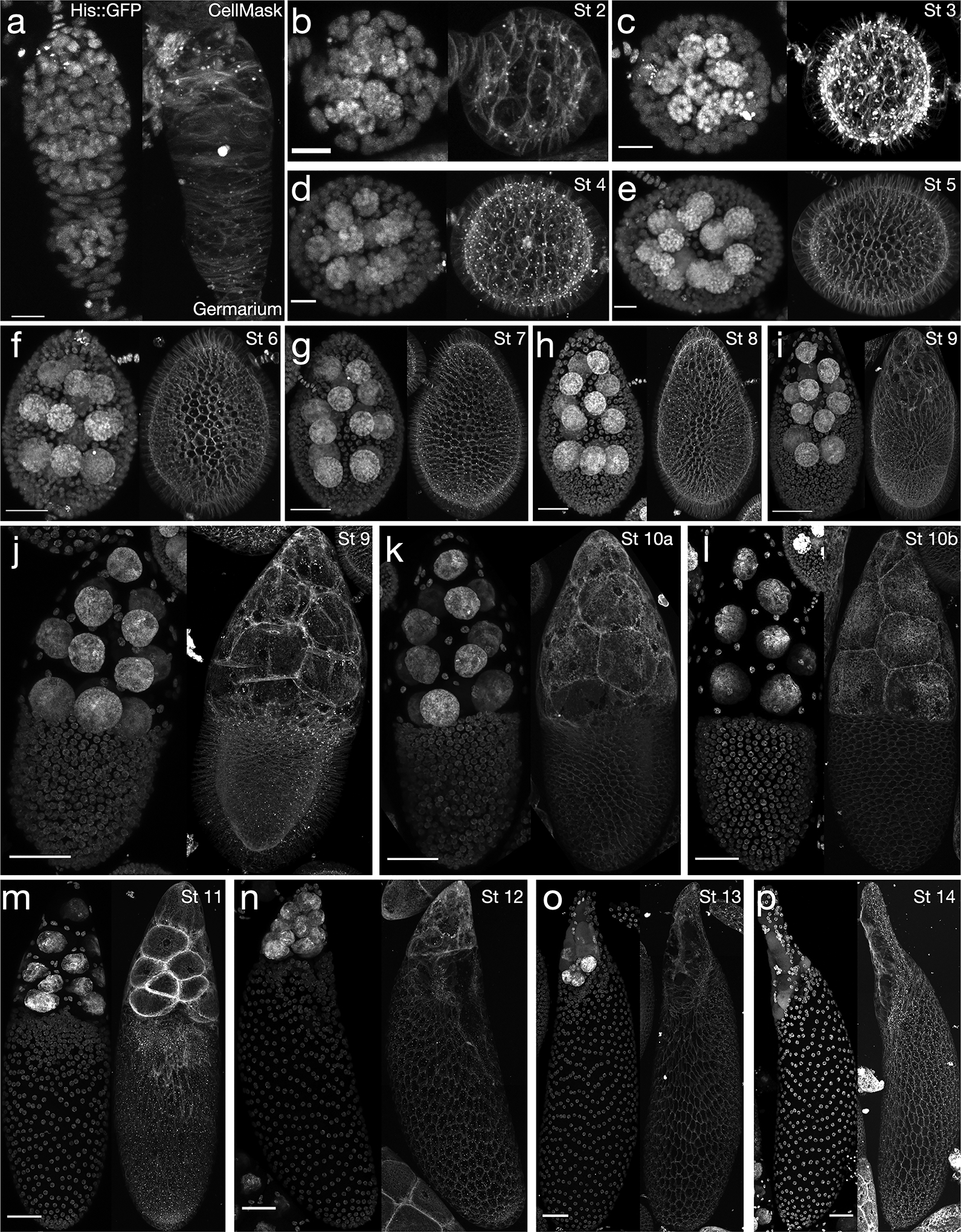
Histone H2A::GFP (left) and CellMask signal (right) for egg chambers from youngest to oldest, with stage (St) for each. A. germarium; B. stage 2; C. stage 3; D. stage 4; E. stage 5; F. stage 6; G. stage 7; H. stage 8; I. early stage 9, just prior to border cell migration; J. later in stage 9, when the border cells are partway to the oocyte; K. stage 10a; L. stage 10b; M. stage 11, relatively early into the nurse cell dumping process; N. stage 12, roughly at the end of nurse cell dumping; O. stage 13; P. stage 14. Note: inhomogeneity in follicle cell layer CellMask signal in M,N is partly a result of uneven staining where the egg chamber was in contact with the glass dish (slight spatial discontinuities in M-P result from imperfect merging of tile scans). Scale bars: 10 μm (A-E), 20 μm (F), 30 μm (G,H), 50 μm (I-P). Stages are determined from a combination of egg chamber dimensions, nuclear size, and other morphological considerations detailed previously [4,23]. Anterior is at the top in all panels except B-E, in which anterior is to the left. Other objects in the corners of images are portions of nearby egg chambers that are partly in the field of view.
