

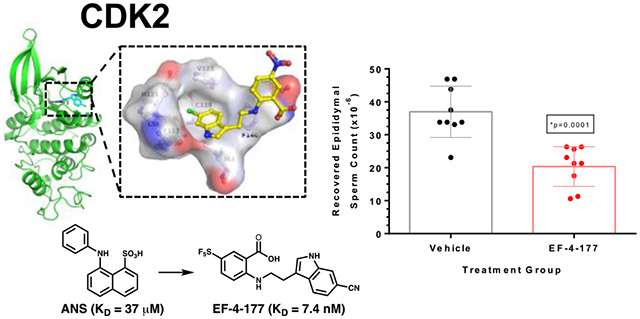
Abstract
While cyclin-dependent kinase 2 (CDK2) is a validated target for both cancer and contraception, developing a CDK2 inhibitor with exquisite selectivity has been challenging due to the structural similarity of the ATP-binding site where most kinase inhibitors bind. We previously discovered an allosteric pocket in CDK2 with potential to bind a selective compound. and then discovered and structurally confirmed an anthranilic acid scaffold that binds this pocket with high affinity. These allosteric inhibitors are selective for CDK2 over structurally similar CDK1 and show contraceptive potential. Herein, we describe the screening and optimization that led to compounds like EF-4-177 with nanomolar affinity for CDK2. EF-4-177 is metabolically stable, orally bioavailable, and significantly disrupts spermatogenesis, demonstrating this series’ therapeutic potential. This work details the discovery of the highest affinity allosteric CDK inhibitors reported and shows promise for this series to yield an efficacious and selective allosteric CDK2 inhibitor.
Graphical Abstract
Introduction
Cyclin-dependent kinase 2 (CDK2) is part of the larger CDK family of kinases that regulate the mitotic cell cycle. It is activated by the binding of E-type cyclins at the G1-S transition and by A-type cyclins in the S phase and involves an additional phosphorylation event. Despite its established role in the cell cycle, surprisingly CDK2 knock-out mice are healthy but sterile, as compensatory mechanisms exist in its absence for mitosis but not meiosis.1-2 However, in certain cancer contexts where E-type and A-type cyclins are overexpressed, CDK2 is hyperactivated, and preclinical animal models show that CDK2 inhibition for these cancers inhibit tumor growth.3-5 Additionally, because CDK2 is necessary for meiosis and not mitosis, it is a validated target for non-hormonal contraceptive development.6 While E- and A-type cyclins also play a role in meiosis, it appears that a cyclin-independent function of CDK2 involving the binding of Speedy1 is crucial in the pachytene stage of prophase I of spermatocytes.7 Overall, developing a selective CDK2 inhibitor could be a great benefit for both cancer patients who cannot tolerate off-target side effects of previously developed CDK2 inhibitors and individuals desiring safe and efficacious non-hormonal male contraception.
While the biochemical role of CDK2 has been studied for decades, an inhibitor with exquisite selectivity for CDK2 (e.g. >1,000-fold over other kinases) has been challenging and none have gained FDA approval.8-9 Efforts to inhibit CDK2 have included the pan-CDK inhibitors such as dinaciclib, roscovitine, and flavopiridol, all of which have reached clinical trials but inhibit a multitude of CDK enzymes.10 Most developed CDK2 inhibitors bind in a type I fashion, meaning they bind the ATP-binding site in CDK2’s active conformation, a conformation highly similar to the active states of other similar kinases like cyclin-dependent kinase 1 (CDK1).9, 11 This leads to undesirable off-target effects, especially considering the necessity of CDK1 function.12-13 As a result, only one type I CDK2 inhibitor exists to our knowledge (PF-07104091) that has 100+-fold selectivity over other CDK family members.14-15 However, a different approach can be pursued to achieve this desired selectivity.
Targeting the inactive state of CDK2 is a promising avenue to develop selective inhibitors. For example, previous work has shown the structural divergence of inactive CDK2 and CDK1 in contrast to their active states.11 One way to achieve the desirable selectivity is by targeting allosteric pockets distal to the ATP-binding site, i.e., developing type III inhibitors. Allosteric pockets are often unique to a particular kinase and can be exploited not only for their selectivity advantage, but also hold promise to overcome therapeutic resistance to type I ligands.9 We previously discovered an unrecognized allosteric pocket within CDK2 that bound the commercial dye 8-anilino-1-naphthalene sulfonic acid (ANS) with moderate affinity.16 We further developed an assay that exploited the environmentally sensitive nature of ANS fluorescence to develop a high-throughput assay to discover more drug-like chemical matter that bound to this allosteric pocket.17 Despite this discovery, the only molecules reported to bind near the ANS pocket have been fragments confirmed by X-ray crystallography18-19 or putative ligands whose allosteric binding mode were not empirically confirmed by such structural studies and whose ability to directly inhibit CDK2 is either weak or unknown.20-22
In contrast, we recently reported an anthranilic acid scaffold that binds this site with high affinity and demonstrates a negative cooperative relationship with cyclin binding to stabilize CDK2 in its inactive state.23 In addition, we confirmed this scaffold has high selectivity for CDK2 over structurally similar CDK1 (no binding to CDK1 measurable in two orthogonal assays) and shows promise as a contraceptive agent by recapitulating the Cdk2−/− and Spdya−/− phenotypes when added to explanted mouse testes.23 Furthermore, the promise of disrupting the CDK2-cyclin A interaction for anticancer purposes has recently been demonstrated by the complex natural product homoharringtonine (HHT), although confirmation of HHT’s binding pose by empirical means such as X-ray crystallography and its selectivity over other kinases have yet to be explored.24 In this paper, we will report our screening and lead optimization efforts that have resulted in a series of nanomolar affinity inhibitors that were crystallographically confirmed to bind the allosteric pocket of CDK2, and possess desirable pharmacokinetic and pharmacodynamic properties that demonstrate their potential as future therapeutics.
Results
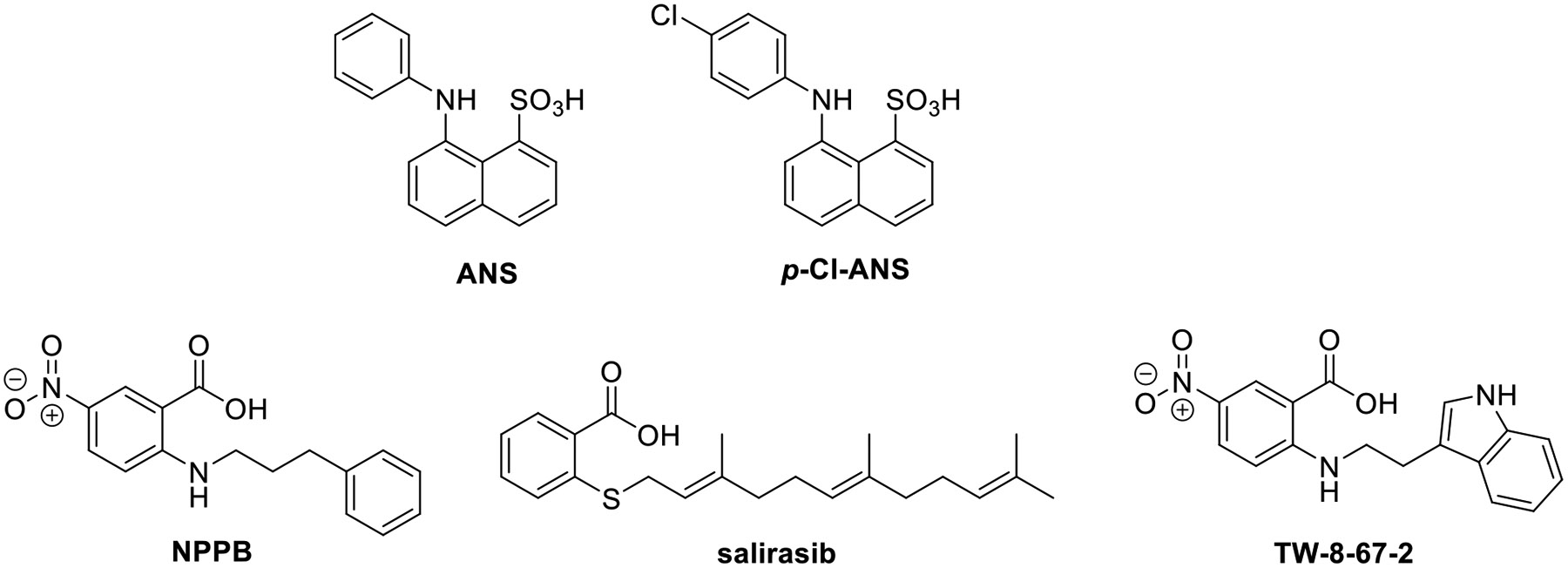
To discover new chemical matter that binds the allosteric site of CDK2, we developed a fluorescent binding assay based on the occupancy of the allosteric site by ANS.17, 25 The high affinity ATP site ligand staurosporine does not substantially affect ANS binding and can be used to differentiate between inhibitors of ANS binding directly at the allosteric site versus those binding to the active site and those cooperatively displacing ANS via conformational change.17 The ANS analog p-Cl-ANS was used as the ligand for the assay instead of ANS due to its improved fluorescent properties and presumed similar binding to CDK2 (structures in Fig. 1).25 In this assay, compound displacement of p-Cl-ANS from the hydrophobic interior of the allosteric site into the aqueous solvent results in a decrease in fluorescence yield due to the environmentally sensitive nature of ANS and its analogs.
Fig. 1. Structures of allosteric CDK2 ligands.
We conducted both a high-throughput screen (HTS) using the p-Cl-ANS displacement assay and a virtual high-throughput screen (vHTS) using the crystal structure of ANS bound to CDK2. In the HTS, we selected ~20k compounds from our in-house compound collection for screening, partly based on their similarity to ANS, particularly due to the presence of a negatively charged carboxylic or sulfonic acid. Hits producing > 50% loss of p-Cl-ANS fluorescence at the 30 μM screening concentration were cherry-picked from DMSO stocks and tested in dose-response in the p-Cl-ANS assay in the absence and presence of staurosporine to distinguish between inhibitors acting at the ATP and allosteric sites. Hits that produced IC50 values < 50 μM with little change in the absence and presence of staurosporine (“staurosporine shift” < 2-fold) were selected for repurchase. Of the compounds newly repurchased after the screen, ten hits decreased the fluorescent signal of p-Cl-ANS in a concentration-dependent manner in the absence and presence of staurosporine (Fig. S-1, Fig. S-2B, Table S-1). All compounds completely inhibited p-Cl-ANS binding with IC50 values ranging from 2-34 μM in the absence of staurosporine. Staurosporine produced ≤ 2-fold reduction in potency for 9 hits but reduced the potency of the ATP-competitive c-Met inhibitor PHA-665752 and the reference CDK2 ATP site inhibitor SU9516 by 2.8- and 234- fold, respectively (Fig. S-1, Fig. S-2A, Table S-1). Although the IC50 ratio for PHA-665752 was surprisingly low, these results indicate that the “staurosporine shift” is useful to detect ATP site inhibitors that allosterically inhibit the binding of p-Cl-ANS to the type III allosteric site.
Simultaneously, a vHTS using ~720k structures from our in-house collection was conducted. Of the 260 vHTS hits selected for purchase, 230 were commercially available and tested for inhibition of p-Cl-ANS binding at 30 μM, but only 7 hits produced > 25% inhibition. Of these, three compounds produced concentration-dependent inhibition of the fluorescent signal production of p-Cl-ANS with IC50 values < 100 μM, and of these the RAS inhibitor salirasib was the most potent (IC50 = 3.1 μM; Fig. 1; Fig. S-2B, Table S-1). Salirasib only partially inhibited binding, possibly due to limited solubility at the higher concentrations tested.
