

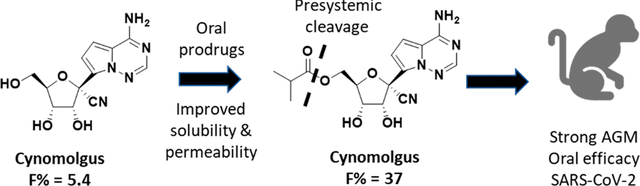
Abstract
Remdesivir 1 is an phosphoramidate prodrug that releases the monophosphate of nucleoside GS-441524 (2) into lung cells, thereby forming the bioactive triphosphate 2-NTP. 2-NTP, an analog of ATP, inhibits the SARS-CoV-2 RNA-dependent RNA polymerase replication and transcription of viral RNA. Strong clinical results for 1 have prompted interest in oral approaches to generate 2-NTP. Here, we describe the discovery of a 5′-isobutyryl ester prodrug of 2 (GS-5245, Obeldesivir, 3) that has low cellular cytotoxicity and 3–7-fold improved oral delivery of 2 in monkeys. Prodrug 3 is cleaved presystemically to provide high systemic exposures of 2 that overcome its less efficient metabolism to 2-NTP, leading to strong SARS-CoV-2 antiviral efficacy in an African green monkey infection model. Exposure-based SARS-CoV-2 efficacy relationships resulted in an estimated clinical dose of 350–400 mg twice daily. Importantly, all SARS-CoV-2 variants remain susceptible to 2, which supports development of 3 as a promising COVID-19 treatment.
Graphical Abstract
INTRODUCTION
Widespread SARS-CoV-2 infection has resulted in the current COVID-19 pandemic, the largest global pandemic since the Spanish Flu, with more than 750 million reported cases and almost 7 million deaths.1 Multiple vaccines have been developed to combat the pandemic, but due to the emergence of SARS-CoV-2 variants and continued transmission of the virus, the risk of hospitalization and severe disease remains a concern. Aside from vaccines, several treatment options have emerged including small molecule antivirals and antibodies that target different SARS-CoV-2 proteins to block viral replication and spread2–4. Small molecule antivirals include the nucleotide analog remdesivir (1, Veklury, Figure 1) and the nucleoside analog molnupiravir (Lagevrio), which both target the viral RNA-dependent RNA polymerase (RdRp) by different mechanisms.5–7 Nirmatrelvir is an inhibitor of the SARS-CoV-2 3CL main protease and is administered in combination with the P450 3A4 inhibitor ritonavir as a pharmacokinetic enhancer (combination product marketed as Paxlovid).8 In the outpatient setting, 5-day oral Paxlovid and 3-day intravenous (iv) 1 treatment have both demonstrated ≥87% effectiveness in reducing the hospitalization of high-risk SARS-CoV-2-infected patients.9,10 Despite the promising efficacy of these treatment options, limitations in their broad application exist. Nucleotide prodrug 1 requires iv administration, molnupiravir has safety concerns due to its mutagenic mechanism of action, and oral Paxlovid is contraindicated in some patients due to drug–drug interactions.7,11 These drawbacks indicate continued exploration to discover novel oral treatment options for COVID-19 is imperative.
Figure 1.
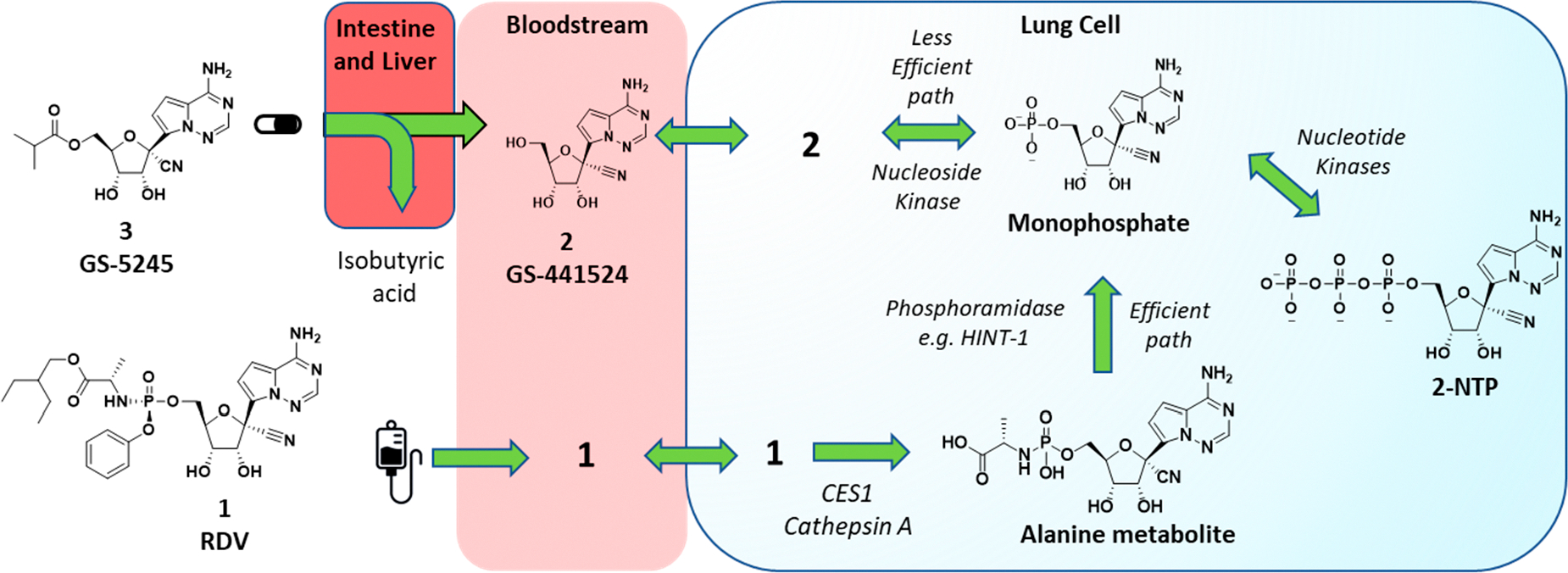
Metabolic activation pathways of prodrug 3 and phosphoramidate 1 to the common active 2-NTP metabolite in lungs. Ester prodrug 3 administered orally is metabolized presystemically in the intestine and liver to parent nucleoside 2 which then distributes into cells, including lung cells, where it is metabolized by a nucleoside kinase to the monophosphate. The monophosphate is then metabolized to the active metabolite, 2-NTP, by the action of nucleotide kinases. These steps are reversible and allow the phosphorylated metabolites generated inside all cells to also be broken down to parent nucleoside 2 and released back into systemic circulation. Phosphoramidate 1, following iv administration, rapidly distributes into many cell types, including lung cells, where it is metabolized irreversibly by the action of hydrolases (CES1, Cathepsin A) to the alanine metabolite and then to the same monophosphate by the action of phosphoramidases, e.g., HINT-1. The metabolism of 1 to the monophosphate inside cells is efficient and is the major pathway for generation of 2-NTP following administration of 1.
Nucleotide 1 is the first approved treatment for COVID-19 and is a chiral phosphoramidate prodrug bearing the 2-ethylbutyl-l-alanine and phenol pro-moieties.12 The promoieties are intracellularly cleaved to generate the monophosphate of the parent nucleoside 2 in different cells and tissues, including lung cells (Figure 1). Initially, the parent nucleoside 2 was designed as a potential treatment for hepatitis C virus but was then found through broad screening to be more potent toward respiratory syncytial virus (RSV).12–15 Phosphate prodrug exploration aimed at improving the formation of the active metabolite 2-NTP in lung cells for RSV then resulted in the initial discovery of 1.12 Following iv administration, the phosphoramidate prodrug 1 rapidly distributes into lung cells, in addition to other tissues, where it is efficiently metabolized by enzymes into the monophosphate nucleotide, effectively bypassing a rate-limiting first phosphorylation step of 2 to the same monophosphate intermediate (Figure 1). The monophosphate is then further metabolized to the active 2-NTP that inhibits the viral replication processes of multiple viral polymerases including HCV, RSV, Dengue, Ebola, Nipah, SARS-CoV, MERS, and more recently SARS-CoV-2.5,6,16,17 Prior to the pandemic, efficacy studies in animal models of RSV and SARS-CoV and MERS coronaviruses supported the potential utility of 1 as a treatment for these respiratory viruses.12,18–20 In the first clinical studies in patients hospitalized with COVID-19, iv administration of 1 was shown to shorten the hospitalization duration by 5 days (ACTT-1 trial) and improve survival of hospitalized patients with COVID-19.21–23 A more recent clinical study in high-risk nonhospitalized patients (PINETREE trial) demonstrated an 87% reduction in hospitalizations following a short 3-day iv administration of 1.9 This result indicates that delivery of the active 2-NTP metabolite within the lung during an early stage of SARS-CoV-2 infection can provide a meaningful clinical benefit for patients.
The clinical evidence for 1 as a COVID-19 treatment has prompted much interest in its potential for oral delivery. Unfortunately, the metabolic process that occurs in lung cells to generate the 2-NTP metabolite also occurs in other tissues, including the liver, which effectively hydrolyzes 1 following oral delivery. In nonhuman primates (NHPs), <1% oral bioavailability of the intact prodrug 1 was observed, suggesting that orally delivered 1 would be ineffective at providing sufficient intact prodrug in systemic circulation that is comparable to iv administration in order to be efficacious.12 Therefore, shortly after the pandemic started, we initiated programs aimed at orally delivering effective concentrations of 2-NTP into lungs leading to the selection of the 5′-isobutyryl ester prodrug 3 (GS-5245, Obeldesivir, ODV) as a clinical candidate (Figure 1). Here, we disclose the chemical structure, synthesis, and in vitro biophysical evaluation of 3 together with its oral properties and efficacy in the SARS-CoV-2 infection model in African green monkeys (AGMs). Importantly, the efficacy of 3 in the AGM model closely matched that of 1 which was dosed at clinically relevant exposures in the same AGM model, suggesting that oral 3 has the potential to match the efficacy of 1 in the clinic.24 In addition to this work, other groups have identified and characterized prodrugs of 2, including a 5′-isobutyryl ester 3.25 Efficacy was reported across several mouse models, but no plasma exposure–efficacy analysis was reported to allow human doses to be estimated. Here, we utilized exposure–efficacy data for 3 from the AGM model together with data from several independent SARS-CoV-2 models with an earlier prodrug 6 to develop an exposure–SARS-CoV-2 efficacy model. This model was then used to estimate a human daily dose of 3 that ranged from 350 to 400 mg twice daily. The results presented here supported the rapid advancement of 3 into the clinic as a potential oral treatment for COVID-19.
RESULTS AND DISCUSSION
In order to generate the 2-NTP metabolite from an orally delivered compound, we considered other monophosphate prodrugs besides 1 that had improved in vitro liver stability in addition to the parent nucleoside 2, which has a long human microsomal half-life of >8 h in vitro (Table 1). Following iv administration in NHPs, 2 demonstrated an in vivo half-life of 2.6–2.7 h, which is significantly longer than the 0.4–0.8 h half-life previously reported for phosphoramidate 1 in NHPs (Table 2).12 Allometric scaling of the iv data resulted in a projected human half-life for 2 of ~4 h, which supported a convenient twice-daily regimen for sustained plasma exposures of 2.
Table 1.
In Vitro Pharmacokinetic and Physical Properties of 2 and Its Prodrugs 3 and 5–7
| property | 2 | 3 | 5 | 6 | 7 |
|---|---|---|---|---|---|
| log D | <0.3 | 0.9 | <0.3 | 3.5 | 1.5 |
| Caco-2 AB (10−6 cm s−1)a | 0.1 | 1.0 | 1.4 | 22 | 4.0 |
| Caco-2 efflux ratiob | 12 | 2.9 | 0.5 | <1 | 3.0 |
| solubility, pH 2 (mg/mL) | ≤1 | ≥40 | 0.6 | ≥40 | |
| solubility, pH 7 (mg/mL) | ≤0.07 | ≥1.0 | 1.0 | <0.01 | 1.4 |
| solution stability, pH 2c | ≥94 | 89 | ≥96 | 82 | |
| solution stability, pH 7c | ≥96 | 96 | ≥98 | 79 | |
| GI S9 r/d/c/h (min) | <2/<2/<2/<2 | <2/226/38/28 | <2/<2/<2/<2 | <2/16/<2/<2 | |
| plasma r/d/c/h (min) | S/S/S/S | <3/412/9.7/29 | 3/564/146/70 | <2/182/7.2/5.5 | <2/111/9.2/4.7 |
| liver S9 r/d/c/h (min) | S/S/S/S | <2/4.3/<2/<2 | 10/25/5.3/19 | 2.9/<2/<2/<2 | <2/<2/<2/<2 |
| hrCES 1b/1c/2 (min) | 4/4.3/<2 |
Caco-2 assay performed in the presence of broad-spectrum esterase inhibitor phenylmethylsulfonyl fluoride for prodrugs 3–7.
Efflux ratio is calculated as basolateral-apical/AB in Caco-2 assay.
Percent remaining at 24 h, 40 °C. AB, apical to basolateral. GI, gasterointestinal; hr, human recombinant; CES, carboxyesterase; S, stable (>500 min); r = rat; d = dog; c = cynomolgus monkey; h = human.
Table 2.
