

Abstract
Nucleic acid amplification tests (NAATs), which amplify and detect pathogen nucleic acids, are vital methods to diagnose diseases, particularly in cases where patients exhibit low levels of infection. For many blood-borne pathogens such as HIV or Plasmodium falciparum, it is necessary to first extract pathogen RNA or DNA from patient blood prior to NAAT analysis. Traditional nucleic acid extraction methods are expensive, resource-intensive and are often difficult to deploy to resource-limited areas where many blood-borne infections are widespread. Here, we describe a portable, paper-and-plastic device, called SNAPflex, for instrument-free nucleic acid extraction from whole blood, which builds upon our previous work for RNA extraction using a pressure-driven extraction system. SNAPflex shows improved HIV RNA extraction from simulated patient samples compared to traditional extraction methods as well as long-term stability of extracted RNA without the need for cold storage. We further demonstrated successful extraction and recovery of P. falciparum DNA from cultured parasites in whole blood. SNAPflex was designed to be easily manufacturable and deployable to resource-limited settings.
Introduction
Nucleic acid amplification tests (NAATs) have become vital tools for disease diagnosis. Amplification and detection of trace amounts of pathogen nucleic acids enables highly sensitive, quantitative diagnosis of infectious diseases. Unlike other diagnostic tests such as portable enzyme-linked immunosorbent assays (ELISA)-based rapid diagnostic tests, NAATs enable quantitative output, even in cases of low pathogenicity.
For blood-borne pathogens, such as HIV or malaria, NAATs require detection of pathogen nucleic acids from patient blood. While some isothermal methods amplify nucleic acids in the presence of small amounts of whole blood,1–3 traditional methods such as quantitative polymerase chain reaction for DNA targets (qPCR) and reverse transcriptase (RT)-qPCR for RNA targets are not compatible with direct blood amplification because of assay inhibition by whole blood components. Whole blood quenches fluorescence readout from passive reference dyes and double stranded DNA intercalating dyes, inhibiting real time amplification readout. Furthermore, immunoglobulin G, haemoglobin, and hematin in whole blood have all been shown to inhibit polymerization by binding to single stranded DNA and/or affecting DNA polymerase activity.4,5 Therefore, directly using whole blood as an input sample is often not possible for routinely used NAATs such as qPCR and RT-qPCR.
An important first step for traditional NAATs, therefore, is the extraction, isolation, and purification of pathogen nucleic acids from whole blood. Commercially-available kits typically use solid phase extraction methods with centrifugation to collect nucleic acids in spin columns with silica membranes.6 Typically, these kits contain a chaotropic lysis buffer to liberate nucleic acids from the sample matrix. Extracted nucleic acids are then driven to bind to the silica membrane in the presence of the chaotrope, further propagated by the introduction of a primary alcohol to the solution.6,7 Wash buffers are then used to purify the nucleic acids on the membrane. A final elution step into an appropriate buffer recovers the nucleic acids from the membrane for use in NAATs.
While spin column-based kits are useful, there are some concerns with using them in resource-limited settings (RLS). One concern is that, depending on the target pathogen, sample pre-processing may be required to isolate the target nucleic acids. For example, in the case of HIV, plasma separation may be required prior to extracting viral RNA from the sample,8,9 while in the case of malaria, red blood cells must be lysed, as the parasites are intracellular to red blood cells.10 Additionally, the requirement for centrifugation to isolate and purify the nucleic acids makes these methods viable only in a clean and well-resourced laboratory environment rather than in field-based clinical settings.
In RLS, it may be required to collect samples from patients which can then be shipped to central testing facilities that are equipped to perform the necessary nucleic acid isolation steps. For blood samples, this shipment process requires cold storage and fast shipping times, as blood coagulation and sample degradation can occur within 24–48 hours of sample collection.11 In some instances, it has been estimated that preparation and transport of blood and plasma samples from clinic and hospital settings in RLS to central testing facilities can be extremely expensive, comprising up to a third of testing costs.12 The lack of cold chain infrastructure and the high cost of sample shipment can therefore make traditional NAATs inaccessible to many patients in RLS.
Dried blood spot (DBS) and dried plasma spot (DPS) samples can be collected as alternatives to whole blood and plasma samples and transitioning to these sample types may be advantageous for RLS. DBS and DPS samples facilitate sample shipment without cold storage, thereby enabling simpler sample transport and delayed sample analysis.13–15 In the case of HIV viral load (VL) monitoring for example, dried samples have the potential to increase testing access by 19% in low-and middle-income countries (LMICs).16 For these samples, finger prick blood volumes are collected on specialized cards and dried for approximately three hours prior to storage and shipment to a central testing facility for nucleic acid extraction and analysis. While DBS and DPS simplify sample collection, there are several downsides to these methods. Extensive sample processing is still required to extract and purify nucleic acids from the samples prior to NAAT analysis, and nucleic acid recovery can often be much lower than from fresh samples. Additionally, particularly RNA in DBS samples has been shown to degrade over time both from contact with water and due to the presence of RNAses in the blood sample and in the environment.17,18 Depending on the drying conditions (i.e. temperature, humidity) and the pathogen loads, RNA could degrade at different rates, thereby resulting in variable and potentially unreliable test results.17 For HIV-1 RNA, for example, evaluation of three commercial assays for RT-qPCR HIV-1 VL analysis from DBS samples showed highly variable diagnostic accuracy.19
It is therefore necessary to improve nucleic acid sample collection to enable highly sensitive NAATs in RLS. It has been shown that extracted RNA stored in dry conditions shows less degradation and more consistent amplification, even when stored at room temperature or elevated temperatures over several weeks.20–23 However, nucleic acid extraction from blood prior to shipment to central testing facility can be resource-intensive and impractical for RLS.24 While some commercial products are available to stabilize RNA when blood is collected, these can be cost-prohibitive, and still require cold storage of the blood sample.25
Here we describe a novel paper-based device for room temperature extraction, purification, and long-term storage of nucleic acids from whole blood. Our instrument-free extraction method is designed for use in RLS. This device is a significant redesign of the pressure-driven system for nucleic acid sample preparation (SNAP) we reported previously.26 In our proposed method, whole blood lysis and nucleic acid precipitation are performed in a sample tube at room temperature, and a flexible paper-and-plastic device (SNAPflex) is used to capture purified nucleic acids on a glass fiber membrane. Nucleic acids eluted from the membrane can be used in routine NAATs such as qPCR or RT-qPCR. We demonstrate the utility of this device with two different types of samples: HIV virions in whole blood were used as a model system for RNA, while P. falciparum parasite-infected red blood cells in whole blood were used as a model system for DNA. Using these model systems, we show that SNAPflex can extract and purify both DNA and RNA samples from whole blood.
Materials and methods
SNAPflex device materials and equipment
The SNAPflex device consists of layers of laminating plastic (Staples thermal laminating plastic, 5mil), adhesive plastic (Fellowes self-laminating sheets, 3mil), a chromatography paper waste pad (Ahlstrom 320), and a paper capture membrane (Millipore 0.7 μm glass fiber without binder). Laminating plastic, chromatography paper, and adhesive sheets were cut to the appropriate dimensions using a Trotec Speedy 100 60 W laser cutter. The glass fiber capture membrane was cut to the appropriate dimensions using a Graphtec FCX2000–60VC cutter plotter. Laminating plastic was sealed using an Apache AL18P laminator. The final device also uses a removable stabilization clip which was printed with polylactic acid using an Ultimaker 3 3D printer.
SNAPflex device design
The main components of the SNAPflex device are a glass fiber capture membrane (Fig. 1A and S1A†) to collect purified nucleic acids and a chromatography paper waste pad (Fig. 1B and S1B†) to provide capillary force for passive wicking of fluid through the glass fiber capture membrane. Consistent contact between the capture membrane and waste pad is necessary to ensure successful sample application; a protrusion is therefore created on the waste pad using a 3D-printed mold (Fig. S1A†).
Fig. 1.
