

Abstract
The neurons of the melanocortin system regulate feeding and energy homeostasis through a combination of electrical and endocrine mechanisms. However, the molecular basis for this functional heterogeneity is poorly understood. Here, a voltage‐gated potassium (Kv+) channel named KCNB1 (alias Kv2.1) forms stable complexes with the leptin receptor (LepR) in a subset of hypothalamic neurons including proopiomelanocortin (POMC) expressing neurons of the Arcuate nucleus (ARHPOMC). Mice lacking functional KCNB1 channels (NULL mice) have less adipose tissue and circulating leptin than WT animals and are insensitive to anorexic stimuli induced by leptin administration. NULL mice produce aberrant amounts of POMC at any developmental stage. Canonical LepR‐STAT3 signaling—which underlies POMC production—is impaired, whereas non‐canonical insulin receptor substrate PI3K/Akt/FOXO1 and ERK signaling are constitutively upregulated in NULL hypothalami. The levels of proto‐oncogene c‐Fos—that provides an indirect measure of neuronal activity—are higher in arcuate NULL neurons compared to WT and most importantly do not increase in the former upon leptin stimulation. Hence, a Kv channel provides a molecular link between neuronal excitability and endocrine function in hypothalamic neurons.
Keywords: hypothalamus, K+ channel, leptin, melanocortin, obesity, POMC, potassium channel
The voltage‐gated K+ (Kv) channel KCNB1 is expressed in the hypothalamus, mainly in the arcuate nucleus, where it forms a complex with the leptin receptor. Mice lacking the KCNB1 channel (NULL) exhibit dysregulated calorie intake—a function mediated by the arcuate nucleus—along with altered leptin metabolism and signaling.
Abbreviations
- AAALAC
Association for Assessment and Accreditation of Laboratory Animal Care
- AgRP
agouti‐related protein
- Akt
protein kinase B
- ARH
Arcuate nucleus
- BSA
bovine serum albumin
- CART
cocaine and amphetamine‐regulated transcript
- c‐Fos
protein c‐Fos
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DEE
developmental and epileptic encephalopathy
- DIV
days in vitro
- EDTA
Ethylenediaminetetraacetic acid
- ELISA
enzyme‐linked immunosorbent assay
- ERK
extracellular signal‐regulated kinase
- FOXO1
Forkhead box protein O1
- GABA
γ‐aminobutyric acid
- HCl
hydrochloric acid
- HRP
Streptavidin Horseradish Peroxidase
- IACUC
Animal Care and Use Committee
- IRS
insulin receptor substrate
- JAK2
janus kinase 2
- K(ATP)
ATP‐sensitive potassium channel
- KCNB1
voltage‐gated potassium channel sub‐family 2 member 1
- KCNB2
voltage‐gated potassium channel sub‐family 2 member 2
- Kv
voltage‐gated potassium
- LepR
Leptin receptor
- MACS
magnetic‐activated cell sorting
- Map2
Microtubule‐associated protein 2
- MAPK
mitogen activated protein kinase
- NPY
neuropeptide Y
- OCT
optimal cutting temperature compound
- PBS
Phosphate Buffer Saline
- PFA
paraformaldehyde
- PI3K
Phosphoinositide 3‐kinase
- POMC
proopiomelanocortin
- PVH
paraventricular nucleus of hypothalamus
- RIPA
radioimmunoprecipitation assay
- SDS
sodium dodecyl sulfate
- SH2B1
Sar homology family member
- SK
small conductance calcium‐activated K+ channel
- STAT3
transducer and activator of transcription 3
- Tris
tris(hydroxymethyl)aminomethane
- αMSH
α‐melanocyte‐stimulating hormone
1. INTRODUCTION
The term metabolism describes the countless chemical reactions that sustain the life of an organism. In this context, the brain and, in particular, the hypothalamus play a major role in the control of feeding and energy homeostasis, achieved through the combined action of electrical and endocrine mechanism. 1 , 2 An example is provided by the neurons of the Arcuate nucleus (ARH). Historically, two major populations of hypothalamic neurons, one co‐expressing agouti‐related protein (AgRP), neuropeptide Y (NPY), and γ‐aminobutyric acid (GABA) and the other co‐expressing proopiomelanocortin (POMC) and cocaine and amphetamine‐regulated transcript (CART) neurons, have been considered key players in feeding mechanisms. 