In Vivo Plasma Pharmacokinetic Parameters of 2 and 3a
| 2 |
3 |
2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| route/cpd. | model | doseb | CL(L/h/kg) | Vss(L/kg) | T1/2 (h) | Cmax (μM) | AUC0–24 (μM·h) | AUC0–24 (μM·h) | lung [2-NTP] (nmol/g) | F% | F% (solid) |
| iv 2c | rat | 1 | 1.36 ± 0.02 | 1.66 ± 0.06 | 1.03 ± 0.02 | 1.78 ± 0.28 | 2.50 ± 0.04 | ||||
| dog | 1 | 0.18 ± 0.01 | 0.33 ± 0.06 | 1.82 ±0.11 | 13.5 ± 2.9 | 18.7 ± 1.5 | |||||
| cyno | 20 | 0.56 ± 0.05 | 1.03 ± 0.09 | 2.68 ± 0.07 | 79.2 ± 6.7 | 123 ± 11 | 0.41 ± 0.09 | ||||
| AGM | 20 | 0.49 ± 0.12 | 0.89 ± 0.26 | 2.63 ± 0.19 | 87.3 ± 37.4 | 147 ± 36 | 0.23 ± 0.13 | ||||
| oral 2c | ratd | 5 | 1.87 ± 0.59 | 1.11 ± 0.30 | 3.97 ± 1.19 | 32 ± 10 | |||||
| dog | 5 | 4.18 ± 0.64 | 27.3 ± 3.5 | 83.4 ± 14.4 | 89 ± 15 | ||||||
| cyno | 5 | 3.31 ± 0.65 | 0.54 ± 0.28 | 1.67 ± 0.76 | 5.4 ± 2.5 | ||||||
| AGM | 5 | 6.65 ± 1.82 | 0.36 ± 0.05 | 3.15 ± 0.94 | 8.6 ± 2.5 | ||||||
| oral 3c | rat | 6 | 2.02 ± 0.33 | 2.10 ± 0.49 | 7.57 ± 0.53 | BLQ | 63 ± 4 | 90 ± 15 | |||
| dog | 14.4 | 3.62 ± 0.23 | 57.8 ± 11.3 | 202 ± 44 | 0.40 ± 0.04 | 93 ± 20 | 67 ± 12e | ||||
| cyno | 11.7 | 2.62 ± 0.55 | 7.57 ± 6.07 | 21.6 ± 11.0 | 0.01 ± 0.01 | 37 ± 19 | 18 ± 3e | ||||
| AGM | 14.4 | 3.19 ± 0.19 | 6.81 ± 3.08 | 26.6 ± 12.1 | 0.02 ± 0.02 | 31 ± 14 | |||||
| AGM | 60 | 2.90 ± 0.04 | 22.5 ± 1.3 | 129 ± 2 | 0.03 ± 0.03 | 0.31 ± 0.09 | 36 ± 1 | ||||
| AGM | 86.5 | 2.98 ± 0.22 | 19.6 ± 9.8 | 136 ± 67 | 0.68 ± 0.75 | 27 ± 13 | |||||
Doses and parameters shown are for the solution formulation. All studies are n = 3 animals unless noted.
Units mg/kg.
Formulations described in the Experimental Section.
For n = 6 animals.
Solid dosing was performed with Form III crystalline material with a mean particle size distribution of 203 μm (D90 = 385 μm). Cyno, cynomolgus monkey; AGM, African green monkey; BLQ, below limit of quantification.
Despite the promising liver stability of 2, some significant challenges hindering its oral potential were identified. The first challenge was its inefficient intracellular metabolism from 2 to its monophosphate and then to 2-NTP as described in Figure 1.12 This less efficient metabolism has also been reported by independent groups and manifests as weaker antiviral activity across different lung cells in vitro compared to 1.12,24,26 The reduced 2-NTP formation in vitro was also observed in vivo through analyzing lung tissue samples harvested at 24 h from NHPs and ferrets dosed iv with either 2 or 1.12,24,27 These results suggest that high plasma exposures of 2 are likely necessary to drive sufficient lung 2-NTP formation in vivo to achieve comparable efficacy to 1. The second challenge was the poor physicochemical properties of 2 including a low apical to basolateral permeability in Caco-2 cells, a 12-fold efflux ratio, and low pH 7 solubility (Table 1). These physicochemical properties result in a low and variable oral bioavailability across preclinical species ranging from <8.6% in two species of NHPs to 89% in dogs (Table 2). The high oral bioavailability in dogs was not considered to be a reliable predictor of human oral potential given the low F% in NHPs and the “leaky intestine” phenomena in dogs as a result of their larger and more abundant paracellular junctions compared to human.28 Indeed, the 4′-azido cytidine ribonucleoside analog R1479 had a similar preclinical profile in NHPs and dogs to that of 2, but in humans, only an estimated 6–18% oral bioavailability was observed, consistent with the NHP data.29 Our strategy therefore focused on addressing the oral limitations of 2 with prodrugs that could effectively deliver high concentrations of 2 to overcome its metabolic deficiencies and drive efficacy. Finally, nucleoside analogs such as 2 are challenging and expensive to manufacture due to their structural complexity. The prospect that a prodrug could significantly reduce the proportion of the administered dose that is eliminated without absorption was highly appealing as a strategy to reduce the likely dose required.
Scheme 1 describes the synthesis for several ester-based prodrugs including 5′-mono esters 3 and 5 and the triesters 6 and 7. The latter two prodrugs were prepared prior to the pandemic as potential oral treatments for RSV, and their synthesis has been previously reported.12 The 5′-mono ester prodrugs were readily synthesized from the 2′,3′-acetonide-protected nucleoside 4, a known intermediate within our remdesivir synthetic process.30 Straightforward coupling of the intermediate 4 with alkyl carboxylic acids using carbodiimide was followed by acid-mediated cleavage of the 2′,3′-protecting group to yield the monoesters 3 and 5, respectively. Of note, phosphoramidate 1 is also synthesized from the same intermediate 4 through a similar two-step process but instead utilizes a synthetically complex chiral 5′-monophosphoramidate prodrug reagent 8.30 The reduced synthetic complexity of the ester prodrugs is a favorable feature that may lead to downstream benefits in scalability relative to phosphoramidate 1. The ability to rapidly scale and deliver more cost-effective therapies for COVID-19 is important for broad accessibility. However, despite the reduced complexity of 3, the per mole dose required for efficacy is likely to be significantly higher than the dose of 1, which will limit the impact of its reduced complexity.
Scheme 1.
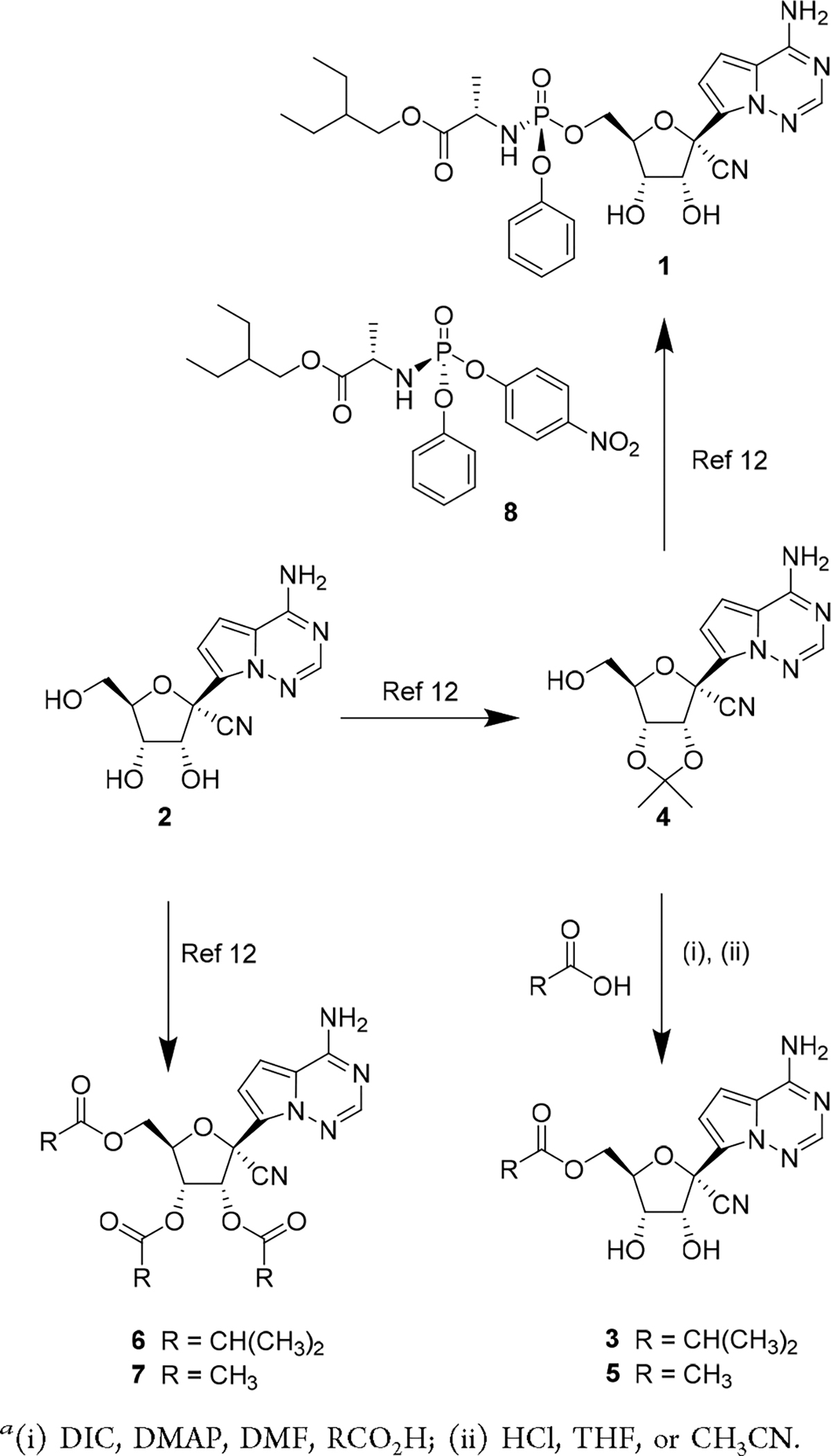
Synthesis of 5′-Ester Prodrugsa
The permeability of the ester prodrugs was compared to parent 2 using a Caco-2 cell assay in the presence of bis(nitrophenol) phosphate, a broad-spectrum carboxyesterase inhibitor to suppress rapid esterase-mediated cleavage of the prodrugs. Mono esters 3 and 5 both demonstrated ~10-fold improved forward permeability over parent 2 and reduced the Pgp-mediated efflux ratio to <3-fold (Table 1). In contrast, the triester prodrug 6 increased the log D above 3 and significantly improved passive permeability with no evidence of efflux. As expected, the permeability properties of the esters trended in line with their log D, with the triesters 6 and 7 demonstrating the highest log D values and more favorable permeability properties over the mono esters.
The thermodynamic solubility of the esters in crystalline or amorphous solid forms was assessed at pH 2 and pH 7. The triester 6, with the highest log D, demonstrated the lowest solubility and was less soluble than 2. In contrast, the less lipophilic isobutyryl ester 3 and acetate esters 5 and 7 all demonstrated >10-fold improved pH 7 solubility over 2 and >100-fold over 6. Overall, the combined solubility and permeability of 3, 5, and 7 was considered more favorable than 6, especially when formulating solid dosage forms where dissolution properties are more critical. Chemical pH-dependent stability was also assessed to determine the relative potential for the prodrugs to remain intact in the stomach and intestine during oral delivery. Ester 3 demonstrated excellent pH 2 and pH 7 stability, while the acetate esters 5 and 7 were both less stable, suggesting 3 was the optimal compound based on overall stability, permeability, and solubility.
Crystallization studies on 3 resulted in the identification of multiple crystalline forms, including stable salt-free Form III (see Supporting Information). The X-ray crystal structure of Form III confirmed the overall chemical structure of 3. In contrast, solid crystalline forms of the triester 6 were challenging to identify, and an extensive salt screening effort was required to identify the crystalline monopyruvate salt. In this example, the pyruvate salt combined with the tri-isobutyryl esters resulted in a significant 1.6-fold increase in molecular weight compared to 3, which was considered a disadvantage with respect to future pill mass. Hydrobromide salt forms of 6 have now been reported in addition to the pyruvate form.31 Taken together, the in vitro assessments indicated that the overall physicochemical properties for the mono 5′-isobutyryl ester 3, including permeability, solubility, pH-dependent stability, and crystalline form isolation, favored its progression as a potential candidate compound.
The cell-based SARS-CoV-2 antiviral activity of 3 and other ester prodrugs was evaluated in A549-hACE2 and NHBE cells but not considered part of our candidate selection process. This was because the intact prodrug was not expected to be present in vivo based on our earlier experience with ester prodrugs on 2 for RSV (see later discussion). The antiviral assays were however useful to dissect the factors governing the metabolism of 2 to 2-NTP by intracellular kinases compared to the metabolism of 1 to 2-NTP, which occurs via a different metabolic pathway (Figure 1). As shown in Table 3, 3 inhibits SARS-CoV-2 with an EC50 value of 1.90 ± 0.61 μM in the A549-hACE2 cell line and has comparable potency to 2. This reflects the facile ability of 3 to break down to 2 in the cell culture experiment, leading to the same potency. However, in NHBE cultures, 3, rather unexpectedly, demonstrated higher potency than 2 with an EC50 value of 0.43 ± 0.09 μM, suggesting that 3 may be facilitating more rapid delivery of 2 into the NHBE cells because of its improved permeability properties. Phosphoramidate prodrug 1 exhibited significantly greater potency than either 2 or 3 against SARS-CoV-2 across both cell lines. In both A549-hACE2 and NHBE cell cultures, neither 2 nor 3 were found to be cytotoxic at concentrations up to 50 μM and phosphoramidate 1 demonstrated at least 300-fold selectivity (CC50/EC50) (Table 3).
Table 3.
SARS-CoV-2 Antiviral Activity and Cytotoxicity Profiling of 3
| 1 | 2 | 3 | puromycin | ||
|---|---|---|---|---|---|
| antiviral activity EC50 (μM)a | |||||
| A549-hACE2b | lung carcinoma | 0.06 ± 0.03 | 2.49 ± 0.62 | 1.90 ± 0.61 | |
| NHBEc | primary | 0.04 ± 0.01 | 3.25 ± 0.8 | 0.43 ± 0.09 | |
| cytotoxicity CC50 (μM)a | |||||
| A549-hACE2 | lung carcinoma | 22.9 ± 6.9 | >50 | >50 | |
| Huh7 | hepatoma | >89 | >44 | 0.43 ± 0.08 | |
| HepG2 | hepatoblastoma (galactose) | 11.1 ± 1.2d | >100d | 71 ± 7.5 | 0.78 ± 0.22 |
| PC-3 | prostate cancer (galactose) | 1.4 ± 0.1d | >100d | >100 | 0.47 ± 0.05 |
| MT-4 | lymphoblast T cell | 1.7 ± 0.4d | 69 ± 26d | >50 | 0.12 ± 0.03 |
| MRC5 | lung fibroblast | >89 | >44 | 0.32 ± 0.03 | |
| NHBE | primary | >50 | >50 | >50 | 0.60 ± 0.2 |
| quiescent PBMCs | primary | >20d | >100d | >100 | 5.05 ± 0.01 |
| stimulated PBMCs | primary | 14.8 ± 5.8d | >100d | >100 | 1.10 ± 0.01 |
| PHH | primary | 2.5 ± 0.6d | >100d | >100 | 1.1 ± 0.4 |
Values are the mean ± standard deviation (SD) of the results from ≥3 independent experiments.
