

Abstract
Objective:
Neisseria meningitidis is an encapsulated, diplococcus, kidney bean-shaped bacteria that causes bacterial meningitis. Our study hopes to advance our understanding of disease progression, the spread frequency of the bacteria in people, and the interactions between the bacteria and human body by identifying a functional protein, potentially serving as a target for meningococcal medicine in the future.
Methods:
A hypothetical protein HP (PBJ89160.1) from N. meningitidis was employed in this study for extensive structural and functional characterization. In the predictive functional role of HP, several constitutive bioinformatics approaches are applied, such as prediction of physiological properties, domain and motif family function, secondary and tertiary structure prediction, energy minimization, quality validation, docking, and ADMET analysis. To create the protein’s three-dimensional (3D) structure, a template protein (PDB_ID: 3GXA) is used with 99% sequence identity by homology modeling technique with the HHpred server. To mitigate the pathogenicity associated with the HP function, it was docked with the natural ligand methionine and five other drug compounds like Verapamil, Loperamide, Thioridazine, Chlorpromazine, and Auranofine.
Results:
The protein is predicted to be acidic, soluble and hydrophilic by physicochemical properties analysis. Subcellular localization analysis demonstrated the protein to be periplasmic. The HP has an ATP-binding cassette transporter (also known as ABC transporter) involved in uptake of methionine (MetQ) that creates nutritional virulence in host. Energy minimization, multiple quality assessments, and validation value determination led to the conclusion that the HP model had a workable and acceptable quality. Following ADMET analysis and binding affinity assessments from the docking studies, Loperamide emerged as the most promising therapeutic compound, effectively inhibiting the ATP transporter activity of the HP.
Conclusion:
Comparative genomic analysis revealed that this protein is specific to N. meningitidis and has no homologs in human proteins, thereby identifying it as a potential target for therapeutic intervention.
Keywords: ATP-binding cassette transporter, ABC transporter superfamily, MET-Q, hypothetical protein, nutritional virulence
Introduction
By colonizing upper respiratory mucosal surfaces, the gram-negative facultative pathogenic bacteria Neisseria meningitides of the pseudomonodonta group can cause invasive meningococcal disease. 1 N. meningitidis colonizes the nasopharynx, crossing mucosal host defenses to bind to human epithelial cells. It can multiply rapidly, allowing it to enter the bloodstream and cause sepsis, and cause meningeal, pericardial, and joint infections. 2 Meningitis is caused by bacteria that can proliferate in cerebrospinal fluid due to crossing the blood-brain barrier.
While healthcare professionals have expressed worry about the susceptibility of meningococci to penicillin and other antibiotics employed in managing meningococcal disease, it has been widely recognized that penicillin remains the preferred antibiotic for treating meningococcal infections. 3 Contrarily, there have recently been numerous reports of penicillin-resistant meningococci from places including Spain, 4 Italy, 5 Greece, 6 the United Kingdom, 7 the United States, 8 and Israel.9,10 Among the main bacterial causes of meningitis, N. meningitidis is distinct because it can spread endemic (sporadic) illness as well as epidemic meningitis. Major meningococcal disease outbreaks still occur often in the African meningitis belt, such as in sub-Saharan Africa. 11 In Sub-Saharan Africa, serogroup A N. meningitidis was responsible for the greatest meningococcal epidemic outbreak ever, which resulted in over 300,000 cases and 30,000 fatalities. 12
Since 1980, significant serogroup B epidemics and/or outbreaks of serogroup A or C meningitis have also occurred in Europe, the United States, Canada, China, Nepal, Mongolia, New Zealand, Cuba, Brazil, Chile, Saudi Arabia, and South Africa.13 -16 In November 2020, the World Health Assembly approved the global roadmap to defeat meningitis, which aims to eliminate bacterial meningitis epidemics, reduce vaccine-preventable cases and deaths by 50% and 70%, respectively, decrease disability, and enhance the quality of life. Key to this initiative is the development of novel, cost-effective vaccines such as NmCV-5. 17
Despite the development of vaccine like NmCV-5, the antibiotic resistance attracts the attention of researchers to consider other pharmacological targets to combat meningococcal disease. Researchers are particularly interested in studying hypothetical proteins (HP) because they lack proper functional annotations and are derived from open reading frames without any experimental evidence of translation. N. meningitidis strain M26503 has 2115 encoded proteins, of which 386 are yet to be characterized. These HPs belong to uncharacterized protein families and domains with unknown functions. 18 The recognition and characterization of HP play a crucial role in the selection of targets for drug design. 19 For instance, Naveed et.al (2017) annotated six HPs from human adenovirus as novel drug targets. 20 Therefore, we tried to address a HP (PBJ89160.1) of N. meningitidis using in silico approach to establish it as a new functional protein that is supposed to be a prospective therapeutic target.
Meanwhile, Bacteria often scavenge vital nutrients, such as amino acids, from their hosts to overcome the host’s natural defenses and/or develop mechanisms which target important host biosynthetic pathways. This strategy, known as “nutritional virulence.” 21 We were concerned whether this HP has the capability to induce nutritional virulence or not. Therefore, by performing molecular docking, it is believed that this protein might play a crucial role in nutritional virulence as well as a methionine transporter in the ABC transport mechanism.
Materials and Methods
Sequence retrieval
In this study, we accessed the genome of Neisseria meningitidis CNQ34_02465 from the database of National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). The chosen focus was a hypothetical protein from this N. meningitidis strain having 287 amino acid residues (accession no. PBJ89160.1). In order to facilitate downstream investigations, the protein sequence was acquired in FASTA format. The complete workflow, the bioinformatics tools and databases have been depicted in Figure 1.
Figure 1.

A complete workflow with used database in this study.
Analysis of physicochemical properties
ProtParam tool (http://web.expasy.org/protparam/) 22 of ExPASy was used to examine the HP’s physical and chemical characteristics, such as its molecular weight, extinction coefficients, aliphatic index (AI), isoelectric point (pI), and GRAVY (grand average of hydropathy).
Subcellular localization and solubility prediction
The subcellular localization of the HP was predicted using CELLO (http://cello.life.nctu.edu.tw/), 23 PSLpred (https://webs.iiitd.edu.in/raghava/pslpred/submit.html) 24 and BUSCA (https://busca.biocomp.unibo.it/). 25 To calculate the average hydrophobicity and estimate the solubility of the protein, SOSUI was employed (http://harrier.nagahama-i-bio.ac.jp/sosui/). 26 Transmembrane areas were identified via the SOSUI server. Additionally, the prediction of the protein’s solubility was carried out using the Protein sol server (http://protein-sol.manchester.ac.uk.). 27
Function prediction by domain and motif analysis
Various tools were utilized for domain analysis, including the NCBI Conserved Domain Search Service (CD Search) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) 28 and InterProScan (http://www.ebi.ac.uk/Tools/services/web/toolform.ebi?tool=iprscan5). 29 The conserved domains of a protein sequence are detected through CD Search. RPS-BLAST (Reverse Position Specific BLAST), which compares a query sequence to position-specific score matrices derived from conserved domain alignments in the Conserved Domain Database (CDD), assesses the query sequence against these alignments. The ScanProsite (https://prosite.expasy.org/scanprosite/) 30 service was utilized to scrutinize the motif in the protein sequence.
Pathway analysis
KEGG pathway data sets were retrieved from KEGG server 31 (https://www.genome.jp/). The KEGG pathway database is a comprehensive collection of manually curated pathway maps that depict our understanding of molecular interactions, reactions, and relationship networks across various biological processes. These include metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems, human diseases, and drug development. The KEGG ID for the HP in this study is K02073.