3 These neurons named respectively ARHAgRP and ARHPOMC, form sub‐populations each modulated by distinct hormones. For example, two groups of ARHPOMC neurons have been identified: one expressing the glucagon receptor and one expressing the leptin receptor. 4 Leptin, which is produced by the adipose tissue, is excitatory of ARHPOMC neurons and inhibitory of ARHAgRP neurons. 5 , 6 However, the relationship is bidirectional as also the electric status of the ARH neurons affects their endocrine function and sensitivity to leptin. 7 One crucial question, still unresolved, is how hypothalamic neurons integrate processes as different as endocrine modulation and neuronal excitability to achieve their heterogenic functionality. We note that both processes originate at the plasma membrane as hormones bind to receptors, which activate opportune biochemical cascades and channels that shape the excitability of the neurons by regulating the diffusion of ions across the membrane. In fact, several ion channels, including voltage‐gated potassium (Kv) channels, have the ability to interact with membrane proteins and affect their functions. 8 , 9
In this study, we report that the Kv channel KCNB1 is expressed in hypothalamic neurons, where its absence causes dysregulated leptin signaling and metabolic anomalies. Hypothalamic NULL neurons—including the ARHPOMC—exhibit elevated levels of c‐Fos protein—an indirect marker of electrical activity—that do not respond to the excitatory effect of leptin. Notably, KCNB1 forms stable complexes with the long isoform of the leptin receptor (LepR). Overall, these data may unveil a mechanism through which macromolecular complexes containing Kv channels and receptors enable hypothalamic neurons to achieve electrical and hormonal synergy.
2. MATERIALS AND METHODS
2.1. Reagents and resources
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| POMC (D3R1U) Rabbit mAb | Cell Signaling Technology | #23499 |
| c‐Fos (9F6) Rabbit mAb | Cell Signaling Technology | #2250 |
| Stat3 (124H6) Mouse mAb | Cell Signaling Technology | #9139 |
| Phospho‐Stat3 (Tyr705) (M9C6) Mouse mAb | Cell Signaling Technology | #4113 |
| FoxO1 (C29H4) Rabbit mAb | Cell Signaling Technology | #2880 |
| Phospho‐FoxO1 (Ser256) (E1F7T) Rabbit mAb | Cell Signaling Technology | #84192 |
| p44/42 MAP kinase (phosphorylated Erk1/2) | Cell Signaling Technology | Cat# 9101 |
| p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb | Cell Signaling Technology | Cat# 4695 |
| Integrin beta‐5 (D24A5) Rabbit mAb | Cell Signaling Technology | Cat# 3629 |
| Integrin beta‐1 antibody | Cell Signaling Technology | Cat #4706 |
| Alpha MSH polyclonal rabbit | Bioss Antibodies | bs‐1848R |
| AgRP polyclonal antibody | Thermo Fisher | PA5‐118934 |
| Leptin receptor polyclonal antibody | Thermo Fisher | PA1‐053 |
| Leptin receptor recombinant Rabbit monoclonal antibody | Thermo Fisher | MA5‐32685 |
| GFP monoclonal antibody (GF28R) biotin | Thermo Fisher | MA5‐15256‐BTIN |
| Mouse leptin R biotinylated antibody | R&D systems | BAF497 |
| IgG (3E8) mouse monoclonal | Santa Cruz Biotechnology | Sc‐69 786 |
| c‐Fos antibody (E‐8) | Santa Cruz Biotechnology | sc‐166 940 |
| Anti‐Kv2.1/KCNB1 antibody [K89/34] | ABCAM | ab192761 |
| Anti‐MAP2 antibody | ABCAM | ab5392 |
| Anti‐actin antibody, clone C4 | Millipore | MAB1501 |
| Streptavidin, Alexa Fluor™ 594 conjugate | Thermo Fisher | S11227 |
| Donkey anti‐mouse IgG (H+L) highly cross‐adsorbed secondary antibody, Alexa Fluor™ Plus 488 | Thermo Fisher | A32766 |
| Donkey anti‐Rabbit IgG (H+L) highly cross‐adsorbed secondary antibody, Alexa Fluor™ Plus 488 | Thermo Fisher | A32790 |
| Goat anti‐Rabbit IgG (H+L) highly cross‐adsorbed secondary antibody, Alexa Fluor™ 594 | Thermo Fisher | A11037 |