Two-day SARS-CoV-2-Nluc antiviral assay.
One-day SARS-CoV-2-Fluc antiviral assay.
Reference 32.
To correlate the antiviral potency with intracellular concentrations of 2-NTP, both cell lines were incubated continuously with 2 or 3 at 10 μM or 1 at 1 μM and the 2-NTP concentrations were determined by LCMS at different time points over 48 h (Figure 2A and 2B). In both human immortalized and primary human lung cells, 2 yielded similar average concentrations of 2-NTP consistent with its comparable antiviral activity in the cell cultures. The 2-NTP concentrations from 2, when normalized to the same incubation concentration of 1, were >17-fold lower reflecting the weaker antiviral activity of 2 relative to 1 and confirming the greater efficiency of the phosphoramidate prodrug for generating the common monophosphate intermediate and ultimately 2-NTP in these cell cultures. For prodrug 3, the average 2-NTP concentrations were also consistent with the antiviral activity profile. Similar average 2-NTP concentrations to those generated by 2 were observed in the A549-hACE2 cells where antiviral activity was comparable, but approximately 3-fold elevated concentrations were noted in the NHBE cells, especially at the early time points, consistent with the increased potency observed. In summary, the average 2-NTP concentrations align very well with the antiviral data and confirm that 2 is rate limited in its first intracellular metabolism step in lung cells to the monophosphate relative to the phosphoramidate prodrug 1. Moreover, poor cell permeability is also implicated as a factor contributing to the lower efficiency for generating 2-NTP from 2.
Figure 2.
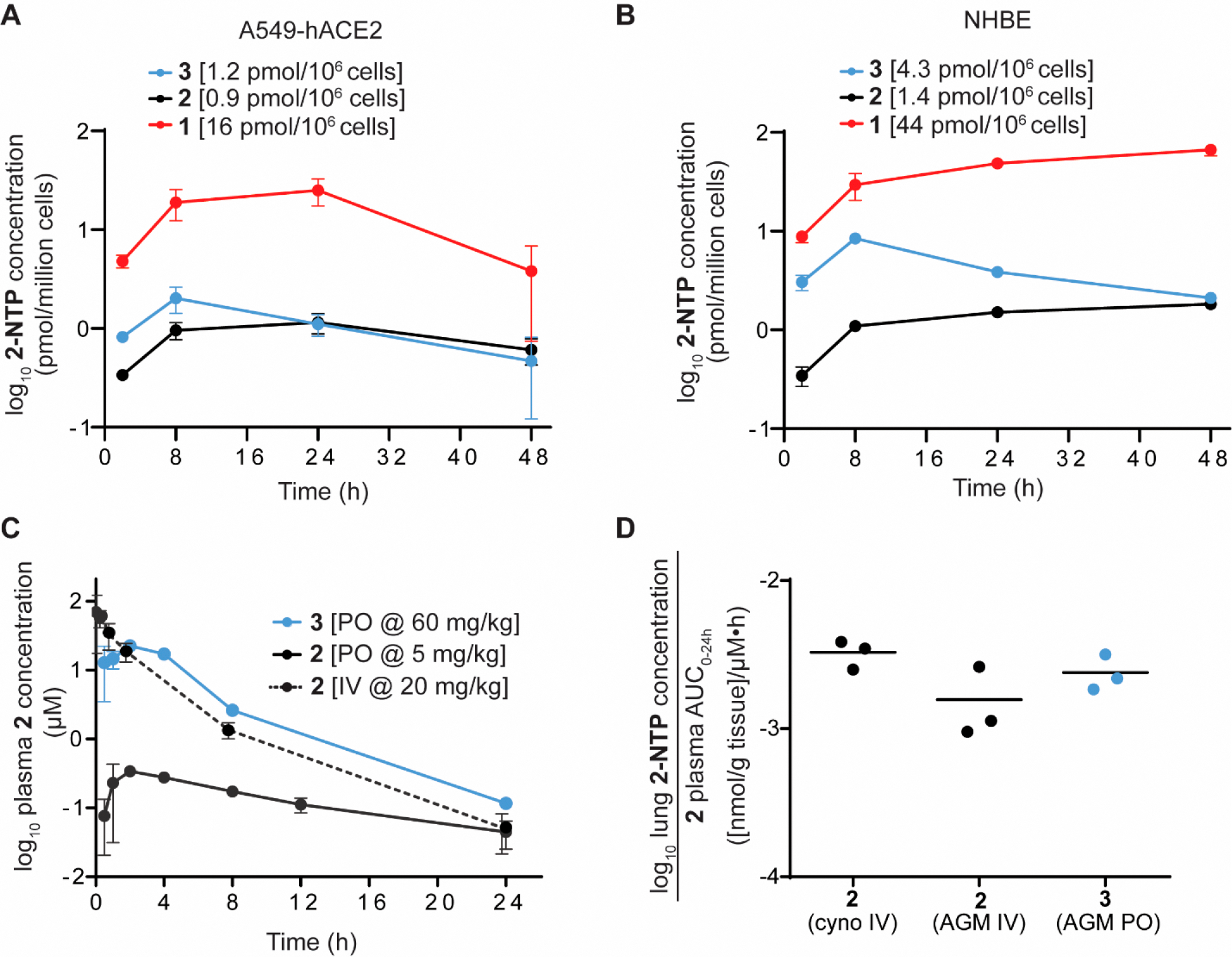
Cellular metabolism and in vivo pharmacokinetics of 3. (A) Intracellular metabolism of 3 (blue), 2 (black), and 1 (red) to the active metabolite 2-NTP in A549-hACE2 cells. Average 2-NTP concentrations are indicated after dose normalization to 1 μM. (B) Intracellular metabolism in NHBE cells. Average 2-NTP concentrations are indicated after dose normalization to 1 μM. (C) Plasma concentration–time profile of 2 following iv and oral administration and oral administration of 3 in AGMs. (D) Lung 2-NTP concentrations at 24 h normalized to exposure of 2 in plasma following iv administration of 2 in both cynomolgus monkeys and AGMs and oral dosing of 3 in AGMs.
Following oral dosing, cells in the intestine, liver, and circulating immune cells are expected to be exposed to varying concentrations of 3 over time in addition to high systemic concentrations of 2. Prodrug 3 and the main metabolite 2 were therefore evaluated for toxicity across a panel of five human transformed cell lines and several primary human cell types, including quiescent and stimulated peripheral blood mononuclear cells (PBMCs) and primary human hepatocytes (PHH) with a 5-day treatment (Table 3). Low toxicity of 3 was observed with CC50 values > 44 μM, consistent with parent 2 that also showed minimal toxicity in all cells tested. Prodrug 3 was also found to be nontoxic in PHH and PBMCs up to 100 μM, and neither 3 nor 2 showed any inhibition of mitochondrial respiration and mitochondrial protein synthesis at the highest concentration tested (100 μM) (see Supporting Information).32 Similarly, a minimal effect was observed on the mitochondrial DNA synthesis when the cells were treated with 0.4–40 μM 3 or 1.0–100 μM 2. Taken together, these data suggest that 3 and its metabolite 2 have low risks for cellular and mitochondrial toxicity in relevant tissues that would be exposed to these compounds upon oral delivery.
Since the start of the pandemic, multiple new variants of SARS-CoV-2 have emerged. Sequence analysis suggests that the majority of amino acid substitutions observed in variants occur in the envelope glycoprotein (spike), while the SARS-CoV-2 RdRp has been highly conserved.33 Due to this high conservation, 1 and 2 and by inference the 2-NTP metabolite, have been shown to retain antiviral activity against emergent variants up to the Omicron variant (B.1.1.529/BA.1).34 Here, we assessed the potency of 3 against a broad panel of new variants of concern (VOC) including Omicron and its subvariants and the Delta variant.35 Activity was assessed in an antiviral ELISA assay performed in the A549-hACE2-TMPRSS2 cell line, and the results are reported in Table 4 along with 1 and 2 as fold change relative to the ancestral WA1 isolate (Lineage A).
Table 4.
Antiviral Activity of 1–3 Against SARS-CoV-2 Variantsa
| 1 |
2 |
3 |
||||
|---|---|---|---|---|---|---|
| variant (replicates)b | EC50 ± SD (μm) | fold change ± SD from WA1c | EC50 ± SD (μm) | fold change ± SD from WA1c | EC50 ± SD (μm) | fold change ± SD from WA1c |
| WA1 (n = 3) | 0.11 ± 0.02 | 1.0 | 4.45 ± 1.03 | 1.0 | 1.01 ± 0.33 | 1.0 |
| Delta (B.1.617.2) (n = 3) | 0.07 ± 0.04d | 0.59 ± 0.20d | 3.26 ± 1.30d | 0.62 ± 0.24d | 0.74 ± 0.38 | 0.72 ± 0.12 |
| WA1 (n = 7) | 0.12 ± 0.03 | 1.0 | 6.52 ± 2.01 | 1.0 | 1.84 ± 0.35 | 1.0 |
| Omicron BA.1 (n = 3) | 0.04 ± 0.01d | 0.45 ± 0.13d | 2.66 ± 0.48d | 0.37 ± 0.10d | 0.65 ± 0.20 | 0.32 ± 0.04 |
| WA1 (n = 2) | 0.11 ± 0.04 | 1.0 | 4.55 ± 0.94 | 1.0 | 1.92 ± 0.26 | 1.0 |
| Omicron BA.2 (n = 2) | 0.02 ± 0.0002 | 0.23 ± 0.07 | 1.33 ± 0.24 | 0.31 ± 0.12 | 0.71 ± 0.10 | 0.37 ± 0.001 |
| WA1 (n = 2) | 0.16 ± 0.0006 | 1.0 | 7.36 ± 3.46 | 1.0 | 1.57 ± 0.21 | 1.0 |
| Omicron BA.2.12.1 (n = 2) | 0.03 ± 0.002e | 0.20 ± 0.01e | 1.79 ± 0.10 | 0.28 ± 0.14 | 0.44 ± 0.13 | 0.29 ± 0.12 |
| Omicron BA.4 (n = 2) | 0.02 ± 0.003 | 0.15 ± 0.02 | 1.63 ± 0.28 | 0.26 ± 0.16 | 0.40 ± 0.02 | 0.26 ± 0.02 |
| Omicron BA.5 (n = 2) | 0.11 ± 0.003 | 0.66 ± 0.02 | 4.80 ± 0.28 | 0.74 ± 0.39 | 1.30 ± 0.13 | 0.83 ± 0.02 |
| WA1 (n = 2) | 0.11 ± 0.006 | 1.0 | 7.73 ± 0.35 | 1.0 | 1.97 ± 0.14 | 1.0 |
| Omicron BA.2.75 (n = 2) | 0.03 ± 0.006 | 0.30 ± 0.07 | 2.58 ± 0.72 | 0.33 ± 0.08 | 0.69 ± 0.14 | 0.35 ± 0.05 |
| WA1 (n = 2) | 0.15 ± 0.02 | 1.0 | 8.56 ± 2.97 | 1.0 | 3.96 ± 0.29 | 1.0 |
| Omicron BA.4.6 (n = 2) | 0.09 ± 0.005e | 0.64 ± 0.13e | 5.40 ± 0.10 | 0.67 ± 0.25 | 2.45 ± 0.28 | 0.62 ± 0.12 |
| Omicron BF.5 (n = 2) | 0.13 ± 0.02e | 0.94 ± 0.31e | 7.78 ± 0.19 | 0.97 ± 0.36 | 3.13 ± 0.36 | 0.80 ± 0.15 |
| WA1 (n = 2) | 0.08 ± 0.0004 | 1.0 | 6.53 ± 0.58 | 1.0 | 1.84 ± 0.14 | 1.0 |
| Omicron XBB (n = 2) | 0.09 ± 0.002 | 1.07 ± 0.01 | 6.30 ± 0.97 | 0.97 ± 0.23 | 2.03 ± 0.56 | 1.10 ± 0.22 |
| Omicron BQ.1.1 (n = 2) | 0.09 ± 0.002 | 1.12 ± 0.03 | 8.77 ± 2.04 | 1.33 ± 0.18 | 2.44 ± 0.09 | 1.33 ± 0.06 |
n represents the number of replicate experiments.
ELISA antiviral assay was assessed at 44 h post-infection for Delta variant and 72 h post-infection for Omicron subvariants.
A fold change was calculated for each experiment, and a mean fold change ± SD was calculated with these values.
Reference 34.
Reference 35.
The Omicron subvariants and the Delta variant evaluated demonstrated similar in vitro susceptibility to compounds 1–3 compared to the ancestral WA1 isolate with a range of 0.15–1.33-fold change. It is anticipated based on these results and the conserved nature of the RdRp that oral delivery of 2-NTP will retain potent antiviral activity against future emergent variants. As such, oral 3 has the potential to be an important and durable treatment option for SARS-CoV-2 infection as it continues to evolve within the human population.
Ester prodrugs are typically broken down rapidly by carboxyesterases that are expressed in the intestine and the liver. Prodrug 3 was evaluated as a substrate for three human carboxyesterases and found to be an excellent substrate for each with a short half-life of <4.3 min (Table 1). Consistent with the enzyme data, rapid hydrolysis was observed for 3 and the triester prodrugs 5 and 6 in intestinal and liver S9 fractions. Monoacetyl ester 5 was notably more stable in all species except rats, and this is presumed to be due to its low log D that likely reduces its ability to be recognized by carboxyesterases. The plasma stability across the higher order species was generally greater than the intestinal and liver S9 stability for all of the prodrugs. The combined in vitro stability results across these matrices suggest that 3 will be efficiently cleaved presystemically during absorption in vivo.