Protein network map
The functional association between two or more proteins that induce a biological effect can be investigated using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database. Accordingly, a STRING 12.0 search was conducted to identify potential functional interaction networks for the HP under study (https://string-db.org/). 32
Protein family and phylogenetic tree analysis
The analysis involved utilizing Protein-BLAST (BLASTp) 33 through NCBI. The search was performed against the non-redundant database using default parameters to locate homologs of HP. This method relies on local protein sequence alignment to identify proteins with comparable functions. To align multiple sequences and construct phylogenetic trees for a limited set of chosen sequences, MEGA 11 version was employed. 34
Secondary structure determination
The self-optimized prediction method with alignment (SOPMA) 35 was employed to predict the secondary structure. The outcomes from PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) 36 and ENDscript (http://endscript.ibcp.fr/ESPript/ENDscript/) 37 were compared and cross-referenced with the results obtained from SOPMA.
Homology modeling
To predict the 3D structure of the hypothetical protein, a comparative analysis was conducted using pairwise comparison of profile hidden Markov models (HMMs) and the HHpred server. Beyond multiple alignments between the query and a set of templates selected from the search results, HHpred has the capability to generate pairwise query-template alignments. The MODELLER software can then use these alignments to generate 3D structural models. 38 A 3D model of the putative protein that displays 99% identity was constructed using the template protein (PDB_ID: 3GXA-C). The structure of the 3D model was visualized using BIOVIA Discovery Studio Visualizer (version 20.1.0.19295). 39 To visualize the detailed protein ligand interaction, another server, PDBsum 40 was used.
Energy minimization of the model structure
The energy minimization of predicted three-dimensional model structure from HHpred server was employed by YASARA force field minimizer. 41 It reduces energy consumption and provides reliable and precise 3D structure of the hypothetical protein.
Active site determination
The web-based tool Computed Atlas of Surface Topography of Protein (CASTp) (http://sts.bioengr.uic.edu/castp) 42 was employed to identify the protein’s active site. CASTp is instrumental in detecting, outlining, and quantifying geometric and topological features crucial for proper protein function, such as surface pockets, interior cavities, and cross channels. Additionally, it facilitates the mapping of functionally annotated residues on protein 3D structures.
Quality assessment
The quality of the HP 3D structure was assessed using PROCHECK, 43 Verify3D (https://servicesn.mbi.ucla.edu/Verify3D/), 44 QMEAN (https://swissmodel.expasy.org/qmean/), 45 programs available on the ExPASy server of the SWISS-MODEL Workspace and ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/). 46 Software from UCSF called Chimera was also used to overlay the hypothetical protein and the template structure and visualize the results. 47 By employing the ProSA-web server, the template and hypothetical proteins Z scores were calculated. 48 To enrich our validation, VoroMQA tool was used. 49
Comparative genomics approach
The proteome datasets of Homo sapiens was searched against the HP using BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) to determine whether this HP, resembles any human proteins. A minimum bit score of 100 and a threshold E-value (expected value) of 0.005 were used to filter the hits.
Molecular docking analysis
AutoDock Vina was used to perform molecular docking to predict probable receptor-substrate interactions. 50 Structural evidence from a previous study indicates that the template protein 3GXA (GNA1946) is an L-methionine binding lipoprotein located in the outer membrane. 51 Thus, methionine is a natural compound that acts as a ligand for the HP. To assess the binding affinity between the ligand and the Hypothetical Protein, as well as a template protein (3GXA) with a 3D structure modeled using HHpred and MODELER, AutoDock Vina was employed. The docking results were subsequently examined using Discovery Studio Visualizer and PyMOL 3.0 softwere. 52 To perform docking analysis, the initial step was to prepare the hypothetical protein and ligand. For protein preparation, ligand and water molecules were removed, and polar hydrogen atoms were added to the HP using AutoDock tool. At the same time, Kollman and Gasteiger charges were differently assigned as partial charge to the receptor and ligand respectively. The protein was then saved in AutoDock assessable format with PDBQT file extension. To prepare the configuration file, a grid box was created by selecting the amino acids that constitute the active site of the protein. However, 0.375 Å spacing was applied and grid box orientation was constructed with −21.808, 60.032 and −30.852 for specifying center of X, Y, and Z coordinates respectively.We selected five additional drug compounds (Verapamil, 53 Loperamide, 54 Thioridazine, 55 Chlorpromazine, 56 and Auranofin 57 ) as putative ligands through literature review due to their potential to inhibit ATP transport activity in multidrug-resistant bacteria. 3D conformations of these compounds with SDF file extension were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). 58 The ligands were then converted to PDB format using Open Babel 59 software and subsequently uploaded to AutoDock tool, where they were converted to PDBQT format. Finally, the prepared compounds were saved as PDBQT file extension and utilized in AutoDock Vina for docking. The docking results were stored accordingly as a log file containing binding affinity score and an output file having different poses of the complex.
Absorption, distribution, metabolism, excretion (ADME) and toxicological properties analysis
Following docking, we assessed the Pharmacokinetic (PK) and ADME properties of our selected compounds using the open-source web-based tool called SwissADME (www.swissadme.ch), 60 which estimate the PK characteristics and drug-likeness of small compounds. Numerous molecular factors were included in this analysis, such as the number of hydrogen bond donors (nHBDs), hydrogen bond acceptors (nHBAs), molecular mass (MM) of the compounds, rule of five violations (nRB), and topological polar surface area (TPSA). Orally administered medications must adhere to drug-likeness properties to be considered pharmaceutically consistent with their bioactivity scores. In addition to ADME properties, the potential toxicity of the compounds to human organs or cells was also evaluated. Considering that perspective, the toxicity level of chosen compounds was assessed using the ProTox-3.0 online server (https://tox.charite.de/protox3/) 61 and the admetSAR web based server. 62
Results
Physiochemical properties and subcellular localization of hypothetical protein
Table 1 summarizes the physiochemical properties of the HP (PBJ89160.1). According to predictions, the protein has 287 amino acids, a molecular weight of 31277.43 Daltons and with a theoretical isoelectric point (pI) of 5.11. Isoelectric point is pH at which the net charge on the protein is zero and at this pH the protein become less soluble, compact and stable that leads to crystallization of protein. The GRAVY index of this HP is −0.272. The negative GRAVY index suggests that the protein is soluble and hydrophilic in nature. It was predicted that the hypothetical protein’s expected instability index was 25.98, indicating that the protein is stable. The protein contains Ala (A) (40, 13.9%), Arg (R) (5, 1.7%), Asn (N) (14, 4.9%), Asp (D) (20, 7.0%), Cys (C) (1, 0.3%), Gln (Q) (8, 2.8%), Glu (E) (21, 7.3%), Gly (G) (17, 5.9%), His (H) (3, 1.0%), Ile (I) (11, 3.8%), Leu (L), (26, 9.1%), Lys (K) (28, 9.8%), Met (M) (4, 1.4%), Phe (F) (14, 4.9%), Pro (P) (13, 4.5%), Ser (S) (17, 5.9%), Thr (T) (13, 4.5%), Trp (W) (4,1.4%), Tyr (Y) (9, 3.1%), Val (V) (19, 6.6%). Alanine (40) was found to be the most prevalent amino acid residue, followed by glutamic acid (21) and aspartic acid (20) whereas Cysteine (1) was found to be the lowest. The protein is made up of 33 positively charged residues (arginine + lysine) and 41 negatively charged residues (aspartic acid + glutamic acid). 4406 atoms make up the atomic makeup, and the protein’s molecular formula is C1410H2198N362O431S5.
Table 1.