To confirm that these hits were not false positives and the result of fluorescence interference, we needed an orthogonal non-fluorescent assay. We chose to use HSQC NMR of N15 labeled CDK2 to accomplish this. We first noticed that ligands that bind the ATP-binding site, such as non-hydrolysable ATP, dinaciclib, and roscovitine, showed a similar set of global chemical shift perturbations upon binding, as described previously.25 Furthermore, at saturating concentrations of roscovitine, we discovered that titration of ANS into CDK2 caused additional distinct global chemical shift perturbations (Fig. S-3A). From this, we were able to discern an NMR “fingerprint” of compounds that bound the ATP-binding site or the allosteric site of interest. Of the 11 hit compounds tested, three did not show any further chemical shift perturbations and were assumed to be either ATP-binding site ligands or non-binders, five compounds showed indeterminate binding due to observed protein aggregation in the assay, and three compounds (NPPB, salirasib, and Sigma S660124) demonstrated binding to the allosteric site (Fig. S-3B-D). Of these three compounds, two (NPPB and salirasib) were ortho-substituted benzoic acids, yielding our lead scaffold (Fig. 1). Although the chloride channel inhibitor NPPB26 was the least potent hit evaluated (IC50 = 34 μM; Fig. S-2B, Table S-1), it was selected for lead optimization due to its chemical tractability, low “staurosporine shift,” and allosteric NMR “fingerprint.” Importantly, we were able to confirm the binding of NPPB to both the allosteric pocket and ATP-binding site by X-ray crystallography (Fig. 2, Table S-2, Table S-3, PDB ID 7RWE). In the allosteric site, the carboxylate on the phenyl ring establishes a salt bridge with the catalytic lysine (Lys33) and engages with the main chain NH groups of the DFG residues Asp145 and Phe146, which are the same residues that interact with the sulfonate of ANS. In the ATP site, the carboxylate establishes an H-bond with the main chain NH of Leu83 of the hinge region, which presumably is the driving force for interaction with the ATP site. Both NPPB molecules lacked contiguous electron density (Fig. S-4), and the crystallographic B-factors using full occupancy refinement were 120 and 130% higher than the average B-factor for all protein atoms, likely reflecting the weak binding potential of NPPB for CDK2. By comparison, the electron density of the more potent indole-containing inhibitors of this study was well defined for almost all ligand atoms, and the B-factor was as low as 79% of the protein average.
Fig. 2. Hit compound NPPB binds to an allosteric pocket in CDK2.
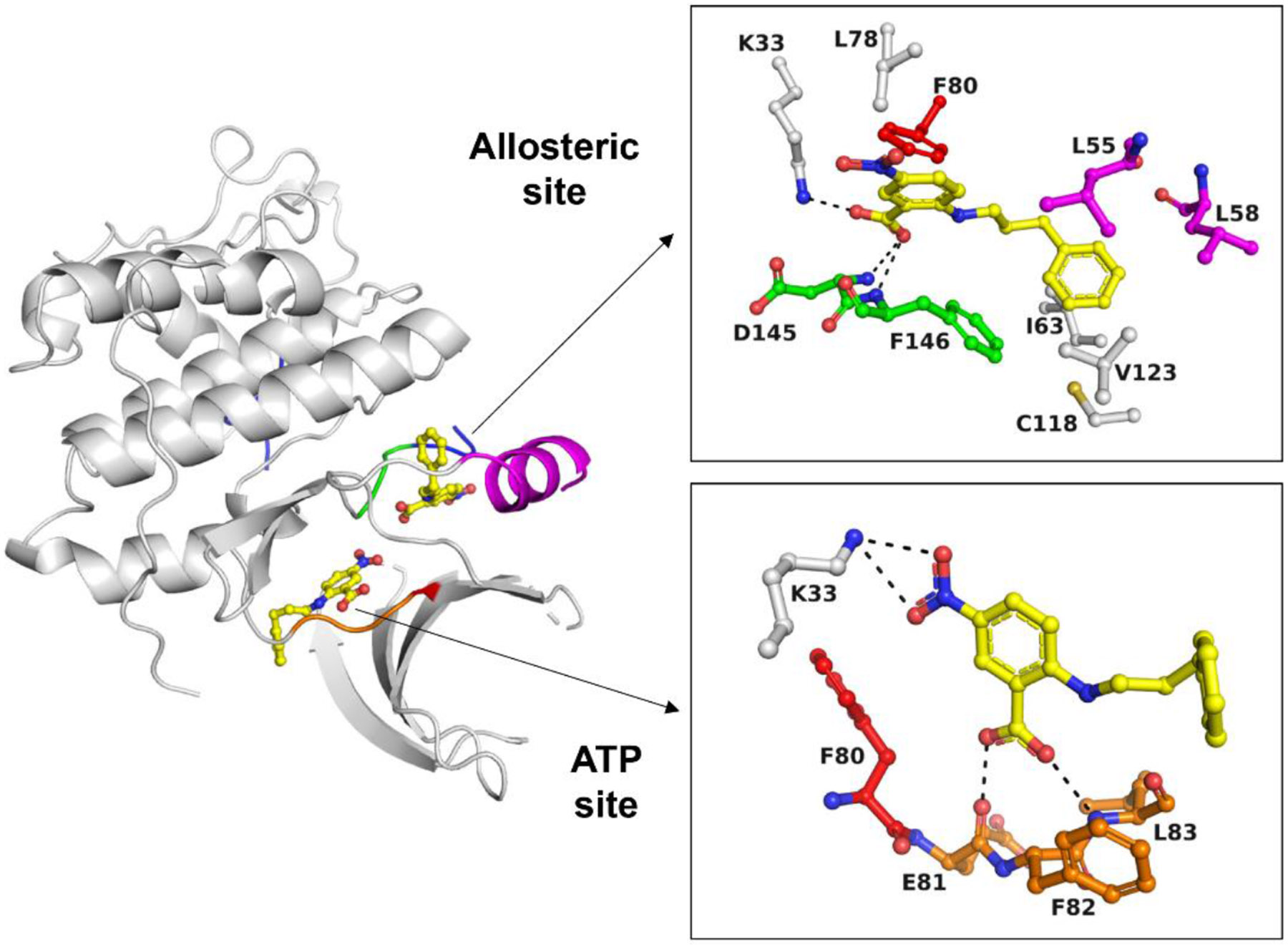
Hit NPPB binds to both the ATP site and the adjacent allosteric pocket of CDK2 (PDB ID 7RWE). For the allosteric site, all residues in the immediate vicinity of NPPB (d < 4 Å) are shown. For the ATP site, only the hinge region and Lys33 are shown. The different colors denote the DFG motif (green), the C-helix (magenta), the gatekeeper residue (red), the hinge region (orange) and the activation loop (blue). H-bonding and polar interactions are indicated as black dotted lines.
Using NPPB as our hit to be optimized, we next prepared TW-8-67-2 (Fig. 1) using nucleophilic aromatic substitution (SnAr) followed by hydrolysis (Scheme 1, method A). Most anthranilic acid analogs were prepared in this manner but due to commercial availability and the desire to explore electron-rich aromatic systems, a one-step Ullmann coupling was employed to make some analogs (Scheme 1, method B). In our p-Cl-ANS assay, TW-8-67-2 has a 10-fold improved affinity for the allosteric site (p-Cl-ANS IC50 = 4.6 ± 1.0 μM) compared to the initial hit compound NPPB. As described previously, the crystal structure of TW-8-67-2 bound to CDK2 revealed an additional H-bond interaction made by the indole nitrogen in addition to the increased rigidification of the otherwise flexible alkyl linker.23 Additionally, TW-8-67-2 only bound the allosteric pocket, unlike NPPB, which bound both the allosteric and ATP-binding site (Fig. 2). A plausible explanation for the lack of binding potential of indole-containing analogs for the ATP site is steric hindrance imposed by residues of the front specificity pocket (Fig. S-5). The less bulky and less restrained alkyl phenyl moiety of NPPB fits this site without steric hindrance. The affinity of TW-8-67-2 was largely unchanged in the presence of staurosporine, the known high affinity ATP-binding site ligand (p-Cl-ANS IC50 = 5.7 ± 1.3 μM), further confirming the allosteric nature of how TW-8-67-2 binds. To orthogonally assess the binding of these compounds, an isothermal titration calorimetry (ITC) assay was developed. For TW-8-67-2, the ITC KD = 5.2 μM,23 comparable to the p-Cl-ANS IC50 = 4.6 ± 1.0 μM (Fig. 3).
Scheme 1.
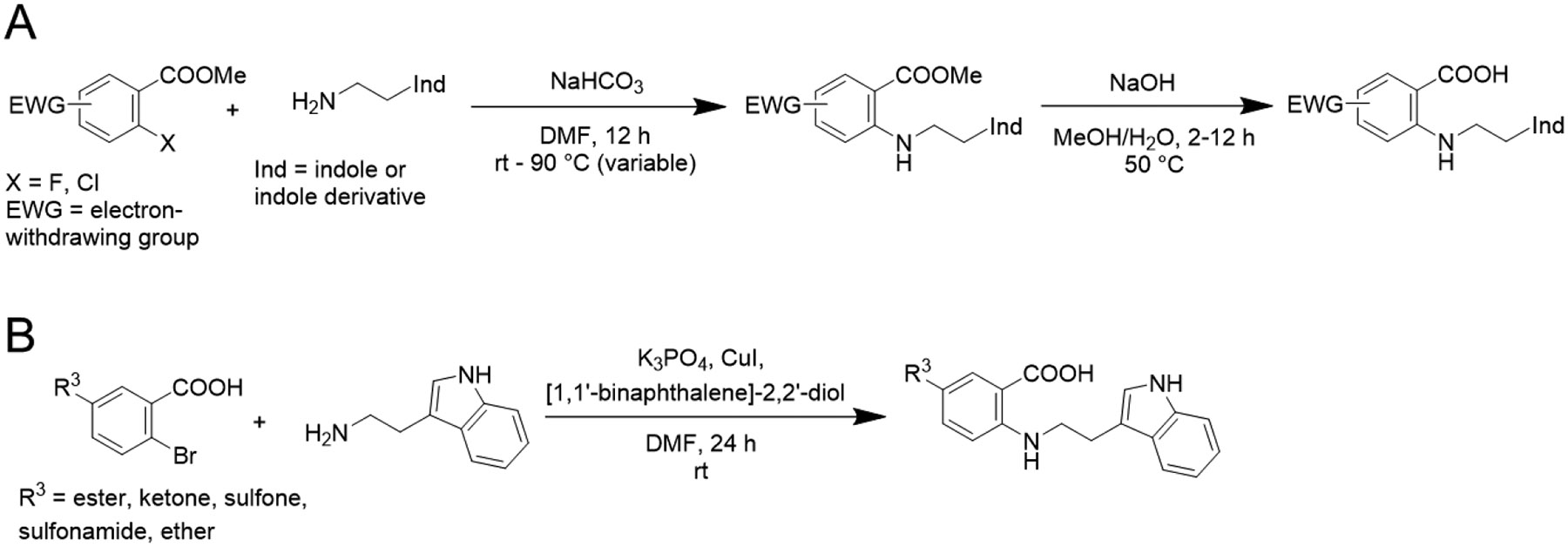
Anthranilic acid analogs were synthesized using nucleophilic aromatic substitution (SnAr) followed by hydrolysis (method A) or by one-step Ullmann coupling (method B)
Fig. 3. Anthranilic acid TW-8-67-2 binds only the allosteric pocket with improved affinity in the p-Cl-ANS assay.