Because of the reproducibility of egg chamber structure, the high number of chambers per fly, the abundance of genetic tools available for Drosophila, and the amenability of egg chambers to ex vivo live imaging, studies of fruit fly oogenesis have provided insight into processes including stem cell regulation, tissue-level shape changes, cell migration, and three-dimensional morphogenesis in epithelia [6–9]. One behavior for which Drosophila oogenesis provides an exciting model is cytoplasmic transport, both within cells, as with cytoplasmic streaming in the oocyte [10,11], and between cells, as has also been observed during mouse oocyte development [12]. Around stages 11 and 12 of oogenesis, nurse cells transfer upwards of 75 percent of their cytoplasmic contents through ring canals into the oocyte over approximately 90 minutes, accompanied by actomyosin-driven cell shape changes. This process, known as ‘nurse cell dumping’, results in rapid growth of the oocyte as it receives the materials necessary to eventually support early embryonic development [13–15]. Fluid transport is largely unidirectional, proceeding towards the oocyte at the posterior of the chamber, and it is hierarchical, with fluid from more anterior nurse cells passing through up to three more posterior nurse cells before entering the oocyte. Nurse cell dumping thus presents an opportunity to study the interplay of cytoplasmic pressure, contractility, and cell geometry during fluid transport within a multicellular network.
In recent years, improvements in imaging modalities and ex vivo culturing techniques have opened the door to longer-term, more quantitative studies of developmental processes during oogenesis [6,9,14,16]. Several excellent protocols for live imaging of egg chambers have been previously published, using various designs for the imaging apparatus and focusing on different stages or developmental processes [16,17–19]. Here, we present a relatively simple protocol for long-term (~2–4 hours) live imaging of late-stage egg chambers (stages 10–14), originally developed for capturing the dynamics of cytoplasmic flow during nurse cell dumping in stage 11/12 chambers [14] but readily adaptable to younger egg chambers. Additionally, we describe the use of confocal reflection microscopy [20,21] to image intra- and intercellular flow of unlabeled cellular contents. Finally, we provide a brief description of two possible types of analysis that can be performed on data collected using this protocol.
2. Materials
2.1. Materials for dissection of individual follicles
Fly stocks (exact stocks used will depend on the goals of the experiment).
Fly vials with food.
Dry yeast.
- Schneider’s insect medium (see Notes 1 and 2).
- Fetal bovine serum (FBS), added to Schneider medium to a final concentration of 15% (vol/vol) (optional for stage 10+ egg chambers, but necessary for stage 9 or younger; see Note 3).
- Insulin, added to Schneider medium to a final concentration of 250 μg/mL (optional for stage 10+ egg chambers, but necessary for stage 9 or younger).
1.5 mL microcentrifuge tubes, to hold dissection medium.
100 μL pipette with tips cut off about ¼ inch from the end, to avoid shearing egg chambers during transfer.
CO2 source and diffusing pad.
Fine brush and/or feather tool for sorting flies.
Straight forceps (110 mm length × 8 mm width, tip diameter ~0.1 mm).
Stereomicroscope with black stage and adjustable light source.
Glass dissecting slide with concave wells.
Fine tungsten needles (tip diameter 0.001 mm).
Metal needle holders.
35mm, no. 1.5 glass-bottomed dish.
Kimwipes.
70% isopropanol, for cleaning forceps and the dissecting slide.
2.2. Imaging
(Optional) Membrane stain (e.g., CellMask Deep Red Plasma membrane stain, Invitrogen).
Laser-scanning confocal (or similar) microscope able to perform both fluorescence and reflection microscopy (see Note 4).
2.3. Image analysis
Computer with image analysis software such as FIJI [22] installed. More specialized software intended for 3D/4D viewing and volume measurements, such as Imaris (Bitplane), is also useful.
3. Methods
Carry out all procedures at room temperature unless otherwise specified.
3.1. Dissection of individual follicles
The day before dissection, transfer females 3–5 days post-eclosion, along with a roughly equal number of males of any age, to a new vial with a thin layer of dry yeast added. Flies fed with yeast will produce more eggs, making it be easier to remove ovaries (see Note 5).
Transfer about 500 μL of medium to an Eppendorf tube. Allow this tube to come to room temperature.
Anesthetize flies on the CO2 pad. Separate out 2–5 females to dissect and return the remaining flies to the vial.
Place a dissecting slide on the stage of the stereomicroscope. Add 80–100 μL of medium to one well of the slide.
Using one pair of forceps, pick up a fly by its legs, transferring it to the filled well of the glass slide, wings down (see Note 6).