| Goat anti‐Chicken IgY (H+L) cross‐adsorbed secondary antibody, Alexa Fluor™ Plus 64 | Thermo Fisher | A32933 |
| Rabbit anti‐mouse IgG (H+L) secondary antibody, HRP | Thermo Fisher | 61‐6520 |
| Goat anti‐Rabbit IgG (H+L) secondary antibody, HRP | Thermo Fisher | 31460 |
| Streptavidin horseradish peroxidase (HRP) conjugate | Thermo Fisher | 434323 |
| Chemicals | ||
| Leptin, recombinant (mouse) | Phoenix Pharmaceuticals, INC. | 003‐13 |
| Recombinant mouse leptin | Leinco Technologies, INC | L.117 |
| D‐(+)‐glucose | Sigma Aldrich | G8270 |
| Bovine serum albumin | Sigma Aldrich | A9418‐5G |
| Normal donkey serum, sterile | Sigma Aldrich | 566460 |
| Protein G‐Agarose | Millipore | 11719416001 |
| Streptavidin agarose resin | Thermo Fisher | 20349 |
| Pierce™ BCA protein assay kits | Thermo Fisher | 23225 |
| TaqMan gene expression assays—leptin | Thermo Fisher | 4331182 |
| TaqMan gene expression assays—GAPDH | Thermo Fisher | 4331182 |
| TaqMan™ fast virus 1‐step master mix for qPCR | Thermo Fisher | 4444434 |
| Protease inhibitor Cocktail Set I | Calbiochem | 539131 |
| RNAscope® intro pack for multiplex fluorescent reagent kit | ACD (Bio‐Techne) | 323280 |
| Mus musculus leptin receptor (Ob‐r) mRNA complete cds | ACD (Bio‐Techne) | 402731‐C2 |
| RNAscope® probe—Mm‐Kcnb1‐C3 | ACD (Bio‐Techne) | 316941‐C3 |
| RNAscope® probe—Mm‐Fos‐C2 | ACD (Bio‐Techne) | 316921‐C2 |
| RNAscope® probe—Mm‐Pomc | ACD (Bio‐Techne) | 314081 |
| Commercial assays | ||
| Leptin mouse ELISA kit | Thermo Fisher | KMC2281 |
| Insulin mouse ELISA kit | Thermo Fisher | EMINS |
| AKT (total) human ELISA kit | Thermo Fisher | KHO0101 |
| AKT (Phospho) [pS473] human ELISA kit | Thermo Fisher | KHO0111 |
| Mouse alpha MSH (Sandwich ELISA) ELISA kit | LifeSpan BioSciences, Inc. | LS‐F55869 |
| Neuronal isolation | ||
| Neuronal tissue dissociation kit—Postnatal neurons | Miltenyi Biotec | 130‐094‐802 |
| Anti‐Biotin MicroBeads | Miltenyi Biotec | 130‐090‐485 |
| MiniMACS™ separator and starting kit | Miltenyi Biotec | 130‐090‐312 |
| HBSS 1X Hanks' balanced salt solution | Gibco | 14025‐092 |
| Organisms/strains | ||
| Mouse: C57BL6/J background | The Jackson Laboratory | Strain #:000664; RRID:IMSR_JAX:000664 |
| Mouse: NULL mouse | Genome Editing Share Resource Core Facility at Rutgers | C57BL/6J‐Kcnb1em2Sesf/J |
| Microscopes | ||
| Zeiss Axiophot | ||
| Olympus FV1000MPE | ||
| Nikon Eclipse Ti2 series | ||
| Leica TCS SP8 tauSTED 3X (SP8tauSTED3X) | ||
| Software and algorithms | ||
| Neuroanatomy Fiji ImageJ | Tiago Ferreira | https://imagej.net/update‐sites/neuroanatomy/ |
| EthoVision XT | N/A | https://www.noldus.com/ethovision‐xt |
| Colocalization Threshold Fiji ImageJ (Coloc 2) | Tony Collins | https://imagej.net/plugins/colocalization‐threshold |
| Prism | GraphPad by Dotmatics | https://www.graphpad.com/scientific‐software/prism/ |
| Image J | NIH | https://imagej.net/software/fiji/ |
2.1.1. Mice
All experiments with mice performed in this study were approved by our institution's Animal Care and Use Committee (IACUC). We used C57BL6/J mice (wild type, WT) and homozygous Kcnb1 NULL KI mice (C57BL/6J‐Kcnb1em2Sesf/J) here called NULL. The Kcnb1 NULL allele bears a nonsense (stop) mutation in the exon coding for the S4 transmembrane domain of the KCNB1 protein. This codes a truncated protein (~37 kDa vs. ~110 kDa of the WT protein) that is not trafficked to the plasma membrane and is quickly degraded. 8 Results from preclinical models of obesity have not underscored a clear sex‐dependence. Therefore, mice of either sex at developmental stages: E13‐14 and 1, 3, 5, and 6 months old were used. Littermates of either sex were randomly assigned (by flipping of a coin) to experimental groups. The animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)‐approved vivarium, under the care of a veterinarian. The mice lived in large cages, with a 12 h light–dark cycle, and were fed ad libitum.