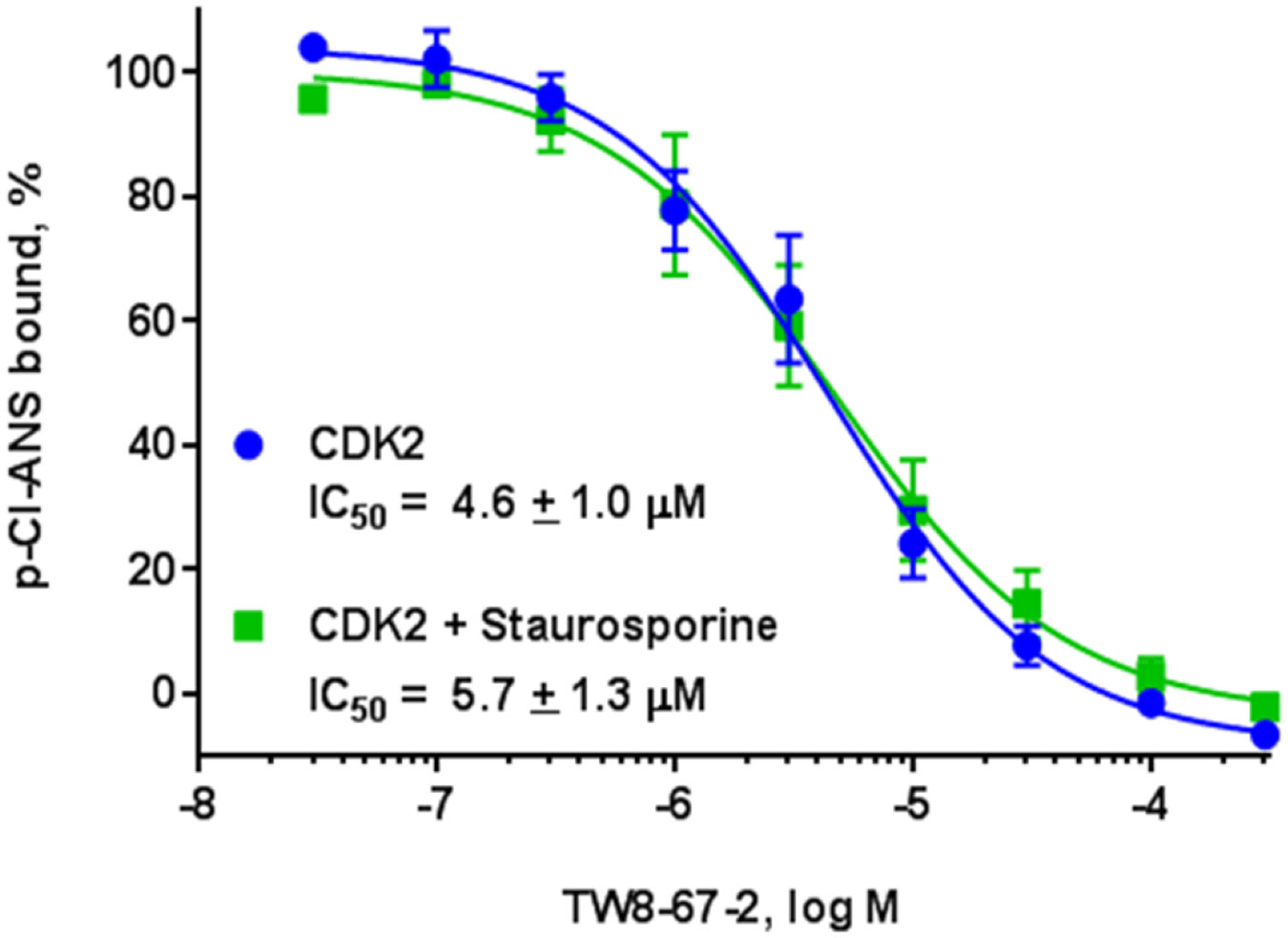
In the presence of ATP-binding site inhibitor staurosporine, its affinity is relatively unchanged.
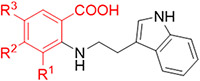
To further explore the structure-activity relationship (SAR) of allosteric binding potential of these anthranilic acids, we first examined how tolerant the different positions on the phenyl ring were to changed substitution (Table 1), and how important substitution was at the R3 position given that only one of the two preliminary screening hits contained a substituent at this position (Fig. 1). We chose substituents to explore at the R1, R2, or R3 positions based on commercial availability of starting materials that are compatible with our developed synthetic methods (Scheme 1). An analog without any substituents at the R1, R2, or R3 positions (compound 1) did not show binding to the allosteric site in the p-Cl-ANS assay and showed weak binding by ITC. Nitro and trifluoromethyl substitutions at the R1 position did not demonstrate significant binding to the allosteric site (compounds 2 and 3). However, a methyl group at the R1 position combined with a nitro group at the R3 position yielded compound 4 with moderate affinity to the allosteric site and 7-fold reduced affinity compared to the parent compound TW-8-67-2. Analogs with substitutions at the R2 position of the phenyl ring (compounds 5-10) showed improved affinity to the allosteric site compared to analogs with similar substitutions at the R1 position, but once again the affinity was strengthened by the presence of a nitro substituent at the R3 position (for example, compare compounds 7 and 8). Despite this, no analog with a substitution at the R2 position showed substantially improved affinity compared to parent compound TW-8-67-2. However, the presence of a substituent at the R3 position in general seemed to drive binding activity.
Table 1.
SAR of R1 and R2 positions of the phenyl ring of the anthranilic acid series
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | IC50 (p-Cl-ANS, μM) |
| TW-8-67-2 | H | H | NO2 | 4.6 |
| 1 | H | H | H | >300 |
| 2 | NO2 | H | H | 38 |
| 3 | CF3 | H | H | >300 |
| 4 | Me | H | NO2 | 38 |
| 5 | H | NO2 | H | 6.1 |
| 6 | H | CF3 | H | 260 |
| 7 | H | Br | H | 13 |
| 8 | H | Br | NO2 | 2.8 |
| 9 | H | OMe | NO2 | 4.6 |
| 10 | H | NH2 | NO2 | 42 |
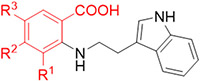
Next, we explored if replacement of the R3 position substituent with groups other than nitro could improve the affinity towards CDK2 (Table 2). A nitrile substituent diminished the affinity (compound 11), as did a methyl ester group more mildly (compound 12). However, the methyl amide, ketone, sulfone, and sulfonamide analogs (compounds 13-16) showed a substantial worsening in affinity. In contrast, large halogen atoms showed near equipotency to the nitro group (compounds 17 and 18). A trifluoromethyl group at the R3 position (compound 19) improved the affinity 4-fold, and a trifluoromethoxy group at this position (compound 20) showed a similar improvement in affinity. Additionally, we crystallographically confirmed the tolerance of functional groups that replace the nitro group, such as the methyl ester, trifluoromethyl, and the trifluoromethoxy group (co-crystal structures of compounds 12 and 20 with CDK2 in Fig. S-6 [PDB ID 7RXO] andS-7 [PDB 7S85] with conditions in Table S-2 and data in Table S-3). Replacing the fluorine atoms in the trifluoromethoxy group with hydrogens, which decreases the size of the substituent and its electron-withdrawing potential, seemed to decrease the affinity substantially (compounds 21 and 22). Because bulky and electron-withdrawing groups seemed to be advantageous for binding, we hypothesized the larger pentafluorosulfanyl (SF5) group may further improve affinity.27 Accordingly, the SF5 analog 23 showed a 10+-fold improvement in affinity compared to TW-8-67-2. We therefore pursued both the CF3 and SF5 groups in future analogs.
Table 2.
SAR of the R3 position of the phenyl ring of the anthranilic acid series
| ||
|---|---|---|
| Compound | R3 | IC50 (p-Cl-ANS, μM) |
| TW-8-67-2 | NO2 | 4.6 |
| 11 | CN | 15 |
| 12 |
|
10 |
| 13 |
|
>300 |
| 14 |
|
47 |
| 15 |
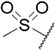
|
>300 |
| 16 |
|
>300 |
| 17 | Cl | 8.0 |
| 18 | Br | 4.0 |
| 19 | CF3 | 1.1 |
| 20 | OCF3 | 0.94 |
| 21 | OCF2H | 8.9 |
| 22 | OCH3 | 45 |
| 23 | SF5 | 0.30 |
We next turned to optimizing the indole ring in a similar manner as we had done with the phenyl ring (Table 3). We first set out to explore how important the NH was in the indole as a hydrogen bond donor by making methylated analog 24 and benzofuran analog 25. In both cases, there was a 4-6-fold loss in affinity, corresponding to a loss of a hydrogen bond at this position as seen crystallographically (Fig. 2). Next, a methyl group at the R2 position (compound 26) did not improve the binding of these compounds to the allosteric pocket. At the R4, R5, and R7 positions, a similar lack of substantial improvement in affinity was observed as methyl and chloro groups were introduced at these positions in a Topliss tree-like fashion (Table 3, compounds 27-32).28 In particular, a methyl group at the R5 position (compound 29) worsened binding substantially, as did methyl and chloro substituents at the R7 position (compounds 31 and 32). The R4 position in the crystal structure faces back towards the linker and phenyl ring so it is unlikely that small hydrophobic groups at this position, like those explored (compounds 27 and 28), would drastically affect the binding affinity towards the protein. However, substituents at the R6 position substantially improved the affinity for CDK2. For example, a R6 methyl group (compound 33) improved the affinity 5-fold while a chloro substituent (compound 34) improved the affinity 10-fold, with other halogens showing similarly large improvements in affinity (compounds 35 and 36). This aligns with the observed unoccupied pocket near this position in the crystal structure of TW-8-67-2 (Fig. 4A-B, Fig. S-7, Table S-2, Table S-3). An overlay of TW-8-67-2 and compound 34 bound to CDK2 reveals a near identical binding pose, despite a 10+-fold difference in affinity (Fig. 4C). The difference in affinity is ostensibly due to the space filling by the chlorine. Overall, halogens on the R6 position on the indole were found to best improve the affinity of these compounds towards CDK2. Other small substituents at this position, like nitrile, also showed improved affinity towards CDK2, such as in EF-4-084 and EF-4-177 (Table 4).
Table 3.
SAR of the indole ring of anthranilic acid series using a Topliss tree-like exploration of all the available synthetic sites
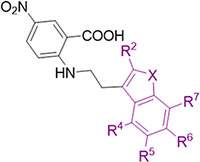
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | X | R2 | R4 | R5 | R6 | R7 | IC50 (p-Cl-ANS, μM) |
| TW-8-67-2 | NH | H | H | H | H | H | 4.6 |
| 24 | NMe | H | H | H | H | H | 21 |
| 25 | O | H | H | H | H | H | 32 |
| 26 | NH | Me | H | H | H | H | 16 |
| 27 | NH | H | Me | H | H | H | 5.0 |
| 28 | NH | H | Cl | H | H | H | 3.9 |
| 29 | NH | H | H | Me | H | H | 21 |
| 30 | NH | H | H | Cl | H | H | 5.8 |
| 31 | NH | H | H | H | H | Me | 24 |
| 32 | NH | H | H | H | H | Cl | 15 |
| 33 | NH | H | H | H | Me | H | 0.92 |
| 34 | NH | H | H | H | Cl | H | 0.44 |
| 35 | NH | H | H | H | F | H | 0.37 |
| 36 | NH | H | H | H | Br | H | 0.25 |
Fig. 4. Unoccupied pocket off the indole.
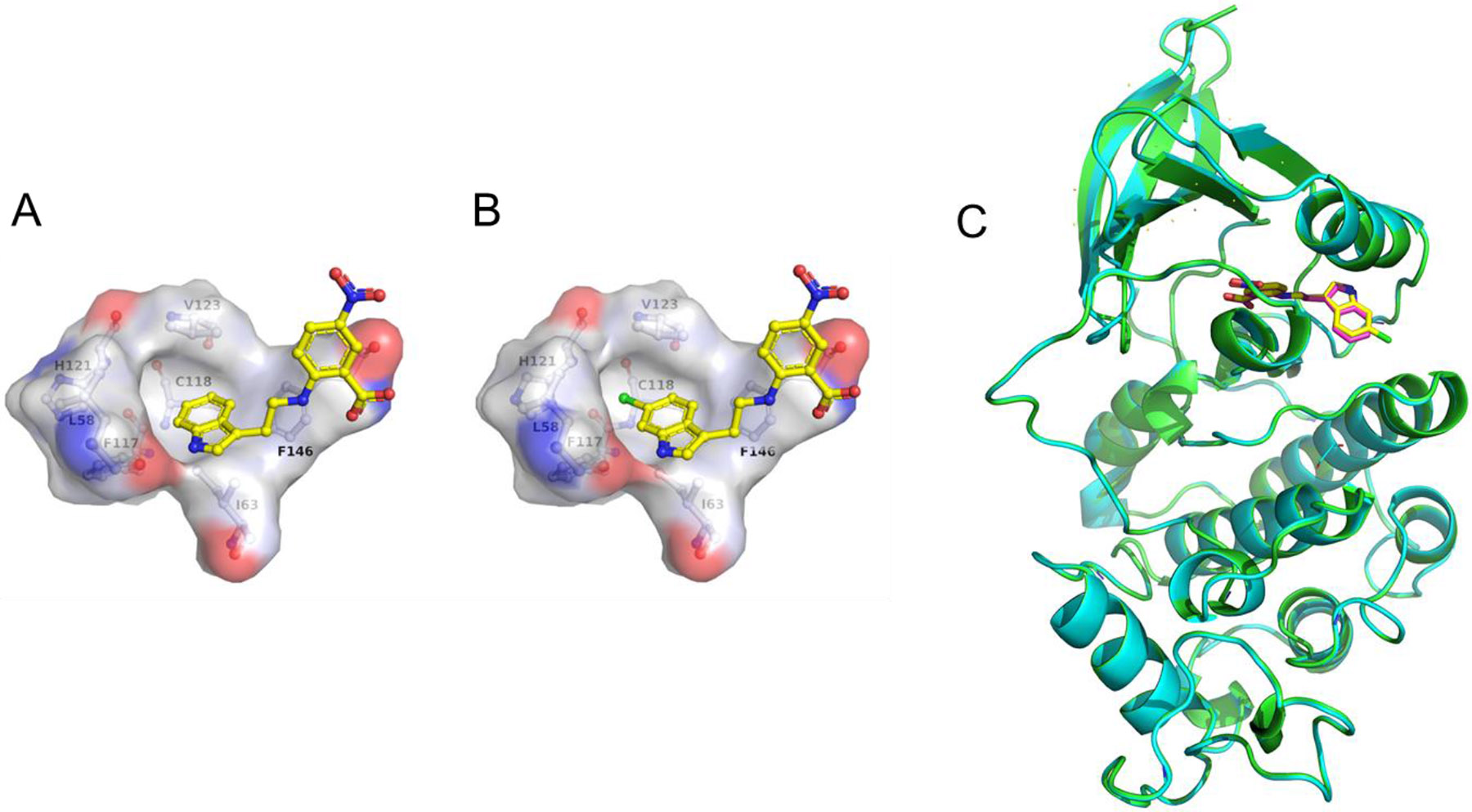
A. The crystal structure of TW-8-67-2 bound to CDK2 reveals an unoccupied hydrophobic pocket off the R6 position of the indole (PDB 7RWF). B. The chlorine atom of compound 34 is nested in this subsite (PDB 7S4T). C. Overlay of crystal structures of CDK2 bound to TW-8-67-2 (pink; CDK2 in cyan; PDB ID 7RWF) and compound 34 (yellow; CDK2 in green; PDB ID 7S4T).