Grasp the fly around the anterior end of the abdomen using the forceps in the non-dominant hand and apply gentle pressure to distend the abdomen slightly. With the pair of forceps in the dominant hand, pinch the abdomen near the ovipositor and pull directly away from the first pair of forceps. The ovaries will usually separate from the fly (Fig. 2A) (see Notes 7–9).
Immediately remove the fly carcass and crush it using a Kimwipe.
If necessary, use the tungsten needles or forceps to separate the two ovaries by severing the oviduct connecting them.
Either pin one ovary down with gentle pressure from a tungsten needle or grasp the ovary carefully with one pair of forceps (see Notes 10 and 11).
While keeping the ovary immobilized with one hand, use a tungsten needle to slide between ovarioles from the posterior to the anterior tip, disrupting the peritoneal muscle sheath to separate ovarioles (Fig. 2B). Ideally, the ovarioles will splay out, allowing easy access to the egg chambers (see Note 12).
- Identify stage 10b-11 egg chambers, shortly before or during nurse cell dumping (Fig. 2C). For a more detailed discussion of stage determination, see [23].
- These are typically near the posterior end of ovarioles, often just anterior to fully mature eggs.
- At stage 10b, the oocyte is ~50% the length of the germline cluster. If it is slightly larger but the nurse cells are still individually visible, dumping has begun relatively recently (Fig. 2D).
Stage 10–12 egg chambers will often be ejected from the epithelial muscle sheath surrounding the ovariole due to contractions of the sheath (see Note 13). If they are not, use both needles in a scissoring motion to cut through the stalk cells between egg chambers and remove egg chambers from either end of the desired one. Ensure there is no remaining epithelial muscle sheath around the egg chamber of interest, or any egg chamber still connected to it, as its contractions will cause the egg chamber to move sporadically while imaging.
Using the side of one needle, move all egg chambers to be imaged to one side of the well and all remaining tissue as far away from them as possible.
Pipette 100–200 μL of medium into the well of the glass-bottomed dish.
Place the cut-off tip of a pipette near the group of stage 10b-11 chambers, on the side away from the remaining, unwanted chambers. Gently pipette up the egg chambers, avoiding the other chambers, and transfer them to a 35mm, no. 1.5 glass-bottomed dish (see Note 14). The total volume of medium in the dish should be at least 200 μL now; if not, add more. Having excess medium is better than having too little.