A potential route of nonproductive metabolism during absorption is metabolism of the adenosine-like C-nucleobase by the action of adenosine deaminase that is highly expressed in intestinal mucosa. Indeed, ester prodrugs of adenosine analogs have been previously designed to potentially alleviate adenosine deaminase metabolism during absorption.36,37 The metabolism of 2 and 3 by adenosine deaminase was therefore investigated in vitro, and both compounds were found to be weak substrates or inhibitors of adenosine deaminase (see Supporting Information). The in vitro data suggests that the oral bioavailability of 2 may not be significantly impacted by undesired intestinal metabolism.
To evaluate the impact of permeability on oral bioavailability, nucleoside 2 and prodrug 3 were initially dosed as solutions in rats, dogs, cynomolgus monkeys, and AGMs (Table 2). The bioavailability of 2 in systemic circulation following oral administration of 3 was calculated based on the exposure of 2 determined from iv dosing normalized to the same milligram-equivalent/kilogram of 2 dosed. Consistent with the in vitro stability data, only transient and low levels of intact prodrug 3, when detectable, were observed at the early time points across all species. This was consistent with the in vitro stability observations in the intestinal and liver S9 fractions and established that the lung tissues in vivo are not exposed to any appreciable concentrations of the intact prodrug but only to the parent metabolite 2. Given the relatively modest Caco-2 permeability and efflux improvements for 3, we were surprised to observe a significant 2-, 3.6-, and 7-fold improvement in the oral exposure of metabolite 2 in rats, AGMs, and cynomolgus monkeys, respectively. Indeed, the bioavailability of 2 that was achieved from oral dosing of 3 in cynomolgus monkeys was comparable to the value reported for the highly lipophilic tri-isobutyryl ester prodrug 6 (F% = 28).12 Interestingly, similar ester-prodrug studies on the 4′-azido cytidine ribonucleoside analog, R1479, in cynomolgus monkeys concluded that a log P > 2.0 was generally required for improved oral bioavailability in monkeys and that less lipophilic 5′-monoesters with log P values below 2.0 were not very effective.29 The significantly improved oral properties of the adenosine analog 3 in cynomolgus monkeys despite its log D < 2.0 may be the result of favorable interactions with intestinal nucleoside transporters which requires further investigation in the future. In dogs, the bioavailability of 2 is very high, and this was maintained by the prodrug 3. Thus, the bioavailability of 2 following oral solution dosing of 3 was improved or maintained across multiple preclinical species tested and averaged 64% across rats, dogs, and cynomolgus monkeys.
For rapid clinical development, a stable crystalline form formulated into tablets was highly preferred. Therefore, the crystalline salt-free Form III of 3 that was identified from crystallization studies was dosed in dogs and cynomolgus monkeys in tablet form. A less than 2-fold drop to 67% oral bioavailability was observed in dogs together with a 2-fold drop in oral bioavailability in cynomolgus monkeys. In rats, suspension dosing was used and showed high oral bioavailability consistent with solution dosing. Despite the lower bioavailability of 2 in cynomolgus monkeys, oral bioavailability was still 3.3-fold superior to the solution dosing of 2, leading to an average oral bioavailability of 2 from tablet dosing of 3 in dogs and cynomolgus monkeys of 42%. The favorable balance of permeability and solubility for 3 therefore resulted in improved oral delivery of 2 in both solution and solid forms. Taken together, these data supported the selection of 3 as the oral candidate.
The in vivo metabolism of 2 to 2-NTP has been reported in lung tissue at 24 h following iv dosing in cynomolgus monkeys, AGMs, and ferrets.11,24,27 In these lung samples, 2-NTP formation has been shown to be 10–15-fold less efficient from equimolar doses of 2 compared to the phosphoramidate 1, consistent with the in vitro metabolism data described earlier. Therefore, both in vitro and in vivo data from iv administration supported the need for high systemic exposures of 2 to drive sufficient 2-NTP formation in lung cells. Oral dosing results in a lower Cmax compared to iv administration, and it was not understood from the current data how the in vivo 2-NTP concentrations in lung would be impacted by the different plasma–time exposure profile of 2 when delivered orally using the prodrug. Therefore, oral dosing of 3 was performed at 60 mg/kg in AGMs to afford comparable AUC0–24h exposures of 2 to those generated in the iv NHP experiments at 20 mg/kg reported in Table 2, and lung tissue samples were collected at 24 h for 2-NTP analysis (Table 2). When normalized to the equivalent AUC0–24h exposure of 2, the data shows that 2-NTP formed from oral administration is comparable to that of iv administration (Figure 2D), suggesting that lung 2-NTP formation is more closely correlated with systemic AUC0–24h exposures of 2 rather than Cmax concentrations. Given this observation, it is also reasonable to expect that antiviral efficacy is also correlated with the systemic exposures of 2 rather than Cmax concentrations.
Recently, we have reported on the efficacy of 1 compared to iv administered 2 in a SARS-CoV-2 infection model in AGMs and observed strong efficacy for the nucleoside 2 at high systemic exposures.24 The same AGM model was then utilized to determine if efficacious concentrations of 2-NTP in lung tissue could be achieved from systemic exposure to 2 through orally administered 3. Oral treatment with 3 was initiated at 8 h post-infection with 60 or 120 mg/kg doses and then continued daily for 5 days. The doses were selected to provide a daily systemic exposure of 2 that bracketed the exposure from iv administered 2 at 20 mg/kg, which demonstrated strong efficacy relative to 1 in the earlier study.24 The viral loads (genomic RNA and infectious virus titer) were evaluated on bronchoalveolar lavage fluid (BALF), nasal, and throat swab samples, collected on 1, 2, 4, and 6 days post-infection (dpi) (Figure 3 and Supporting Information). At the end of the study, terminal respiratory tissues were harvested for evaluation of tissue viral RNA loads.
Figure 3.
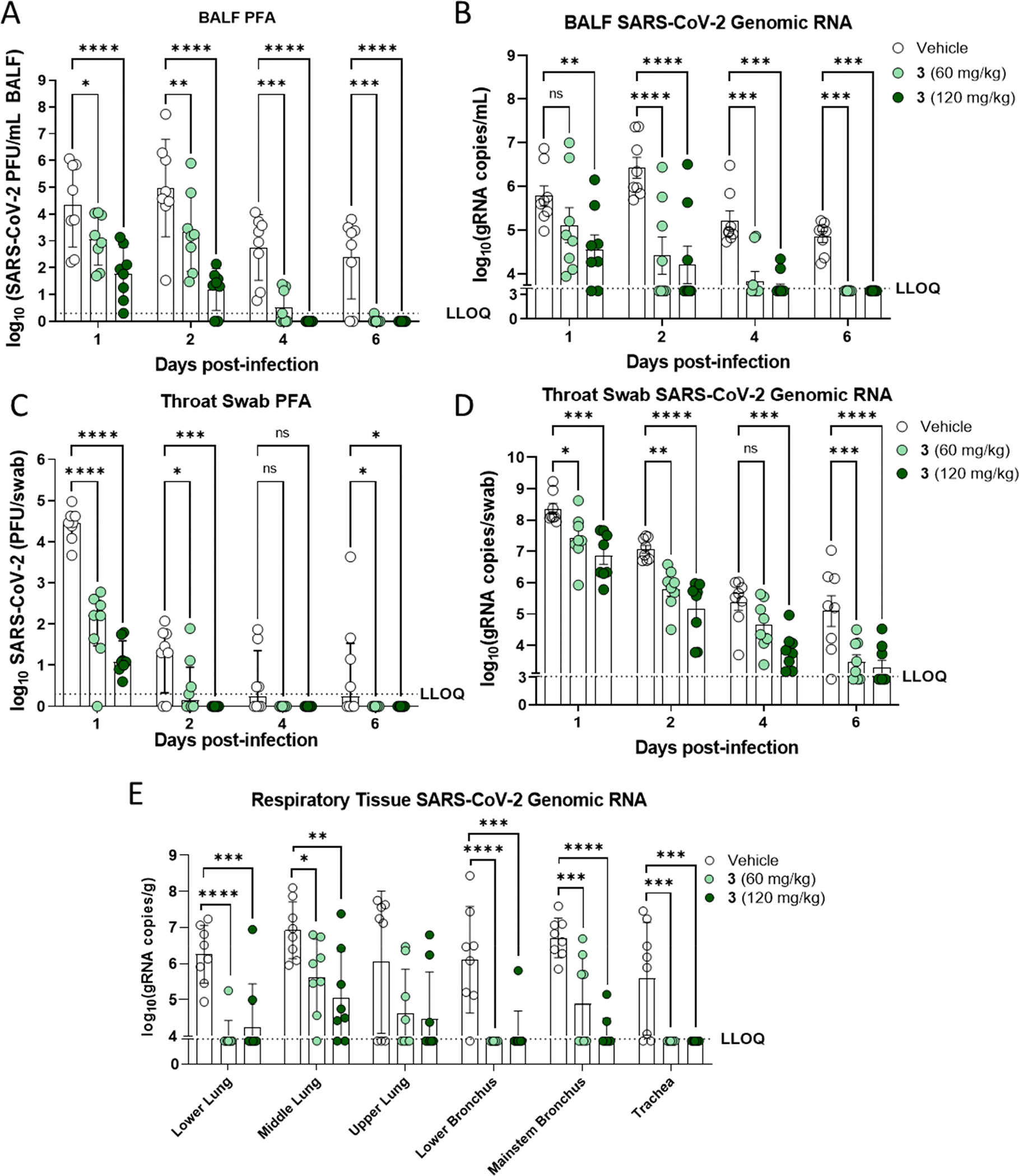
Antiviral effect of oral 3 on respiratory tract lavage, swab, and tissue samples in the African green monkey SARS-CoV-2 infection model. (A) Infectious virus in bronchoalveolar lavage fluid (BALF). (B) Genomic RNA in BALF. (C) Infectious viral titer in throat swab. (D) Genomic RNA in throat swab. (E) Genomic RNA in respiratory tissue samples harvested day 6 across the respiratory tract. LLOQ, lower limit of quantification. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Treatment of infected AGMs with either 60 or 120 mg/kg 3 resulted in statistically significant decreases in infectious viral loads in the BALF throughout infection (Figure 3A). Treatment with 3 resulted in only 1 animal (in the 60 mg/kg group) with infectious SARS-CoV-2 in the BALF at or above the limit of quantification at day 6 post-infection; meanwhile, 6 of the 8 vehicle animals had quantifiable infectious viral loads at this time point (Figure 3A). The extent of infectious viral load reductions was dose dependent, with larger reductions of infectious viral load observed for the group receiving 3 at 120 mg/kg over the sampling period. In line with reductions in infectious viral loads in the BALF, genomic RNA loads in BALF of 3 treated animals were also found to be significantly reduced at nearly all study time points assessed, with the one exception being the 60 mg/kg 3 treatment group at day 1 post-infection (Figure 3B). The results from RNA and infectious viral loads in the BALF highlights the significant effect of treatment with 3 in the lower airway.
In the upper airway, infectious viral titers and RNA were quantified from throat (Figure 3C and 3D) and nasal swabs (Supporting Information). In throat swabs, significant reductions in SARS-CoV-2 infectious (Figure 3C) and RNA loads (Figure 3D) were observed from the throat swabs at 1, 2, and 6 dpi from both dose groups. Furthermore, animals in the 120 mg/kg group showed additional reductions in viral RNA at day 4 post-infection. Notably, in both throat and nasal swabs, no detectable infectious virus was observed for any animal in either group receiving 3 after day 4 post-infection. There was a large observed difference in the SARS-CoV-2 genomic RNA levels and the infectious viral loads from the nasal swab analysis (Supporting Information). Vehicle control animals at day 1 post-infection had a higher viral RNA load than that observed in BALF, but only one-half of the vehicle control animals had detectable levels of infectious virus. This difference highlights that viral RNA levels do not always correlate with infectious virus and that viral RNA levels in the nasal cavity may, in part, be due to noninfectious viral RNA released from dead or dying cells. On study day 6, terminal respiratory tissues were assessed for SARS-CoV-2 RNA loads and both doses of 3 resulted in significant reductions of viral RNA levels in 5 out of the 6 respiratory tissues evaluated (Figure 3E). Collectively, these data demonstrate that 60 and 120 mg/kg doses of oral 3 are both highly efficacious at reducing infectious virus and viral RNA loads throughout the upper and lower respiratory tract of AGMs and similarly efficacious to 1.24
Having established efficacy from oral dosing in AGMs, we considered various methods to estimate the human efficacious dose of 3 for the clinic. One human dose estimation method employed Cmax/Ctrough exposures of 2 relative to the in vitro SARS-CoV-2 antiviral EC50 potency of ~1–2 μM in Vero E6 cells and primary lung cultures.38 However, a limitation of this method is that the relationship of plasma concentrations and in vitro antiviral EC50 has not been clearly established for SARS-CoV-2 efficacy in the clinic. Consideration was also given to a lung pharmacokinetic model that targeted doses of 3 that would generate equivalent lung 2-NTP concentrations to that of iv administered 1 at its clinically efficacious dose. This resulted in an unfeasible multigram predicted dose of 3 and was inconsistent with the AGM efficacy study data. The 60 and 120 mg/kg doses in the AGM model were both highly efficacious despite the lung 2-NTP concentrations at 24 h being several-fold lower than the levels reported following iv administration of 1.12,24 A potential reason for this disconnect is the 2-NTP concentrations from the gross tissue analysis at a single time point do not accurately reflect the concentration and distribution of 2-NTP across infected cell types in the lungs. This is consistent with the different activation pathways of 3 and 1 to the common monophosphate metabolite, which are likely cell dependent and impacted by differences in the expression levels of the key enzymes.39 This is nicely illustrated by the fact that 1 is not effectively metabolized in Vero E6 cells, for example.26 Given the limited lung 2-NTP data and the challenges in using this parameter to estimate efficacy, we elected to use the plasma exposures of 2 (AUC0–24h) and correlate this PK parameter to in vivo SARS-CoV-2 efficacy. In constructing an exposure–efficacy relationship model, we combined all of the efficacy data generated from the SARS-CoV-2 model in AGMs for both oral 3 and iv 2 in addition to the efficacy data reported in the ferret and mouse SARS-CoV-2 models using prodrug 6 (Table 5).24,27,40
Table 5.