Physicochemical properties of HP PBJ89160.1 estimated by ProtParam tool.
| No. of amino acid | MW(Da) | Half life (hr) | PI | (Asp + Glu) | (Arg + Lys) | Instability index | GRAVY |
|---|---|---|---|---|---|---|---|
| 287 | 31,277.43 | 30 | 5.11 | 41 | 33 | 25.98 | −0.272 |
Bioinformatics-based predictions of protein function and genome annotation include predictions of protein subcellular localization, a crucial aspect aiding in the identification of potential therapeutic targets. The CELLO and PSLpred subcellular localization techniques identified the periplasmic region as the preferred subcellular location of our hypothetical protein, while the BUSCA server indicated localization in the extracellular region (Table 2). This finding correlates with the ProtParam analysis, where a GRAVY index value of −0.272 suggests that the protein is soluble. The subcellular localization predictions for our protein of interest show some inconsistency, with CELLO and PSLpred predicting a periplasmic location while BUSCA suggests an extracellular location. Upon examining the confidence scores, both CELLO and PSLpred provide high confidence in their periplasmic predictions. Additionally, features such as signal peptides identified using SignalP support a secretory pathway, which aligns with periplasmic localization in Gram-negative bacteria. However, BUSCA’s integration of multiple predictors highlights potential extracellular secretion, which could occur under certain conditions or in specific strains. Experimental evidence from similar proteins in related species indicates a primary periplasmic localization, supporting the predictions by CELLO and PSLpred. Given that cytoplasmic and periplasmic proteins are typically soluble, we infer that our subcellular predictions by CELLO, PSLpred are accurate. The SOSUI server determines that the protein is soluble and authenticated its solubility by the Protein Sol service to be 0.669 (Supplementary Figure 1).
Table 2.
Analysis of subcellular localization of HP PBJ89160.1.
| Tools | Subcellular location |
|---|---|
| CELLO | Periplasmic |
| PSLpred | Periplasmic |
| BUSCA | Extracellular |
Domain and motif analysis
To uncover the conserved domain and potential functions of the protein, NCBI-CD search, ScanProsite, and InterProscan were employed. The NCBI CD search results suggest that the HP possesses a conserved membrane-associated lipoprotein domain in BLAST output. Gna1946 appeared as the most similar hit and therefor homology of PBJ89160.1. Moreover, Gna1946 shares a great deal of structural and sequence homology with the periplasmic substrate-binding domain of the ATP-binding cassette (ABC) transporter, which is in charge of absorbing methionine (MetQ) at positions 44-270. This substrate-binding domain is a member of the PBP2 superfamily of type 2 periplasmic binding fold proteins. 63 The PBP2 proteins act like Venus flytraps by binding their ligand in the space between their two globular subdomains, which are usually connected by a flexible hinge. These proteins are mostly involved in the absorption of small soluble substances in eubacteria and archaea. According to ScanProsite, the HP PBJ89160.1 has one motif that can be found in positions 1 through 20 of the bacterial membrane lipoprotein. These proteins are produced from a precursor signal peptide by a specific signal peptidase called signal peptidase II, which is present in lipoproteins. The peptidase identifies the cysteine residue as a component of a conserved sequence and removes upstream from the glyceride-fatty acid lipid.64 -66 It contains several antigenic elements that could increase the pathogenicity of the bacteria. MetQ is a part of a D-methionine permease that facilitates ATP-driven transport by binding proteins. ABC transporters’ uncharacterized substrate-binding components have been found in other members of this family, namely NlpA. It has been established that the inner-membrane-anchored lipoprotein NlpA has a comparatively small function in methionine import. Pfam database identifies a domain as NlPA lipoprotein in 1-281 position, but PANTHER database predicts a domain D-methionine binding lipoprotein MetQ in 1-279 places. Methionine and ATP-driven transport systems are both used in each of these domains (Table 3).
Table 3.
Analysis of domain, motif, and protein family of HP PBJ89160.1.
| Database | Description | Interval | E-value |
|---|---|---|---|
| NCBI CD search | PBP2_lipoprotein_Gna1946 Superfamily | 44-270 | 3.39e−144 |
| InterProscan | Lipoprotein NlPA family | 1-281 | N.A. |
| Pfam | NLPA lipoprotein; this family of bacterial lipoproteins contains several antigenic members | 45-280 | 2.25e−105 |
| PANTHER | D-methionine binding lipoprotein MetQ | 1-279 | NA |
| ScanProsite (motif) | Prokaryotic membrane lipoprotein | 1-20 | NA |
KEGG metabolic pathway
KEGG pathway analysis reveals that the HP is likely involved in ATP hydrolysis for the active transport of a diverse range of substrates, including ions, sugars, lipids, sterols, peptides, proteins, and drugs. Specifically, the protein is implicated in D-amino acid metabolism, particularly in cysteine and methionine metabolism (Supplementary Figure 2).
Phylogenetic and protein family analysis
BLASTp was used to search the non-redundant database; the results showed sequence similarities (up to 96%) with other MetQ/NlpA family ABC transporter substrate-binding regions (Table 4). To see the conserved and different residues among the homologs, multiple sequence alignments were performed using the BLASTp (Figure 2) which revealed a detailed
Table 4.
Analysis of BLASTp showing similarity among HP PBJ89160.1 and other proteins.
| Accession no. | Organism name | Protein name | Score | Percentage identity | E-value |
|---|---|---|---|---|---|
| PBJ89160.1 | Neiserria meningitidis | Hypothetical protein CNQ34_02465c | NA | NA | NA |
| WP_002221629.1 | Neiserria meningitidis | MetQ/NlpA family ABC transporter substrate-binding protein | 583 | 100 | 0 |
| WP_101087969.1 | Neiserria meningitidis | MetQ/NlpA family ABC transporter substrate-binding protein | 583 | 99.65 | 0 |
| WP_002218060.1 | Neiserria meningitidis | MetQ/NlpA family ABC transporter substrate-binding protein | 580 | 99.30 | 0 |
| WP_042743643.1 | Neiserria meningitidis | MetQ/NlpA family ABC transporter substrate-binding protein | 579 | 99.30 | 0 |
| AAF42636.1 | Neiserria meningitidis | Outer membrane lipoprotein GNA1946 | 583 | 100 | 0 |
| EEZ72030.1 | Neiserria cinerea | NLPA lipoprotein | 288 | 95.49 | 0 |
| KIC05807.1 | Morococcus cerebrosus | Membrane protein | 497 | 89.79 | 1e−175 |
| EFC88029.1 | Neisseria mucosa ATCC 25996 | NLPA lipoprotein | 497 | 89.79 | 1e−175 |
| TCP01958.1 | Uruburuella suis | D-methionine transport system substrate-binding protein | 479 | 85 | 2e−168 |
| UOO81404.1 | Uruburuella testudinis | MetQ/NlpA family ABC transporter substrate-binding protein | 481 | 86.41 | 2e−169 |
Figure 2.

Multiple sequences alignment analysis.
Source: For the sequences: Rows 2, 3, 4, 5, 6, Neisseria meningitidis; Row 7, Neisseria cinerea; Row 8, Morococcus cerebrosus; Row 9, Neisseria mucosa; Row 10, Uruburuella testudinis; Rows 11, Uruburuella suis. Version 8 of CLC Sequence Viewer was used.
Multiple sequences alignment of various methionine uptake system proteins, with the target protein in the top row.
Comparative analysis of the hypothetical protein from Neisseria meningitidis against various homologous and related proteins from different bacterial species. PBJ89160.1 is highly homologous to several other MetQ/NlpA family ABC transporter substrate-binding proteins within Neisseria meningitidis, indicating that it likely performs a similar role in substrate binding and transport. The high sequence identity to other ABC transporter proteins supports the hypothesis that PBJ89160.1 is involved in methionine transport, contributing to the bacterium’s virulence by aiding in nutrient uptake. Proteins from other species show varying degrees of similarity, with the closest homologs being from other Neisseria species and more distant homologs from unrelated bacteria. This further suggests that while PBJ89160.1 is well- conserved within the Neisseria genus, it has more divergent homologs in other genera. The same data was used to construct a phylogenic tree (Figure 3). Phylogenetic analysis revealed that the HP appears to share a common ancestor with WP002221629.1 and AAF42636.1 proteins of N. meningitidis.