Clean the dissecting slide and forceps with 70% isopropanol and dry with a Kimwipe.
Figure 2: Ovary dissection and identification of egg chambers undergoing nurse cell dumping.
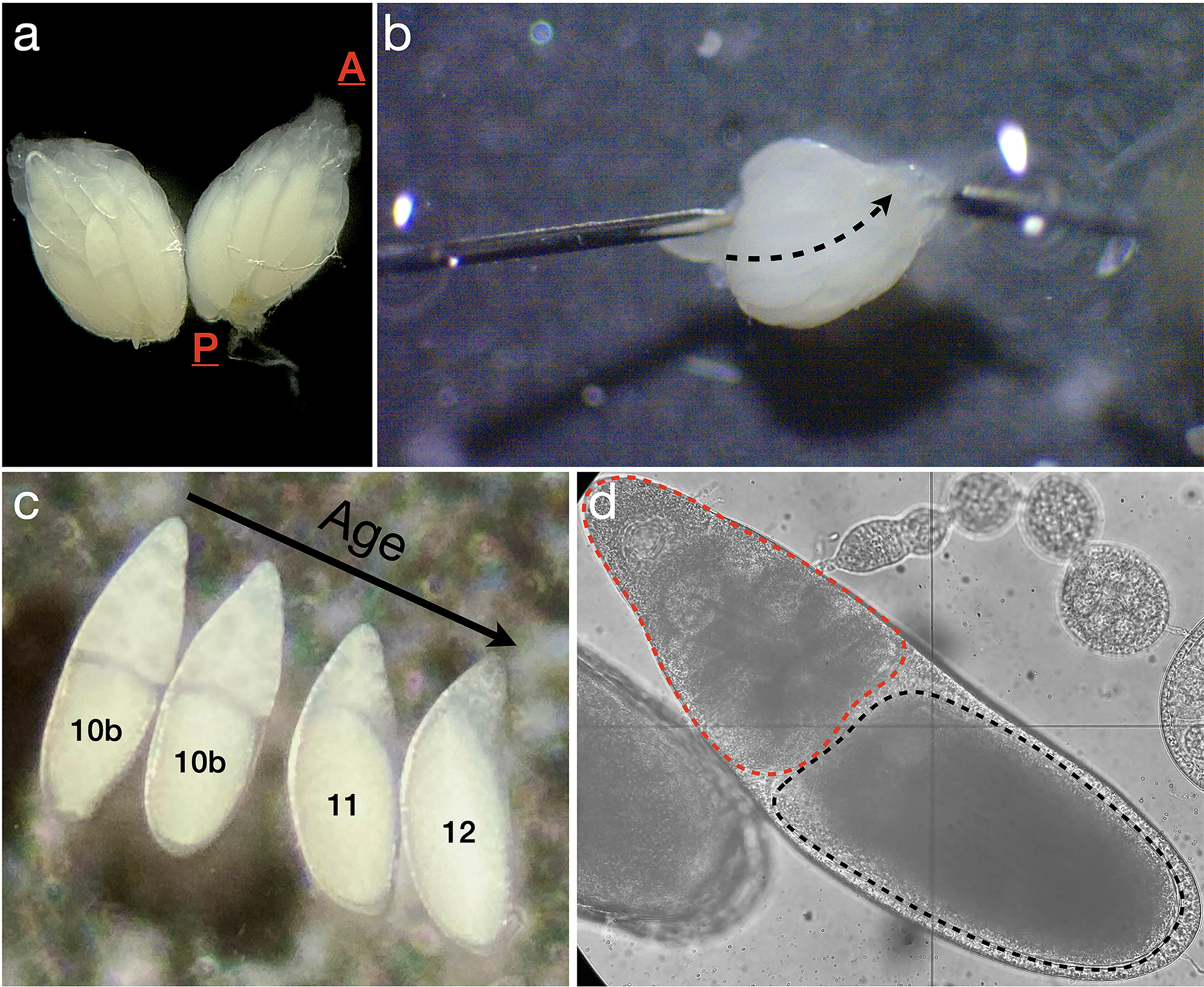
A. One pair of ovaries removed from a fruit fly; A is the anterior end, where the germaria are located, and P is the posterior end, where mature stage 14 eggs reside. B. Disruption of the peritoneal muscle sheath. The needle on the left is used to immobilize the ovary, while the needle on the right is moved gently along the dashed arrow, between ovarioles, to remove the muscle sheath holding the ovarioles together. C. Four egg chambers in stages 10b-12, with the youngest (stage 10b) just beginning the nurse cell dumping process at left, and the oldest (stage 12) shortly after dumping is completed at right. D. Brightfield image of an egg chamber approximately 10–20 minutes into the dumping process with the oocyte (black dashed outline) and the nurse cell cluster (red dashed outline) highlighted.
3.2. Imaging
(Optional) If desired, add CellMask to the dish to a final concentration of 5 μg/mL, or use a different membrane dye (see Note 15). Swirl the dish gently to mix and allow egg chambers to incubate in CellMask 5–10 minutes before imaging (see Note 16).
Place the dish, with its lid on to prevent drying of the medium and sample, on an inverted microscope.
For fluorescence microscopy, image z-stacks as usual.
- To adapt a fluorescence microscopy setup to a reflection microscopy setup (see Notes 17 and 18):
- Replace the dichroic beamsplitter normally used in the beam path for fluorescence microscopy with a partial mirror. This will allow some incident light to reach the sample and some reflected light of the same wavelength to reach the detector (see Note 19).
- Set the detection wavelength equal to the excitation wavelength (see Note 20).
Set up time-lapse z-stack imaging. Egg chambers can be cultured and imaged for ~2–4 hours with no noticeable ill effects depending on intensity and frequency of irradiation (see Notes 21 and 22).
When finished imaging, dispose of the glass-bottomed dish in the sharps disposal.
3.3. Image analysis
This section describes two examples of the type of analysis possible with images acquired using the protocol above. The first involves determining nurse cell size trajectories from fluorescence images (see Fig. 3A), and the second describes more rapid cytoplasmic flow behaviors observed using reflection microscopy [14].
Figure 3: Analysis of cell volume changes and reflection microscopy images of intracellular flow during nurse cell dumping.