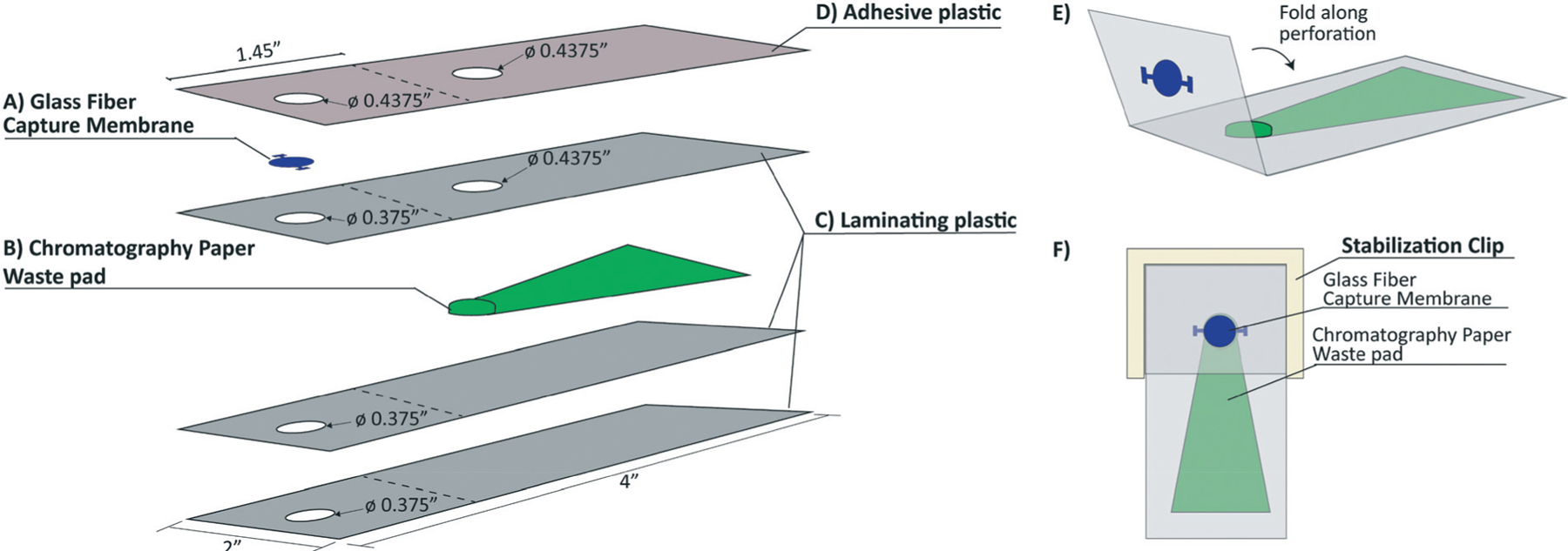
An exploded view of the layers in the SNAPflex device and final device assembly. A) Glass fiber membrane captures nucleic acids, B) chromatography paper waste pad provides capillary force for fluid flow through the capture membrane, C) three layers of laminating plastic encase the waste pad, and D) one layer of adhesive plastic secures the capture membrane to the waste pad. E) Prior to sample application, the device is folded along the perforation to bring the capture membrane in contact with the waste pad, and F) 3-D printed stabilization chip secures contact between the capture membrane and the waste pad.
The base of the SNAPflex device sandwiches the waste pad between two layers of laminating plastic with 0.375″ holes for sample application and one layer of laminating plastic with a 0.375″ sample application hole and 0.4375″ hole to accommodate the waste pad protrusion (Fig. 1C). This 4-layer base unit is heated to 320 °F with a laminator to seal the waste pad within the laminating plastic. The glass fiber capture membrane is secured to the device using an adhesive plastic with two 0.4375″ holes for sample application and contact with the waste pad (Fig. 1D).
The capture membrane portion of the device is divided from the waste pad by a perforation in the laminating plastic and adhesive plastic layers. Prior to sample application, the top (capture) section is folded over along the perforation to bring the capture membrane into contact with the waste pad (Fig. 1E). A reusable 3D-printed stabilization clip is used to keep the capture membrane in contact with the waste pad throughout the sample application and washing steps (Fig. 1F). The stabilization clip does not come into contact with either the sample or lysis buffer, and therefore can be cleaned and used with multiple devices.
For RNA capture experiments (HIV samples), the glass fiber capture membrane was washed three times in nuclease free water (Sigma-Aldrich) and dried overnight at room temperature. For DNA capture experiments (Plasmodium falciparum samples), the glass fiber capture membrane was submerged for 6–8 hours in trifluoroacetic acid (TFA, Sigma-Aldrich), and dried at room temperature overnight before assembly. The TFA-treated membrane was stored in an airtight container with desiccant until use.
Nucleic acid extraction chemistry
The custom lysis buffer we developed contains guanidine thiocyanate, N-lauroylsarcosine, and 2-mercaptoethanol to lyse cells and virions and denature proteins including RNAses. To prepare the lysis buffer, 5.8 M guanidine thiocyanate (Sigma-Aldrich) supplemented with 0.1 M MOPS (pH 7.0, titrated from free acid with 10 M NaOH, Sigma-Aldrich) was dissolved in nuclease-free water; the solution was heated at 50 °C for 10 minutes and vortexed for 1 minute to enable dissolution. 1.36 mL (2.7% v/v) of 20% N-lauroylsarcosine (sodium salt) solution (Sigma-Aldrich) were added to the solution. The solution was cooled to room temperature, 360 μL (0.72% v/v) 2-mercaptoethanol (Sigma-Aldrich) was added and the final volume was adjusted to 50 mL with nuclease free water.
Prior to sample lysis, a complete lysis buffer was prepared containing 68% (v/v) custom lysis buffer, 29% (v/v) 10% nonyl phenoxypolyethoxylethanol (NP-40, Sigma-Aldrich), and 3% (v/v) GlycoBlue co-precipitant (15 mg mL−1 blue glycogen, Applied Biosystems). GlycoBlue was included in the complete lysis buffer as a hydrophilic carrier particle which associates with nucleic acids released during sample extraction, increasing the effective particle size and enabling capture on the glass fiber capture membrane.
Each blood sample was lysed at a ratio of 1 : 2 (sample: complete lysis buffer) at room temperature for 15 minutes with inversion to mix the blood and lysis buffer together. After lysis, 35% (v/v) 1-butanol was added to the solution as a precipitating agent, increasing the hydrophobicity of the solution and causing the hydrophilic glycogen–nucleic acid complexes to form precipitates while the denatured proteins in the sample remain in the hydrophobic phase. To test reduced precipitation solution volume, some samples were also precipitated with 25% (v/v) 1-butanol mixed with chloroform (90 μL 1-butanol, 10 μL chloroform). The lysed sample was subsequently applied to the SNAPflex device to isolate and purify the precipitated nucleic acid particles.
Sample application to SNAPflex and sample elution
After the addition of precipitating buffer, the lysed blood solution was inverted to mix and immediately applied to the capture membrane on the device in 40 μL increments to capture precipitated nucleic acid–glycogen particles (Fig. 2A). An initial “pre-wash” buffer (70% ethanol, 12.5% custom lysis buffer, 17.5% nuclease free water) was applied to the capture membrane to further solubilize and remove remnant proteins and cell debris from the capture membrane; 400 μL “prewash” buffer was applied in 40 μL increments to ensure there was no overflow along the edges of the device (Fig. 2B). This wash step was followed with 200 μL 70% ethanol in nuclease free water to wash residual salts from the capture membrane (Fig. 2C). The final wash was 100 μL 95% ethanol to enable rapid drying of the preserved nucleic acids on the membrane (Fig. 2D). The 3D printed clip was then removed from the SNAPflex device, and the center circle of the capture membrane was removed for drying (Fig. 2E).
Fig. 2.

Sample application on the SNAPflex device, demonstrated with colored water for visualization. A) The entire sample is first applied to the glass fiber membrane, followed by B) 400 μL pre-wash buffer to solubilize remnant blood components, C) 200 μL 70% ethanol to wash residual salts, and D) 95% ethanol to enable rapid drying. E) The membrane is removed from the device and dried at ambient conditions. Nucleic acids can either be F) stored on the membrane in dry conditions and G) eluted from the membrane.
Once the capture membrane was completely dry (approximately 10 minutes at room temperature), the sample was either stored in a zip-top Mylar bag with a silica packet (Fig. 2F) or immediately eluted (Fig. 2G). Sample elution was performed by transferring the capture membrane to a 0.2 mL PCR tube, submerging the membrane in 100 μL elution buffer (10 mM Tris–Cl, pH 8.5, QIAGEN) and heating to 50 °C for 10 minutes. The eluted sample was collected from the membrane and stored at the appropriate storage temperature prior to analysis by RT-qPCR (HIV RNA studies) or qPCR (P. falciparum DNA studies). Eluted RNA samples were stored at −80 °C, while eluted DNA samples were stored at −20 °C.
In vitro transcribed HIV RNA recovery with SNAPflex
We first investigated isolation, purification, and recovery of RNA from blood using in vitro transcribed RNA for the HIV gag gene. We isolated DNA for the HIV gag gene from the pLAIΔmls plasmid (Addgene, plasmid #24594) and cloned the isolated DNA into a pGEM-T Easy plasmid vector (Promega) with standard cloning protocols. This purified plasmid was used as a template for in vitro transcription with a Ribomax transcription kit (Promega) to generate RNA of ∼1488 nt. The RNA stocks were diluted into elution buffer to concentrations of 1 × 107 cp mL−1, 1 × 106 cp mL−1, 1 × 105 cp mL−1, and 1 × 102 cp mL−1 to test; a subsequent 1 : 10 dilution of each stock was performed into the final sample matrix.
For experiments using in vitro transcribed RNA, 10 μL diluted RNA was first introduced directly into 200 μL of the complete lysis buffer and 90 μL whole blood (Research Blood Components, Cambridge, MA) was subsequently added to the mixture and mixed by manual inversion (1 : 2 ratio of sample : lysis buffer). In vitro transcribed RNA was introduced directly to the lysis buffer to protect purified RNA from degradation by RNAses in the blood samples. After 15 minutes of room temperature lysis, 129 μL 1-butanol (35% (v/v)) was added to each sample and immediately mixed by inversion to induce RNA–glycogen precipitation. Each sample was then applied to the capture membrane on the SNAPflex device and eluted as described above. Eluted RNA was stored at −80 °C until RT-qPCR analysis. Samples were quantified using HIV RT-qPCR and compared to an internal standard curve to quantify RNA copies recovered in each sample. Percent recovery was calculated compared to expected values based on the input RNA concentration. Six independent experiment replicates were performed for each RNA concentration, using blood from three separate donors. Six negative control replicates were also performed, using blood from three separate donors, but without introducing RNA.