Table 4.
Combining features from the phenyl ring SAR and indole ring SAR yield high affinity allosteric inhibitors of CDK2
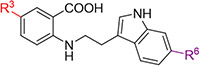
| ||||
|---|---|---|---|---|
| Compound | R3 | R6 | IC50 (p-Cl-ANS, nM) |
KD (ITC, nM) |
| TW-8-67-2 | NO2 | H | 4,600 | 5,200 |
| 19 | CF3 | H | 1,100 | 2,300 |
| EF-3-006 | CF3 | Br | 160 | 18 |
| EF-4-084 | CF3 | CN | 100 | 23 |
| 23 | SF5 | H | 300 | 260 |
| EF-4-177 | SF5 | CN | 87 | 7.4 |
With potency elements discovered on both the phenyl and indole rings, we next combined these elements to yield compounds with higher affinity. Because some high affinities were approaching the sensitivity limit of the p-Cl-ANS assay, ITC was used to measure and rank the relative affinities of compounds with KD < 100 nM (Table 4, Fig. S-8, or previously reported23). By combining some of the potency elements found from both the phenyl and indole ring SAR, ITC revealed that high affinity compounds EF-3-006, EF-4-084, and EF-4-177 had a KD < 50 nM. Combining the SF5 group at the R3 position of the benzene ring with the nitrile at the R6 position of the indole ring (EF-4-177) leads to the highest affinity compound by ITC of the series (KD = 7.4 nM) and is at the sensitivity limit of the instrument. This demonstrates the interchangeability of these features to modify the physicochemical properties of lead compounds.
We have previously shown that this series, in particular EF-3-006, binds CDK2 in a manner that is negatively cooperative with cyclin binding. EF-3-006 was selective for CDK2 over structurally similar CDK1, showing the promise of targeting the allosteric pocket to achieve selectivity.23 Lastly, in a cyclin-independent contraceptive context, EF-3-006 recapitulated the desired phenotype of Cdk2−/− and Spdya−/− spermatocytes, showing the therapeutic potential of this series.23 From these promising results, we next explored the pharmacokinetic and pharmacodynamic properties of these compounds for future study in animals and ultimately humans.
The metabolic stability of lead compound EF-3-006 had a short ½ life in both human and mouse microsomes. However, higher affinity compound EF-4-177 demonstrated a ½ life that was 8+-fold longer in humans and 7+-fold longer in mice (Fig. 5A) compared to EF-3-006. These long ½ lives were desirable given the therapeutic indications of anticancer and contraceptive purposes we are targeting. Additionally, oral administration of EF-4-177 in mice demonstrated a corresponding long exposure in both plasma and more specifically, testis for contraceptive uses (Fig. 5B). Coupled with observed low clearance, the overall stability of EF-4-177 demonstrates potential for multiple therapeutic uses that require selective targeting of CDK2 and the potential of this series as oral medications. To test this hypothesis, we conducted a longitudinal study with this compound to analyze sperm counts in a manner analogous to studies of other non-hormonal male contraceptive agents.29 After a 28 day exposure, EF-4-177 significantly decreased sperm count in CD-1 mice compared to vehicle treatment by approximately 45% (20.4 ± 6.1 x 106 versus 37.0 ± 7.8 x 106) with only slight toxicity observed in the first week of dosing (Fig. 5C, Fig. S-9). Coupled with the optimal pharmacokinetic properties of EF-4-177, this desirable phenotypic result affirms the potential of this series as future therapeutics.
Fig. 5. Metabolic profiles of high affinity allosteric CDK2 inhibitors.
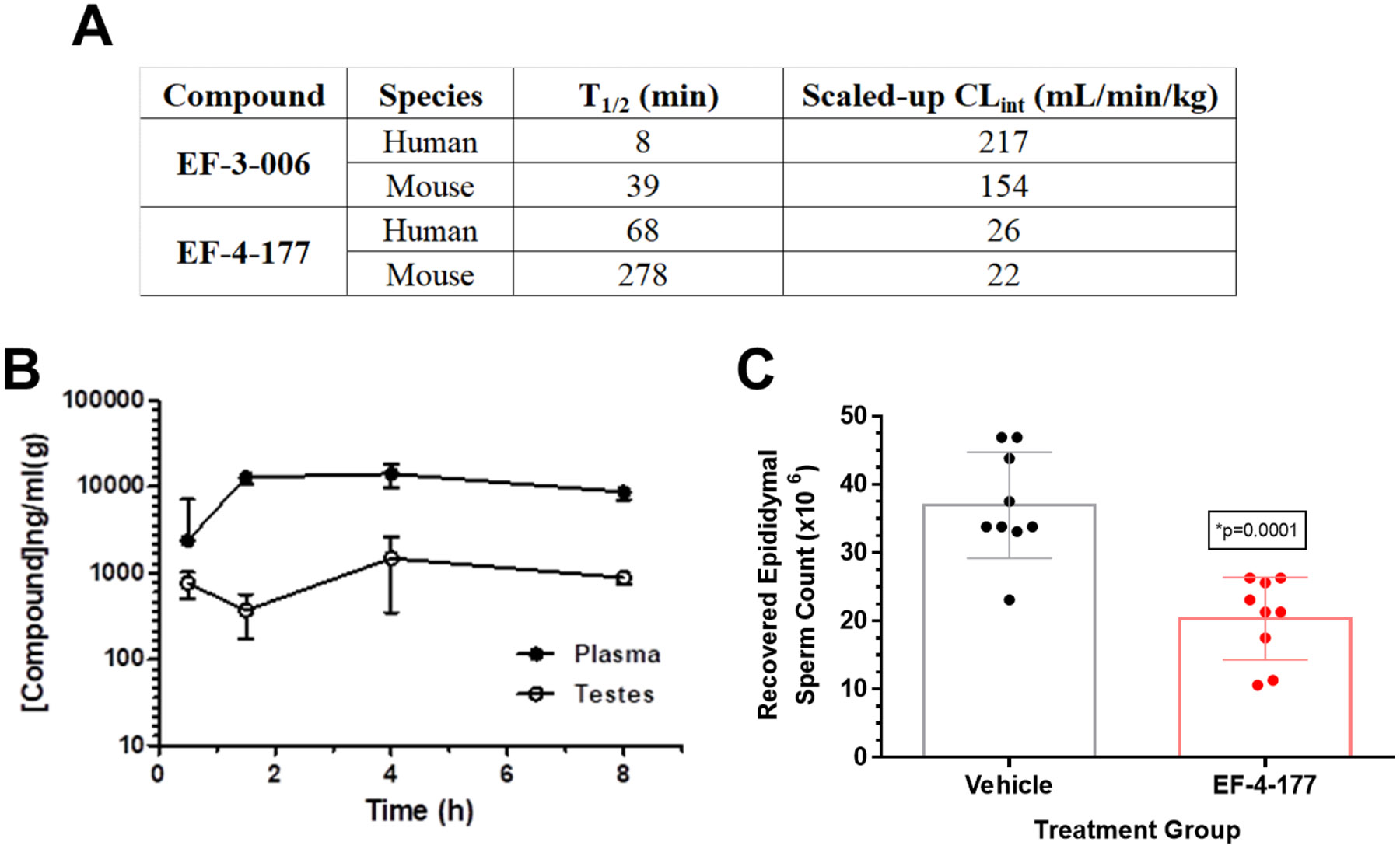
A. Metabolic stability in human and mouse microsomes for EF-3-006 and EF-4-177 (data provided by Pharmaron). B. EF-4-177 shows a long ½ life in mice and distribution into the testis. C. EF-4-177 significantly reduces sperm count after 28-day exposure in mice. Individual sperm counts are displayed in the scatter plot and summary statistics are expressed as the mean (x 106) ± S.D.
Discussion and Conclusion
We have developed and confirmed by crystallography, NMR, and biochemical methods the highest affinity allosteric CDK inhibitors known to date8, 30 with first-in-class compounds possessing low nanomolar affinities for CDK2. To discover the new anthranilic acid scaffold, we employed a combination of HTS and vHTS. Apart from the ANS scaffold, our anthranilic acid series is only the second scaffold to be structurally confirmed by crystallography to bind an allosteric pocket of CDK2 in a type III fashion. The anthranilic acid scaffold has the additional advantage of replacing the sulfonate in ANS that engages with the catalytic Lys33 with a more drug-like carboxylate. The improvement in affinity from ANS to EF-4-177 is 5,000-fold. This makes EF-4-177 the highest affinity known allosteric CDK inhibitor, spurring promise for other efficacious and selective inhibitors towards other CDKs involved in disease processes.
While our lead compounds are certainly more lipophilic compared to our starting compound TW-8-67-2, the lipophilic ligand efficiencies (pKD – log D7.4) of the compounds improved from 4.58 for TW-8-67-2 to 5.62 for EF-4-177. While EF-4-177 replaces the nitro group in TW-8-67-2 with a more hydrophobic SF5 group and has an additional nitrile at the R6 position of the indole, the improvement in affinity is substantial at 600+-fold. Crystal structures of these compounds reveal these allosteric ligands stabilize CDK2 in an inactive conformation of the kinase incompatible with cyclin binding.
We have previously described the mechanism of action and selectivity of EF-3-006 for CDK2 over other closely related kinases.23 While lead compound EF-3-006 showed promise in cellular studies as a contraceptive agent,23 its short ½ life was undesirable considering its therapeutic indication. However, by replacing the CF3 group with a larger SF5 group on the benzene ring and substituting a nitrile for a bromine on the indole, the metabolic stability improved substantially both in microsome incubation studies as well as in mouse pharmacokinetic studies for EF-4-177. Additionally, this compound adequately distributes into testis tissue and significantly reduced sperm count in a longitudinal mouse study as desired, in agreement with prior mechanistic studies of this series in spermatocyte chromosome spreads affecting CDK2 function.23 Combined, our findings show promise for further optimization of this series to yield a safe and efficacious allosteric CDK2 inhibitor for contraception and other therapeutic purposes.
Experimental Section
The expression and purification of CDK2 for the p-Cl-ANS displacement assay, crystallography, and ITC have been described previously.23 The liver microsome studies were completed by Pharmaron (Beijing, China).
p-Cl-ANS Displacement Assay
The p-Cl-ANS fluorescent binding assay was conducted as described previously.25 Compounds in DMSO were added to 384-well black clear bottom plates (Greiner 781091) using the Echo 550 (Beckman). SU9516 (Tocris; 50 μM final) and DMSO were used as controls defining 100% and 0% inhibition of p-Cl-ANS binding, respectively. For HTS, ~21,000 compounds (final 30 μM) were tested in the presence of staurosporine (Enzo Life Sciences; DMSO 0.3% final). 20 μL of assay buffer (150 mM NaCl, 50 mM HEPES, pH 7.5 containing 0.01% Triton and 2 mM DTT) was added to each well, the plate was shaken for 1 min, centrifuged at 2000 rpm for 5 min intervals until air bubbles were eliminated and then read on a CLARIOstar plate reader (BMG; Ex 388 nm, Em 455 nm) to obtain background fluorescence measurements for correction of intrinsic compound fluorescence. Absorbance was then measured at both the excitation and emission wavelengths for correction of the inner filter effect. Next, 5 μL of buffer containing p-Cl-ANS (synthesis reported previously;31 15 μM final) and staurosporine (final 5 μM) was added. Lastly, 5 μL of CDK2 (final 0.5 μM) in buffer was added (final volume of 30 μL) and the plate was shaken, centrifuged, incubated for 2 h at rt, and read as described above. The data were analyzed by correcting the raw fluorescence data using the pre-read fluorescence and absorbance data to minimize test compound fluorescence interference and inner filter effect as described previously.25 For HTS, hit confirmation was conducted with cherry-picked compounds from DMSO stocks (10 mM; DMSO 1% final) in 8-point dose response in duplicate in the absence and presence of staurosporine. Selected hits were newly repurchased and tested in the same manner (purchasing information in SI). IC50 values were calculated using the four-parameter logistic equation using Prism 6.0 (GraphPad). For all compounds in Tables 1-4, a method with 8-point dose response in duplicate, analogous to that done for HTS hit confirmation, was employed. The p-Cl-ANS IC50 values in those tables are reflective of single (n=1) independent experiments. For Table S-1, the number of independent replicates is noted next to each compound in the table.