2.2. Behavioral protocols
2.2.1. Leptin injection
Lyophilized, recombinant leptin, was dissolved in sterile 20 mM Tris–HCl (pH 8.0) at a 1.0 mg/mL stock concentration as per manufacturer's instructions. For intraperitoneal (i.p.) injection, stock leptin was freshly diluted in saline (0.9% NaCl) to a final volume of 100 μL. Mice were sacrificed 90 min post‐injection.
2.2.2. Glucose load
Mice were single‐caged and fed normally (ad libitum) for one day and then fasted for 16 h. A measurement of blood glucose was taken before fasting. At t = 0 min, mice were i.p. injected with 2.0 g/kg glucose body weight dissolved in water. Blood samples for glucose measurement were taken with a Metene TD‐4116 monitor at t = 0 (fasting), 15, 30, 60, and 120 min. Blood glucose was specifically measured by tipping the tail and massaging to promote blood flow, the first drop was discarded before every measurement. At the end of the experiment (t = 120 min) mice were fed ad libitum.
2.2.3. Hormone measurements
For leptin and insulin assays, submandibular blood was obtained from ad libitum chow‐fed mice at 4 and/or 20 weeks of age. Serum was collected by centrifugation and assayed by ELISA.
2.2.4. Feeding and adipose tissue extraction
Littermates were single‐caged (post‐weaning), and their weight was recorded on the same day twice a week at noon starting from the second (p14) to the ninth week of age and at the twelfth, sixteenth, and twentieth week afterward. Pups in the same litter were identified by a mark on the tail. Food was weighted on the same day twice a week at noon starting from the fourth to the ninth week of age. Mice were sacrificed at 20 weeks of age and visceral fat pads were collected and weighted.
2.2.5. Chronic control/leptin fasting protocol
Habituated single‐caged 8‐ to 12‐week‐old mice were injected with 3 mg/kg body weight leptin for 4 days. 50 g of food was provided in a plastic container. Food consumption was measured by weighting the food that remained in the plastic container; for food spilling, we used a strainer to collect crumbs. Food consumed and animals' weight were measured before leptin injection every 24 h.
2.2.6. Acute control/leptin fasting protocol
Habituated single‐caged 8‐ to 12‐week‐old mice were fasted for 24 h in new clean cages, then allowed ad libitum food (50 g of regular chow) for 24 h. Food consumption and body weight were recorded every hour for the first 7 h post‐fasting. Blood glucose was measured before and at the end of the fasting period. At the end of the 24 h resting period, the mice were injected with 1.0 mg/kg body weight leptin, immediately transferred to a new clean cage, and fasted for 24 h. At the end of the fasting period, blood glucose was measured, and immediately after, the mice were injected again with 1.0 mg/kg body weight leptin. After an hour, the mice were allowed ad libitum food. Food consumption and body weight were recorded every hour for the first 7 h post‐fasting. The cumulative food consumed post‐fasting normalized to body weight, C(h), was calculated as:
| (1) |
where F h is the food consumed in grams at hour h, and W 0 is the weight of the mouse at the end of the fasting period. The weight gained or lost, expressed as percentage of body weight, B, was calculated as:
| (2) |
where W i and W f are the initial and final mouse's weights expressed in grams.
2.3. Biochemistry
2.3.1. Tissue lysis
Brains were extracted from WT or homozygous NULL mice of either sex similarly to as described in ref. [10]. Hypothalami were dissected as described. 11 Samples were stored at −80°C, if not immediately used. Half sagittal brains or hypothalami were homogenized with a plastic tissue grinder in lysis buffer (0.32 M sucrose, 5 mM Tris‐Cl pH 6.8, 0.5 mM EDTA) or RIPA Buffer (150 mM NaCl, 1% Nonidet p‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris pH 7.4) supplemented with protease inhibitor cocktail. The samples were sonicated for 90 s and then centrifuged at 14 000 rpm for 15 min at 4°C. The lysates were used immediately or stored at −80°C for further analyses.