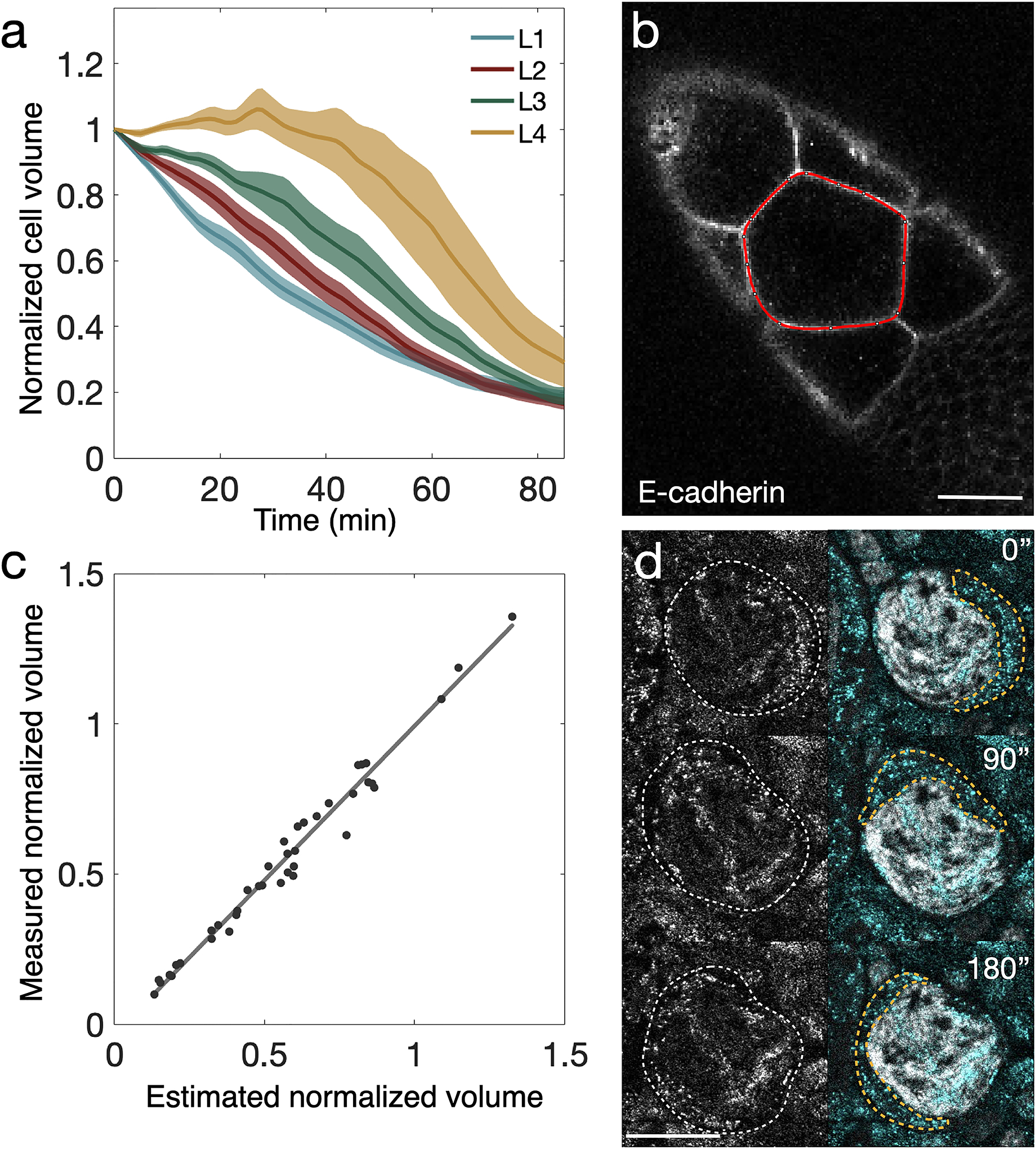
A. Averaged, normalized cell volume trajectories from 41 nurse cells. t = 0 is the onset of nurse cell dumping. Lines show averages, envelopes standard error; colors correspond to rows of cells at different distances from the oocyte. Layer 1 (L1) cells are connected directly to the oocyte, while each successive layer is one ring canal farther away from the oocyte. (n = 15, 12, 9, and 5 cells for layers 1, 2, 3, and 4, respectively). B. Example of one midplane outline (red) in an E-cadherin::GFP-expressing egg chamber shortly after dumping onset. Scale bar: 40 μm. C. Plot showing correlation between normalized volume measured from Bitplane’s Imaris and normalized volume estimated from midplane area measurements in FIJI. Solid line shows best fit (R2 = 0.98, with slope 1.03). D. Left column: reflection microscopy signal from a histone H2A::GFP-expressing egg chamber. Right column: merge of the same reflection signal (cyan) and H2A signal (gray), showing a pocket of cytoplasm (orange outline) flowing counterclockwise around the nucleus. Scale bar: 20 μm. A-C reproduced from [14].
3.3.1. Size trajectories
Open a 4D (x, y, z, t) movie in FIJI, preferably in which at least one channel includes a cortical or cell body marker. If the file size is larger than the memory available to FIJI, use the Bio-Formats importer to open the movie as a virtual stack instead.
Determine the approximate frame around which dumping begins. This is most easily accomplished by observing the boundary between the oocyte anterior and the posterior side of the nurse cell cluster, as the frame of dumping onset corresponds to the sudden increase in the rate at which this boundary moves towards the anterior (see Note 23).
For each nurse cell of interest, at each time point to use for analysis, step through the z-dimension to locate the plane with maximal cross-sectional area for that nurse cell (see Notes 24 and 25).