Virtual High Throughput Screening
Two sets of vHTS were performed against the most interior allosteric ANS site in CDK2 (PDB ID 3PXQ): a first using the original in-house GPHR collection (224k compounds, 400k structures after LigPrep) and the second using newer additions to the GPHR collection (120k compounds, 320k structures after LigPrep). The GPHR collections include 10k compounds from various commercial kinase inhibitor libraries, 195k compounds from ChemBridge (DiverSet-CL library and EXPRESS-Pick collection), 56k from ChemDiv, 5k polymerase-targeting compounds, 7k peptidomimetics, and 10k natural products. Protein preparation, ligand preparation, and docking screening were performed using Protein Preparation Wizard, LigPrep, and Glide in Maestro (Schrödinger, LLC), respectively. Using HTVS in Glide, the top 12.5% scored compounds were advanced to SP docking, where the subsequent top 10% scored compounds were advanced to XP docking, and then the subsequent top 40% scored compounds were advanced to MM-GBSA scoring. MM-GBSA calculations were performed using Prime (Schrödinger, LLC). The top 10% scored compounds from the MM-GBSA analysis were selected (154 compounds from the first GPHR collection and 106 compounds from the second) and the 230 compounds that were commercially available for repurchase at the time of screening were tested in the p-Cl-ANS displacement assay as described above.
Crystallization
Purified CDK2 was buffer exchanged into 100 mM Na/K phosphate, pH 6.2, 2 mM DTT using PD10 columns (GE Life Sciences) and concentrated to 9 mg/mL. Crystallization was performed at 19 °C in hanging droplets by vapor diffusion, and crystals grew from 5% to 7.5% (v/v) PEG3350, 50 mM HEPES, pH 7.5 and 4.5 mg/mL CDK2. Each structure had only 1 crystal each. Most cocrystals were generated by in-diffusion of ligand-free crystals with inhibitors as detailed in Table S-2.
Data Collection and Refinement
X-ray diffraction data of CDK2 liganded with NPPB, 20, and 34 were collected on a Rigaku Micro-Max 007-HF X-ray generator, equipped with a CCD Saturn 944 system located in the Chemical Biology Core of the Moffitt Cancer Center. Data of CDK2 liganded with 12 were collected at the GM/CA beamline (23ID-B with Dectris PILATUS 3-6M detector, 23ID-D with Dectris EIGER 16M detector) at Argonne National Laboratory. Collection wavelength was 1.54178 Å for NPPB, 20, and 34 and was 1.03318 Å for 12. All data were collected at 93.2 K and processed with XDS and Aimless of the CCP4 suite.32 Initial phasing of all structures was done by molecular replacement using PDB deposited structure 4KD1 as the model in PHASER.33 The inhibitor geometrical restraints were obtained with Phenix Elbow.34 All structures were refined with Phenix,35 and model building was performed with Coot.36 Data collection and refinement statistics are shown in Table S-3.
HSQC NMR
The method of expression and purification of N15 labeled CDK2 have been described previously.25 NMR samples were prepared with 50 μM CDK2 and 500 μM roscovitine, plus 1 mM of allosteric compound. Trosy-HSQC spectra were collected on a Bruker Avance III 850 MHz spectrometer, using 2048 points in the proton dimension and 96 points in the indirect dimension. Data were analyzed with NMRPipe37 and SPARKY.38
Isothermal Titration Calorimetry (ITC)
The ITC method for CDK2 has been described previously.23 The inhibitor concentration for 23 and EF-4-177 in the syringe was 75 μM, and the CDK2 concentration in the sample cell was 5 μM. As before, a one-site binding model was used to fit the ITC data.
Pharmacokinetic Studies in Mice
All animal procedures in this study were approved by the University of Minnesota Institutional Animal Care and Use Committee. Animals were allowed to acclimate in their cages after arrival for at least 72 h and were allowed access to rodent chow and water ad libitum. The in-life portion of the study consisted of dosing 12 male CD-1 mice (6-8 weeks, Envigo, Indianapolis, IN) PO with 10 mg/kg of EF-4-177. At specified time points (30 min, 90 min, 2 h and 4 h) sets of 3 animals were bled, euthanized and testes harvested. Resulting plasma and tissue were frozen at ≤ −20 °C until processing and analysis by LC/MS/MS. Processing of testes consisted of weighing samples and adding 2x volume of cold H2O. The tissue mixtures were then homogenized using a Polytron PT 2500E (Kinematica AG, Luzern, Switzerland) with a 5 mm probe (PT-DA 05/2 EC-E85, Kinematica AG). Homogenates were stored at ≤ −20 °C until analysis.
LC/MS/MS Quantitation of Animal Samples
LC/MS/MS analysis was performed using a Quattro Ultima triple quadrupole mass spectrometer (Waters, Milford, MA) coupled with a Waters Acquity Ultra Performance Liquid Chromatography system by operating in electrospray in the negative ion mode. Mass-to-charge ratio (m/z) transitions for the analyte (EF-4-177, MW = 431.4 Da) and internal standard (MW = 485.3 Da) were determined to be 429.84 > 126.76 and 484.64 > 126.81, respectively. For liquid chromatographic separation, an isocratic elution (30% H2O/0.1% formic acid, 70% acetonitrile/0.1% formic acid) at was performed using a Synergi Polar-RP column (75 × 2 mm, 4 μm; Phenomenex, Torrance, CA) with a flow rate of 0.5 mL/min. The total run time was 4.0 min. Standard curve working solutions were prepared by serial dilution of stock solutions (made from 1 mg/mL master stock in DMSO) with 100% acetonitrile to obtain working concentrations at 0.21, 0.62, 1.86, 5.56, 16.67, 50, 150 and 450 ng/μl. Calibration standards were prepared by spiking 50 μL of blank with freshly prepared working solutions to achieve standards with concentrations of 0.23, 0.7, 2.1, 6.2, 18.6, 55.6, 166.7, 500, 1500 and 4500 ng/mL. Quality control samples were prepared by spiking 50 μL of blank with freshly prepared working solutions of 0.21, 0.62, 5.56 and 450 ng/μL to obtain the limit of quantitation, low quality control, medium quality control, and high-quality control. Plasma and tissue samples (50 μL) were placed in a 1.5 mL Eppendorf tube, 100 μL 100% acetonitrile was added with a spiked amount of internal standard, and the mixture was vortexed. Samples were left to stand at 4 °C for at least 30 min and then microcentrifuged at 16,000 x g for 5 min and the supernatant was recovered. Supernatants (100 μL) were transferred to a clean tube and evaporated to dryness under a nitrogen stream at 25 °C for approximately 10-15 min. Samples were reconstituted with 100 μL of HPLC elution buffer (70% H2O/0.1% Formic Acid, 30% acetonitrile/0.1% Formic Acid). Processed samples were then transferred to 12 x 32 mm glass vials with 400 μL inserts and loaded into 96-vial auto-sampler plates from where 7.5 μL was injected into the LC/MS/MS system. Sample analysis was performed using MassLynx Software (v4.1, Waters, Milford, MA). Data are expressed as the mean ± standard deviation from the 3 replicate animals.
Sperm Count Analysis
EF-4-177 was formulated in 25% Carbitol (2-(2-Ethoxyethoxy) ethanol, Sigma-Aldrich, St. Louis, MO) in water and orally gavaged (10 mg/kg) daily into mice (CD-1, male, 31.6 ± 2.1 g, Envigo, Indianapolis, IN) for 28 consecutive days (n=9). Vehicle control animals received 25% Carbitol only (n=9). Body weights were recorded just prior to dosing.
One day following the final dose of test compound, animals were euthanized and all components of the epididymis (cauda, corpus and caput) were separately harvested. The epididymal tissue was incubated in 1 mL Whitten’s Solution (100 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.7 mM CaCl2, 0.8 mM pyruvic acid, 4.8 mM sodium lactate, 20 mM Hepes; pH 7.2-7.4) at 37 °C for 15-20 min to allow viable sperm to swim out. 50 μL of the resulting sperm suspension was then diluted into 0.45 mL 10% neutral buffered formalin. Spermatozoa were counted in a hemocytometer and results are expressed as sperm count (x 106). Statistical analysis comparing data from the vehicle and EF-4-177 groups was performed using unpaired Student’s t-tests (GraphPad Prism, San Diego, CA).
Compound synthesis
All tested compounds were determined to have at least 95% purity as determined by qHNMR. The synthesis of compound p-Cl-ANS was published previously,31 as were the syntheses of TW-8-67-2, 19, EF-3-006, and EF-4-084.23 Most anthranilic acids were prepared using SnAr followed by hydrolysis using commercial methyl esters and tryptamine derivatives. Compounds 13, 23, and EF-4-177 required additional synthesis to produce the desired methyl ester. The synthesis to produce the desired tryptamine derivative for EF-4-177 has been described previously.39 NMR spectra and purity data of previously uncharacterized compounds can be found in the Supporting Information.
General method of SnAr followed by hydrolysis to yield anthranilic acids compounds 1-11, 17-18, 24-36
Commercial electron-deficient methyl 2-chloro- or 2-fluorobenzoic esters (0.30 mmol) were dissolved in DMF (5 mL) together with commercial tryptamine derivatives (1.1 equiv, in their neutral form or as HCl salts) and sodium bicarbonate (3-4 equiv). Potassium carbonate was often an acceptable substitute for NaHCO3 and was sometimes used instead. The reaction temperature employed varied from room temperature to 90 °C (room temperature for 2, 4, 8, 24-36; 70 °C for 3, 5-7, 9-11, 17-18; 90 °C for 1) and was dependent on the degree of electron-deficiency of the benzene ring and whether the substitution patterns around the benzene ring stabilize the Meisenheimer complex formed as a reactive intermediate in SnAr. The solution was stirred for 12 h and then 1 M HCl (aq) was added until pH < 7 and extracted two times into DCM. The DCM solvent was removed via rotary evaporation and the crude product was dissolved in MeOH to proceed to the hydrolysis step without purification. Water was added to the MeOH mixture until a 1:1 ratio of solvents was reached. A solution of 5 M NaOH (aq, 1 mL) was added, and the hydrolysis proceeded at 50 °C for 12 h and monitored by TLC for completion. The mixture was cooled to room temperature and the solvent was removed by rotary evaporation. 1 M HCl (aq) was added until pH < 4 and the solution was extracted two times into EtOAc. The EtOAc layer was dried with MgSO4 and loaded onto Celite®. Subsequent purification was accomplished by silica gel flash column chromatography with a gradient of EtOAc in hexanes of 0-100%, eluting around 60% EtOAc as the free carboxylic acid. The solvent was removed by rotary evaporation to furnish the final product.
Ullmann coupling to yield compounds 12, 14-16, 20-22
The respective 2-bromobenzoic acid (0.9 mmol), tryptamine (1.5 equiv), potassium phosphate (2 equiv), copper (I) iodide (0.1 equiv), [1,1'-binaphthalene]-2,2'-diol (0.2 equiv) were added to anhydrous DMF (8 mL) and stirred at rt for 24 h under nitrogen. The reaction was then filtered through Celite®, whereby 1M HCl (aq) was added dropwise until pH < 4. The product was extracted three times into EtOAc. The EtOAc layer was then dried with MgSO4, filtered, and concentrated under reduced pressure. The remaining residue was purified by silica flash column chromatography with a gradient of EtOAc in hexanes of 0-100% over 7 min, eluting around 60% EtOAc as the free carboxylic acid. The solvent was removed by rotary evaporation to furnish the final product.