HIV-1 virion RNA recovery with SNAPflex
We next investigated virion lysis and HIV RNA recovery from simulated patient samples using cultured HIV-1 virions spiked into whole blood. AccuSpan HIV-1 RNA linearity panel cultured virions (SeraCare) were used for simulated patient samples. SeraCare HIV-1 linearity panel members 2–5 were used as input samples (virion concentrations of 3.6 × 107 cp mL−1, 2.8 × 106 cp mL−1, 2.7 × 105 cp mL−1, 3.8 × 104 cp mL−1, respectively). 10 μL of each virion sample was diluted directly into 90 μL whole blood, and 200 μL of the complete lysis buffer (1 : 2 ratio of sample : lysis buffer) was subsequently added and mixed by inversion. 129 μL (35% (v/v)) of 1-butanol was added to the sample to induce nucleic acid precipitation, and the solution was applied to the SNAPflex device and eluted as described. Eluted RNA was stored at −80 °C until RT-qPCR analysis. Samples were amplified using RT-qPCR and compared to an internal standard curve to quantify recovered HIV virion RNA. Percent recovery was calculated in comparison to quantification values provided in the Certificate of Analysis, calculated using Roche COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 version 2.0 assay prior to purchase from SeraCare. Six independent experiment replicates were performed for each RNA concentration, using blood from three separate donors. Six negative control replicates were also performed, using blood from three separate donors, but without introducing virions.
For comparison to a gold standard extraction method, each SeraCare panel member was diluted 1 : 10 in human plasma to a final volume of 100 μL and processed with the QIAamp Viral Mini kit (QIAGEN). The protocol was followed as specified by the manufacturer, but the input volume was adjusted to 100 μL plasma, and the elution volume was changed to 100 μL to allow for more direct comparison with SNAPflex. Six independent experiment replicates were performed for each virion concentration, using plasma from three separate donors. Three negative control replicates were also performed, using plasma from three separate donors, but without introducing virions. All eluted RNA samples were stored at −80 °C until RT-qPCR analysis.
Long-term stability of HIV RNA on SNAPflex membrane
We investigated the utility of SNAPflex for long-term storage of extracted RNA at two different temperatures. We first processed in vitro transcribed RNA sample extraction and capture to quantify RNA degradation over time at room temperature (∼25 °C) and at an elevated temperature (37 °C). In vitro transcribed RNA for the HIV gag gene was diluted in elution buffer to a final concentration of 1 × 105 cp mL−1 in enough volume for 14 samples. For each sample, 10 μL of the diluted RNA was added directly to 200 μL lysis buffer and 90 μL whole blood was subsequently added to the mixture. After 15 minutes of room temperature lysis, 129 μL 1-butanol (35% (v/v)) was added to each sample and immediately mixed by inversion to induce RNA-glycogen precipitation. Each sample was then applied to the capture membrane on the SNAPflex device as described above.
For each experiment, two samples were eluted for each time point at day 0, day 1, day 7, and day 14 after sample extraction. For day 0 and for two negative control conditions, samples were eluted immediately after extraction and stored at −80 °C until RT-qPCR analysis. For all other time point samples (days 1–14), the dried capture membranes were stored in Mylar zip-top bags containing silica packets. The samples were randomized and divided into two desiccant-containing storage containers. One container was left in a laminar flow hood at room temperature (approx. 25 °C) and the other was stored in a dry incubator at 37 °C. At days 1, 7, and 14 after extraction, two samples from each container were eluted as described above and the eluted RNA was stored at −80 °C until RT-qPCR analysis. Six independent experiment replicates were performed for each time point, using blood from three separate donors. Six negative control replicates were also performed, using blood from three separate donors, but without introducing RNA.
For long-term stability experiments only, RT-qPCR samples were analysed using a “master” standard curve. The standard curve (Fig. S2†) was compiled from 9 separate standard curves performed on 9 days. CT values from each concentration across all replicates for the standard curves were compiled, and a line of fit was determined using least squares analysis. For the stability samples, RNA eluted at each time point was analysed with RT-qPCR and the resulting CT values were compared to the line of fit from the “master” standard curve to determine the final concentration.
HIV RT-qPCR analysis
One-step brilliant II RT-qPCR core kit (Agilent) was used for internal quantitative analysis of extracted HIV RNA recovery. The RT-qPCR assay used here was developed in-house based on the available HIV sequence information from the Los Alamos HIV database.27,28 Briefly, forward and reverse primers with melting temperature of ∼60 °C were identified, and primers were optimized to enable both first-strand synthesis and PCR amplification without non-specific amplification. The assay was performed on a QuantStudio 5 thermocycler and primers and probes were obtained from integrated DNA technologies (Commercial Park, Coralville, IA). Gene-specific primers for the HIV gag gene (forward: 5′-GGCTACACTAGAAGAAATGATGACAGCAT-3′, reverse: 5′-CCCT TCTTTGCCACAATTGAAACACTT-3′) initiated PCR amplification, with the reverse PCR primer also initiating first strand synthesis for the reverse transcription step. A Cy5 and black hole quencher dual-labeled species-specific probe was included for fluorescence quantification (5′-Cy5-AGTAGG AGGACCCGGCCATA-IAbRQSp-3′). In vitro transcribed RNA for the HIV gag gene was used for standard curve analysis with input concentration ranging from 1 × 104 cp mL−1 to 1 × 107 cp mL−1. 10 μL sample was added to 15 μL master mix for a final reaction volume of 25 μL (final concentrations: 1X SureStart Core RT-PCR buffer, 3 mM MgCl2, 0.2 μM forward primer, 0.2 μM reverse primer, 0.2 μM probe, 0.8 mM dNTPs, 30 nM ROX reference dye, 0.5 U mL−1 Taq polymerase, 1 μL reverse transcriptase). Samples were incubated at 50 °C for one hour for reverse transcription, followed by an initial denaturation step at 95 °C for 10 minutes, and amplified at 40 cycles of 95 °C for 30 seconds and 57.5 °C for 1.5 minutes.
Plasmodium falciparum DNA recovery with SNAPflex
We investigated the capability of SNAPflex to extract and purify DNA using Plasmodium falciparum as a model system. We first used purified P. falciparum genomic DNA (BEI resources, MRA-102G) inoculated into whole blood to investigate successful isolation, purification, and elution of DNA with the SNAPflex device. P. falciparum genomic DNA (gDNA) was diluted into elution buffer (10 mM Tris–HCl, pH 8.5, QIAGEN) to stocks of 1 × 105 fg μL−1, 1 × 104 fg μL−1, 1 × 103 fg μL−1, and 1 × 102 fg μL−1. Each of these stocks was then diluted 1 : 10 directly into whole blood. 100 μL of each diluted blood sample was lysed with 200 μL complete lysis buffer (1 : 2 ratio sample : lysis buffer) at room temperature for 15 minutes. 129 μL of 1-butanol was added to induce precipitation of DNA–glycogen complexes, the sample was applied to the SNAPflex and eluted as described. Eluted DNA was stored at −20 °C until qPCR analysis; each sample was quantified with qPCR and compared against an internal standard curve. Six independent replicates were performed for each concentration using whole blood from a different, non-parasite-infected donor. Six negative control blood samples with blood from three separate donors but without introducing P. falciparum DNA were also processed.
P. falciparum infected red blood cell DNA recovery with SNAPflex
We next investigated the capability of SNAPflex to extract and purify DNA from P. falciparum parasites. P. falciparum parasites grow intracellular to red blood cells; therefore, the system must successfully lyse two membranes in addition to the proteinaceous whole blood sample to isolate the target DNA for these samples.
P. falciparum parasites (3D7 strain, BEI Resources, MRA-102), were cultured in human red blood cells using standard culturing procedures for malaria parasites.29 Briefly, P. falciparum parasite culture was maintained in 5% human red blood cells purified from whole blood (Research Blood Components) with complete RPMI medium (Gibco) supplemented with hypoxanthine (Sigma-Aldrich); culture was maintained at 37 °C, 5% CO2, 3% O2. Parasite culture was maintained at high parasitaemia levels (>10%), with daily media change. The parasites were synchronized to ring stage cultures once per week using 5% d-sorbitol (Sigma-Aldrich) to ensure that the cultures were in a single growth stage for experiments.
For experimental conditions, two 3 μL samples of each culture were collected and stained with Giemsa stain (Sigma-Aldrich), and visualized with a 100× oil immersion objective. It was determined that the parasites were primarily in a single, synchronized culture stage, the percentage of infected red blood cells in 10 fields of view on each slide was quantified, and the parasitaemia was determined as an average of the percentage of infected red blood cells in all 20 fields of view. The infected red blood cell cultures were serially diluted to 10%, 5%, 1%, and 0.1% final parasitaemia (percent infected red blood cells) directly into whole blood.
100 μL of each of the diluted samples were combined with 200 μL of the complete lysis buffer (1 : 2 ratio of sample : lysis buffer), mixed by inversion, and lysed at room temperature for 15 minutes. 129 μL 1-butanol was added to the sample SNAPflex as described above. To compare sample recovery to a commercial DNA extraction method, 100 μL of each sample was also processed with QIAGEN Blood & Tissue kits according to the manufacturer’s instructions. For each experiment, a sample of whole blood without parasites was also processed with both methods as a negative control. The eluted DNA was stored at −20 °C until qPCR analysis; each sample was quantified using qPCR compared against an internal standard curve. A total of 9 independent cultures at ring stage and 3 independent cultures at schizont stage were processed as described. Six process control samples using whole blood but no parasites were also performed.