Use the selection tool to trace the cell membrane (Fig. 3B), then measure the cell’s cross-sectional area. Assuming the cells are roughly spherical, volume can be approximated by (see Note 26 and Fig. 3C regarding validation of this estimate using the full 3D data).
Repeat for each cell and timepoint to use for analysis. Ensure that one time point includes the initial frame corresponding to dumping onset (t = 0).
For each nurse cell, normalize the size measurements to the size at dumping onset (thus the normalized volume at time t is ), where is the measured cross-sectional area at time t and is the cross-sectional area at dumping onset.
Size trajectories can now be plotted for each cell if desired, showing the fractional change in volume over time (see Note 27).
3.3.2. Cytoplasmic flow behaviors
Open a reflection-mode microscopy movie in FIJI. Cell boundaries should be discernible from the reflection signal, as should a darker region in the cell interior corresponding to the nucleus (Fig. 3D).
In wild-type egg chambers, during the first ~45–60 minutes of dumping, flow of bright spots through ring canals should be visible, manifesting as bright puncta moving in a straight line between cells.
Once cell shape contractions begin, large-scale cytoplasmic movements can be tracked by searching for coordinated movement of large groups of puncta.
Flow of reflective cytoplasmic contents is in theory amenable to measurement via methods such as particle image velocimetry (PIV, see Note 28).
4. Notes
Egg chambers stage 10 or older can be cultured in Schneider’s medium without supplementation, but younger egg chambers require Schneider’s supplemented with insulin and fetal bovine serum.
Store Schneider’s medium and insulin at 4°C and FBS in aliquots at −20°C. If supplementing with insulin and FBS, add Schneider’s medium and insulin to the FBS aliquot the day of the experiment and keep at 4°C when not in use.
Adding FBS to the dissection medium can also reduce the chance of egg chambers sticking to the pipette tip or to the glass dish during transfer.
Many commercial systems can perform both reflection and fluorescence microscopy. For those that cannot, the dichroic beam-splitter used for fluorescence microscopy must be replaced by a mirror.
For egg chamber studies, flies are kept at room temperature (22–23°C). Higher temperatures can also be used, e.g., to increase efficiency of GAL4/UAS-driven RNA interference [24]. In that case, flies are allowed to eclose and are then transferred to the desired temperature to develop for at least 4 days.
Surface tension in the pool of dissecting medium in the well can pull the fly to the side of the pool and occasionally turn it on its side. Step 6 can still be performed in this case, or the fly can be turned using the other pair of forceps and held in place.
If the ovaries remain inside the abdomen after pulling, use one pair of forceps to make a slit in the ventral side of the abdomen from the anterior end of the ovaries to the tear made in Step 6. Carefully use the forceps to then remove the ovaries. This may require using the forceps to pull away connective tissue just anterior to the tip of the ovary.
Other tissues, especially part of the posterior cuticle and intestines, usually come with the ovaries when pulling with the forceps. In this case, use the tungsten needle probes to separate the ovaries from all remaining tissue, then remove everything else from the dissecting medium using forceps.
Avoid rupturing the fat body or any part of the intestinal tract when performing Step 6. Doing so will fill the dissection medium pool with debris; in this case, it is often best to start over with fresh medium in another well on the slide to avoid any possible effects of the released contents on the remaining egg chambers.
To avoid damaging stage 10/11 egg chambers, grasp or pin down the ovary as close to its posterior end as possible. If flies are well-fed, there should be several mature eggs posterior to the stage 10/11 egg chambers, which will allow pinning the ovary down without damaging the egg chambers of interest.
If at any point an egg chamber is ruptured, move it as far away from the remaining ones as possible to prevent its contents from leaking and surrounding the intact chambers, which can potentially affect the development of the intact chambers.
If the method in Steps 9 and 10 is difficult or if the egg chambers of interest are too close to the posterior end, anchor the ovary by inserting one needle through the egg chambers at the anterior end. Slide the other probe from anterior to posterior to disrupt the peritoneal sheath, or place both probes at the anterior end and pull them apart in the direction perpendicular to the anterior-posterior axis.
Occasionally an egg chamber will slide only partly out of the epithelial muscle sheath and the sheath will contract around the middle of the egg chamber, deforming it for a prolonged period. To be safe, avoid using these egg chambers as this contraction can cause subtle damage that leads to a failure of nurse cell dumping.
Avoid adding too many stage 10b or later egg chambers to the medium in the dish. Doing so can deplete nutrients from the medium too quickly and reduce the time egg chambers remain healthy in the culture medium, and it will also crowd the dish and cause chambers to contact one another.
A dye such as CellMask can be used to assess integrity of egg chambers and to non-specifically label membranes. If a cell is damaged such that membrane integrity is compromised, the cell interior will appear bright, in contrast to the usual pattern of a dark interior with a few bright puncta and a bright membrane.