Procedure to yield compound 13
Compound 13 was made via amide coupling of methylammonium chloride (1.1 equiv) with the commercial 4-fluoro-3-(methoxycarbonyl)benzoic acid (0.5 mmol) using hexafluorophosphate azabenzotriazole tetramethyl uronium (1 equiv) and triethylamine (3 equiv) in DMF (5 mL) at room temperature, stirred for 1 h. Then 1 M HCl (aq) was added until pH < 7 and the intermediate was extracted two times into EtOAc, dried over MgSO4, and loaded onto Celite®. Purification of the intermediate methyl ester was accomplished by silica gel flash column chromatography with a gradient of EtOAc in hexanes of 0-100% over 7 min, eluting at 70% EtOAc. Next, the methyl ester and tryptamine (1.1 equiv) were dissolved in DMF (5 mL) and NaHCO3 (3 equiv) was added. The reaction mixture was stirred for 12 h at 70 °C in an SnAr step as described above for the “General method of SnAr followed by hydrolysis to yield anthranilic acids.” After extraction into EtOAc from 1 M HCl (aq), a similar hydrolysis step was performed to yield compound 13, which was worked up and purified as the free carboxylic acid via flash column chromatography in 0-30% MeOH in DCM in 5 min, eluting at 25% MeOH.
Preparation of methyl 2-fluoro-5-(pentafluoro-λ6-sulfaneyl)benzoate for the synthesis of compounds 23 and EF-4-177
To 2-Fluoro-5-(pentafluoro-λ6-sulfaneyl)benzoic acid (0.50 mmol) was added methyl iodide (5 equiv) in DMF (5 mL) under anhydrous conditions together with K2CO3 (4 equiv) and was stirred at 40 °C for 12 h. The reaction was extracted into DCM twice and dried over MgSO4. Subsequent purification was accomplished by silica gel flash column chromatography using a gradient of EtOAc in hexanes of 0-100% over 7 min, eluting around 30% EtOAc in 71% yield as an oil. Next, the general SnAr/hydrolysis method was employed to yield 23 and EF-4-177.
2-((2-(1H-Indol-3-yl)ethyl)amino)benzoic Acid (compound 1).
7% yield. Colorless solid. m.p. 158-161 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.23 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 6.53 (t, J = 7.5 Hz, 1H), 3.46 (t, J = 7.1 Hz, 2H), 3.03 (t, J = 7.0 Hz, 2H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-3-nitrobenzoic Acid (compound 2).
47% yield. Yellow solid. m.p. 75-77 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.70 (s, 1H), 8.05 (d, J = 7.6 Hz, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.54 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.16 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.73 (t, J = 7.9 Hz, 1H), 3.16 – 3.09 (m, 2H), 3.00 (t, J = 6.8 Hz, 2H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-3-(trifluoromethyl)benzoic Acid (compound 3).
52% yield. Yellow solid. m.p. 86-89 °C. 1H NMR (400 MHz, acetone-d6) δ 10.05 (s, 1H), 8.17 (d, J = 9.8 Hz, 1H), 7.77 (d, J = 9.8 Hz, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.22 (s, 1H), 7.09 (t, J = 7.6 Hz, 1H), 7.01 (t, J = 6.9 Hz, 1H), 6.89 (t, J = 8.2 Hz, 1H), 3.61 (t, J = 7.9 Hz, 2H), 3.09 – 3.14 (m, 2H). 19F NMR (376 MHz, acetone-d6) δ 120.3. 13C NMR (101 MHz, acetone-d6) δ 169.7, 151.2, 137.8, 137.0, 134.12, 134.06, 128.4. 123.8, 123.6, 122.2, 119.5, 119.2, 118.3, 117.3, 112.6, 112.2, 49.6, 27.4.
2-((2-(1H-Indol-3-yl)ethyl)amino)-3-methyl-5-nitrobenzoic Acid (compound 4).
81% yield. Yellow solid. m.p. 210-211 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.40 (s, 1H), 10.86 (s, 1H), 8.47 (d, J = 2.9 Hz, 1H), 8.01 (d, J = 2.9 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.07 (t, J = 7.6 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 3.82 – 3.77 (m, 2H), 3.00 (t, J = 7.0 Hz, 2H), 2.43 (s, 3H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-4-nitrobenzoic Acid (compound 5).
68% yield. Yellow solid. m.p. 200-202 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.59 (s, 1H), 8.00 (d, J = 8.6 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.43 (s, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 8.8 Hz, 1H), 7.24 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 3.54 (t, J = 7.2 Hz, 2H), 3.06 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.7, 150.6, 136.3, 133.1, 127.0, 123.1, 121.0, 118.3, 118.2, 111.4, 111.3, 107.5, 104.7, 43.0, 24.4.
2-((2-(1H-Indol-3-yl)ethyl)amino)-4-(trifluoromethyl)benzoic Acid (compound 6).
42% yield. Yellow solid. m.p. 191-196 °C. 1H NMR (400 MHz, acetone-d6) δ 11.27 (s, 1H), 10.06 (s, 1H), 8.21 (s, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.29 (s, 1H), 7.13 – 7.06 (m, 2H), 7.03 (t, J = 8.0 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 3.61 – 3.69 (m, 2H), 3.15 – 3.21 (m, 2H). 19F NMR (376 MHz, acetone-d6) δ 113.2. 13C NMR (101 MHz, acetone-d6) δ 169.6, 152.1, 137.8, 134.0, 128.4, 126.4, 123.8, 122.2, 119.5, 119.2, 113.2, 112.9, 112.2, 110.63, 110.60, 108.65, 108.61, 44.1, 25.6.
2-((2-(1H-Indol-3-yl)ethyl)amino)-4-bromobenzoic Acid (compound 7).
66% yield. Colorless solid. m.p. 201-202 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 1H), 10.87 (s, 1H), 8.01 (s, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.23 (s, 1H), 7.08 (t, J = 7.8 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.93 (s, 1H), 6.70 (dd, J = 8.4, 1.9 Hz, 1H), 3.48 (t, J = 7.0 Hz, 2H), 3.03 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.4, 151.5, 136.3, 133.4, 128.6, 127.0, 123.1, 121.0, 118.3, 116.8, 113.4, 111.4, 111.2, 109.0, 42.7, 24.4.
2-((2-(1H-Indol-3-yl)ethyl)amino)-4-bromo-5-nitrobenzoic Acid (compound 8).
56% yield. Yellow solid. m.p. 218-220 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.55 (s, 1H), 10.89 (s, 1H), 8.70 (s, 1H), 8.54 (s, 1H), 7.59 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 7.15 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 3.65 (q, J = 6.4 Hz, 2H), 3.05 (t, J = 6.9 Hz, 2H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-4-methoxy-5-nitrobenzoic Acid (compound 9).
21% yield. Yellow solid. m.p. 220-222 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 10.87 (s, 1H), 8.76 (s, 1H), 8.51 (s, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.22 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.21 (s, 1H), 3.84 (s, 3H), 3.64 (q, J = 6.5 Hz, 2H), 3.07 (t, J = 6.8 Hz, 2H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-4-amino-5-nitrobenzoic Acid (compound 10).
69% yield. Yellow solid. m.p. 221-223 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H), 10.89 (s, 1H), 8.61 (s, 1H), 8.39 (s, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.54 (s, 2H), 7.35 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 6.05 (s, 1H), 3.45 (q, J = 6.6 Hz, 2H), 3.06 (t, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 153.5, 149.9, 136.3, 132.6, 127.0, 123.1, 122.2, 121.0, 118.34, 118.31, 111.4, 111.1, 103.0, 93.2, 43.0, 24.0.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-cyanobenzoic Acid (compound 11).
24% yield. Light yellow solid. m.p. 247 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.12 (s, 1H), 10.87 (s, 1H), 8.53 (s, 1H), 8.07 (s, 1H), 7.68 (d, J = 11.1 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.23 (d, J = 2.3 Hz, 1H), 7.07 (t, J = 8.2 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 9.0 Hz, 1H), 3.63 – 3.53 (m, 2H), 3.04 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.5, 152.8, 136.9, 136.5, 136.3, 127.0, 123.2, 121.0, 119.6, 118.30, 118.28, 112.3, 111.4, 111.0, 110.2, 95.0, 42.6, 24.3.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(methoxycarbonyl)benzoic Acid (compound 12).
56% yield. Colorless solid. m.p. 233-234 °C. 1H NMR (400 MHz, acetone-d6) δ 10.09 (s, 1H), 8.63 (s, 1H), 8.55 (s, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.28 (s, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.91 (d, J = 9.0 Hz, 1H), 3.83 (s, 3H), 3.70 – 3.63 (m, 2H), 3.18 (t, J = 7.1 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 170.0, 166.8, 155.1, 137.8, 136.1, 135.4, 128.4, 123.8, 122.2, 119.5, 119.3, 116.5, 112.8, 112.2, 111.9, 109.8, 51.8, 44.1, 25.6.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(methylcarbamoyl)benzoic Acid (compound 13).
43% yield across three steps. Colorless solid. m.p. 245-248 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 8.55 (s, 1H), 8.36 (d, J = 2.3 Hz, 1H), 8.19 – 8.13 (m, 1H), 7.82 (dd, J = 8.8, 2.4 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.77 (d, J = 8.9 Hz, 1H), 3.51 (t, J = 7.1 Hz, 2H), 3.04 (t, J = 7.0 Hz, 2H), 2.73 (d, J = 4.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.1, 152.2, 136.2, 132.4, 131.6, 127.0, 123.1, 120.9, 119.7, 118.3, 111.4, 110.2, 42.8, 26.1, 24.6.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-acetylbenzoic Acid (compound 14).
54% yield. Light yellow solid. m.p. 228-229 °C. 1H NMR (400 MHz, acetone-d6) δ 10.09 (s, 1H), 8.60 (s, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 8.9 Hz, 1H), 7.40 (d, J = 7.5 Hz, 1H), 7.28 (s, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.4 Hz, 1H), 6.92 (d, J = 9.0 Hz, 1H), 3.68 (q, J = 6.7 Hz, 2H), 3.15–3.21 (m, 2H), 2.48 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 195.1, 170.2, 155.0, 137.8, 135.3, 134.6, 128.4, 125.1, 123.8, 122.2, 119.5, 119.3, 112.8, 111.8, 109.6, 44.1, 25.9, 25.7.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(methylsulfonyl)benzoic Acid (compound 15).
58% yield. Colorless solid. m.p. 224-226 °C. 1H NMR (400 MHz, acetone-d6) δ 11.38 (s, 1H), 10.08 (s, 1H), 8.61 (s, 1H), 8.45 (s, 1H), 7.84 (d, J = 9.2 Hz, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.29 (s, 1H), 7.12 (t, J = 6.9 Hz, 1H), 7.07 – 6.99 (m, 2H), 3.74 – 3.64 (m, 2H), 3.19 (t, J = 7.0 Hz, 2H), 3.05 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 169.5, 155.0, 137.8, 133.9, 133.4, 128.4, 126.8, 123.8, 122.3, 119.6, 119.3, 112.7, 112.6, 112.2, 109.7, 44.9, 44.1, 25.6.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(N-methylsulfamoyl)benzoic Acid (compound 16).
48% yield. Colorless solid. m.p. 233-237 °C. 1H NMR (400 MHz, acetone-d6) δ 10.09 (s, 1H), 8.52 (s, 1H), 8.38 (s, 1H), 7.77 (d, J = 11.3 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.29 (s, 1H), 7.11 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 6.9 Hz, 1H), 6.99 (d, J = 9.0 Hz, 1H), 6.11 (s, 1H), 3.72 – 3.63 (m, 2H), 3.19 (t, J = 7.0 Hz, 2H), 2.55 (d, J = 5.3 Hz, 3H). 13C NMR (101 MHz, acetone-d6) δ 169.6, 154.4, 137.8, 133.9, 133.1, 128.4, 125.0, 123.8, 123.7, 122.2, 119.5, 119.3, 112.8, 112.3, 112.2, 109.6, 44.1, 25.6.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-chlorobenzoic Acid (compound 17).
19% yield. Colorless solid. m.p. 206-210 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 7.70 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.41 – 7.31 (m, 2H), 7.22 (s, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.83 (d, J = 9.1 Hz, 1H), 3.47 (t, J = 7.0 Hz, 2H), 3.02 (t, J = 6.9 Hz, 2H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-bromobenzoic Acid (compound 18).