Plasmodium falciparum qPCR analysis
A previously reported qPCR assay was used for quantitative analysis of extracted P. falciparum DNA.30 Amplification was performed with SureStart Taq polymerase (Agilent) with primers and probes synthesized by Integrated DNA Technologies. Gene-specific primers for the P. falciparum 18S rRNA gene (forward: 5′-CTTTTGAGAGGTTTTGTTACTTTGAG TAA-3′, reverse: 5′-TATTCCATGCTGTAGTATTCAAACACAA-3′) initiated amplification and a HEX and black hole quencher dual-labeled species-specific probe was included for fluorescence quantification (5′-HEX-TGTTCATAACAGACGG GTAGTCATGATTGAGTTCA-IAbFQ-3′). Purified P. falciparum genomic DNA was used for standard curve analysis with input concentration ranging from 1 × 101 fg μL−1 to 1 × 105 fg μL−1. To quantify DNA, 5 μL of sample was added to 20 μL of master mix for a final reaction volume of 25 μL (final concentrations: 1X SureStart 10X buffer, 3 mM MgCl2, 0.3 μM forward primer, 0.3 μM reverse primer, 0.2 μM probe, 0.8 mM dNTPs, 30 nM ROX reference dye, 0.025 U mL−1 Taq polymerase). Samples were incubated for an initial denaturation step at 95 °C for 10 minutes and amplified at 45 cycles of 95 °C for 15 seconds and 60 °C for 1 minute.
Results
The SNAPflex device design is compatible with whole blood
The core components of the SNAPflex device are two paper membranes: a 0.7 μm pore-size glass fiber membrane disc which serves as a nucleic acid capture membrane, and a chromatography paper waste pad that wicks liquid through the glass fiber membrane via capillary forces. The device design uses three layers of laminating plastic to secure the chromatography paper waste pad via heat sealing, and an adhesive plastic layer to secure the glass fiber capture membrane in place, aligned with the chromatography paper (Fig. 1A–D). In the operational configuration, the capture membrane is first folded over to create a contact point with the chromatography paper wicking pad; the chromatography paper features a protrusion to ensure sufficient contact (Fig. 1E). A 3D printed, reusable stabilization clip is used to mechanically anchor the sandwich structure throughout the course of nucleic acid collection and purification (Fig. 1F).
Prior to application on the device, a sample of whole blood is first lysed at room temperature (lysis ratio 1 : 2 blood : lysis buffer) for 15 minutes using a custom lysis buffer containing hydrophilic glycogen as a carrier particle to associate with liberated nucleic acids. A hydrophobic primary alcohol, 1-butanol, is introduced to the sample to induce precipitation of glycogen–nucleic acid complexes. The lysed sample is then applied to the SNAPflex device to isolate and purify nucleic acids on the glass fiber membrane. To begin sample application, the entire sample volume is applied to the glass fiber capture membrane (Fig. 2A). Any remaining blood components are solubilized and cleared from the membrane by a “pre-wash” buffer containing 12.5% lysis buffer and 70% ethanol (Fig. 2B), and excess salts are removed with two ethanol washes (Fig. 2C and D). The entire sample application process takes approximately 13 minutes for 100 μL sample input, and approximately 30 minutes for 200 μL sample input. The capture membrane is then removed from the device and dried at ambient conditions for 10 minutes (Fig. 2E). From this point forward, the precipitated nucleic acids could either be stored in a dried state on the membrane for an extended time period or eluted from the membrane (Fig. 2F and G).
When whole blood is applied onto SNAPflex, persistent contact between the capture membrane and waste pad ensures that cell debris or blood-specific PCR inhibitors are fully washed from the membrane after lysis (Fig. 3A and B). Observation of the dried capture membrane shows that nearly all lysed blood components are removed from the capture membrane, on both the top and bottom of the membrane, by the wash steps. We included GlycoBlue (Applied Biosystems), a blue-tinted co-precipitant, to qualitatively identify sample capture prior to quantitative analysis of nucleic acid capture. The blue tint on top of the membrane indicates the presence of captured glycogen–nucleic acid particles (Fig. 3C). Complete removal of blood components is important for successful amplification of captured nucleic acids, as blood inhibitors co-eluted from the membrane showed significant qPCR assay inhibition (data not shown).
Fig. 3.

Demonstration of SNAPflex with whole blood samples. A) Application of lysed, precipitated blood (100 μL blood sample input). B) Purified capture membrane after application of three wash buffers. C) The top and bottom of the capture membrane shows significant removal of blood components, with blue glycogen precipitant remaining on top of the capture membrane.
RT-qPCR-amplifiable HIV RNA can be extracted from whole blood
In order to evaluate the performance of the SNAPflex device, we used in vitro transcribed HIV RNA and cultured HIV-1 virions. To test successful capture, purification, and elution of RNA, we first assessed the recovery of in vitro transcribed RNA for the HIV gag gene in the presence of whole blood (Fig. 4A). Our data suggest that the SNAPflex device was able to recover >70% of the inoculated RNA across four logs of concentration from 103 to 106 RNA copies per mL. The successful quantification of extracted RNA by RT-qPCR suggests that, for 90 μL whole blood input, purification on the device was successful and PCR inhibitors from blood were not present in high concentrations in the final eluent.
Fig. 4.
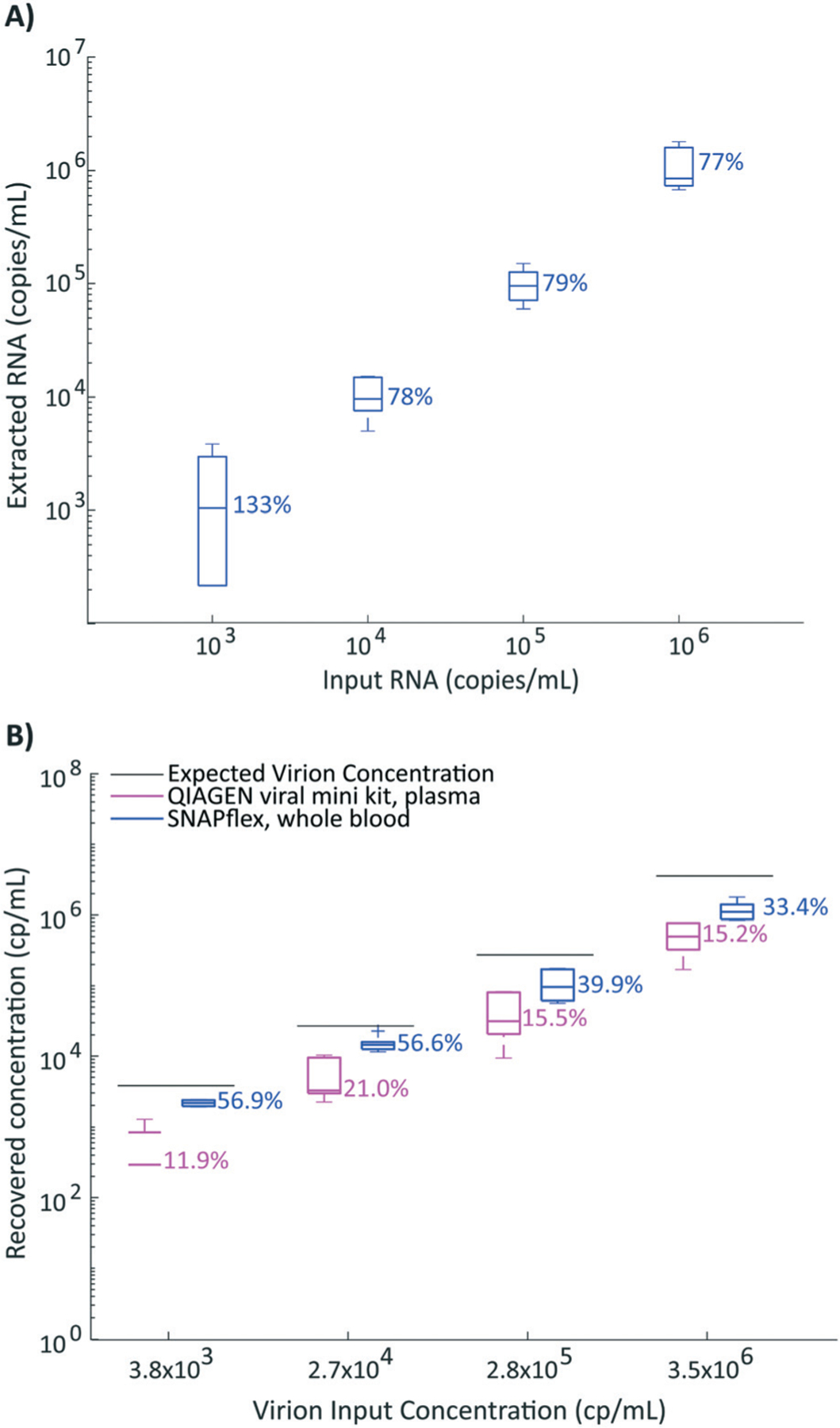
Performance of SNAPflex with RNA. A) Recovery if in vitro transcribed RNA from SNAPflex with whole blood over 4 logs; B) recovery of RNA from simulated patient samples, HIV-1 virions spiked into whole blood extracted with SNAPflex (blue) or spiked into plasma extracted with QIAGEN viral mini kits (pink). Error bars, standard deviation, N = 6 replicates.