CellMask, if added directly to the imaging medium, will continue to bind the cell membranes, which will increase in brightness over time. Alternatively, the egg chambers can be incubated in medium supplemented with CellMask before being washed and imaged in fresh medium without CellMask.
Reflection microscopy can be used to image flow of unlabeled cytoplasmic contents through ring canals. To do so, adjust the focal plane until it bisects a ring canal, visible as a small gap between cells through which bright puncta move. Set the acquisition time to ~0.5–2 seconds per stack, and flow should be visible when playing the acquired movie. A fluorescent marker for membranes or ring canals can be used to locate ring canals before switching to reflection mode.
Depending on the software, it is possible to perform both reflection and fluorescence microscopy in the same movie acquisition. However, doing so requires switching between a beam-splitter and a mirror during acquisition of each z-stack, which in our experience greatly decreases temporal resolution.
We use the Zen Black software, which has a preset box that can be checked to switch to reflection mode.
Reflection microscopy is not wavelength-specific. In our experience, 488 nm excitation works well, as it provides slightly better resolution than longer wavelengths, and shorter wavelengths often cause tissue damage more rapidly.
Because the egg chamber is approximately 150–175 μm in width at this point in oogenesis, imaging through the entire tissue is not generally possible at laser powers that are safe for long-term imaging of the egg chamber.
- Reflection microscopy is the simplest modality for imaging flow in this stage. Some fluorescently-labeled proteins useful for imaging the changes in cell size and shape accompanying cytoplasmic flow include:
- Clip170::GFP [27], a microtubule-binding protein, which in these tissues labels cell bodies fairly uniformly when imaged at low laser power. This label can also be used to image cytoplasmic streaming in the oocyte in stages 10 and 11.
- Pavarotti::GFP [28], a kinesin-like-protein. This label localizes to ring canals, although they only appear clearly in the latter part of dumping.
- PCNA::GFP [29], which changes location from nucleus to cytoplasm during nurse cell dumping shortly before the onset of cell-shape contractions.
If it is difficult to identify the onset of nurse cell dumping by eye, an alternative approach involves a kymograph. Using a marker with a clear distinction between the oocyte and nurse cell cluster, create a kymograph showing intensity along the anterior-posterior axis of the egg chamber. There should be a clear timepoint in the kymograph at which the feature corresponding to the posterior edge of the nurse cells abruptly begins moving quickly; this is the time at which dumping begins.
If there is not a slice with a local maximum of cross-sectional area for a cell, then the cell’s midplane is out of the field of view. Such a cell can no longer be analyzed using this method from this time point onward.
Because the nurse cells are quite large and there is a physical limit on imaging depth, most cells will have at least a portion of their volume outside the field of view. For this reason, volume measurements are often not possible.
Volume measurements have been performed using Bitplane’s Imaris to validate the use of cross-sectional area in volume estimates (Fig. 3C) [14]. Overall, changes in normalized volume measured directly correlated well with changes in normalized, estimated volume from midplane measurements; however, cells must be roughly spherical and not change shape significantly for the correlation to hold.
Nurse cells show a size gradient along the anterior-posterior axis, with cells nearest the oocyte being largest [30]. Because of this, normalizing to the initial volume allows comparisons between different nurse cells. For applications in which knowledge of the absolute cell size is important, the normalization step can be skipped.
There are several free software packages available for performing PIV analysis. Two such examples are PIVlab (for MATLAB) [31] and a PIV plugin for ImageJ [32].
Acknowledgments
This work was supported by NIH grant R01GM125646 to A.C.M. We would like to thank members of the Martin lab for helpful discussions regarding this project.