Plasma 2 Exposures and SARS-CoV-2 Antiviral Efficacy In Vivo
| 2 | |||||||
|---|---|---|---|---|---|---|---|
| cpd. | SARS-CoV-2 model | dose (mg/kg) | route, regimen | Cmax (μM) | 2AUC0–24h (μM·h) | Log10 gRNA decreasea | ref |
| 1 | AGM | 10/5b | iv, once daily | 1.23c | 9.1c | −1.38 | 24 |
| 2 | AGM | 7.5 | iv, once daily | ~33d | ~55d | −1.02 | 24 |
| 2 | AGM | 20 | iv, once daily | 87 | 147 | −1.79 | 24 |
| 3 | AGM | 60 | oral, once daily | 21e | 111e | −1.41 | this work |
| 6 | ferret | 10 | oral, twice daily | 5 | 54 | −1.98f | 27 |
| 6 | mouse | 30 | oral, twice daily | 13 | 95 | −3.29g | 40 |
Decrease in genomic RNA (gRNA) relative to vehicle control in bronchioalveolar lavage sample on day 6 post-infection unless noted otherwise.
Day 1 dose is 10 mg/kg; then, daily maintenance doses of 5 mg/kg were given.
Data shown is for metabolite 2 following the day 1 10 mg/kg dose. The mean Cmax and AUC0–24h of plasma 1 at this dose were 12.6 μM and 7.9 μM·h, respectively.
Estimated based on dose proportionality to PK results obtained at 20 mg/kg.
Calculated AUC0–24h at 60 mg/kg from the average of 14.4, 60, and 86.5 mg/kg AGM PO experiments in Table 2.
Nasal lavage sample on day 4 post-infection.
Lung tissue plaque forming unit (PFU) titer on final necropsy day 4 post-infection.
Including the data from the AGM iv study that evaluated both 1 and 2 in the same study was a critical aspect of the analysis because it is the only study, published up to now, that has directly compared the efficacy of 1 and 2 at clinically relevant exposures of 1. Reported in Table 5 are the Cmax and AUC0–24h exposures of 2, calculated using healthy animal PK, at the efficacious dose across each model. The efficacy in Table 5 from the ferret and mouse SARS-CoV-2 models is from the highest doses evaluated in those respective models following a treatment-based protocol.27,40 Robust efficacy was achieved at plasma AUC0–24h for 2 ranging from 54 to 111 μM·h following oral dosing of prodrugs 3 or 6. Thus, as anticipated, the systemic exposures of 2 that drive preclinical efficacy are >7-fold higher than the plasma exposure of 1 at 10 mg/kg in the AGM model (AUC0–24h = 7.9 μM·h) and >20-fold higher than the exposure of 1 at its 100 mg maintenance dose in humans (AUC0–24h = 2.6 μM·h).24,41 Moreover, the efficacious exposure of 2 following oral dosing of 3 at 60 mg/kg in AGMs is 12-fold higher than the exposure of 2 that is observed as a metabolite following the iv 10 mg/kg dose of 1 (AUC0–24h = 9.1 μM·h), yet a comparable reduction in the bronchoalveolar lavage gRNA was observed (Table 5). It can be concluded that amidate prodrug 1 is very efficient at generating efficacious 2-NTP concentrations in lungs from a relatively brief and low systemic exposure but has the disadvantage of iv administration. In contrast, 2 is much less efficient at metabolism to 2-NTP, so it requires a high systemic exposure and consequently higher doses for comparable preclinical efficacy to that of 1 in the AGM model. However, the higher plasma exposures of 2 can be readily achieved orally using prodrug 3.
Scaling of the preclinical plasma PK parameters to humans using allometry and employing an average ~42% oral bioavailability of 2 (average F% from the tablet dosing in dogs and cynomolgus monkeys) resulted in a projected twice-daily dose of 3 of 350–400 mg. This dose range is estimated to result in a human exposure of 2 that matches the efficacious exposure range in the mouse and AGM models (AUC0–24h = 95–111 μM·h). The dose is considerably higher on a per mole basis than the 100 mg maintenance dose of 1, which effectively reduces any benefit of the more facile synthesis of 3 due to its lower synthetic complexity. Nevertheless, the dose is attainable and would be considerably higher for oral 2 without the benefit of the prodrug improvements. The simulated human PK at this dose affords a Cmax ≈ 7.5 μM and Cave ≈ 4.6 μM, several-fold higher than the antiviral EC50 range of 2.5–3.3 μM reported for 2 in Table 3. We were delighted to find that the observed Phase 1 PK was in good agreement with the estimated human PK from the preclinical species.42 Based on the human phase 1 PK and safety data, a 350 mg twice-daily dose for 5 days is currently being assessed in two Phase 3 clinical trials (ClinicalTrials.gov Identifiers: NCT05603143 and NCT05715528) in people with or without risk factors for progression to severe disease.
CONCLUSION
The development of convenient oral COVID-19 treatment options is critically important as SARS-CoV-2 infections persist and new variants can emerge. Consequently, a great deal of discussion has emerged in the public arena regarding the oral development of 2, the parent nucleoside of 1. However, our previous work on 2 had rigorously established that significant hurdles for oral 2 existed, centered on its low bioavailability in nonhuman primates and the likely requirement for high systemic exposures to overcome its inefficient metabolism to 2-NTP. A prodrug approach was therefore employed to lower the oral dose, reduce drug mass, and avoid loss of precious unabsorbed drug, leading to the selection of a 5′-isobutyryl ester prodrug 3. The ester prodrug 3 balances solubility, stability, and permeability and efficiently breaks down presystemically following oral delivery to provide high plasma concentrations of 2. Despite only a modest improvement in permeability relative to 2, improved oral bioavailability of 2 from 3 was found across multiple species following solution dosing and most importantly solid tablet dosing. Oral administration of 3 was shown to generate the same common active 2-NTP metabolite in the lungs of AGMs as 1 and drive a comparable antiviral effect to 1 in the AGM SARS-CoV-2 infection model. An efficacy–exposure relationship model was then constructed to estimate a human daily dose in the range of 350–400 mg twice daily. We also confirmed in vitro the potent activity of 1, 2, and 3, against recent SARS-CoV-2 variants of concern. Oral administration of prodrug 3 effectively delivers the same active 2-NTP metabolite as intravenous 1 and is currently being evaluated at a dose of 350 mg twice daily in two global Phase 3 COVID-19 trials. The broad activity established for 2 and 3, and by inference the common active metabolite 2-NTP, toward many RNA viruses, also supports the potential to deploy 3 in a rapid-response scenario to combat future emerging RNA viruses with pandemic potential.
EXPERIMENTAL SECTION
All organic compounds were synthesized at Gilead Sciences, Inc. (Foster City, CA) unless otherwise noted. Commercially available solvents and reagents were used as received without further purification. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 400 MHz at rt, with tetramethyl silane as an internal standard. Proton nuclear magnetic resonance spectra are reported in parts per million (ppm) on the δ scale and are referenced from the residual protium in the NMR solvent (CHCl3-d1, δ 7.26; MeOH-d4, δ 3.31; DMSO-d6, δ 2.50; CH3CN-d3, δ 1.93). Data is reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet), coupling constants (J) in Hertz]. LCMS was conducted on an Agilent 1260 Infinity G6125B LCMS equipped with a Phenomenex Kinetex 2.6 μm C18 110 Å column (50 mm × 2.1 mm) eluting with 0.1% acetic acid in water (buffer A) and 0.1% acetic acid in CH3CN (buffer B); gradient, 0–1.0 min 10–100% buffer B, 1.0–1.35 min 100% buffer B, then 1.35–1.36 min 100–10% CH3CN at 1.0 mL/min. Preparative normal phase silica gel chromatography was carried out using a Teledyne ISCO Combi Flash Companion instrument with silica gel cartridges. Purities of the final compounds were determined by high-performance liquid chromatography (HPLC) and were greater than 95%. HPLC purity was determined on an Agilent 1100 Series HPLC equipped with a Gemini column 2.6 μm 100 Å column (100 mm × 4.6 mm) eluting with a 2–98% gradient of 0.1% trifluoroacetic acid in water and 0.1% trifluoroacetic acid in acetonitrile at a flow rate of 1.5 mL/min.
Compounds 1, 2, 6, and 7 have been reported previously.12
Synthesis of ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl Isobutyrate (3).
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f ][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile 4 (2000 mg, 6.0 mmol)30 and isobutyric acid (638 mg, 7.2 mmol) in DMF (5 mL), N,N′-diisopropylcarbodiimide (914 mg, 7.2 mmol) was slowly added followed by 4-(dimethylamino)pyridine (737 mg, 6.0 mmol). The reaction mixture was stirred for 4 h and then diluted with ethyl acetate, washed with water and brine, dried over sodium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 20% MeOH in CH2Cl2 to provide the intermediate ((3aR,4R,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]-dioxol-4-yl)methyl isobutyrate. LCMS m/z = 402.2 (M + 1). To a solution of the intermediate (1500 mg) in THF (10 mL), conc. HCl (2 mL) was added, and the mixture was stirred at rt for 4 h. The reaction mixture was diluted with CH2Cl2, washed with water, saturated aqueous bicarbonate, and brine, dried over sodium sulfate, concentrated, and subjected to silica gel chromatography, eluting with 30% MeOH in CH2Cl2 to afford the title compound (660 mg, 50%). 1H NMR (400 MHz, MeOH-d4) δ 7.88 (s, 1H), 6.96–6.85 (m, 2H), 4.50–4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H). LCMS m/z: 362.1 (M + 1). 13C NMR (400 MHz, CHCl3-d3) δ 175.9, 155.6, 147.9, 123.5, 110.2, 100.8, 116.9, 116.6, 81.3, 79.0, 74.0, 70.2, 62.9, 33.2, 18.7, 18.6. HRMS m/z: 362.14615, C16H20N5O5 362.14644.
((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f ][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl Acetate (5).
The title compound was prepared from 4 (100 mg, 0.3 mmol) using the same method described above except acetic acid was used instead of isobutyric acid and acetonitrile instead of THF in the acid deprotection step to yield the title product (40% overall yield). 1H NMR (400 MHz, DMSO-d6) δ 8.03–7.96 (m, 3H), 6.92 (d, J = 4.5 Hz, 1H), 6.81 (d, J = 4.5 Hz, 1H), 6.31 (d, J = 6.0 Hz, 1H), 5.39 (d, J = 5.9 Hz, 1H), 4.70 (t, J = 5.5 Hz, 1H), 4.33 (dd, J = 11.9, 2.8 Hz, 1H), 4.23 (m, 1H), 4.14 (dd, J = 12.0, 5.9 Hz, 1H), 3.94 (q, J = 5.9 Hz, 1H), 2.02 (s, 3H). LCMS m/z = 334.1 (M + 1).
SARS-CoV-2-NLuc A549-hACE2 Assay.
Tested compounds are prepared in 100% DMSO in 384-well polypropylene plates (Greiner, Monroe, NC, Cat. no. 784201) as 4 replicates of 10 serially diluted concentrations (1:3). The serially diluted compounds were transferred to low dead volume Echo plates (Labcyte, Sunnyvale, CA, Cat. no. LP-0200). The test compounds were then spotted to 384-well assay plates (Greiner, Monroe, NC, Cat. no. 781091) at 200 nL per well using an Echo acoustic dispenser (Labcyte, Sunnyvale, CA). A549-hACE2 cells were harvested, suspended in DMEM (supplemented with 2% FBS and 1× penicillin-streptomycin-l-glutamine), and seeded to the prespotted assay plates at 10 000 cells per well in 30 μL. SARS-CoV-2-NLuc virus43 was diluted in DMEM (supplemented with 2% FBS and 1× penicillin-streptomycin-l-glutamine) at 350 000 plaque forming units (PFU) per mL, and 10 μL per well was added to the assay plates containing cells and compounds (MOI 0.35). The assay plates were incubated for 2 days at 37 °C and 5% CO2. At the end of incubation, Nano-Glo reagent (Promega, Madison, WI, Cat. no. N1150) was prepared. The assay plates and Nano-Glo reagent were equilibrated to room temperature for at least 30 min. A 40 μL per well amount of Nano-Glo reagent was added, and the plates were incubated at room temperature for 30 min before reading the luminescence signal on an EnVision multimode plate reader (PerkinElmer, Waltham, MA). Compound 1 was used as the positive control, and DMSO was used as the negative control. Values were normalized to the positive and negative controls (as 0% and 100% replication, respectively), and data was fitted using nonlinear regression analysis by Gilead’s dose response tool. The EC50 value for each compound was defined as the concentration reducing viral replication by 50%.
SARS-CoV-2 NHBE Cell Assay.
Compounds were evaluated for SARS-CoV-2 potency as described previously.27
SARS-CoV-2 Nucleoprotein ELISA in A549-hACE2-TMPRSS2 Cells.