Next, we further probed the capability of the SNAPflex lysis buffer and device to lyse intact virions and extract, isolate, and purify HIV RNA from simulated patient samples. To simulate a panel of human patient samples infected with HIV, we inoculated a 4 log-linear panel worth of commercial cultured HIV-1 virions (SeraCare AccuSpan HIV-1 Linearity panel) directly into whole blood and lysed 100 μL of each titrated stock and isolated virion RNA with the SNAPflex device. We observed an overall RNA extraction efficiency from virions in whole blood ranging from approximately 33% to 57% (Fig. 4B, blue boxes) when compared to the manufacturer’s provided concentrations. A natural follow-up question we asked was whether SNAPflex extraction efficiencies were comparable to the efficacy of commercial kits that are specifically designed to isolate blood-borne virions. To address this, we extracted HIV-1 virion RNA with commercially available QIAGEN viral mini extraction kits. It is important to note, the viral extraction kit used here only specifies plasma as an input sample;31 therefore, we introduced virions into isolated plasma rather than whole blood. We created a new 4 log panel by titrating the commercial cultured HIV-1 virions into fresh human plasma to serve as simulated human plasma derived from HIV-infected patients and extracted the samples using the commercial extraction kit, with sample volumes comparable to SNAPflex. We quantified extracted RNA by RT-qPCR. When we compare the QIAGEN-extracted RNA yield to the theoretical input concentration provided by the vendor, we found that the commercial kit unilaterally performed worse than SNAPflex data (Fig. 4B, pink boxes, p < 0.001 across all four concentrations).
These initial data suggest that SNAPflex can be used to extract RT-qPCR amplifiable RNA from whole blood with comparable performance to a more resource-intensive commercial kit.
SNAPflex enables purified RNA to be stored at 25 °C for 14 days
We next investigated the capability to stabilize SNAPflex-extracted RNA on the glass fiber membrane without the need for cold storage. In order to demonstrate long-term stability of RNA on paper, we investigated in vitro transcribed RNA in whole blood to quantify sample degradation over time (Fig. 5). We stored the SNAPflex-extracted RNA on the glass fiber capture membrane in a dry environment for up to two weeks prior to elution at either room temperature (∼25 °C) or at an elevated temperature (∼37 °C). At the specified time points (day 1, 7, or 14 after initial sample processing), RNA was eluted and stored at −80 °C; all samples were quantified by RT-qPCR. Our results showed that, at both temperatures, RNA showed minimal degradation over two weeks compared to samples collected immediately after SNAPflex processing (p = 0.14).
Fig. 5.
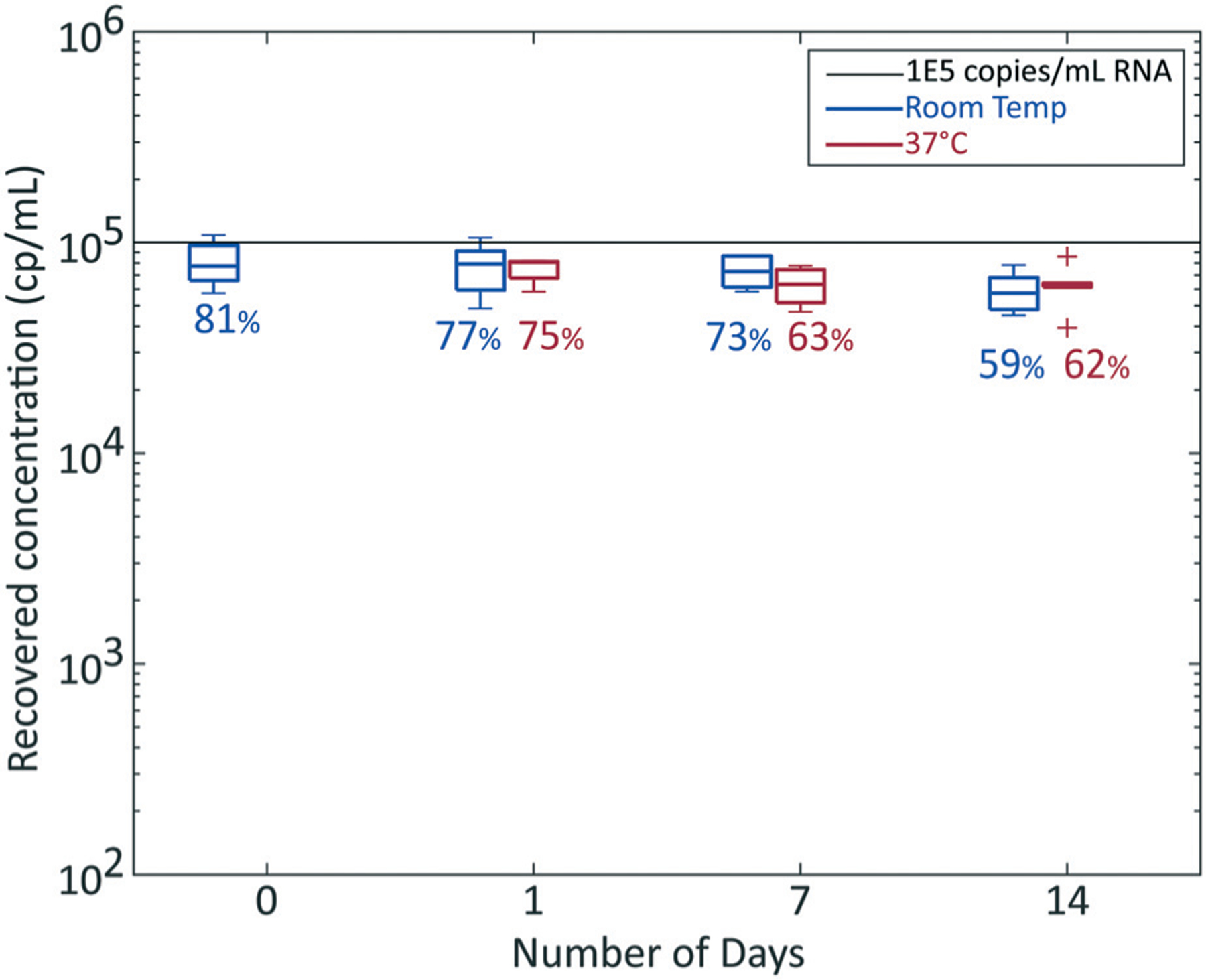
Two week stability of RNA on SNAPflex membranes stored at room temperature (∼25 °C, blue) and 37 °C (red). Sample concentration, 1 × 105 cp mL−1 in vitro transcribed RNA from blood. Error bars, standard deviation, N = 6 replicates.
SNAPflex can recover purified P. falciparum gDNA from blood
In order to demonstrate successful sample isolation, purification, and elution of DNA from blood using the SNAPflex system, we first used purified P. falciparum genomic DNA directly introduced into whole blood (Fig. 6). 100 μL of sample was processed with the SNAPflex device for each concentration; both input gDNA and extracted gDNA were quantified by qPCR and percent recovery was determined by comparing extracted DNA concentration to the input concentration. Our data suggest that the SNAPflex device was able to recover 37% to 73% of the introduced DNA across four orders of magnitude of concentration from 101 to 104 fg gDNA per μL sample. For these data, we also did not observe qPCR, indicating that inhibitors were not co-eluted with the gDNA.
Fig. 6.