27% yield. Colorless solid. m.p. 204-209 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 7.83 (d, J = 2.5 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.48 (dd, J = 9.0, 2.6 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.21 (s, 1H), 7.07 (t, J = 6.9 Hz, 1H), 6.98 (t, J = 8.0 Hz, 1H), 6.78 (d, J = 9.1 Hz, 1H), 3.47 (t, J = 7.0 Hz, 2H), 3.02 (t, J = 6.9 Hz, 2H).
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(trifluoromethoxy)benzoic Acid (compound 20).
78% yield. Colorless solid. m.p. 177-179 °C. 1H NMR (400 MHz, acetone-d6) δ 11.22 (s, 1H), 10.05 (s, 1H), 8.07 (s, 1H), 7.78 (s, 1H), 7.64 (s, 1H), 7.38 (t, J = 9.0 Hz, 2H), 7.27 (s, 1H), 7.11 (t, J = 7.6 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.93 (d, J = 9.3 Hz, 1H), 3.58 – 3.64 (m, 2H), 3.16 (t, J = 7.1 Hz, 2H). 19F NMR (376 MHz, acetone-d6) δ 118.1. 13C NMR (101 MHz, acetone-d6) δ 169.3, 151.3, 137.8, 129.1, 128.4, 125.2, 123.7, 122.2, 110.1, 119.5, 119.3, 113.4, 113.0, 112.2, 110.2, 44.3, 25.7.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(difluoromethoxy)benzoic Acid (compound 21).
69% yield. Colorless solid. m.p. 179-180 °C. 1H NMR (400 MHz, acetone-d6) δ 11.11 (s, 1H), 10.06 (s, 1H), 7.97 (s, 1H), 7.71 (s, 1H), 7.65 (d, J = 7.0 Hz, 1H), 7.40 (d, J = 8.9 Hz, 1H), 7.31 – 7.24 (m, 2H), 7.11 (t, J = 6.9 Hz, 1H), 7.03 (t, J = 6.9 Hz, 1H), 7.01 – 6.60 (m, 2H), 3.59 (t, J = 7.2 Hz, 2H), 3.15 (t, J = 7.1 Hz, 2H). 19F NMR (376 MHz, acetone-d6) δ 95.7, 95.5. 13C NMR (101 MHz, acetone-d6) δ 169.6, 150.5, 140.2, 137.8, 128.5, 124.0, 123.7, 122.2, 119.5, 119.3, 118.0, 113.3, 113.1, 112.2, 110.3, 44.3, 25.8.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-methoxybenzoic Acid (compound 22).
89% yield. Yellow solid. m.p. 170-173 °C. 1H NMR (400 MHz, acetone-d6) δ 10.05 (s, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.45 (s, 1H), 7.39 (d, J = 7.5 Hz, 1H), 7.25 (s, 1H), 7.13 – 7.07 (m, 2H), 7.03 (d, J = 7.9 Hz, 1H), 6.83 (d, J = 9.0 Hz, 1H), 3.74 (s, 3H), 3.54 (t, J = 7.2 Hz, 2H), 3.12 (t, J = 7.2 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 170.1, 150.1, 147.6, 137.8, 128.5, 123.9, 123.6, 122.1, 119.4, 119.3, 115.7, 113.6, 113.4, 112.2, 110.4, 56.0, 44.6, 26.0.
2-((2-(1H-Indol-3-yl)ethyl)amino)-5-(pentafluoro-λ6-sulfaneyl)benzoic Acid (compound 23).
40% yield over three steps. Colorless solid. m.p. 237-238 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.31 (s, 1H), 10.89 (s, 1H), 8.40 (s, 1H), 8.15 (d, J = 2.9 Hz, 1H), 7.80 (dd, J = 9.5, 2.9 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 7.08 (t, J = 6.9 Hz, 1H), 6.98 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 9.5 Hz, 1H), 3.56 (t, J = 7.1 Hz, 2H), 3.05 (t, J = 7.0 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ 91.2, 90.8, 90.4, 90.0, 66.2, 65.8. 13C NMR (101 MHz, DMSO-d6) δ 168.53, 152.08, 136.25, 131.26, 129.29, 126.99, 123.15, 120.98, 118.29, 111.36, 111.27, 111.01, 108.46, 42.74, 24.28.
2-((2-(1-Methyl-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 24).
44% yield. Yellow solid. m.p. 220-223 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.45 (s, 1H), 8.87 (s, 1H), 8.63 (d, J = 2.8 Hz, 1H), 8.18 (dd, J = 9.4, 2.9 Hz, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.24 (s, 1H), 7.15 (t, J = 7.1 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 9.5 Hz, 1H), 3.75 (s, 3H), 3.64 (q, J = 6.7 Hz, 2H), 3.06 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.5, 136.7, 134.5, 129.4, 128.6, 127.6, 127.3, 121.2, 118.6, 118.4, 111.6, 110.2, 109.6, 109.3, 43.1, 32.3, 24.1.
2-((2-(Benzofuran-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 25).
18% yield. Yellow solid. m.p. 205-207 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.48 (s, 1H), 8.91 (s, 1H), 8.63 (d, J = 2.8 Hz, 1H), 8.18 (dd, J = 9.4, 2.9 Hz, 1H), 7.90 (s, 1H), 7.74 (d, J = 6.5 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.32 (td, J = 7.2, 1.4 Hz, 1H), 7.26 (td, J = 7.4, 1.1 Hz, 1H), 7.00 (d, J = 9.5 Hz, 1H), 3.72 (q, J = 6.7 Hz, 2H), 3.05 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.6, 154.5, 142.8, 134.7, 129.4, 128.6, 127.5, 124.4, 122.5, 119.9, 116.9, 111.7, 111.3, 41.7, 22.5.
2-((2-(2-Methyl-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 26).
79% yield. Yellow solid. m.p. 119-120 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.44 (s, 1H), 10.76 (s, 1H), 8.78 (s, 1H), 8.61 (d, J = 2.9 Hz, 1H), 8.14 (dd, J = 9.4, 2.9 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 6.99 (t, J = 6.8 Hz, 1H), 6.93 (t, J = 6.8 Hz, 1H), 6.88 (d, J = 9.5 Hz, 1H), 3.61 – 3.52 (m, 2H), 3.01 (t, J = 6.7 Hz, 2H), 2.34 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.5, 135.3, 134.4, 132.8, 129.3, 128.6, 128.0, 120.1, 118.2, 117.3, 111.6, 110.4, 106.5, 42.9, 23.2, 11.2.
2-((2-(4-Methyl-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 27).
93% yield. Yellow solid. m.p. 253-256 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.45 (s, 1H), 10.91 – 10.86 (m, 1H), 8.90 (s, 1H), 8.64 (d, J = 2.8 Hz, 1H), 8.17 (dd, J = 9.5, 2.9 Hz, 1H), 7.21 – 7.14 (m, 2H), 7.05 – 6.89 (m, 2H), 6.72 (d, J = 7.0 Hz, 1H), 3.65 (q, J = 6.6 Hz, 2H), 3.23 (t, J = 6.9 Hz, 2H), 2.65 (s, 3H), 2.62 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.5, 136.7, 134.5, 129.4, 128.6, 125.4, 123.4, 121.0, 120.0, 111.8, 111.6, 109.4, 44.1, 26.0, 20.1.
2-((2-(4-Chloro-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 28).
68% yield. Yellow solid. m.p. 170-174 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H), 8.67 (s, 1H), 8.00 (dd, J = 9.2, 2.6 Hz, 1H), 7.37 – 7.31 (m, 2H), 7.09 – 6.98 (m, 2H), 6.72 (d, J = 9.5 Hz, 1H), 3.57 (q, J = 6.6 Hz, 2H), 3.25 (t, J = 7.2 Hz, 2H).
2-((2-(5-Methyl-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 29).
33% yield. Yellow solid. m.p. 247-251 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.42 (s, 1H), 10.74 (s, 1H), 8.87 (s, 1H), 8.63 (d, J = 2.9 Hz, 1H), 8.17 (dd, J = 9.5, 2.8 Hz, 1H), 7.35 (s, 1H), 7.23 (d, J = 8.2 Hz, 1H), 7.19 (s, 1H), 6.96 (d, J = 9.5 Hz, 1H), 6.90 (d, J = 9.1 Hz, 1H), 3.63 (q, J = 6.6 Hz, 2H), 3.03 (t, J = 6.8 Hz, 2H), 2.37 (s, 3H).
2-((2-(5-Chloro-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 30).
37% yield. Yellow solid. m.p. 272-275 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.42 (s, 1H), 11.10 (s, 1H), 8.88 (s, 1H), 8.63 (d, J = 2.9 Hz, 1H), 8.16 (dd, J = 9.5, 2.9 Hz, 1H), 7.66 (s, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.34 (s, 1H), 7.07 (dd, J = 8.6, 2.1 Hz, 1H), 6.96 (d, J = 9.6 Hz, 1H), 3.66 – 3.60 (m, 2H), 3.04 (t, J = 7.1 Hz, 2H).
2-((2-(7-Methyl-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 31).
47% yield. Yellow solid. m.p. 253-257 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.42 (s, 1H), 10.86 (s, 1H), 8.86 (s, 1H), 8.63 (d, J = 2.8 Hz, 1H), 8.18 (dd, J = 9.5, 2.9 Hz, 1H), 7.42 (d, J = 7.2 Hz, 1H), 7.24 (s, 1H), 6.96 (d, J = 9.6 Hz, 1H), 6.92 – 6.86 (m, 2H), 3.67 – 3.61 (m, 2H), 3.06 (t, J = 6.9 Hz, 2H), 2.44 (s, 3H).
2-((2-(7-Chloro-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 32).
37% yield. Yellow solid. m.p. 275-279 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1H), 11.28 (s, 1H), 8.86 (s, 1H), 8.63 (d, J = 2.8 Hz, 1H), 8.18 (dd, J = 9.5, 2.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.34 (s, 1H), 7.17 (d, J = 7.5 Hz, 1H), 7.04 – 6.94 (m, 2H), 3.66 (q, J = 6.7 Hz, 2H), 3.08 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.5, 134.5, 133.0, 129.4, 129.0, 128.6, 124.7, 120.6, 119.4, 117.5, 115.8, 112.4, 111.7, 42.9, 24.2.
2-((2-(6-Methyl-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 33).
48% yield. Yellow solid. m.p. 222-224 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.42 (s, 1H), 10.72 (s, 1H), 8.85 (s, 1H), 8.63 (d, J = 2.9 Hz, 1H), 8.18 (dd, J = 9.4, 2.9 Hz, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.17 – 7.11 (m, 2H), 6.95 (d, J = 9.5 Hz, 1H), 6.82 (dd, J = 8.1, 1.5 Hz, 1H), 3.63 (q, J = 6.7 Hz, 2H), 3.03 (t, J = 7.0 Hz, 2H), 2.38 (s, 3H).
2-((2-(6-Chloro-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 34).
31% yield. Yellow solid. m.p. 249-251 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1H), 11.05 (s, 1H), 8.85 (s, 1H), 8.62 (s, 1H), 8.17 (d, J = 7.3 Hz, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.39 (s, 1H), 7.30 (s, 1H), 7.03 – 6.92 (m, 2H), 3.64 (q, J = 6.6 Hz, 2H), 3.05 (t, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 154.5, 136.6, 134.5, 129.4, 128.6, 125.9, 125.8, 124.5, 119.8, 118.7, 111.7, 111.3, 111.0, 109.1, 42.9, 24.1.
2-((2-(6-Fluoro-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 35).
42% yield. Yellow solid. m.p. 249-250 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.96 (s, 1H), 8.85 (t, J = 5.2 Hz, 1H), 8.62 (d, J = 2.9 Hz, 1H), 8.16 (dd, J = 9.5, 3.0 Hz, 1H), 7.58 (dd, J = 8.8, 5.4 Hz, 1H), 7.24 (s, 1H), 7.11 (dd, J = 10.1, 2.4 Hz, 1H), 6.94 (d, J = 9.5 Hz, 1H), 6.83 (td, J = 9.2, 2.4 Hz, 1H), 3.63 (q, J = 6.6 Hz, 2H), 3.04 (t, J = 7.0 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −122.3 (td, J = 9.7, 5.5 Hz).
2-((2-(6-Bromo-1H-indol-3-yl)ethyl)amino)-5-nitrobenzoic Acid (compound 36).
91% yield. Yellow solid. m.p. 253-257 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1H), 11.05 (s, 1H), 8.85 (s, 1H), 8.63 (d, J = 2.8 Hz, 1H), 8.17 (dd, J = 9.4, 2.9 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.53 (s, 1H), 7.33 – 7.26 (m, 1H), 7.11 (dd, J = 8.4, 1.8 Hz, 1H), 6.96 (d, J = 9.6 Hz, 1H), 3.64 (q, J = 6.6 Hz, 2H), 3.05 (t, J = 7.0 Hz, 2H).