SARS-CoV-2 isolates for the ELISA antiviral assays were acquired through the World Reference Center for Emerging Viruses and Arboviruses at UTMB (Delta) and BEI Resources, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). Isolates obtained from BEI Resources were deposited by the CDC (WA1 reference). Omicron variants obtained from BEI were deposited by Viviana Simon (BA.2.12.1), Andrew S. Pekosz (BA.4.6, BF.5, BQ.1.1), and Mehul Suthar (XBB). Omicron isolates BA.2, BA.4, BA.5, and BA.2.75 were obtained from the Gale laboratory, University of Washington. A total of 3 × 104 A549-hACE2-TMPRSS2 cells in 100 μL of DMEM (supplemented with 10% FBS and 1× penicillin–streptomycin) were seeded into each well of a 96-well plate and incubated overnight. The following day, medium was aspirated, and 100 μL of DMEM containing 2% FBS was added to each well. Three-fold serial dilutions of compound (in triplicate) were added to each well using an HP D300e digital dispenser with a final volume of 200 μL per well. Immediately after compound addition, cells were infected with 1.5 × 103 PFU of the relevant SARS-CoV-2 variant (diluted in 100 μL of DMEM supplemented with 2% FBS), resulting in MOI 0.05. Plates were centrifuged for 2 min at 200g and then incubated at 37 °C with 5% CO2 for 48 h (or 72 h for Omicron strains and the WA1 reference), after which medium was aspirated and cells were fixed with 100% MeOH for 10 min at rt. The MeOH was removed, and plates were air dried for 10 min at rt, followed by 1 h of incubation with 100 μL per well of blocking buffer (phosphate-buffered saline (PBS) with 10% FBS, 5% nonfat dry milk, and 0.1% Tween 20) for 1 h at 37 °C. The blocking buffer was then aspirated, and 50 μL of a 1:4000 dilution of rabbit anti-SARS-CoV-2 nucleocapsid antibody (Invitrogen, Waltham, MA, Cat. no. MA536086) in blocking buffer was added and incubated for 2 h at 37 °C. Plates were washed 5 times with 100 μL per well of PBS containing 0.1% Tween 20 prior to addition of 50 μL per well of horseradish peroxidase-conjugated goat antirabbit IgG (ImmunoReagents, Raleigh, NC, Cat. no. GtxRb-003-FHRPX) diluted 1:4000 in blocking buffer. Plates were again incubated for 1 h at 37 °C and then washed 5 times with 100 μL of PBS with 0.1% Tween 20. A 100 μL amount of 3,3′,5,5′-tetramethylbenzidene reagent (Thermo Scientific, Waltham, MA, Cat. no. ENN301) was added to each well and allowed to incubate at rt until visible staining of the positive-control wells, usually 5–10 min. The reaction was stopped with addition of 100 μL per well of 3,3′,5,5′-tetramethylbenzidene stop solution (SeraCare, Milford, MA, Cat. no. 5150–0021). The absorbance was then read at 450 nm using an EnVision plate reader. Fold change for variants was calculated for each experiment with comparison to the relevant WA1 reference. Fold change across from each replicate for all experiments was then averaged to obtain the final reported values. EC50 is defined as the compound concentration at which there was a 50% nucleoprotein expression relative to infected cells with DMSO alone (0% inhibition) and uninfected control cells (100% inhibition). EC50 values were determined using GraphPad Prism 8.1.2 with nonlinear regression curve fits. Constraints were used when required to ensure the bottom and top of the fit curves were close to 0 and 100, respectively.
Cytotoxicity Assay.
Compounds (200 nL) were spotted onto 384-well black Greiner plates prior to seeding 5000 either A549-hACE2 or NHBE cells per well in a volume of 40 μL of culture medium. The plates were incubated at 37 °C for 48 h with 5% CO2. On day 2, 40 μL of CellTiter-Glo (Promega) was added and mixed 5 times. Plates were read for luminescence on an EnVision, and the CC50 (compound concentration for reducing 50% of luminescence signal as a measure of cell viability) values were calculated using a nonlinear regression model (four parameters).
The 5-day CC50 assays for MRC5 and NHBE are described in the Supporting Information. The 5-day CC50 assays for PC-3, MT-4, quiescent PBMC, stimulated PBMC, and freshly isolated PHH are conducted as reported.32
In Vitro Metabolism.
A549-hACE2 or NHBE cells were seeded in a 12-well plate at 2.5 × 105 cells per well. Twenty-four hours later, cell-culture medium was replaced with medium containing compound, as indicated, and incubated at 37 °C with 5% CO2. At the indicated time post compound addition, cells were washed 3 times with ice-cold tris-buffered saline, scraped into 0.5 mL of ice-cold 70% MeOH, and stored at −80 °C. Extracts were centrifuged at 15 000g for 15 min, and supernatants were transferred to clean tubes for evaporation in a miVac Duo concentrator (Genevac, Painter, NY). Dried samples were reconstituted in mobile phase A containing 3 mM ammonium formate (pH 5) with 10 mM dimethylhexylamine in water for analysis by LCMS/MS using a multistage linear gradient from 10% to 50% acetonitrile in mobile phase A at a flow rate of 360 μL/min. Analytes were separated using a 50 × 2 mm, 2.5 μm Luna C18(2) HST column (Phenomenex, Torrance, CA) connected to an LC-20ADXR (Shimadzu, Kyoto, Japan) ternary pump system and an HTS PAL autosampler (LEAP Technologies, Carrboro, NC). Detection was performed on a Qtrap 6500+ (AB Sciex, Redwood City, CA) mass spectrometer operating in positive ion and multiple reaction monitoring modes. Analytes were quantified using a 7-point standard curve ranging from 0.624 to 160 pmol per million cells prepared in extracts from untreated culture wells that were counted for each time point.
Stability in pH 2 and pH 7 Buffered Solutions.
The aqueous stability of compounds was assessed over a time of not less than 24 h at 40 °C. Stability was determined in 50 mM phosphate-buffered solutions at both pH 2 and pH 7 with 150 mM NaCl. For each compound and pH condition, 50 μg/mL samples were prepared with not more than 50% acetonitrile as a cosolvent. Samples were analyzed by UPLC using a Waters Acquity UPLC with a PDA UV detector.
Thermodynamic Solubility in pH 2 Water.
The aqueous solubility of compounds was assessed over a time of not less than 72 h. Solubility was determined at ambient temperature in water adjusted to pH 2 with 1 N HCl. Solids were added to 0.5 mL of pH 2 water in 1.5 mL Eppendorf tubes, vortexed, and then agitated for 24 h in an Eppendorf ThermoMixer C at 1400 rpm. To determine the concentration in solution, the suspensions were centrifuged for 3 min at 15 000 rpm. Supernatants were diluted by a factor of 100 with 50:50 v/v acetonitrile:water. All diluted supernatants were analyzed by UPLC using a Waters Acquity UPLC with a PDA UV detector.
Thermodynamic Solubility in pH 7 Buffered Solution.
The aqueous solubility of compounds was assessed over a time of 24 h. Solubility was determined at ambient temperature in a 50 mM phosphate-buffered pH 7 solution with 150 mM NaCl. Solids were added to the buffered solution in 1.5 mL Eppendorf tubes, vortexed for 1 min, and then agitated for 24 h in an Eppendorf ThermoMixer C. To determine the concentration in solution, the suspensions were centrifuged for 15 min at 15 000 rpm. Supernatants were diluted to a volume of 1 mL with 30:70 v/v acetonitrile:water. All diluted supernatants were analyzed by UPLC using a Waters Acquity UPLC with a PDA UV detector.
Carboxyesterase (CES) Stability.
Test compounds or positive control substrates (oseltamivir for CES1 enzymes or procaine for CES2) were incubated with individual Supersome preparations (Corning Life Sciences, Corning, NY; final CES concentration 1.5 mg/mL) in 0.1 M potassium phosphate buffer (pH 7.4) at 37 °C. Substrates were added to a final concentration of 2 μM to initiate the reaction. The final incubation volume was 250 mL. Aliquots were removed after incubation for 0, 10, 30, 60, and 120 min. The reactions were stopped by the addition of quench solution (90/10 (v/v) CH3CN/MeOH with 0.1% formic acid) containing internal standard. Following protein precipitation and centrifugation, supernatant was diluted with an equal volume of water prior to analysis. For procaine, supernatant was dried down and reconstituted with water. All samples were analyzed by LCMS/MS, and peak area ratios were used for quantification. Analysis by LCMS/MS was performed on a Thermo Q-Exactive mass spectrometer coupled to a Dionex UltiMate 3000 HPLC with a Leap Technologies HTC PAL autosampler. Separation of test compounds was accomplished using a Waters Acquity BEH C18, 50 × 2.1 mm, 1.7 μm column using a multistage linear gradient with mobile phases containing 0.1% formic acid in either water or acetonitrile at a flow rate of 0.2 mL/min.
Intestinal and Liver S9 Stability.
The stability of the compounds was assessed in both intestinal and liver S9 fractions (BioIVT, Baltimore, MD) from select species. For S9 stability, duplicate aliquots of test compound or positive control substrate (GS-7340) were added to either PMSF-free intestinal or liver S9 stock diluted with 100 mM phosphate-buffered saline, pH 7.4, to obtain a protein concentration of either 1.0 or 2.4 mg/mL, respectively. The S9 metabolic reactions were initiated by the addition of the substrates to the S9 reaction mixture to a final concentration of 2 μM. At 0, 10, 20, 30, 60, and 120 min (intestinal S9) or at 2, 12, 25, 45, 65, and 90 min (liver S9), 25 μL aliquots of the reaction mixture were transferred to plates containing 225 μL of internal standard in quenching solution (acetonitrile). After quenching, the plates were centrifuged at 3000g for 30 min, and 150 μL aliquots of each supernatant were diluted with 150 μL of water. Aliquots (10 μL) of the diluted supernatant were analyzed by LCMS/MS as described for CES stability assessment.
Plasma Stability.
The stability of the compounds was assessed in plasma from select species. Duplicate aliquots of plasma were warmed to 37 °C, and the metabolic reactions were initiated by the addition of test compound (6 μL of 0.1 mM DMSO stock) or plasma stability standard (GS-7340) to obtain a final substrate concentration of 2 μM. At 0.05, 0.5, 1, 2, 3, and 4 h plasma, 25 μL aliquots of the reaction mixture were treated and analyzed following the S9 stability method described above.
Caco-2 Permeability.
The bidirectional permeability of the tested compounds was assessed in preplated Caco-2 cells (clone C2BBe1), obtained from Sigma-Aldrich, Inc. (Atlanta, GA). Cell monolayers were grown to confluence on collagen-coated, microporous, polycarbonate membranes in 24-well transwell plates for 21 days. The permeability assay buffer in donor wells was Hanks’ balanced salt solution (HBSS) containing 10 mM HEPES and 15 mM glucose at a pH of 6.5 containing 200 μM BNPP. The receiver wells used HBSS buffer containing 10 mM HEPES and 15 mM glucose at a pH of 7.4 and supplemented with 1% BSA. After an initial equilibration with transport buffer, TEER values were read to test membrane integrity. The experiment was started by the addition of buffers containing test compounds, 200 and 1000 μL in the apical and basolateral chambers, respectively, to determine forward (A to B) and reverse (B to A) permeability. At 0 and 2 h post dose, 10 μL was sampled from the donor compartment and was diluted in 190 μL of 20% MeOH. At 1 and 2 h post dose, 100 μL of solution was taken from the receiver compartments and was diluted in 100 μL of 20% MeOH. Removed buffer was replaced with fresh buffer, and a correction was applied to all calculations for the removed material. Each compound was tested in 2 separate, replicate wells for each condition. All samples were then extracted with 400 μL of 100% acetonitrile containing internal standard to precipitate protein. To test for nonspecific binding and compound instability, the total amount of drug was quantitated at the end of the experiment and compared to the material present in the original dosing solution as a percent recovery. Samples were analyzed by LCMS/MS for quantitation of both prodrug and parent in each chamber on an API 6500+ MS system (Sciex, Framingham, MA) with Shimadzu LC-20ADXR ternary pump system (Shimadzu, Columbia, MD) and an HTC PAL autosampler from LEAP Technologies (LEAP Technologies, Carrboro, NC). Compounds were separated and eluted on a Synergi 4 μm Polar-RP 2.0 × 150 mm column (Phenomenex, Torrance, CA) at a flow rate of 0.6 mL/min and using a multistage gradient with mobile phases containing either 1% or 99% CH3CN in 0.2% formic acid.
In Vivo Pharmacokinetics.
In vivo pharmacokinetic (PK) studies were performed at either Covance (rats, dogs, and cynomolgus monkeys; Madison, WI) or Lovelace Biomedical Research Institute (AGM; LBRI, Albuquerque, NM) in accordance with local IACUC guidelines. Rats, dogs, cynomolgus monkeys, or AGMs received a single intravenous (2 as a 30 min infusion) or oral (2 or 3) administration at doses indicated and in vehicles noted below. Formulation of 2 for iv administration in rats, cynomolgus monkeys, and AGMs was 5% ethanol (EtOH), 30% propylene glycol (PG), 45% polyethylene glycol-300 (PEG300), and 20% water, pH 2–3, or in dog was 5% EtOH, 30% PG, 45% polyethylene glycol-400 (PEG400), and 20% water with 1 equiv of hydrochloric acid (HCl). Formulation of 2 for oral dosing in rats was 5% EtOH, 30% PG, 45% PEG400, and 20% water plus 1 equiv of HCl or 2.5% dimethyl sulfoxide (DMSO), 10% Kolliphor HS-15, 10% Labrasol, 2.5% PG, and 75% water, pH 2. Formulation of 2 for oral dosing in dogs and cynomolgus monkeys was 5% EtOH, 30% PG, 45% PEG400, and 20% water with 1 equiv of HCl. Formulation of 2 for oral dosing in AGMs was 5% EtOH, 45% PEG300, 30% PG, and 20% water, pH 2 hand-compressed 3. Formulation of 3 for oral dosing in rats, cynomolgus monkeys, and AGMs was 2.5% DMSO, 10% Kolliphor HS-15, 10% Labrasol, 2.5% PG, and 75% water, pH 2–3. Formulation of 3 for oral dosing in dogs was 0.5% DMSO, 2% Kolliphor HS-15, 2% Labrasol, 0.5% PG, and 95% water. Formulation of 3 for solid oral dosing in rats was 0.5% methyl cellulose, 99.5% water, pH 6. Formulation of 3 for solid oral dosing in pentagastrin-pretreated dogs (at a 175 mg fixed dose) was a hand-compressed tablet with a composition of 50% 3 freebase Form III, 44.5% microcrystalline cellulose, 4% crospovidone, and 1.5% magnesium stearate. Formulation of 3 for solid oral dosing in cynomolgus monkeys (at a 124 mg fixed dose) was a hand-compressed tablet with a composition of 50.8% 3 freebase Form III, 43.7% microcrystalline cellulose, 4% crospovidone, and 1.5% magnesium stearate.