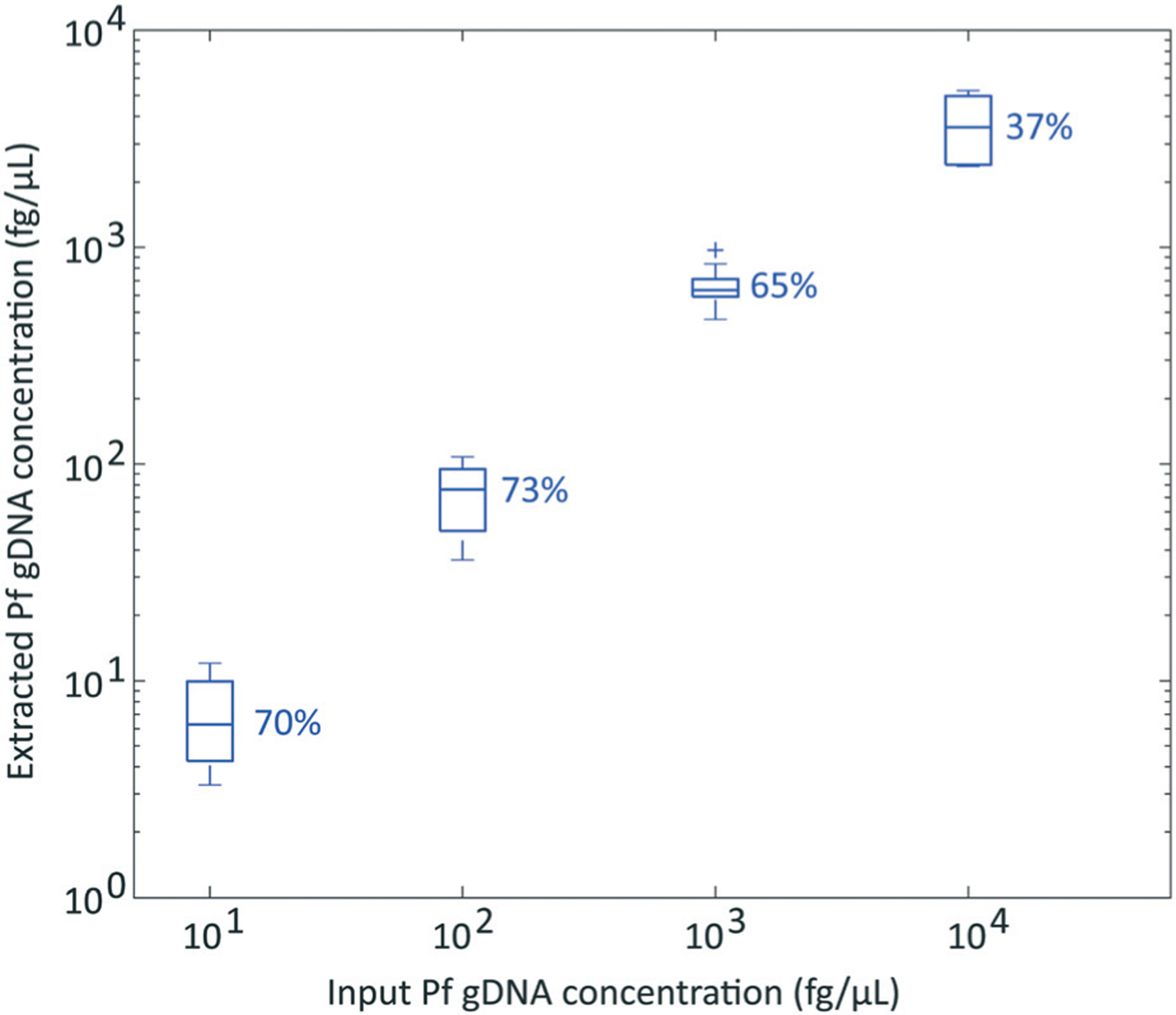
P. falciparum gDNA recovery from whole blood with SNAPflex over 4 logs. Error bars, standard deviation, N = 6.
SNAPflex successfully lyses and purifies P. falciparum DNA from parasites in whole blood
We next investigated the capability of SNAPflex to process simulated Plasmodium patient samples. For this study, we cultured P. falciparum parasites in red blood cells and diluted infected red blood cells into whole blood to four concentrations (0.1–10% infected red blood cells in whole blood). The malaria parasite life cycle consists of several growth stages once the parasite enters red blood cells. Here, parasites were first synchronized to the early ring stage, at which a single parasite copy is present in each red blood cell. For a small number of experiments, we also showed successful lysis of parasites in the schizont stage, where multiple parasites are present within each red blood cell. In each case, the parasite stage and parasite-infected cell concentration of each culture was quantified by microscopy prior to sample dilution into whole blood.
We performed SNAPflex DNA extraction and isolation on 100 μL of each simulated patient blood sample, and DNA was eluted into 100 μL of standard elution buffer (10 mM Tris–HCl, pH 8.5). To compare SNAPflex extraction efficiency to standard extraction methods, the same simulated patient blood samples were also extracted with a commercial DNA extraction method, QIAGEN Blood & Tissue kit. Eluted DNA from both extraction methods was quantified by qPCR, and the extracted DNA concentrations were compared in a pair-wise analysis (Fig. 7). Within each experiment, our data demonstrate that SNAPflex extracted samples showed improved recovery compared to QIAGEN extraction, particularly at higher parasitaemia (i.e. samples fell above the dotted line). Notably, we found that this was true across several concentrations of infected red blood cells (0.1%, yellow, 1%, green, 5%, purple, 10%, red) and across two parasite stages: early ring stage parasites (filled circles), and late schizont stage parasites (open squares).
Fig. 7.
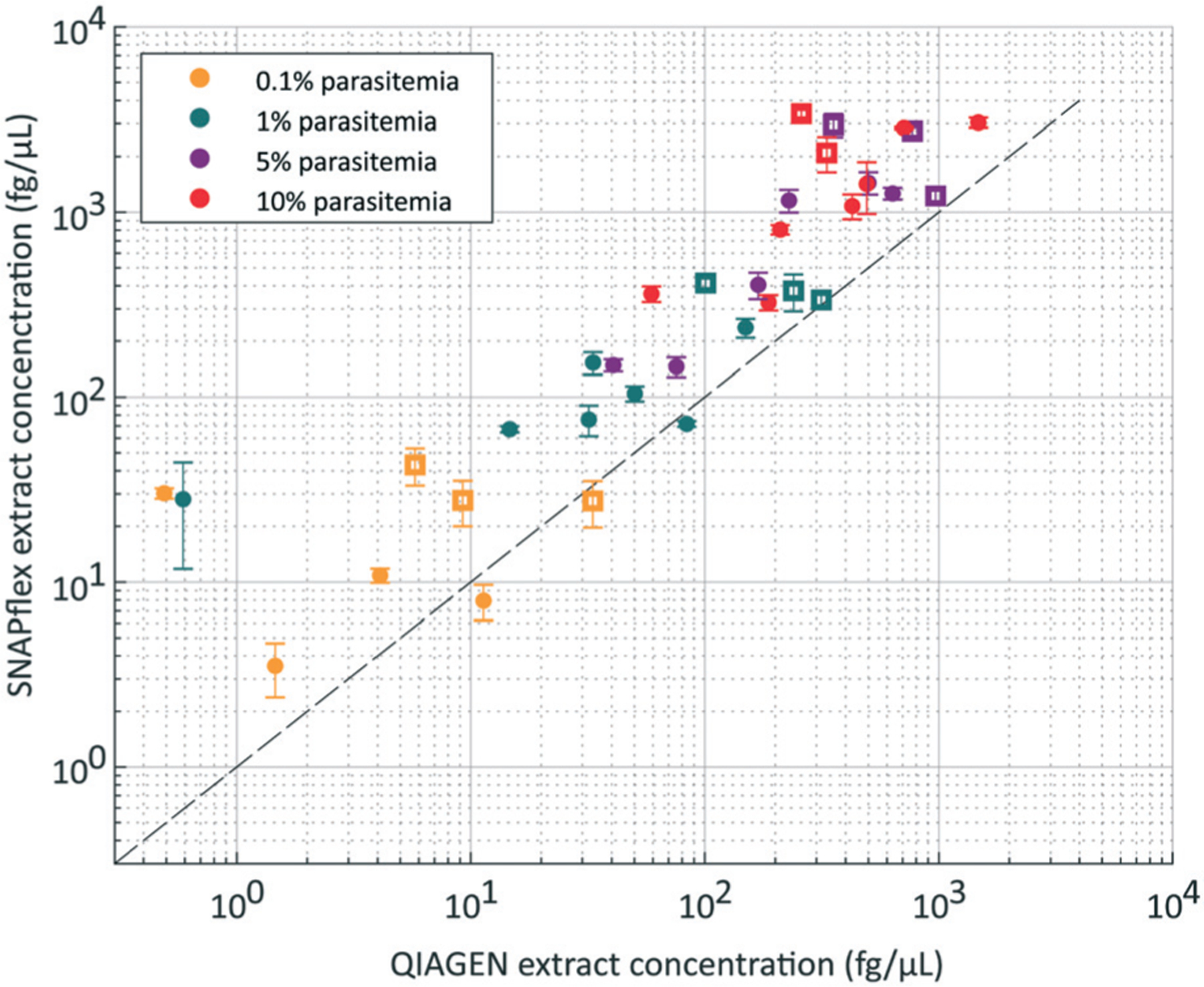
P. falciparum-infected red blood cells in whole blood, extracted by QIAGEN and SNAPflex. Comparison of paired samples supports that SNAPflex extraction results in improved DNA recovery for both late-stage schizont parasites (open squares) and early ring-stage parasites (filled circles). Samples with four parasitaemia levels were tested (yellow, 0.1%, green, 1%, purple, 5%, red, 10%).
Discussion
The SNAPflex device design enables instrument-free, point of care nucleic acid extraction In this work, we developed a paper-and-plastic device for instrument-free, room temperature extraction, isolation, and purification of nucleic acids from whole blood. This device represents an improvement over our previous method,26 requiring only passive wicking for device functionality rather than multiple mechanical steps, and representing a significant improvement in manufacturability and portability with it’s simple, paper-based design. We envision the SNAPflex device to be useful for field-based collection of DNA and RNA for diagnostics in low-resource settings. In particular, we designed this device to process blood samples from patients in field settings for subsequent shipment to central testing facilities for sample analysis with gold standard NAATs. Isolated, purified nucleic acids processed by SNAPflex are stabilized on a glass fiber membrane to enable more affordable sample shipment compared to whole blood shipment, which requires fast shipping times and cold storage. Instrument-free processing will simplify sample processing at the point of collection, rather than requiring extensive processing at the central testing facility, as with DBS samples. The method we describe can be used for both RNA and DNA extraction, making it applicable for several blood-borne infectious diseases.
We designed the SNAPflex device (Fig. 1) to be compatible with a roll-to-roll manufacturing processes, enabling mass production of the devices (Fig. S3†). To prepare the device components for roll-to-roll manufacturing, each of the plastic layers would be fabricated on a continuous spool of the repeated pattern (Fig. S3B†). For manufacturing, the proposed assembly of the device (Fig. S3C†), involves four main process steps. At the end of the process, the completed SNAPflex devices will be wound into a large product spool. The capability for roll-to-roll manufacturing increases the device’s potential for distribution and use at the point of care.