Figure 3.

Phylogenetic tree with genuine distance to HP PBJ89160.1.
MEGA 11 version was used to create the tree. The line segment with the number (0.02) on it represents the amount of genetic alteration, and the scale bar in this case estimates the degree of sequence divergence.
Protein interaction map
A STRING 12.0 search was conducted to identify the potential functional interaction network of the HP PBJ89160.1 (Figure 4). The analysis revealed several significant interactions with other proteins, including NMB1947 (score: 0.999), NMB1948 (score: 0.999), frpC (score: 0.649), NMB0938 (score: 0.527), NMB1483 (score: 0.465), PilB (score: 0.436), metG (score: 0.435), metF (score: 0.562), metK (score: 0.765), and metH (score: 0.589). Among these, NMB1946 predominantly interacts with NMB1947 and NMB1948, which are involved in methionine import. Other identified partners include an iron-regulated protein, two hypothetical proteins, L-methionine-(S)-S-oxide reductase, methionyl-tRNA synthetase, 5,10-methylenetetrahydrofolate reductase, S-adenosylmethionine synthetase, and 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase (Figure 4). The analysis encompassed parameters such as the number of nodes (12), edges (32), average node degree (3.56), average local clustering coefficient (0.536), and protein-protein interaction enrichment p-value (0.034).
Figure 4.

Protein-protein interaction network analysis by STRING.
Here, NMB1946 is the HP PBJ89160.1 under study in this STRING network analysis.
Secondary structure prediction
The SOPMA server was used to determine the percentages of the HP secondary structures including alpha helix (42.51%), random coil (33.80%), extended strand (16.03%), and beta turn (7.67%). A similar outcome was obtained in the PSIPRED study (Figure 5).
Figure 5.

Secondary structure of HP PBJ89160.1.
PSI-PRED server predicted the target protein’s secondary structure. Four distinct components make up this graphic illustration. Bars in the first part are varied heights. The confidence score is proportional to the length of the bar height. In the second section, the alpha helix is represented by the pink color, the beta sheets or strands are represented by the yellow color, and the coils are represented by the gray color. A coil links a specific beta sheet to a specific alpha helix. The secondary structure of a protein is depicted alphabetically in the third section; here, the letters E, H, and C stand in for beta sheets, alpha helixes, and coils, respectively. The order of amino acids is listed alphabetically in the final section.
Determination of the three-dimensional structure
3D structure of the hypothetical protein was ascertained using the template outer membrane lipoprotein GNA1946 (PDB_ID: 3GXA), which showed 99% identity with our HP in the HHpred server. To visualize the structure, BIOVIA Discovery Studio Visualizer was utilized (Figure 6A). Utilizing UCSF Chimera software, the superimposition of the HP PBJ89160.1 and template protein 3GXA was conducted, as illustrated in (Figure 6B). LIGPLOT analysis from PDBsum server showed the detailed protein ligand interactions (Supplementary Figure 3).
Figure 6.

Three-dimensional structure prediction of HP PBJ89160.1 and superimposition with template protein. Here, (A) HHpred server’s prediction of the hypothetical protein’s three-dimensional structure (shown by BIOVIA Discovery Studio Visualizer version 20.1.0.19295). (B) Superimposition of the HP (PBJ89160.1) and template protein by UCSF chimera software.
Energy minimization of predicted hypothetical protein 3D structure
The three-dimensional structure of the HP was subjected to energy minimization using the YASARA force field minimizer. As a result of this process, the energy level was reduced from −108,282 kj/mol to −141,099.1 kj/mol. Despite this significant energy reduction, the final value changed from −0.8 to 0.4, indicating a stable structure.
Evaluation of the model quality
After energy minimization, the quality of the stable 3D structure of HP was assessed using PROCHECK analysis. Specifically, the Ramachandran plot indicated that 94% of the amino acid residues are located in the most favored regions, signifying a high level of structural acceptability (Table 5, Figure 7A). Upon analysis of the Verify 3D plot, we determined that 88.70% of residues exhibited an average 3D-1D ⩾ 0.1. It is inferred that at least 80% of the amino acids have scored ⩾0.1 in the 3D-1D profile for being the structure of the protein to be stable. Therefore, the HP in the study is stable. The ERRAT software assigned an overall quality factor of 92.3404 to the predicted protein model, and the QMEAN4 score further gauges model reliability by comparing its structure to previously established experimental structures of similar sizes. The QMEAN4 global score for our hypothetical protein is −0.75, indicating a favorable model quality (Figure 7B). The Z score of a model serves as an indicator of its overall quality and is used to assess whether the input structure falls within the typical range observed for native proteins of similar size. In this study, a Z score of −8.48 for the model HP (Figure 8A) and −8.82 for the template (Figure 8B), suggesting significant homology between the template and the modeled structure. The root-mean-square deviation (RMSD) between 239 pruned atom pairs was measured at 0.449 angstroms, consistently observed across all 239 pairs. The protein is further validated with VoroMQA server in which the validation score is 0.507 that indicates a good protein quality as per global VoroMQA score (Supplementary Figure 4).
Table 5.
Ramachandran plot statistics of HP PBJ89160.1.
| Statistics | No. of AA residues (%) |
|---|---|
| Residues in the most favored regions [A, B, L] | 203 (94) |
| Residues in additional allowed regions [a, b, I, p] | 13 (6) |
| Residues in generously allowed regions [~a, ~b, ~l, ~p] | 0 (0) |
| Residues in disallowed regions | 0 (0) |
| No. of non-glycine and non-proline residues | Total 216 (100) |
| No. of end residues (excl. Gly and Pro) | 2 |
| No. of glycine residues (shown in triangles) | 13 |
| No. of proline residues | 12 |
| Total no. of residues | 243 |
Figure 7.
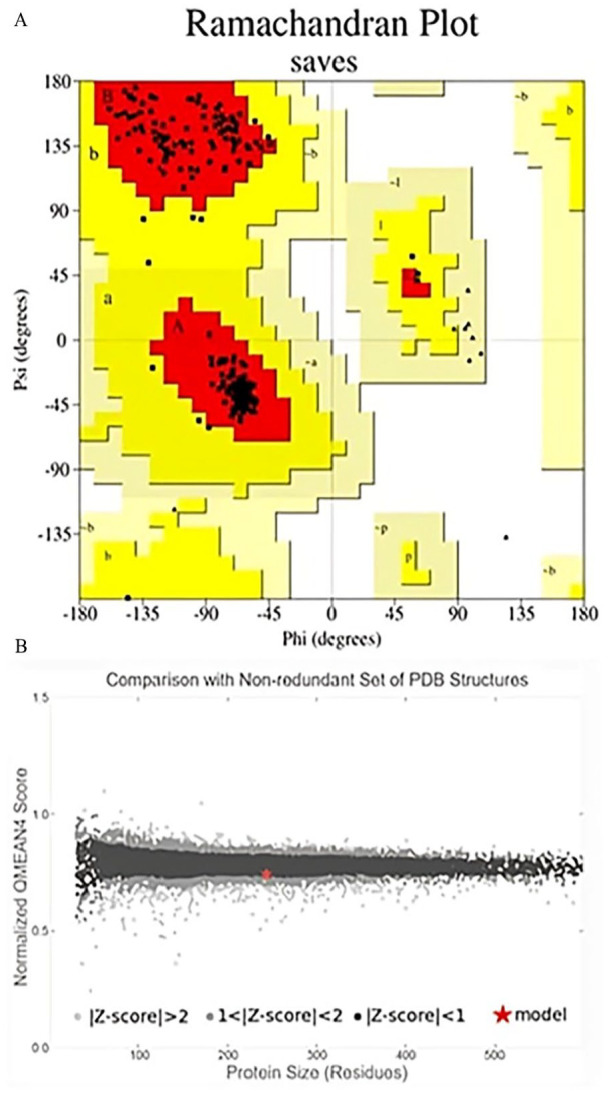
Evaluation of the model’s quality of HP PBJ89160.1 three-dimensional structure. Here, (A) The PROCHECK program-validated Ramachandran plot of the model structure and (B) the graphic representation of the model’s QMEAN result show that the model structure and experimental structures of comparable size (−0.75) are in good agreement.