Serial blood collection was performed from 3 animals and processed to plasma at iv predose, 0.25, 0.48, 0.58, 0.75, 1.5, 3, 6, 8, 12, 24 h or oral predose, 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 h postdose. Aliquots of 20–50 μL of plasma were added to a mixture containing 250 μL of MeOH and 25 μL of internal standard solution and centrifuged, and 250 μL of resulting supernatant was then transferred, dried under a stream of nitrogen at 40 °C, and reconstituted in a mixture of 5% acetonitrile and 95% water. Plasma concentrations of 3 and/or 2 were determined using 8-point calibration curves spanning at least 3 orders of magnitude with quality control samples to ensure accuracy and precision, all prepared in plasma. Analytes were separated on a 50 × 3.0 mm, 2.5 μm Synergi Polar-RP column (Phenomenex, Torrance, CA) using a multistage linear gradient with mobile phases containing 0.1% formic acid in either water or acetonitrile at a flow rate of 0.7 mL/min. Pharmacokinetic parameters were calculated using Phoenix (v8, Certara, Princeton, NJ).
AGM lung tissue concentrations were determined as previously described.12 Briefly, lung tissues were collected as a nonsurvival surgical procedure at 24 h postdose. Animals were sedated with ketamine administered via an intramuscular injection prior to surgery. A section of the lower lung was dissected and immediately placed into liquid nitrogen less than 5 min from the start of surgery. Frozen tissues were pulverized using a cell crusher and transferred into preweighed conical tubes, all performed and maintained on dry ice. Tissues were weighed, and a volume of dry ice-cold extraction buffer was added. Resulting mixtures were then promptly homogenized. Undosed control tissues were used for quantification of the metabolites in lung tissue, generated by spiking an appropriate amount of metabolite standard solution into control tissues. An aliquot of the homogenate was filtered, evaporated to dryness, and reconstituted with 1 mM ammonium phosphate buffer for analysis by LCMS/MS. Analysis was performed using similar methods as previously described.15
AGM SARS-CoV-2 Antiviral Efficacy Study.
The in vivo efficacy study was conducted at LBRI. All studies were conducted under an IACUC-approved protocol in compliance with the Animal Welfare Act, PHS policy, and other federal statutes and regulations relating to animals and experiments involving research animals. The efficacy study, which involved animals experimentally infected with SARS-CoV-2, was conducted in an animal biosafety level 3 (ABSL-3) laboratory. Wild-caught AGMs (St. Kitts origin) were sourced through Worldwide Primates Inc. (Florida, USA) for all studies. AGMs were housed in adjacent individual cages within a climate-controlled room with a fixed light/dark cycle (12-h light/12-h dark). AGMs were monitored at least twice daily for the duration of the study by trained personnel. Commercial monkey chow, treats, and fruit were provided twice daily. Water was available to the AGMs ad libitum. The antiviral efficacy of 3 was evaluated in 24 animals (12 males and 12 females) in 3 cohorts of 8 animals each staggered by 1 day. Animals were infected under anesthesia with 3 × 106 TCID50 of SARS-CoV-2 virus (WA1 isolate) via intranasal (0.5 mL each nostril = 1 mL total) and intratracheal (2 mL) instillation. Animals were randomly placed into one of three groups (vehicle control, 60 mg/kg 3, or 120 mg/kg 3) each with N = 8. Per os (PO) dosing for all groups was initiated via oral gavage at 8 h post-infection and then once daily thereafter through day 5 post-infection. Compound 3 was formulated at either 30 or 60 mg/mL solutions in 2.5% dimethyl sulfoxide (DMSO), 2.5% propylene glycol, 10% Labrasol, 10% Kolliphor HS-15, and 75% water at pH 2–3. Blood samples were collected from a femoral, saphenous, or cephalic vein daily into K2EDTA tubes. A daily blood sample was collected from each animal immediately prior to gavage to monitor dosing trough plasma PK levels. Samples were centrifuged at 1700–1800g at 4 °C for 10 min; plasma was isolated, aliquoted, frozen immediately on dry ice, and stored at −70 °C until inactivation by organic solvent sterilization and subsequent bioanalysis. Nasal and throat swabs were collected at 1, 2, 4, and 6 days post-infection (dpi) using a cotton-tipped applicator presoaked in sterile saline. Swabs were placed in a tube containing 0.5 mL of sterile saline, frozen immediately on dry ice, and stored at −70 °C until further processing. Bronchioalveolar lavage fluid (BALF) was collected from left and right caudal lung lobes at 1, 2, 4, and 6 dpi. For BALF collection, a pediatric bronchoscope (Olympus XP-40) was advanced into the caudal lung lobe, 10 mL of sterile saline was infused, and the maximum volume was aspirated. BALF was centrifuged at 1000g for 10 min at 4 °C. The resulting cell pellet and 1 mL aliquots of supernatant were flash frozen and stored at −70 °C until further processing for viral titer analysis or inactivation by organic solvent sterilization and subsequent bioanalysis. Animals were monitored daily for any clinical evidence of disease and body weights, and temperature measurements were recorded on days 1, 2, 4, and 6 post-infection. Animals were euthanized for tissue collection and necropsy at day 6 post-infection. Sections of the right lung were flash frozen in liquid nitrogen and stored at −70 °C for analysis of viral RNA loads. Reverse transcription quantitative PCR (RT-qPCR) and plaque forming assay (PFA) methods are described in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
We would like to extend the following acknowledgments: Lovelace Biomedical support teams for contributions to the execution of the SARS-CoV-2 AGM efficacy model. The following reagents were deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate hCoV-19/USA/NY-MSHSPSP-PV56475/2022 (Lineage BA.2.12.1; Omicron Variant), NR-56781, deposited by Dr. Viviana Simon; SARS-Related Coronavirus 2, Isolate hCoV-19/USA/MD-HP35538/2022 (Lineage BA.4.6; Omicron Variant), NR-58715 and SARS-Related Coronavirus 2, Isolate hCoV-19/USA/MD-HP34985/2022 (Lineage BF.5; Omicron Variant), NR-58716, contributed by Dr. Andrew S. Pekosz. We thank Kenneth S. Plante, Jessica A. Plante, and David S. Blakeman for coordination of virus stocks from the World Reference Center for Emerging Viruses and Arboviruses at the University of Texas Medical Branch. This study was funded by Gilead Sciences, Inc. and supported by the Infectious Diseases Clinical Research Consortium through the National Institute for Allergy and Infectious Diseases of the National Institutes of Health, under award number UM1AI148684. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
ABBREVIATIONS USED
- AB
apical to basolateral
- AGM
African green monkey
- ATP
adenosine triphosphate
- BALF
bronchioalveolar lavage fluid
- BEGM
bronchial epithelial growth medium
- CES
carboxyesterase
- cyno
cynomolgus monkey
- DIC
N,N′-diisopropylcarbodiimide
- DMAP
4-(dimethylamino)pyridine
- DMEM
Dulbecco’s modified eagle medium
- dpi
day post-infection
- FBS
fetal bovine serum
- GI
gasterointestinal
- gRNA
genomic RNA
- Hep
hepatocytes
- hr
human recombinant
- IACUC
International Animal Care and Use Committee
- iv
intravenous
- LLOQ
lower limit of quantification
- MERS
Middle East Respiratory Syndrome
- MOI
multiplicity of infection
- NHBE
normal human bronchial epithelial
- NHP
nonhuman primate
- NTP
nucleotide triphosphate
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate-buffered saline
- PFA
plaque forming assay
- PFU
plaque forming unit
- PHH
primary human hepatocyte
- PMSF
phenylmethylsulfonyl fluoride
- RdRp
RNA-dependent RNA polymerase
- RSV
respiratory syncytial virus
- SARS
severe acute respiratory syndrome
- VOC
variant of concern
- WHO
World Health Organization
Footnotes
The authors declare the following competing financial interest(s): Some authors are current or former employees of Gilead Sciences and may own company stock.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00750.
Molecular formula strings (CSV)
Additional experimental details, supporting data tables and figures, NMR and HPLC spectral data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.3c00750
Contributor Information
Richard L. Mackman, Medicinal Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States; Present Address: R.M.: 360 Ashton Avenue, Millbrae, California 94030, United States
Rao V. Kalla, Medicinal Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States
Darius Babusis, Drug Metabolism, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Jared Pitts, Discovery Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Kimberly T. Barrett, Formulation and Process Development, Gilead Sciences Incorporated, Foster City, California 94404, United States
Kwon Chun, Medicinal Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Venice Du Pont, Discovery Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Lauren Rodriguez, Clinical Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Jasmine Moshiri, Clinical Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Yili Xu, Biochemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Michael Lee, Biology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Gary Lee, Biology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Blake Bleier, Formulation and Process Development, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Anh-Quan Nguyen, Formulation and Process Development, Gilead Sciences Incorporated, Foster City, California 94404, United States.
B. Michael O’Keefe, Process Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Andrea Ambrosi, Process Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States; Present Address: A.A.: 1 DNA Way, South San Francisco, California 94080, United States.
Meredith Cook, Process Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Joy Yu, Process Chemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Kassibla Elodie Dempah, Process Development, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Elaine Bunyan, Process Development, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Nicholas C. Riola, Discovery Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States
Xianghan Lu, Discovery Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Renmeng Liu, Drug Metabolism, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Ashley Davie, Drug Metabolism, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Tien-Ying Hsiang, Center for Innate Immunity and Immune Disease, Department of Immunology, School of Medicine, University of Washington, Seattle, Washington 98109, United States.
Justin Dearing, Lovelace Biomedical Research Institute, Albuquerque, New Mexico 87108, United States; Present Address: J.D.: Department of Microbiology and Immunology, University of North Carolina, 125 Mason Farm Road, Chapel Hill, North Carolina 27599, United States..
Meghan Vermillion, Lovelace Biomedical Research Institute, Albuquerque, New Mexico 87108, United States; Present Address: M.V.: Gilead Sciences, Incorporated, Foster City, California 94404, United States..
Michael Gale, Jr., Center for Innate Immunity and Immune Disease, Department of Immunology, School of Medicine, University of Washington, Seattle, Washington 98109, United States
Anita Niedziela-Majka, Biology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Joy Y. Feng, Biochemistry, Gilead Sciences Incorporated, Foster City, California 94404, United States
Charlotte Hedskog, Clinical Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
John P. Bilello, Discovery Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States
Raju Subramanian, Drug Metabolism, Gilead Sciences Incorporated, Foster City, California 94404, United States.
Tomas Cihlar, Discovery Virology, Gilead Sciences Incorporated, Foster City, California 94404, United States.
REFERENCES
- (1).https://covid19.who.int/ (Accessed Feb 26, 2023).
- (2).V’kovski P; Kratzel A; Steiner S; Stalder H; Thiel V Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19 (3), 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Taylor PC; Adams AC; Hufford MM; de la Torre I; Winthrop K; Gottlieb RL Neutralizing Monoclonal Antibodies for Treatment of COVID-19. Nat. Rev. Immunol. 2021, 21 (6), 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gil C; Ginex T; Maestro I; Nozal V; Barrado-Gil L; Cuesta-Geijo MÁ; Urquiza J; Ramírez D; Alonso C; Campillo NE; Martinez A. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63 (21), 12359–12386. [DOI] [PubMed] [Google Scholar]
- (5).Gordon CJ; Tchesnokov EP; Woolner E; Perry JK; Feng JY; Porter DP; Götte M Remdesivir is a Direct-Acting Antiviral that Inhibits RNA-Dependent RNA Polymerase From Severe Acute Respiratory Syndrome Coronavirus 2 with High Potency. J. Biol. Chem. 2020, 295 (20), 6785–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tchesnokov EP; Gordon CJ; Woolner E; Kocinkova D; Perry JK; Feng JY; Porter DP; Götte M Template-dependent Inhibition of Coronavirus RNA-dependent RNA Polymerase by Remdesivir Reveals a Second Mechanism of Action. J. Biol. Chem. 2020, 295 (47), 16156–16165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kabinger F; Stiller C; Schmitzová J; Dienemann C; Kokic G; Hillen HS; Höbartner C; Cramer P Mechanism of Molnupiravir-Induced SARS-CoV-2 Mutagenesis. Nat. Struct. Mol. Biol. 2021, 28 (9), 740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y An oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science. 2021, 374 (6575), 1586–1593. [DOI] [PubMed] [Google Scholar]
- (9).Gottlieb RL; Vaca CE; Paredes R; Mera J; Webb BJ; Perez G; Oguchi G; Ryan P; Nielsen BU; Brown M; Hidalgo A; Sachdeva Y; Mittal S; Osiyemi O; Skarbinski J; Juneja K; Hyland RH; Osinusi A; Chen S; Camus G; Abdelghany M; Davies S; Behenna-Renton N; Duff F; Marty FM; Katz MJ; Ginde AA; Brown SM; Schiffer JT; Hill JA GS-US-540–9012 (PINETREE) Investigators. Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients. N. Engl. J. Med. 2022, 386 (4), 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hammond J; Leister-Tebbe H; Gardner A; Abreu P; Bao W; Wisemandle W; Baniecki M; Hendrick VM; Damle B; Simón-Campos A; Pypstra R; Rusnak JM EPIC-HR Investigators. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Engl. J. Med. 2022, 386 (15), 1397–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/ritonavir-boosted-nirmatrelvir--paxlovid-/paxlovid-drug-drug-interactions/ (Accessed Feb 26, 2023).