References
- 1.Bastock R, St. Johnston D (2008) Drosophila oogenesis. Curr. Biol 18(23):1082–1087 [DOI] [PubMed] [Google Scholar]
- 2.López-Schier H, St. Johnston D. (2001) Delta signaling from the germ line controls the proliferation and differentiation of the somatic follicle cells during Drosophila oogenesis. Genes Dev. 15(11):1393–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godt D, Tepass U (1998) Drosophila oocyte localization is mediated by differential cadherin-based adhesion. Nature 395:387–391 [DOI] [PubMed] [Google Scholar]
- 4.Spradling AC (1993) The developmental genetics of oogenesis. Cold Spring Harbor Laboratory Press; Plainview, N.Y. pp 3–6 [Google Scholar]
- 5.McLaughlin JM, Bratu D (2015) Drosophila melanogaster Oogenesis: An Overview. Methods Mol. Biol 1328:1–20 [DOI] [PubMed] [Google Scholar]
- 6.Morris LX, Spradling AC (2011) Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development 138(11):2207–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haigo SL, Bilder D (2011) Global Tissue Revolutions in a Morphogenetic Movement Controlling Elongation. Science 331:1071–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai D, Chen S-C, Prasad M, et al. (2014) Mechanical Feedback through E-Cadherin Promotes Direction Sensing during Collective Cell Migration. Cell 157:1146–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osterfield M, Du X, Schüpbach T et al. (2013) Three-dimensional epithelial morphogenesis in the developing Drosophila egg. Dev. Cell 24(4):400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganguly G, Williams LS, Palacios IM, Goldstein RE (2012) Cytoplasmic streaming in Drosophila oocytes varies with kinesin activity and correlates with the microtubule cytoskeleton architecture. Proc. Nat. Acad. Sci 109(38):15109–15114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theurkauf WE (1994) Premature microtubule-dependent cytoplasmic streaming in cappuccino and spire mutant oocytes. Science 265(5181):2093–2096 [DOI] [PubMed] [Google Scholar]
- 12.Lei L, Spradling AC (2016) Mouse oocytes differentiate through organelle enrichment from sister cyst germ cells. Science 352(6281):95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutzeit HO, Koppa R (1982) Time-lapse film analysis of cytoplasmic streaming during late oogenesis of Drosophila. J. Embryol. Exp. Morph 67:101–111 [Google Scholar]
- 14.Imran Alsous J, Romeo N, Jackson JA, et al. (2021) Dynamics of hydraulic and contractile wave-mediated fluid transport during Drosophila oogenesis. Proc. Nat. Acad. Sci 118(10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahajan-Miklos S, Cooley L (1994) Intercellular Cytoplasm Transport during Drosophila Oogenesis. Dev. Biol 165(2):336–351 [DOI] [PubMed] [Google Scholar]
- 16.Peters NC, Berg C (2016) In Vitro Culturing and Live Imaging of Drosophila Egg Chambers: A History and Adaptable Method. Methods Mol. Biol 1457:35–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prasad M, Jang AC-C, Starz-Gaiano M, et al. (2007) A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nat. Protocols 2(10):2467–2473 [DOI] [PubMed] [Google Scholar]
- 18.Wilcockson SG, Ashe HL (2021) Live imaging of the Drosophila ovarian germline stem cell niche. STAR Protocols 2(1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cetera M, Lewellyn L, Horne-Badovinac S (2016) Cultivation and Live Imaging of Drosophila Ovaries. Methods Mol. Biol 1478:215–226 [DOI] [PubMed] [Google Scholar]
- 20.Gáspár I, Szabad J (2009) In vivo analysis of MT-based vesicle transport by confocal reflection microscopy. Cell Motil. Cytoskeleton 66(2):68–79 [DOI] [PubMed] [Google Scholar]
- 21.Guggenheim EJ, Lynch I, Rappoport JZ (2017) Imaging In focus: Reflected light imaging: Techniques and applications. Int. J. Biochem. Cell Biol 83:65–70 [DOI] [PubMed] [Google Scholar]
- 22.Schindelin J, Arganda-Carreras I, Frise E, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9(7):676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia D, Xu Q, Xie Q, et al. (2016) Automatic stage identification of Drosophila egg chamber based on DAPI images. Sci. Reports 6:18850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duffy JB (2002) GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34(1–2):1–15 [DOI] [PubMed] [Google Scholar]
- 25.Martin AC, Gelbart M, Fernandez-Gonzalez R, Kaschube M, Wieschaus EF (2010) Integration of contractile forces during tissue invagination. J. Cell Biol 188(5):735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oda H, Tsukita S (2001) Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J. Cell Sci 114:493–501 [DOI] [PubMed] [Google Scholar]
- 27.Beaven R, Dzhindzhev NS, Qu Y, et al. (2015) Drosophila CLIP-190 and mammalian CLIP-170 display reduced microtubule plus end association in the nervous system. Mol. Biol. Cell 26(8):1491–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minestrini G, Máthé E, Glover DM (2002) Domains of the Pavarotti kinesin-like protein that direct its subcellular distribution: effects of mislocalisation on the tubulin and actin cytoskeleton during Drosophila oogenesis. J. Cell Sci 115:725–736 [DOI] [PubMed] [Google Scholar]
- 29.Kisielewska J, Lu P, Whitaker M (2012) GFP-PCNA as an S-phase marker in embryos during the first and subsequent cell cycles. Biol. Cell 97(3):221–229 [DOI] [PubMed] [Google Scholar]
- 30.Imran Alsous J, Villoutreix P, Berezhkovskii AM, Shvartsman SY (2017) Collective Growth in a Small Cell Network. Curr. Biol 27(17):2670–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thielicke W, Sonntag R (2021) Particle Image Velocimetry for MATLAB: Accuracy and enhanced algorithms in PIVlab. J. Open Res. Softw 9(12) [Google Scholar]
- 32.Tseng Q. https://sites.google.com/site/qingzongtseng/piv. Accessed 25 Feb 2022.