Figure 8.

Z scores of the Model HP PBJ89160.1. Here, (A) is structure of HP PBJ89160.1 and (B) template protein.
The locations of the two structures were typical for natural proteins of similar size that have been determined experimentally (by NMR and X-ray).
Active site analysis
In the development of a drug or inhibitor to target a protein, it is crucial to identify the protein’s active site. The CASTp server was employed to determine the active site in the 3D structure (Figure 9). One of the largest pockets revealed the most active site, with a total volume of 58.983 amino acids and a solvent-accessible (SA) surface area of 92.084, respectively. The pocket revealed key active residues are THR79, ASP80, ASP80, TYR81, TYR81, TYR81, TYR81, VAL82, TYR103, TYR103, PRO148, ASN149, ASP150, ASP150, ASP150, ASN153, ASN153, GLU196, GLU196, GLU196, GLU196, GLU196, GLU196, ALA197, ALA197, ALA198, ALA198, ASN213, and TYR216.
Figure 9.
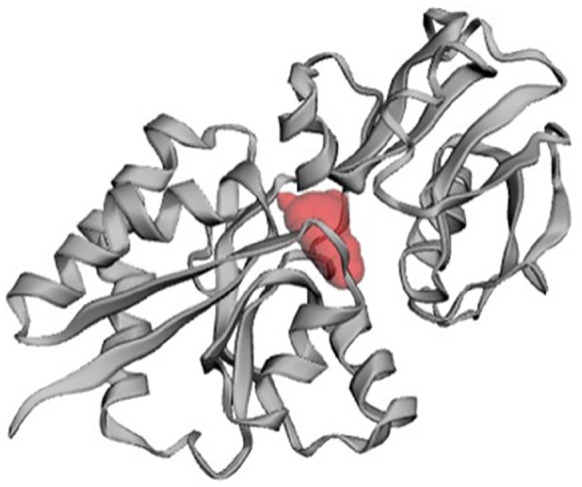
Prediction of active site of HP PBJ89160.1.
The HP’s PBJ89160.1 active site is determined by CASTp server. The largest active site was found in the areas with 92.084 and volume of 58.983 amino acids.
Molecular docking
Molecular docking results explore the binding affinity among five tentative drug compounds aforementioned earlier and to the selective hypothetical protein. Interestingly, the natural compound, Methionine was exhibited the lowest binding affinity (−4.9 kcal/mol) where the highest binding affinity was calculated for Loperamide (−7.6 kcal/mol). Thus, molecular docking analysis successfully anticipated a receptor-ligand complex having the strongest binding affinity. However, rests of the calculated energies of other drug compounds with the same hypothetical protein through docking analysis are as summarized in Table 6. Visualization of the top scored complex was done by accessing docked structures having nine different poses. More the hydrogen bonds are present in the complex meaning the docking complex is more preferable as well as fitted for drug discovery. 67 The highest number of interacting residues was observed in the interaction of Loperamide and HP where a single hydrogen bond interacted to His111 with an oxygen radical (from 2.2 Å) of Loperamide. Moreover, Val82 was interacted via pi-alkyl bond combining with cyclohexane ring (from 4.7 Å) and benzene ring (from 5.3 Å). Similarly, Pro151 were interacted via pi-alkyl bond with nitrogen atom found in the middle of the Loperamide structure distancing from 4.4 Å. Tyr103 and Glu110 were contracted with pi-pi- stacked and pi-anion bond respectively. In this case, another benzyl group found in ligand compound is the interacting structure that is connected with two amino acid residues distancing from 4.6 Å to 4 Å respectively (Figure 10).
Table 6.
Binding affinity among five therapeutic compounds and HP PBJ89160.1.
| Target protein | Ligand(s) | PubChem ID | Binding affinity (kcal/mol) |
|---|---|---|---|
| Hypothetical protein | Methionine (MetQ) | −4.9 | |
| Verapamil | 2520 | −6.8 | |
| Loperamide | 3955 | −7.6 | |
| Thioridazine | 5452 | −6.4 | |
| Chlorpromazine | 2726 | −5.4 | |
| Auranofine | 24199313 | −5.1 |
Figure 10.

Molecular docking analysis showing the best pose of HP with Loperamide. (A) Protein-ligand interaction with surface representation. Red and blue color surrounding to ligand (yellow) depicted polar and non-polar residues, respectively. (B) An enhanced 3D illustration showed ligand-binding pocket. (C) 2D image showing interactions between receptor and substrate via one conventional hydrogen bond distancing 2.29 Å and two Pi-Alkyl bonds. A single pi-anion and a pi-pi stacked found interacting with a benzene ring of Loperamide.
ADMET analysis and toxicity prediction of the selected drug compounds
The canonical SMILES of the five compounds were analyzed using the SwissADME web tool. Table 7 presents the physicochemical properties of these compounds, indicating their potential as pharmaceuticals. Table 8 details the predicted toxicological properties, derived using the admetSAR server. The findings reveal that CID 24199313 is toxic according to the Ames test. However, all compounds were identified as weak inhibitors of the human ether-a-go-go-related gene (hERG) and exhibited low rat acute toxicity, with a median lethal dose (LD50) of 2.35 mol/kg. Based on the ADMET prediction profile, compounds were categorized into four groups based on acute oral toxicity: Group I (LD50 ⩽ 50 mg/kg), Group II (50 < LD50 ⩽ 500 mg/kg), Group III (500 < LD50 ⩽ 5000 mg/kg), and Group IV (LD50 > 5000 mg/kg). According to the predicted acute oral toxicity values, compounds CID 2520, CID 3955, and CID 2726 fall within Class II, while compounds CID 5452 and CID 24199313 fall within Class III. These classifications indicate that the compounds are generally suitable for drug development, with LD50 values less than 5000 mg/kg.
Table 7.
Pharmacokinetic properties of selected five therapeutic compounds.
| PubChem ID | CID 2520 | CID 3955 | CID 5452 | CID 2726 | CID 24199313 | |
|---|---|---|---|---|---|---|
| Physiochemical properties | Molecular weight (g/mo) | 454.60 | 477.04 | 370.57 | 318.86 | 678.48 |
| Heavy atoms | 33 | 34 | 25 | 21 | 32 | |
| Arom. heavy atoms | 12 | 18 | 12 | 12 | 0 | |
| Rotatable bond | 13 | 8 | 4 | 4 | 12 | |
| H-bond acceptor | 6 | 3 | 1 | 1 | 9 | |
| H-bond donor | 0 | 1 | 0 | 0 | 0 | |
| Lipophilicity | Log Po/w | 4.50 | 4.06 | 4.03 | 3.47 | 0.00 |
| Water solubility | Log S (ESOL) | −4.46 | −5.82 | −5.95 | −5.25 | −4.55 |
| Pharmacokinetics | GI absorption | High | High | High | High | High |
| Drug likeliness | Lipinski | Yes | Yes | Yes | Yes | Yes |
| Medical chemistry | Synthetic accessibility | Easy | Easy | Easy | Easy | Easy |
Table 8.