2-((2-(6-Cyano-1H-indol-3-yl)ethyl)amino)-5-(pentafluoro-λ6-sulfaneyl)benzoic Acid (EF-4-177).
9.8% yield over five steps.39 Colorless solid. m.p. 210-212 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.31 (s, 1H), 11.50 (d, J = 2.6 Hz, 1H), 8.39 (s, 1H), 8.15 (d, J = 2.8 Hz, 1H), 7.85 (s, 1H), 7.84 – 7.75 (m, 2H), 7.57 (s, 1H), 7.31 (d, J = 8.3 Hz, 1H), 6.92 (d, J = 9.5 Hz, 1H), 3.58 (q, J = 6.2 Hz, 2H), 3.07 (t, J = 7.0 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ 91.5, 91.1, 90.7, 90.3, 89.9, 66.2, 65.7. 13C NMR (101 MHz, DMSO-d6) δ 168.5, 152.1, 139.0, 134.9, 131.2, 130.1, 129.3, 128.0, 121.0, 120.7, 119.6, 116.3, 112.4, 111.3, 108.5, 102.3, 42.7, 23.9.
Supplementary Material
Acknowledgment
Funding for this project was provided by NICHD: 5 R01 HD080431 and 1 R61 HD099743 and R01 GM121515. E.B.F. was supported by NIH/NIGMS (through training Grant T32 GM008244 and T32 GM132029), as well as by a NIH/NCI fellowship (F30 CA232303). We acknowledge support by the GM/CA beamlines at the Advanced Photon Source (APS), funded by the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006, P30GM138396, S10OD012289). The APS is a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-06CH11357. We thank the Moffitt Chemical Biology Core for use of the crystallization and X-ray facilities (National Cancer Institute grant P30-CA076292). We thank Pharmaron for conducting liver microsome metabolic stability assays and thank Reaction Biology for conducting kinase activity data for SI Table 2. We thank Dr. Tim Ward for his initial contributions to the project.
Abbreviations Used
- ANS
8-anilino-1-naphthalene sulfonic acid
- CDK1
cyclin-dependent kinase 1
- CDK2
cyclin-dependent kinase 2
- CLint
clearance
- D7.4
distribution constant at pH 7.4
- Em
emission wavelength
- EtOAc
ethyl acetate
- Ex
excitation wavelength
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ITC
isothermal titration calorimetry
- KD
dissociation equilibrium constant
- MeOH
methanol
- MM-GBSA
molecular mechanics with generalized Born and surface area solvation
- NPPB
5-nitro-2-(3-phenylpropylamino)benzoic acid
- p-Cl-ANS
8-(4-chloroanilino)-1-naphthalene sulfonic acid
- qHNMR
quantitative 1H NMR
- SnAr
nucleophilic aromatic substitution
- SP
standard precision
- vHTS
virtual high-throughput screen
- XP
extra precision
Footnotes
Supporting Information
Representative p-Cl-ANS data for screening hits, HSQC NMR data for allosterically bound ligands, additional crystal structures, ITC data, body weight of CD-1 mice exposed to vehicle and EF-4-177, crystallography conditions table and PDB codes, NMR spectra and qHNMR purity data of newly described compounds, molecular formula strings
PDB ID Codes
7RWE, 7RWF, 7S84, 7S85, 7S4T, 7RXO. Authors will release the atomic coordinates upon article publication.
Conflict of Interest Disclosure
E.B.F., N.W., J.E.H., and G.I.G. are listed as inventors for a US provisional patent application covering the compounds described in this paper.
References
- 1.Berthet C; Aleem E; Coppola V; Tessarollo L; Kaldis P, Cdk2 knockout mice are viable. Curr. Biol 2003, 13 (20), 1775–85. [DOI] [PubMed] [Google Scholar]
- 2.Ortega S; Prieto I; Odajima J; Martin A; Dubus P; Sotillo R; Barbero JL; Malumbres M; Barbacid M, Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet 2003, 35 (1), 25–31. [DOI] [PubMed] [Google Scholar]
- 3.Au-Yeung G; Lang F; Azar WJ; Mitchell C; Jarman KE; Lackovic K; Aziz D; Cullinane C; Pearson RB; Mileshkin L; Rischin D; Karst AM; Drapkin R; Etemadmoghadam D; Bowtell DD, Selective targeting of cyclin E1-amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clin. Cancer Res 2017, 23 (7), 1862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tadesse S; Anshabo AT; Portman N; Lim E; Tilley W; Caldon CE; Wang S, Targeting CDK2 in cancer: challenges and opportunities for therapy. Drug Discov. Today 2020, 25 (2), 406–13. [DOI] [PubMed] [Google Scholar]
- 5.Akli S; Van Pelt CS; Bui T; Meijer L; Keyomarsi K, Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res. 2011, 71 (9), 3377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faber EB; Wang N; Georg GI, Review of rationale and progress toward targeting cyclin-dependent kinase 2 (CDK2) for male contraception. Biol. Reprod 2020, 103 (2), 357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mikolcevic P; Isoda M; Shibuya H; del Barco Barrantes I; Igea A; Suja JA; Shackleton S; Watanabe Y; Nebreda AR, Essential role of the Cdk2 activator RingoA in meiotic telomere tethering to the nuclear envelope. Nat. Commun 2016, 7, 11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie Z; Hou S; Yang X; Duan Y; Han J; Wang Q; Liao C, Lessons learned from past cyclin-dependent kinase drug discovery efforts. J. Med. Chem 2022, 65 (9), 6356–89. [DOI] [PubMed] [Google Scholar]
- 9.Tadesse S; Caldon EC; Tilley W; Wang S, Cyclin-dependent kinase 2 inhibitors in cancer therapy: an update. J. Med. Chem 2019, 62 (9), 4233–51. [DOI] [PubMed] [Google Scholar]
- 10.Roskoski R Jr., Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res 2019, 139, 471–88. [DOI] [PubMed] [Google Scholar]
- 11.Wood DJ; Korolchuk S; Tatum NJ; Wang LZ; Endicott JA; Noble MEM; Martin MP, Differences in the conformational energy landscape of CDK1 and CDK2 suggest a mechanism for achieving selective CDK inhibition. Cell Chem. Biol 2018, 26, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diril MK; Ratnacaram CK; Padmakumar VC; Du T; Wasser M; Coppola V; Tessarollo L; Kaldis P, Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (10), 3826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santamaria D; Barriere C; Cerqueira A; Hunt S; Tardy C; Newton K; Caceres JF; Dubus P; Malumbres M; Barbacid M, Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448 (7155), 811–5. [DOI] [PubMed] [Google Scholar]
- 14.Behenna DC; Freeman-Cook KD; Hoffman RL; Nagata A; Ninkovic S; Sutton SC Preparation of aminopyrazolylcyclopentyl carbamate derivatives for use as CDK2 inhibitors. WO 2020157652, 2020. [Google Scholar]
- 15.Hoffman RL, PF-07104091: A CDK2 selective inhibitor of cyclinE amplified cancers. In AACR Annual Meeting 2021, Session DDT02—New Drugs on the Horizon: Part 3, 2021. [Google Scholar]
- 16.Betzi S; Alam R; Martin M; Lubbers DJ; Han H; Jakkaraj SR; Georg GI; Schonbrunn E, Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem. Biol 2011, 6 (5), 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin MP; Alam R; Betzi S; Ingles DJ; Zhu JY; Schonbrunn E, A novel approach to the discovery of small-molecule ligands of CDK2. Chembiochem 2012, 13 (14), 2128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludlow RF; Verdonk ML; Saini HK; Tickle IJ; Jhoti H, Detection of secondary binding sites in proteins using fragment screening. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (52), 15910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood DJ; Lopez-Fernandez JD; Knight LE; Al-Khawaldeh I; Gai C; Lin S; Martin MP; Miller DC; Cano C; Endicott JA; Hardcastle IR; Noble MEM; Waring MJ, FragLites-minimal, halogenated fragments displaying pharmacophore doublets. An efficient approach to druggability assessment and hit generation. J. Med. Chem 2019, 62 (7), 3741–52. [DOI] [PubMed] [Google Scholar]
- 20.Rastelli G; Anighoro A; Chripkova M; Carrassa L; Broggini M, Structure-based discovery of the first allosteric inhibitors of cyclin-dependent kinase 2. Cell Cycle 2014, 13 (14), 2296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y; Li S; Liu F; Geng L; Shu X; Zhang J, Discovery of novel nonpeptide allosteric inhibitors interrupting the interaction of CDK2/cyclin A3 by virtual screening and bioassays. Bioorg. Med. Chem. Lett 2015, 25 (19), 4069–73. [DOI] [PubMed] [Google Scholar]
- 22.Carlino L; Christodoulou MS; Restelli V; Caporuscio F; Foschi F; Semrau MS; Costanzi E; Tinivella A; Pinzi L; Lo Presti L; Battistutta R; Storici P; Broggini M; Passarella D; Rastelli G, Structure-activity relationships of hexahydrocyclopenta[c]quinoline derivatives as allosteric inhibitors of CDK2 and EGFR. ChemMedChem 2018, 13 (24), 2627–34. [DOI] [PubMed] [Google Scholar]
- 23.Faber EB; Tang J; Roberts E; Ganeshkumar S; Sun L; Wang N; Rasmussen D; Majumbar A; John K; Yang A; Khalid H; Hawkinson JE; Levinson NM; Schonbrunn E; Chennathukuzhi V; Harki DA; Georg GI, Development of allosteric, selective cyclin-dependent kinase 2 (CDK2) inhibitors that are negatively cooperative with cyclin binding and show potential as contraceptive agents. bioRxiv 2022. [Google Scholar]
- 24.Zhang J; Gan Y; Li H; Yin J; He X; Lin L; Xu S; Fang Z; Kim BW; Gao L; Ding L; Zhang E; Ma X; Li J; Li L; Xu Y; Horne D; Xu R; Yu H; Gu Y; Huang W, Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat. Commun 2022, 13 (1), 2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faber EB; Tian D; Burban D; Levinson NM; Hawkinson JE; Georg GI, Cooperativity between orthosteric inhibitors and allosteric inhibitor 8-anilino-1-naphthalene sulfonic acid (ANS) in cyclin-dependent kinase 2. ACS Chem. Biol 2020, 15 (7), 1759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fryklund J; Mattsson JP; Berglund ML; Helander HF; Larsson H, Effects of chloride transport inhibitors on intestinal fluid and ion transport in vivo and in vitro. Acta Physiol. Scand 1993, 149 (3), 365–76. [DOI] [PubMed] [Google Scholar]
- 27.Sowaileh MF; Hazlitt RA; Colby DA, Application of the pentafluorosulfanyl group as a bioisosteric replacement. ChemMedChem 2017, 12 (18), 1481–90. [DOI] [PubMed] [Google Scholar]
- 28.Topliss JG, A manual method for applying the Hansch approach to drug design. J. Med. Chem 1977, 20 (4), 463–9. [DOI] [PubMed] [Google Scholar]
- 29.Chung SS; Wang X; Roberts SS; Griffey SM; Reczek PR; Wolgemuth DJ, Oral administration of a retinoic Acid receptor antagonist reversibly inhibits spermatogenesis in mice. Endocrinology 2011, 152 (6), 2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Łukasik P; Baranowska-Bosiacka I; Kulczycka K; Gutowska I, Inhibitors of cyclin-dependent kinases: types and their mechanism of action. Int. J. Mol. Sci 2021, 22 (6), 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang N; Faber EB; Georg GI, Synthesis and spectral properties of 8-anilinonaphthalene-1-sulfonic acid (ANS) derivatives prepared by microwave-assisted copper(0)-catalyzed Ullmann reaction. ACS Omega 2019, 4 (19), 18472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 2011, 67 (Pt 4), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 2007, 40 (Pt 4), 658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriarty NW; Grosse-Kunstleve RW; Adams PD, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr 2009, 65 (Pt 10), 1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–32. [DOI] [PubMed] [Google Scholar]
- 37.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6 (3), 277–93. [DOI] [PubMed] [Google Scholar]
- 38.Lee W; Tonelli M; Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31 (8), 1325–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z; Cohen MP; Fisher MJ; Giethlen B; Gillig JR; McCowan JR; Miller SC; Schaus JM Preparation of N-(2-Arylethyl)benzylamines as antagonists of the 5-HT6 receptor. WO 2002078693, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.