The SNAPflex device was designed with a final capacity of 1 mL, including lysed sample and wash buffers. Keeping this constraint in mind, we have designed the SNAPflex lysis buffer to enable nucleic acid extraction from up to 200 μL whole blood, as this volume can be obtained from pooled finger prick volumes (routine finger prick blood sampling yields 50–100 μL (ref. 32 and 33)). Although larger sample volumes may improve detection, we constrained the device volume for volumes that could be collected with a simple finger prick without the need for venepuncture.
In the development of the buffer, we combined components of the guanidine thiocyanate-based lysis buffers previously reported by Boom et al.34 and Chomczynski and Sacchi.35 In addition to guanidine thiocyanate, we also included components from the original buffer recipes in Chomczynski: the anionic surfactant N-lauryl sarcosine enables complete denaturation of proteins and white blood cells, and 2-mercaptoethanol prevents significant protein precipitation by keeping the lysate in a reduced state. In addition, we found that adding a small amount of NP-40 further enables solubilization of cell membranes and fats. Depending on the implementation setting, it may be necessary to eliminate 2-mercaptoethanol from the lysis solution if hazardous waste disposal is not possible.
For this work, we adjusted the sample : lysis buffer ratio for suitability with the SNAPflex system for capture and purification of nucleic acid–glycogen complexes on the glass fiber membrane. Blood was lysed at a sample : lysis buffer ratio of 1 : 2 to enable complete degradation of blood components, resulting in a homogenous solution without aggregated protein or cell debris on the membrane surface (Fig. S4†). To mitigate protein aggregation, we explored the commonly-used combination of proteinase K digestion in the presence of guanidine hydrochloride at 37 °C to 50 °C; however, we found that significant protein precipitation from the heating process overcame any benefits that came from using the protease itself (data not shown).
Successful SNAPflex capture of nucleic acids relies on the formation of hydrophilic nucleic acid–glycogen complexes, which form precipitates in the solution mediated by the introduction of a hydrophobic primary alcohol. In this work, nucleic acid precipitation was induced by the addition of the primary alcohol 1-butanol. While isopropanol (50% v/v final) or ethanol (70% v/v final) are often used as precipitating agents, 1-butanol (35% v/v final) allows us to minimize the total alcohol volume necessary to induce nucleic acid precipitation from the solution, as we reported in previous work.36,37 An important note for the use of 1-butanol is the necessity for mixing by inversion rather than vortexing; aggressive mixing in the presence of 1-butanol may result in large protein aggregates at the aqueous-organic interface, which would clog the membrane and impede fluid flow.
It is possible to further reduce the volume of the precipitating reagent by introducing chloroform (2.9% v/v final) in combination with 1-butanol (26.5% v/v final) to enhance the hydrophobicity of the alcohol mixture. Because chloroform requires specialized hazardous waste disposal and hazardous sample handling, we did not include it in the final sample treatment described in this work. However, in well-resourced lab environments, including chloroform in the final precipitation buffer may be a viable option to further reduce the necessary volume of precipitating acid. We have performed the RNA experiments we described above with chloroform in the precipitation buffer to further demonstrate that RNA sample recovery remains consistent in the presence of a lower volume of precipitation buffer with chloroform (Fig. S5†).
We next determined that successful purification of the extracted nucleic acid requires a “pre-wash” step which includes 12.5% of lysis buffer in addition to 70% ethanol to solubilize and discard any captured protein and cell debris on the surface of the capture membrane. This is akin to a similar “pre-wash” buffer that is used in commercial silica-based nucleic acid extraction columns, where traces of guanidine-thiocyanate are added to a 70% ethanol mixture to minimize the chance of protein aggregates clogging the silica pores. Two further ethanol washes (70% and 95%) enable removal of residual salts and rapid drying of the capture membrane post-processing.
The SNAPflex sample application method described here used several pipetting steps. However, moving forward, we propose simplifying sample application by storing lysis and wash buffers in convenient, squeezable containers with dropper caps to enable sample application without a pipette. Devices would be developed to control the droplet size to ensure that the sample drop is contained to the surface of the membrane, ensuring that there is no sample loss due to overflow over the sides of the capture membrane.
MakerHealth has extensive experience developing field-applicable kits for healthcare settings in low-resource areas, and our subsequent studies will focus on developing such kits with feedback from healthcare workers in these settings. Similarly, we will expand on the proof of principle demonstrated in this work to understand device performance with finger prick blood samples in field settings.
SNAPflex shows successful recovery of HIV RNA from whole blood
Our device demonstrated successful extraction and elution of HIV RNA using both in vitro transcribed RNA and HIV-1 virions in whole blood to simulate patient samples.
We first investigated the recovery of in vitro transcribed RNA from whole blood (Fig. 4A). Because whole blood contains RNases which could degrade unprotected RNA, the in vitro transcribed RNA was first introduced to the lysis buffer and whole blood was added to the lysis buffer after RNA addition. The lysis buffer contains guanidine thiocyanate and 2-mercaptoethanol, which aggressively denature proteins in the solution (including RNases, which usually contain numerous disulphide bonds) thereby preventing degradation of RNA in solution. The results from in vitro transcribed RNA recovery experiments indicate that the lysis buffer accomplishes the necessary functions: (1) complete lysis of whole blood components (proteins and cell debris), (2) precipitation and capture of RNA onto the capture membrane, (3) purification of captured RNA on the paper membrane and removal of PCR inhibitors from paper, and (4) successful elution of captured RNA from the paper membrane. The results show successful recovery of RNA across four logs. At the lowest RNA concentration, we observed that some samples exhibited greater than 100% recovery concentrations. We hypothesize this is likely due to stochasticity at the lower limit of quantification for the RT-qPCR assay (approximately 1 × 103 copies per mL).
We next showed successful extraction of HIV-1 RNA from simulated patient samples using cultured HIV virions diluted directly into whole blood. In this case, the virion glycoprotein envelope should protect the viral RNA from nuclease attack, making RNA degradation in whole blood less likely. The results from these experiments (Fig. 4B) show successful recovery and quantification of HIV RNA from simulated patient samples, but with lower RNA recovery values. The percent recovery in this case was calculated by comparing RNA concentration calculated by an internal RT-qPCR assay to estimates from the commercial virion Certificate of Analysis. Therefore, the use of two different methods for quantification of virion RNA may explain the lower percent recovery compared to in vitro transcribed RNA recovery results. Because we did not have the capability to verify the vendor-provided concentration using the same assay, we instead performed RNA extraction with a commercial kit and quantified RNA with RT-qPCR as a “gold standard” method.
We were also able to show that, over two weeks, HIV RNA remained stable on the glass fiber matrix both at room temperature and at elevated temperature without significant sample degradation (Fig. 5). We stored samples with desiccant to ensure that the RNA remained dry, particularly because contact with water has been shown to accelerate RNA degradation.17 For this preliminary study, we did not challenge the storage conditions with increased humidity because we sought to determine the initial baseline degradation due to temperature alone. Furthermore, we investigated two-week stability because we estimated this to be a reasonable shipping time for samples from field settings to central testing facilities based on informational interviews. The next step for this work is to investigate the effect of elevated humidity and to fully characterize the shelf life of extracted RNA on this matrix.
Together, these data serve as an initial proof of concept that HIV-1 virion RNA can be successfully extracted using SNAPflex, and that purified RNA remains stable over an extended time period. Because our device can be used at room temperature and without any specialized equipment, the technology we describe represents an important step forward in sample collection for HIV viral load monitoring. The extracted RNA samples can be quantified using routine RT-qPCR analysis, making the sample collection method simpler to integrate into existing testing infrastructure using standard NAATs.
SNAPflex shows successful recovery of P. falciparum malaria gDNA from whole blood
We next investigated recovery of DNA with the SNAPflex system using P. falciparum gDNA as a model system. We first used purified P. falciparum gDNA in whole blood in order to investigate successful DNA precipitation, capture, and elution. In this case, purified gDNA was introduced directly into whole blood because DNA should not be susceptible to degradation by whole blood proteins. The results from these studies showed successful recovery and quantification of purified P. falciparum gDNA from whole blood samples using SNAPflex (Fig. 7). While our results showed >65% recovery up to 103 fg μL−1 DNA, we observed much lower recovery (37%) at a higher DNA concentration, 104 fg μL−1. Further investigation is required to understand why DNA recovery decreased at the highest concentration of P. falciparum DNA. However, this concentration corresponds to a high parasite load (∼426 parasites per μL, assuming conversion factor of 23.5 fg per 1 parasite genome38) which is much higher than the limits of detection for routine malaria testing with NAATs.39 Therefore, we anticipate that, although sample recovery is imperfect, diagnosis would still be possible at this recovery with high parasite concentrations.
We further tested SNAPflex extraction of DNA from P. falciparum parasite-infected red blood cells in whole blood (Fig. 8). Here, our results demonstrate that the lysis buffer is capable of extracting DNA from within membrane-bound parasites in red blood cells. Furthermore, since the SNAPflex extraction was more efficient at recovering parasite DNA than extraction with a centrifuge dependent QIAGEN Blood & Tissue kit, we believe SNAPflex represents an improvement over current point of care methods in blood extraction kits.