Toxicological properties of predicted five therapeutic compounds.
| Compound (PubChem ID) | hERG inhibition | RAT (LD50) | AMES test | Carcinogens | Acute oral toxicity | Carcinogenicity (three class) |
|---|---|---|---|---|---|---|
| 2520 | Weak | 3.4137 | No | No | II | Not required |
| 3955 | Weak | 3.6560 | No | No | II | Not required |
| 5452 | Weak | 2.5395 | No | No | III | Not required |
| 2726 | Weak | 3.3196 | No | No | II | Not required |
| 24199313 | Weak | 2.7163 | Toxic | No | III | Not required |
To further validate the toxicity profiles, the ProTox 3 web server was employed (Table 9). The analysis identified three compounds (CID 2520, CID 5452, and CID 2726) as immunotoxic. After evaluating all the properties, CID 3955 (Loperamide) emerged as the best fit against our hypothetical protein HP (PBJ89160.1). The graphical interaction between HP and Loperamide is depicted in Figure 10.
Table 9.
Toxicity profiling of top five predicted therapeutic compounds through ProTox-3.0 server.
| Compound (PubChem ID) | Cytotoxicity | Immunotoxicity | Mutagenecity | Carcinogenecity |
|---|---|---|---|---|
| 2520 | Inactive | Active | Inactive | Inactive |
| 3955 | Inactive | Inactive | Inactive | Inactive |
| 5452 | Inactive | Active | Inactive | Inactive |
| 2726 | Inactive | Active | Inactive | Inactive |
| 24199313 | Inactive | Inactive | Inactive | Inactive |
Discussion
Neisseria meningitidis is the causative agent of major meningococcal disease outbreaks and high morbidity rates. Additionally, the emergence of multiple antibiotic-resistant strains due to improper antibiotic use has made treating meningococcal disease more challenging. Therefore, identifying alternative therapeutic targets is essential to combat this bacterial infection. In many studies it was reported that HPs of an organism could serve as valuable sources for alternative therapeutic targets.68 -71 Therefore, characterizing HP (PBJ89160.1) from N. meningitidis can enhance our understanding of bacterial metabolic regulation, aid in formulating disease control strategies, and contribute to the development of effective therapeutics. The characterization of this hypothetical protein involved the sequential application of various bioinformatics tools. Functional analysis, conducted using NCBI CD Search, ScanProsite, InterProscan, Pfam, and PANTHER, suggests that the protein may exhibit D-methionine absorption activity and possess several antigenic characteristics. The 3D structure of the protein was modeled using the HHpred server, employing a template (3GXA; GNA1946 outer membrane lipoprotein) with over 99% similarity. GNA1946 is a type of lipoprotein and one of the first ABC transporter family members. It has a strong affinity and specificity towards L-methionine, and it is believed to bind to it instead of D-methionine. Pathogens, through nutritional assimilation, such as scavenging amino acids for protein synthesis, adapt and survive within their niches. They employ “nutritional virulence” mechanisms to augment the host’s acquisition of limited nutrients by specifically targeting key host biosynthetic pathways or nutrient-rich sources. 72 The amino acid uptake function is a major research target. The protein is a substrate binding protein (SBP) that interacts with microvascular endothelial cells in the human brain, possibly functioning as an adhesion that is periplasmically localized and absorbs methionine. 73 This nutritional virulence is caused by ABC Transporter family proteins and our HP belongs to this family. Meanwhile, ABC transporters are used as (1) Target for antimicrobials-by designing antibiotics that mimic transporter substrates specifically that have broad specificity for substrate binding, (2) target for drug development by designing inhibitors that target components of the transporters, essential for survival and act as nutritional virulence factors, and (3) target for vaccine development-by designing vaccine against transporter proteins. 74
The main focus of the study is to advance the understanding of the HP from Neisseria meningitidis, particularly in terms of its structural and functional characteristics, and to explore its potential as a target for therapeutic intervention in treating bacterial meningitis. Therefore, it is a prerequisite to know about the function of domain and motif of the protein. The NCBI CD search analysis revealed that HP (PBJ89160.1) is composed of multiple enzyme families that serve as substrate-binding domains and are members of the type 2 periplasmic binding fold protein superfamily (PBP2). These enzyme families share structural similarities. The bulk of PBP2 proteins work in eubacteria and archaea to take up tiny soluble substrates. The HP has one motif, which is bacterial membrane lipoprotein, according to ScanProsite. Previous studies demonstrated that a precursor signal peptide is used in the synthesis of these proteins. Signal peptidase II, a key component of lipoproteins, is responsible for cleaving the signal peptide. 65
The CLC sequence viewer was utilized for generating multiple sequence alignment, and MEGA software facilitated phylogenetic tree analysis. The HP is homologous to two proteins, one of which functions as ABC transporter SBP (WP002221629.1) and another is outer membrane lipoprotein GNA1946 (AAF42636.1) in N. meningitidis. Protein-protein interaction is commonly used to detect the interactive functional proteins in a signaling pathway which reveals the cellular mechanism of an organism. 75 In STRING analysis it was observed that HP PBJ89160.1 protein interacts with ten other proteins. Among them higher interaction was observed with two proteins (NMB1947, NMB1948) that has methionine importer activity. According to STRING database, NMB1947 is involves in D-methionine transmembrane transport and NMB1948 is an ABC transporter, ATP-binding protein; Part of the ABC transporter complex. MetNIQ involved in methionine import, responsible for energy coupling to the transport system and belongs to the ABC transporter superfamily. The HP PBJ89160.1 protein also showed comparable function with NMB1947 and NMB1948.
In the quality analysis, the ProCheck server revealed reliable scores for the Ramachandran plot, Verify 3D, ERRAT, and Qmean value, indicating that the structure is likely significant. 76 According to the global VoroMQA score, protein’s having more than 0.4 score validated as good quality protein and our protein’s VoroMQA score is 0.507 predicted as good protein quality. The modeled protein’s 3D structure was refined using Discovery Studio, which removed all water molecules and docked the protein with its natural ligand, methionine. The binding affinity between the hypothetical protein and methionine was found to be −4.9 kcal/mol.
There are some reports where ADMET and Pharmacophore characterization approaches were used to predict the best drug candidate. 77 Aiming to identify potential therapeutic compounds that could inhibit the molecular function of the HP (PBJ89160.1), we selected five candidate drugs for further analysis. These compounds were docked with the modeled HP (PBJ89160.1) to assess their binding affinities. Subsequent ADME and toxicological properties analyses led to the elimination of four drugs due to their toxic properties. Only one drug, Loperamide, met all the criteria. Loperamide demonstrated a binding affinity of −7.6 kcal/mol with the HP (PBJ89160.1), making it the most promising drug candidate against the protein. P-glycoprotein (P-gp) is an ATP-dependent efflux transporter, and loperamide is found to be inhibiting its function. P-gp plays a crucial part in the absorption and disposal of drugs by expelling different substances, including drugs, from cells. 78 The quick uptake and efflux of medications across the cell membrane is facilitated by the P-gp, one of the proteins that make up the ATP-binding cassette transporter family. 79 Therefore, our candidate drug Loperamide could play potential vital role in the treatment of meningococcal disease.
Finally, human homologous research using BLAST revealed that the chosen protein does not homologous to humans. As a result, there are no longer any odds of an adverse consequence, proving that this protein may be a good therapeutic target. It’s expected that additional functional and structural research on the ABC transporters of Gram-negative bacteria will shed light on their as-yet-unidentified transport mechanism. Despite this, our findings are helpful in creating broad-spectrum vaccinations to protect against the bacterium N. meningitides. 80
Conclusion
In conclusion, this study has successfully characterized a new HP, which is believed to play a crucial role in nutritional virulence and the ABC transport mechanism as a methionine transporter. However, further experimental validation and functional characterization are necessary to confirm its predicted structural and functional attributes. There are several in vitro studies like (Gene expression analysis, protein-protein interaction, enzyme assay, and cell based assay) and in vivo studies like (animal model, disease model, histopathological studies) can be conducted to verify the protein’s function and potential as drug target. To build on these findings, we recommend conducting additional immune-informatics and structural biology methodologies, alongside in vitro and in vivo studies, to thoroughly understand and validate the drug targets against meningitis. Nonetheless, this discovery lays a strong foundation for further research aimed at understanding the genetic and proteomic profile of N. meningitidis and identifying potential drug targets. Such comprehensive studies are essential due to the inherent limitations of in silico models, which struggle to fully capture the complexity of biological systems, individual genetic variations, and environmental influences on drug response.