- (12).Mackman RL; Hui HC; Perron M; Murakami E; Palmiotti C; Lee G; Stray K; Zhang L; Goyal B; Chun K; Byun D; Siegel D; Simonovich S; Du Pont V; Pitts J; Babusis D; Vijjapurapu A; Lu X; Kim C; Zhao X; Chan J; Ma B; Lye D; Vandersteen A; Wortman S; Barrett KT; Toteva M; Jordan R; Subramanian R; Bilello JP; Cihlar T Prodrugs of a 1’-CN-4-Aza-7,9-dideazaadenosine C-Nucleoside Leading to the Discovery of Remdesivir (GS-5734) as a Potent Inhibitor of Respiratory Syncytial Virus with Efficacy in the African Green Monkey Model of RSV. J. Med. Chem. 2021, 64 (8), 5001–5017. [DOI] [PubMed] [Google Scholar]
- (13).Cho A; Saunders OL; Butler T; Zhang L; Xu J; Vela JE; Feng JY; Ray AS; Kim CU Synthesis and Antiviral Activity of a Series of 1′-Substituted 4-Aza-7,9-dideazaadenosine C-nucleosides. Bioorg. Med. Chem. Lett. 2012, 22 (8), 2705–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cihlar T; Mackman RL Journey of Remdesivir From the Inhibition of Hepatitis C virus to the Treatment of COVID-19. Antiviral Ther. 2022, 27 (2), 13596535221082773. [DOI] [PubMed] [Google Scholar]
- (15).Mackman RL; Cihlar T Veklury® (Remdesivir), A Nucleotide Prodrug Approved for the treatment of COVID-19. 2022 Medicinal Chemistry Reviews; Medicinal Chemistry Reviews; MEDI, Inc. Published by American Chemical Society: Washington, DC, 2022; Vol. 57, pp 545–569;. [Google Scholar]
- (16).Warren TK; Jordan R; Lo MK; Ray AS; Mackman RL; Soloveva V; Siegel D; Perron M; Bannister R; Hui HC; Larson N; Strickley R; Wells J; Stuthman KS; Van Tongeren SA; Garza NL; Donnelly G; Shurtleff AC; Retterer CJ; Gharaibeh D; Zamani R; Kenny T; Eaton BP; Grimes E; Welch LS; Gomba L; Wilhelmsen CL; Nichols DK; Nuss JE; Nagle ER; Kugelman JR; Palacios G; Doerffler E; Neville S; Carra E; Clarke MO; Zhang L; Lew W; Ross B; Wang Q; Chun K; Wolfe L; Babusis D; Park Y; Stray KM; Trancheva I; Feng JY; Barauskas O; Xu Y; Wong P; Braun MR; Flint M; McMullan LK; Chen SS; Fearns R; Swaminathan S; Mayers DL; Spiropoulou CF; Lee WA; Nichol ST; Cihlar T; Bavari S Therapeutic Efficacy of the Small Molecule GS-5734 Against Ebola Virus in Rhesus Monkeys. Nature 2016, 531, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gordon CJ; Lee HW; Tchesnokov EP; Perry JK; Feng JY; Bilello JP; Porter DP; Götte M Efficient Incorporation and Template-Dependent Polymerase Inhibition are Major Determinants for the Broad-Spectrum Antiviral Activity of Remdesivir. J. Biol. Chem. 2022, 298 (2), 101529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sheahan TP; Sims AC; Graham RL; Menachery VD; Gralinski LE; Case JB; Leist SR; Pyrc K; Feng JY; Trantcheva I; Bannister R; Park Y; Babusis D; Clarke MO; Mackman RL; Spahn JE; Palmiotti CA; Siegel D; Ray AS; Cihlar TC; Jordan R; Denison MR; Baric RS Broad-Spectrum Antiviral GS-5734 Inhibits Both Epidemic and Zoonotic Coronaviruses. Sci. Trans. Med. 2017, 9 (396), No. eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sheahan TP; Sims AC; Leist SR; Schäfer A; Won J; Brown AJ; Montgomery SA; Hogg A; Babusis D; Clarke MO; Spahn JE; Bauer L; Sellers S; Porter D; Feng JY; Cihlar T; Jordan R; Denison MR; Baric RS Comparative The rapeutic Efficacy of Remdesivir and Combination Lopinavir, Ritonavir, and Interferon Beta Against MERS-CoV. Nat. Commun. 2020, 11 (1), 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).de Wit E; Feldmann F; Cronin J; Jordan R; Okumura A; Thomas T; Scott D; Cihlar T; Feldmann H Prophylactic and Therapeutic Remdesivir (GS-5734) Treatment in the Rhesus Macaque Model of MERS-CoV Infection. Proc. Nat. Acad. Sci. 2020, 117 (12), 6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Beigel JH; Tomashek KM; Dodd LE; Mehta AK; Zingman BS; Kalil AC; Hohmann E; Chu HY; Luetkemeyer A; Kline S; Lopez de Castilla D; Finberg RW; Dierberg K; Tapson V; Hsieh L; Patterson TF; Paredes R; Sweeney DA; Short WR; Touloumi G; Lye DC; Ohmagari N; Oh M.-d.; Ruiz-Palacios GM; Benfield T; Fatkenheuer G; Kortepeter MG; Atmar RL; Creech CB; Lundgren J; Babiker AG; Pett S; Neaton JD; Burgess TH; Bonnett T; Green M; Makowski M; Osinusi A; Nayak S; Lane HC for the ACTT-1 Study Group Members. Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chokkalingam AP; Hayden J; Goldman JD; Li H; Asubonteng J; Mozaffari E; Bush C; Wang JR; Kong A; Osinusi AO; Gottlieb RL Association of Remdesivir Treatment With Mortality Among Hospitalized Adults With COVID-19 in the United States. JAMA Netw. Open. 2022, 5 (12), No. e2244505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mozaffari E; Chandak A; Zhang Z; Liang S; Thrun M; Gottlieb RL; Kuritzkes DR; Sax PE; Wohl DA; Casciano R; Hodgkins P; Haubrich R Remdesivir Treatment in Hospitalized Patients With Coronavirus Disease 2019 (COVID-19): A Comparative Analysis of In-hospital All-cause Mortality in a Large Multicenter Observational Cohort. Clin. Infect. Dis. 2022, 75 (1), e450–e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pitts J; Babusis D; Vermillion MS; Subramanian R; Barrett K; Lye D; Ma B; Zhao X; Riola N; Xie X; Kajon A; Lu X; Bannister R; Shi PY; Toteva M; Porter DP; Smith BJ; Cihlar T; Mackman R; Bilello JP Intravenous Delivery of GS-441524 is Efficacious in the African Green Monkey Model of SARS-CoV-2 Infection. Antiviral Res. 2022, 203, 105329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cao L; Li Y; Yang S; Li G; Zhou Q; Sun J; Xu T; Yang Y; Liao R; Shi Y; Yang Y; Zhu T; Huang S; Ji Y; Cong F; Luo Y; Zhu Y; Luan H; Zhang H; Chen J; Liu X; Luo R; Liu L; Wang P; Yu Y; Xing F; Ke B; Zheng H; Deng X; Zhang W; Lin C; Shi M; Li CM; Zhang Y; Zhang L; Dai J; Lu H; Zhao J; Zhang X; Guo D The Adenosine Analog Prodrug ATV006 is Orally Bioavailable and has Preclinical Efficacy Against Parental SARS-CoV-2 and Variants. Sci. Transl. Med. 2022, 14 (661), No. eabm7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tao S; Zandi K; Bassit L; Ong YT; Verma K; Liu P; Downs-Bowen JA; McBrayer T; LeCher JC; Kohler JJ; Tedbury PR; Kim B; Amblard F; Sarafianos SG; Schinazi RF Comparison of Anti-SARS-CoV-2 Activity and Intracellular Metabolism of Remdesivir and its Parent Nucleoside. Curr. Res. Pharm. Drug Discovery 2021, 2, 100045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cox RM; Wolf JD; Lieber CM; Sourimant J; Lin MJ; Babusis D; DuPont V; Chan J; Barrett KT; Lye D; Kalla R; Chun K; Mackman RL; Ye C; Cihlar T; Martinez-Sobrido L; Greninger AL; Bilello JP; Plemper RK Oral Prodrug of Remdesivir Parent GS-441524 is Efficacious Against SARS-CoV-2 and a Variant of Concern in Ferrets. Nat. Commun. 2021, 12, 6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).He YL; Murby S; Warhurst G; Gifford L; Walker D; Ayrton J; Eastmond R; Rowland M Species Differences in Size Discrimination in the Paracellular Pathway Reflected by Oral Bioavailability of Poly(ethylene glycol) and D-peptides. J. Pharm. Sci. 1998, 87 (5), 626–633. [DOI] [PubMed] [Google Scholar]
- (29).Klumpp K; Smith DB In Discovery and Clinical Evaluation of the Nucleoside Analog Balapiravir (R1626) for the Treatment of HCV Infection, 1st ed.; Kazmierski WM, Ed.; John Wiley & Sons Inc., 2011. [Google Scholar]
- (30).Siegel D; Hui HC; Doerffler E; Clarke MO; Chun K; Zhang L; Neville S; Carra E; Lew W; Ross B; Wang Q; Wolfe L; Jordan R; Soloveva V; Knox J; Perry J; Perron M; Stray KM; Barauskas O; Feng JY; Xu Y; Lee G; Rheingold AL; Ray AS; Bannister B; Strickley R; Swaminathan S; Lee WA; Bavari S; Cihlar T; Lo MK; Warren TK; Mackman RL Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f ] [triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60, 1648–1661. [DOI] [PubMed] [Google Scholar]
- (31).Wei D; Hu T; Zhang Y; Zheng W; Xue H; Shen J; Xie Y; Aisa HA Potency and Pharmacokinetics of GS-441524 Derivatives Against SARS-CoV-2. Bioorg. Med. Chem. 2021, 46, 116364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xu Y; Barauskas O; Kim C; Babusis D; Murakami E; Kornyeyev D; Lee G; Stepan G; Perron M; Bannister R; Schultz BE; Sakowicz R; Porter D; Cihlar T; Feng JY Off-Target In Vitro Profiling Demonstrates that Remdesivir Is a Highly Selective Antiviral. Antimicrob. Agents Chemother. 2021, 65 (2), No. e02237–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Martin R; Li J; Parvangada A; Perry J; Cihlar T; Mo H; Porter D; Svarovskaia E Genetic Conservation of SARS-CoV-2 RNA Replication Complex in Globally Circulating Isolates and Recently Emerged Variants from Humans and Minks Suggests Minimal Pre-Existing Resistance to Remdesivir. Antiviral Res. 2021, 188, 105033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Pitts J; Li J; Perry JK; Du Pont V; Riola N; Rodriguez L; Lu X; Kurhade C; Xie X; Camus G; Manhas S; Martin R; Shi PY; Cihlar T; Porter DP; Mo H; Maiorova E; Bilello JP Remdesivir and GS-441524 Retain Antiviral Activity against Delta, Omicron, and Other Emergent SARS-CoV-2 Variants. Antimicrob. Agents Chemother. 2022, 66 (6), No. e00222–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brown SM; Katz MJ; Ginde AA; Juneja K; Ramchandani M; Schiffer JT; Vaca C; Gottlieb RL; Tian Y; Elboudwarej E; Hill JA; Gilson R; Rodriguez L; Hedskog C; Chen S; Montezuma-Rusca JM; Osinusi A; Paredes R Consistent Effects of Early Remdesivir on Symptoms and Disease Progression Across At-Risk Outpatient Subgroups: Treatment Effect Heterogeneity in PINETREE Study. Infect. Dis. Ther 2023, 12, 1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Moorman AR; Chamberlain SD; Jones LA; de Miranda P; Reynolds DJ; Peoples ME; Krenitsky TA 5′ -Ester Prodrugs of the Varicella-Zoster Antiviral Agent, 6-Methoxypurine Arabinoside. Anti. Chem. & Chemother 1992, 3 (3), 141–146. [DOI] [PubMed] [Google Scholar]
- (37).Baker DC; Haskell TH; Putt SR Prodrugs of 9-Beta-D-Arabinofuranosyladenine. 1. Synthesis and Evaluation of some 5′-(O-Acyl) Derivatives. J. Med. Chem. 1978, 21 (12), 1218–1221. [DOI] [PubMed] [Google Scholar]
- (38).Wang AQ; Hagen NR; Padilha EC; Yang M; Shah P; Chen CZ; Huang W; Terse P; Sanderson P; Zheng W; Xu X Preclinical Pharmacokinetics and In Vitro Properties of GS-441524, a Potential Oral Drug Candidate for COVID-19 Treatment. Front. Pharmacol. 2022, 13, 918083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Li R; Liclican A; Xu Y; Pitts J; Niu C; Zhang J; Kim C; Zhao X; Soohoo D; Babusis D; Yue Q; Ma B; Murray BP; Subramanian R; Xie X; Zou J; Bilello JP; Li L; Schultz BE; Sakowicz R; Smith BJ; Shi PY; Murakami E; Feng JY Key Metabolic Enzymes Involved in Remdesivir Activation in Human Lung Cells. Antimicrob. Agents Chemother. 2021, 65 (9), No. e00602–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schäfer A; Martinez DR; Won JJ; Meganck RM; Moreira FR; Brown AJ; Gully KL; Zweigart MR; Conrad WS; May SR; Dong S; Kalla R; Chun K; Du Pont V; Babusis D; Tang J; Murakami E; Subramanian R; Barrett KT; Bleier BJ; Bannister R; Feng JY; Bilello JP; Cihlar T; Mackman RL; Montgomery SA; Baric RS; Sheahan TP Therapeutic Treatment with an Oral Prodrug of the Remdesivir Parental Nucleoside is Protective Against SARS-CoV-2 Pathogenesis in Mice. Sci. Transl. Med. 2022, 14 (643), No. eabm3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Humeniuk R; Mathias A; Kirby BJ; Lutz JD; Cao H; Osinusi A; Babusis D; Porter D; Wei X; Ling J; Reddy YS; German P Pharmacokinetic, Pharmacodynamic, and Drug-Interaction Profile of Remdesivir, a SARS-CoV-2 Replication Inhibitor. Clin. Pharmacokinet. 2021, 60 (5), 569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Anoshchenko O; Abdelghany M; Hyland RH; Davies S; Mkaya C; Shen G; Xiao D; Winter H; Llewellyn J; Humeniuk R Pharmacokinetics, Safety, and Tolerability of GS-5245 in Healthy Participants. 33rd European Congress of Clinical Microbiology & Infectious Diseases, Copenhagen (Hovedstaden), Denmark, Apr 15–18, 2023; ESCMID, 2023. [Google Scholar]
- (43).Xie X; Muruato AE; Zhang X; Lokugamage KG; Fontes-Garfias CR; Zou J; Liu J; Ren P; Balakrishnan M; Cihlar T; Tseng CK; Makino S; Menachery VD; Bilello JP; Shi PY A Nanoluciferase SARS-CoV-2 for Rapid Neutralization Testing and Screening of Anti-Infective Drugs for COVID-19. Nat. Commun. 2020, 11, 5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.