An important consideration for this work was the necessity of an additional treatment of the glass fiber capture membrane for DNA samples. While DNA precipitation was induced by the lysis and precipitation buffer, we found poor recovery (<30%) of DNA from glass fiber membranes washed only with DI water (used in RNA studies). In order to address this problem, we performed an additional treatment of the glass fiber capture membrane with trifluoroacetic acid, dried the membrane at room temperature, and stored the membrane in a dry environment. We found that this treatment significantly improved DNA recovery (Fig. S6A†). We hypothesize that the trifluoroacetic acid treatment enabled the successful elution of DNA from glass fiber by depositing a layer of hydroxyl groups onto the surface of the membrane, to which DNA could bind reversibly to enable more successful elution.40 We hypothesize that this layer helps combat irreversible binding of DNA to silica particles in the presence of a chaotrope, enabling more successful recovery. Because the RNA samples tested are much shorter than genomic nucleic acids, we expect that they did not form as many interactions with the paper in the presence of the chaotropic agent, enabling improved recovery compared to DNA without the need for additional treatment with trifluoroacetic acid.7 We further found that storing the TFA-treated membrane in a dry environment was necessary to maintain high percentage recovery (Fig. S6B†). Further studies with this device will focus on understanding the specific binding and elution mechanism of DNA from TFA-treated glass fiber membranes.
Given the large difference in DNA recovery compared to RNA recovery in untreated glass fiber membranes (<20% recovery for DNA, >70% recovery, RNA), it may be possible to take advantage of the differential recovery to elute RNA alone without DNA. This would be particularly useful, for example, in the case of HIV viral load monitoring. Circulating proviral DNA can be amplified by PCR, even in the absence of active HIV infection. Therefore, elution of proviral DNA and HIV RNA together is known to lead to false positives for viral load monitoring, and to inaccurate quantification, particularly at low viral loads.41,42 Testing whether preferential recovery of HIV RNA without interfering proviral DNA is possible requires patient samples and therefore was not investigated in this work. However, this would constitute a promising future direction.
Conclusions
In this work, we have presented a paper-and-plastic device for room temperature, instrument-free extraction of nucleic acids from whole blood. Our device aims to fit the need for simple methods that could be adapted to the point of care but remain easily adaptable to existing testing infrastructure and gold-standard diagnostic methods used in LMICs. Therefore, we focused on creating a platform technology for nucleic acid extraction that can be used with standard NAATs like qPCR and RT-qPCR. The method is agnostic to the downstream assay used and we expect that it is adaptable to isothermal nucleic acid amplification methods or other applications where purified nucleic acids are a necessary input.
The SNAPflex system successfully extracts and purifies amplifiable pathogen nucleic acids (RNA and DNA) from whole blood without the need for instrumentation. The purified nucleic acids are stable at room temperature on the capture membrane, providing a feasible alternative to conventional dried blood spots. SNAPflex was designed to be manufactured at scale using roll-to-roll manufacturing, facilitating its deployment in the field. We have designed this system to be integrated easily into existing workflows and represents a promising step toward expanding the diagnostic capacity of NAATs in resource-limited settings.
Supplementary Material
Acknowledgements
This work was funded by a Bill and Melinda Gates Foundation grant to CMK and JGM (GCE Phase I # OPP1182168) and was supported by the Center for AIDS Research Basic Science Core (P30-AI042853) and the Boston University Precision Diagnostics Center.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00277a
Conflicts of interest
The authors do not have any conflicts of interest to report.
References
- 1.Hayashida K, Kajino K, Hachaambwa L, Namangala B and Sugimoto C, PLoS Neglected Trop. Dis, 2015, 9, e0003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayashida K, Kajino K, Simukoko H, Simuunza M, Ndebe J, Chota A, Namangala B and Sugimoto C, Parasites Vectors, 2017, 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu Y and Zheng Z, Biosens. Bioelectron, 2017, 79, 593–599. [DOI] [PubMed] [Google Scholar]
- 4.Sidstedt M, Hedman J, Romsos EL, Waitara L, Wadsö L, Steffen CR, Vallone PM and Rådström P, Anal. Bioanal. Chem, 2018, 410, 2569–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson IG, MINIREVIEW Inhibition and Facilitation of Nucleic Acid Amplification, 1997, vol. 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM and Van Der Noordaa J, J. Clin. Microbiol, 1990, 28, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melzak K, J. Colloid Interface Sci, 1996, 181, 635–644. [Google Scholar]
- 8.Aptima®, Aptima® HIV-1 Quant Assay. [Google Scholar]
- 9.Monleau M, Montavon C, Laurent C, Segondy M, Montes B, Delaporte E, Boillot F and Peeters M, J. Clin. Microbiol, 2009, 47, 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolluri N, Klapperich CM and Cabodi M, Lab Chip, 2017, 18, 75–94. [DOI] [PubMed] [Google Scholar]
- 11.Huang L-H, Lin P-H, Tsai K-W, Wang L-J, Huang Y-H, Kuo H-C and Li S-C, PLoS One, 2017, 12, e0184692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols BE, Girdwood SJ, Crompton T, Stewart-Isherwood L, Berrie L, Chimhamhiwa D, Moyo C, Kuehnle J, Stevens W and Rosen S, J. Int. AIDS Soc, 2018, 21, e25206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gous N, Scott L, Berrie L and Stevens W, PLoS One, 2016, 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carmona S, Seiverth B, Magubane D, Hans L and Hoppler M, J. Clin. Microbiol, 2019, DOI: 10.1128/JCM.01336-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johannessen A, Troseid M and Calmy A, J. Antimicrob. Chemother, 2009, 64, 1126–1129. [DOI] [PubMed] [Google Scholar]
- 16.Nichols BE, Girdwood SJ and Shibemba A, et al. , Clin. Infect. Dis, 2020, 70(6), 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim MD, Am. J. Trop. Med. Hyg, 2018, 99, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlsson H, Guthenberg C, von Döbeln U and Kristenssson K, Clin. Chem, 2003, 49, 979–981. [DOI] [PubMed] [Google Scholar]
- 19.Pannus P, Claus M, Gonzalez MMP, Ford N and Fransen K, Medicine, 2016, 95, e5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasan MR, Tan R, Al-Rawahi GN, Thomas E and Tilley P, J. Clin. Microbiol, 2012, DOI: 10.1128/JCM.02659-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson BJ, Melde BJ, Dinderman MA and Lin B, PLoS One, 2012, 7, e50356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Relova D, Pérez LJ, Ríos L, Coronado L, Hinojosa Y, Acevedo AM, Frías MT and Perera CL, Span. J. Agric. Res, 2017, 15, e05SC03. [Google Scholar]
- 23.Muller R, Betsou F, Barnes MG, Harding K, Bonnet J, Kofanova O and Crowe JH, Biopreserv. Biobankingt, 2016, 14, 89–98. [DOI] [PubMed] [Google Scholar]
- 24.Lou JJ, Mirsadraei L, Sanchez DE, Wilson RW, Shabihkhani M, Lucey GM, Wei B, Singer EJ, Mareninov S and Yong WH, Clin. Biochem, 2014, 47, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grölz D, et al. , Curr. Pathobiol. Rep, 2018, 6(4), 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrnes S, Fan A, Trueb J, Jareczek F, Mazzochette M, Sharon A, Sauer-Budge AF and Klapperich CM, Anal. Methods, 2013, 5, 3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Los Alamos HIV Database, in HIV Sequence Compendium 2011, 2011, pp. 9–149. [Google Scholar]
- 28.Sanders-buell E, Salminen MO and Mccutchan FE, Sequencing Primers for HIV-1, 1995. [Google Scholar]
- 29.Moll K, Ljungström I, Perlmann H, Scherf A and Wahlgren M, Methods in Malaria Research, 2013. [Google Scholar]
- 30.Perandin F, Manca N, Calderaro A, Piccolo G, Galati L, Ricci L, Medici MC, Arcangeletti MC, Snounou G, Dettori G and Chezzi C, J. Clin. Microbiol, 2004, 42, 1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.QIAGEN, QIAamp RNA Blood Mini Handbook, 2010, pp. 1–44. [Google Scholar]
- 32.Bond MM and Richards-Kortum R, Am. J. Clin. Pathol, 2015, 144, 885–894. [DOI] [PubMed] [Google Scholar]
- 33.Sauer-Budge AF, Brookfield SJ, Janzen R, McGray S, Boardman A, Wirz H and Pollock NR, PLoS One, 2017, 12, e0183625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boom R, Sol C, Beld M, Weel J, Goudsmit J and Wertheim-van Dillen P, J. Clin. Microbiol, 1999, 37(3), 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chomczynski P and Sacchi N, Anal. Biochem, 1987, 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 36.Linnes JC, Fan A, Rodriguez NM, Lemieux B, Kong H and Klapperich CM, RSC Adv, 2014, 4, 42245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez NM, Wong WS, Liu L, Dewar R and Klapperich CM, Lab Chip, 2016, 16, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolluri N, Klapperich CM and Cabodi M, Lab Chip, 2018, 18, 75–94. [DOI] [PubMed] [Google Scholar]
- 39.Bell D, Fleurent AE, Hegg MC, Boomgard JD and McConnico CC, Malar. J, 2016, 15, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodard D, Howard AJ and Down J. US Pat, 5606046, 1997. [Google Scholar]
- 41.Drain PK, Dorward J and Bender A, et al. , Clin. Microbiol. Rev, 2019, 32(3), e00097–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guichet E, Serrano L, Laurent C, Eymard-Duvernay S, Kuaban C, Vidal L, Delaporte E, Ngole EM, Ayouba A and Peeters M, J. Virol. Methods, 2018, 251, 75–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.