Supplemental Material
Supplemental material, sj-docx-1-evb-10.1177_11769343241298307 for In silico Characterization of a Hypothetical Protein (PBJ89160.1) from Neisseria meningitidis Exhibits a New Insight on Nutritional Virulence and Molecular Docking to Uncover a Therapeutic Target by Israt Jahan Asha, Shipan Das Gupta, Md. Murad Hossain, Md. Nur Islam, Nurun Nahar Akter, Mohammed Mafizul Islam, Shuvo Chandra Das and Dhirendra Nath Barman in Evolutionary Bioinformatics
Acknowledgments
The authors acknowledge the Department of Biotechnology and Genetic Engineering, Noakhali Science and Technology University for providing support to conduct the research work.
Footnotes
Author Contributions: Conceptualization: IJA, SDG & DNB; Data curation: IJA & DNB; Formal analysis: IJA, SDG, MMH & DNB; Supervision: DNB; Software: IJA, MNI & NNA; Resources: DNB; Writing original draft: IJA, SDG, NNA & DNB; Writing, review and editing: SDG, MMH, MMI, SCD, MNI & DNB.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID iDs: Md. Murad Hossain  https://orcid.org/0009-0002-0852-9737
https://orcid.org/0009-0002-0852-9737
Dhirendra Nath Barman
https://orcid.org/0000-0002-7086-976X
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Yazdankhah SP, Caugant DA. Neisseria meningitidis: an overview of the carriage state. J Med Microbiol. 2004;53:821-832. [DOI] [PubMed] [Google Scholar]
- 2. Manchanda V, Gupta S, Bhalla P. Meningococcal disease: history, epidemiology, pathogenesis, clinical manifestations, diagnosis, antimicrobial susceptibility and prevention. Indian J Med Microbiol. 2006;24:7-19. [DOI] [PubMed] [Google Scholar]
- 3. Batista RS, Gomes AP, Luiz J, Gazineo JLD, Paulo S, Miguel PSB, Santana LA, Olivera L, Geller M. Meningococcal disease, a clinical and epidemiological review. Asian Pac J Trop Med. 2017;10:1019-1029. [DOI] [PubMed] [Google Scholar]
- 4. Lacey RW, Reeves DS, Emilio P, Aldamiz-echeverria L, Eduardo GP. Meningococci with increased resistance to penicillin. Lancet. 1989;23:8220. [DOI] [PubMed] [Google Scholar]
- 5. Mastrantonio TSMECMO and P. Meningococcal disease in Italy. J Infect. 1989;19:69-74. [DOI] [PubMed] [Google Scholar]
- 6. Tzanakakp G, Blackwell CC, Kremastinou J. Antibiotic sensitivities of Neisseria meningitidis isolates from patients and carriers in Greece. Epidemiol Infect. 1992;108:449-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greenhouse P, Howell PR. Penicillin-insensitive Meningococci in the UK. Lancet. 1988;331:657-658. [DOI] [PubMed] [Google Scholar]
- 8. Woods CR, Smith AL, Wasilauskas BL, Campos J, Givner LB. Invasive disease caused by Neisseria meningitidis relatively resistant to Penicillin in North Carolina. J Infect Dis. 1994;170:453-456. [DOI] [PubMed] [Google Scholar]
- 9. Oppenheim BA. Antibiotic resistance in Neisseria meningitidis. Clin Infect Dis. 1991;24:98-101. [DOI] [PubMed] [Google Scholar]
- 10. Khoosal M. Susceptibility of Neisseria meningitidis in Israel to penicillin and other drugs of interest. J Antimicrob Chemother. 1993;32:166-168. [DOI] [PubMed] [Google Scholar]
- 11. Greenwood BM, Blakebrou GH, Dagger IS, Bradley AK, Wali S, Whittle HC. Meningococcal disease and season in Sub-Saharan Africa. Lancet. 1984;323:1339-1342. [DOI] [PubMed] [Google Scholar]
- 12. Stephens DS, Sherman AC, Stephens DS, Sherman AC, Serogroup A. Serogroup A meningococcal conjugate vaccines: building sustainable and equitable vaccine strategies. Expert Rev Vaccines. 2020;19:455-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. ling Tzeng Y, Stephens DS. Epidemiology and pathogenesis of Neisseria meningitidis. Int J Epidemiol. 2000;29:687-700. [DOI] [PubMed] [Google Scholar]
- 14. Jackson LA, Schuchat A, Reeves MW, Wenger JD. Serogroup C meningococcal outbreaks in the United States. An emerging threat. JAMA. 1995;273:383-389. [PubMed] [Google Scholar]
- 15. Jafri RZ, Ali A, Messonnier NE, et al. Global epidemiology of invasive meningococcal disease. Popul Health Metr. 2013;11:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore PS, Schwartz B, Reeves MW, Gellin BG, Broome CV. Intercontinental spread of an epidemic group a Neisseria meningitidis strain. Lancet. 1989;334:260-263. [DOI] [PubMed] [Google Scholar]
- 17. WHO. Defeating Meningitis by 2030 a Global Road Map. 2021. [DOI] [PubMed] [Google Scholar]
- 18. Mills CL, Beuning PJ, Ondrechen MJ. Biochemical functional predictions for protein structures of unknown or uncertain function. CSBJ. 2015;13:182-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siddiqui Q, Ali MSM, Leow ATC, Oslan SN, Mohd Shariff F. In silico identification and characterization of potential druggable targets among hypothetical proteins of Leptospira interrogans serovar Copenhageni: a comprehensive bioinformatics approach. J Biomol Struct Dyn. 2023;41:10347-10367. [DOI] [PubMed] [Google Scholar]
- 20. Naveed M, Tehreem S, Usman M, Chaudhry Z, Abbas G. Structural and functional annotation of hypothetical proteins of human adenovirus: prioritizing the novel drug targets. BMC Res Notes. 2017;10:706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Passalacqua KD, Charbonneau M, Riordan MXDO. Bacterial metabolism shapes the host – pathogen interface. In: Kudva IT, Cornick NA, Plummer PJ, et al. , eds. Virulence Mechanisms of Bacterial Pathogens. Wiley; 2016:15-41. [Google Scholar]
- 22. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, ed. The Proteomics Protocols Handbook. Humana Press; 2005;112:531-552. [DOI] [PubMed] [Google Scholar]
- 23. Sheng YC, Jen LC. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n -peptide compositions. Protein Sci. 2004;13:1402-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhasin M, Garg A, Raghava GPS. PSLpred: prediction of subcellular localization of bacterial proteins. Nucleic Acids Res. 2005;21:2522-2524. [DOI] [PubMed] [Google Scholar]
- 25. Savojardo C, Martelli PL, Fariselli P, Profiti G, Casadio R. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018;46:459-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378-379. [DOI] [PubMed] [Google Scholar]
- 27. Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33:3098-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu S, Wang J, Chitsaz F, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265-D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quevillon E, Silventoinen V, Pillai S, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:116-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cuche A, Sigrist CJA, De Castro E, et al. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41:344-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353-D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein – protein association networks and functional enrichment analyses for any sequenced genome of interest. 2023;51:638-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, Mcginnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38:3022-3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geourjon C, Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics. 1995;11:681-684. [DOI] [PubMed] [Google Scholar]
- 36. McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404-405. [DOI] [PubMed] [Google Scholar]
- 37. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:320-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1-15. [DOI] [PubMed] [Google Scholar]
- 39. Biovia DS. Discovery Studio Visualizer. Vol. 936. Dassault Systèmes; 2017:240-249. [Google Scholar]
- 40. Thornton JM. PDBsum: structural summaries of PDB entries. Protein Sci. 2018;27:129-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:244-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363-W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477-486. [DOI] [PubMed] [Google Scholar]
- 44. Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. In: Lüthy R, Bowie J, Eisenberg D, eds. Methods in Enzymology. Academic Press; 1997;277:396-404. [DOI] [PubMed] [Google Scholar]
- 45. Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 48. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:407-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Olechnovi K. VoroMQA web server for assessing three-dimensional. Nucleic Acids Res. 2019;47:437-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang X, Wu Z, Wang X, Yang C, Xu H, Shen Y. Crystal structure of lipoprotein GNA1946 from Neisseria meningitidis. J Struct Biol. 2009;168:437-443. [DOI] [PubMed] [Google Scholar]
- 52. Schrodinger LL. The PyMOL Molecular Graphics System. Version 1. 2015:8. [Google Scholar]
- 53. Umar F, Hatta M, Husain DR, et al. Verapamil as an efflux inhibitor against drug resistant Mycobacterium tuberculosis: a review. Syst Rev Pharm. 2019;10:S43-S48. [Google Scholar]
- 54. Heel RC, Brogden RN, Speight TM, Avery GS. Loperamide: a review of its pharmacological properties and therapeutic efficacy in diarrhoea. Drugs. 1978;15:33-52. [DOI] [PubMed] [Google Scholar]
- 55. Amaral L, Martins M, Viveiros M, Molnar J, Kristiansen J. Promising therapy of XDR-TB/MDR-TB with Thioridazine an inhibitor of bacterial efflux pumps. Curr Drug Targets. 2008;9:816-819. [DOI] [PubMed] [Google Scholar]
- 56. Machado D, Pires D, Perdigão J, et al. Ion channel blockers as antimicrobial agents, efflux inhibitors, and enhancers of macrophage killing activity against drug resistant mycobacterium tuberculosis. PLoS One. 2016;11:1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamashita M. Auranofin: past to present, and repurposing. Int Immunopharmacol. 2021;101:108272. [DOI] [PubMed] [Google Scholar]
- 58. Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44:1202-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boyle NMO, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminf 2011;3:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Nat Publ Gr. 2017;7:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024;52:513-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cheng F, Li W, Zhou Y, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099-3105. [DOI] [PubMed] [Google Scholar]
- 63. Gál J, Szvetnik A, Schnell R, Kálmán M. The metD D-methionine transporter locus of Escherichia coli is an ABC transporter gene cluster. J Bacteriol. 2002;184:4930-4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mattar S, Scharf B, Kent SB, Rodewald K, Oesterhelt D, Engelhard M. The primary structure of halocyanin, an archaeal blue copper protein, predicts a lipid anchor for membrane fixation. J Biol Chem. 1994;269:14939-14945. [PubMed] [Google Scholar]
- 65. Hayashi S, Wu C. Mini-review lipoproteins in bacteria. J Bioenerg Biomembr. 1990;22:451-471. [DOI] [PubMed] [Google Scholar]
- 66. Klein P, Somorjai RL, Lau PC. Distinctive properties of signal sequences from bacterial lipoproteins. Protein Eng. 1988;2:15-20. [DOI] [PubMed] [Google Scholar]
- 67. Sarkar T, Bharadwaj KK, Salauddin M, Pati S, Chakraborty R. Phytochemical characterization, antioxidant, anti-inflammatory, anti-diabetic properties, molecular docking, pharmacokinetic profiling, and network pharmacology analysis of the major phytoconstituents of raw and differently dried Mangifera indica (Himsagar cultivar): an in vitro and in silico investigations. Appl Biochem Biotechnol. 2022;1:1-38. [DOI] [PubMed] [Google Scholar]
- 68. Chakma V, Barman DN, Das SC, et al. In silico analysis of a novel hypothetical protein (YP_498675.1) from Staphylococcus aureus unravels the protein of tryptophan synthase beta superfamily (Try-synth-beta_ II). J Genet Eng Biotechnol. 2023;21:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Naveed M, Makhdoom SI, Abbas G, et al. The virulent hypothetical proteins: the potential drug target involved in bacterial pathogenesis. Mini-Reviews Med Chem. 2022;22:2608-2623. [DOI] [PubMed] [Google Scholar]
- 70. Jeba SH, Hasan MA, Faisal AS, et al. In silico approach of exploring the structural and functional characteristics of a hypothetical protein from monkeypox virus: a potential insight for antiviral therapeutics. Microb Infect Diseas. Published Online 23 March 2024. doi: 10.21608/mid.2024.272174.1818 [DOI] [Google Scholar]
- 71. Rabbi MF, Akter SA, Hasan MJ, Amin A. In silico characterization of a hypothetical protein from Shigella dysenteriae ATCC 12039 reveals a pathogenesis-related protein of the type-VI secretion system. Bioinform Biol Insights. 2021;15:11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kwaik YA, Bumann D. Microreview microbial quest for food in vivo: ‘nutritional virulence’ as an emerging paradigm. Cellular Microbiol. 2013;15:882-890. [DOI] [PubMed] [Google Scholar]
- 73. Sharaf NG, Shahgholi M, Kim E, et al. Characterization of the ABC methionine transporter from Neisseria meningitidis reveals that lipidated MetQ is required for interaction. Elife. 2021;10:1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Soni DK, Dubey SK, Bhatnagar R. ATP-binding cassette (ABC) import systems of Mycobacterium tuberculosis: target for drug and vaccine development. Emerg Microbes Infect. 2020;9:207-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Naveed M, Imran K, Mushtaq A, Mumtaz AS, Janjua HA, Khalid N. In silico functional and tumor suppressor role of hypothetical protein PCNXL2 with regulation of the Notch signaling pathway. RSC Adv. 2018;8:21414-21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hasan MA, Mazumder MHH, Chowdhury AS, Datta A, Khan MA. Molecular-docking study of malaria drug target enzyme transketolase in Plasmodium falciparum 3D7 portends the novel approach to its treatment. Source Code Biol Med. 2015;10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Naveed M, Ali I, Aziz T, et al. Assessment of Melia azedarach plant extracts activity against hypothetical protein of Mycobacterium tuberculosis via GC-MS analysis and in silico approaches. J Comput Biophys Chem. 2024;23:299-320. [Google Scholar]
- 78. Wanek T, Römermann K, Mairinger S, et al. Factors governing P-Glycoprotein-mediated drug-drug interactions at the blood-brain barrier measured with positron emission tomography. Mol Pharm. 2015;12:3214-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Leung K. [N- methyl-11C]4-(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenyl- N-dimethyl-butanamide. In: Leung K, ed. Molecular Imaging and Contrast Agent Database. National Center for Biotechnology Information; 2010. [PubMed] [Google Scholar]
- 80. van de Beek D, Brouwer M, Hasbun R, Koedel U, Whitney CG, Wijdicks E. Community-acquired bacterial meningitis. Nat Rev Dis Prim. 2016;2:16074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-evb-10.1177_11769343241298307 for In silico Characterization of a Hypothetical Protein (PBJ89160.1) from Neisseria meningitidis Exhibits a New Insight on Nutritional Virulence and Molecular Docking to Uncover a Therapeutic Target by Israt Jahan Asha, Shipan Das Gupta, Md. Murad Hossain, Md. Nur Islam, Nurun Nahar Akter, Mohammed Mafizul Islam, Shuvo Chandra Das and Dhirendra Nath Barman in Evolutionary Bioinformatics