2.3.2. Immunoprecipitations
To minimize non specific binding, lysates were pre‐incubated with pre‐washed Protein A/G PLUS‐Agarose/ streptavidin beads for 30 min at room temperature. The beads were then collected by centrifugation at 5000 RPM for 1 min and discharged. The lysates were incubated with the primary antibody overnight at 4°C. Pre‐washed beads with 1X phosphate buffer saline (PBS) 0.32 M sucrose, 5 mM Tris‐Cl pH 6.8, and 0.5 mM EDTA were blocked with 3% BSA for 1 h. Then, the beads were added to the lysate and incubated at 4°C for 8 h. The beads were washed four times with PBS and collected by centrifugation at 5000 RPM for 2 min. The pellet was resuspended in 4× Sample Buffer (SDS 10%; bromophenol blue 0.02%; glycerol 30%; Tris–HCL 0.5 M) containing 2% beta‐mercaptoethanol and heated at 95–100°C for 5 min. The samples were centrifuged at 10 000 RPM for 1 min, and the supernatant was resolved in an SDS‐PAGE for Western blot analysis.
2.3.3. Western blotting
For Western blot analysis, protein content, typically 60 μg, was quantified with the Bradford colorimetric assay. Actin was used as loading control. Samples were dissolved in 5× Sample buffer and 5% beta‐mercaptoethanol; heated at 95–100°C for 5 min and then centrifuged at 10 000 RPM for 1 min. The proteins were resolved in 10%–15% SDS‐PAGE and transferred into a nitrocellulose membrane. Membranes were blocked in 5% solution of nonfat dry milk in Tris‐buffered saline with Tween® 20 (TBST) for 1 h. Then, membranes were stained with the primary antibody diluted at 1:1000 at 4°C overnight. After washing 1 h with TBST, the membrane was incubated with the secondary antibody (Goat Anti‐Rabbit IgG Antibody, (H+L) HRP conjugate at 1:2000 dilution or Anti‐Mouse IgG antibody at 1:2000 dilution) for 1 h at room temperature. Membranes were washed for 1 h in TBST, exposed with SuperSignal™ West Pico PLUS Chemiluminescent Substrate and visualized with a Bio‐Rad ChemiDoc.
2.3.4. Nitrocellulose membrane stripping
Coimmunoprecipitation experiments were performed using the same antibody‐stripped membrane. After the first chemiluminescence exposure, nitrocellulose membranes were stripped with Mild Stripping Buffer (1.5% glycine, 0.1% SDS, 1% Tween 20, pH to 2.2) twice for 10 min, with PBS twice for 10 min and TBST twice for 5 min at room temperature. The membranes were blocked with 5% solution of nonfat dry milk in TBST for 1 h and then stained with the primary antibody as described in the Western Blotting section.
2.3.5. ELISA
ELISA assays were performed according to the manufacturer's instructions and measured using an Infinite M Nano+ (Tecan, Männedorf, Switzerland) plate reader.
2.4. Transcriptome analysis
2.4.1. RNA extraction for transcriptome analysis
The hypothalami were extracted from WT or NULL mice brains similarly to as described before. The tissue was homogenized with a plastic tissue grinder in TRI Reagent RT (50–100 mg tissue per 1.0 mL TRI Reagent RT. Molecular Research Center Inc. Cincinnati OH). The homogenate was supplemented with 25 μL bromoanisole, covered and shaken vigorously for 15 s; and centrifuged at 12 000 g for 15 min at 4°C. The RNA was collected from the aqueous phase and transferred into a new centrifuge tube, precipitated with isopropanol, stored at room temperature for 10 min and then centrifuged for 12 000 g for 5 min at room temperature. The RNA white pellet was first washed with 75% ethanol and centrifuged at 6000 g for 5 min at room temperature; afterward, it was resuspended in dH2O. The RNA concentration was measured with a nanodrop and adjusted to a ~300 ng/μL final concentration.
2.4.2. Transcriptome analysis
Raw transcriptome reads were assessed for quality control (FASTQC v0.11.8) and trimmed for quality and adapter contaminant (cutadapt v2.5). Trimmed reads were aligned to the mouse genome (GRCm38) using STAR (v2.6.1), followed by transcript abundance calculation and hit count extraction with StringTie (v2.0) and featureCounts (v1.6.4), respectively. Hit count normalization and differential gene expression group cross‐comparisons were performed using DESeq2 (v1.26.0). Significant differentially expressed gene thresholds were set at FDR‐adjusted p < .05.
2.5. Immunofluorescence
2.5.1. Adult brain
Mouse's organs were first washed with 0.9% filtered NaCl and then perfused with 4% paraformaldehyde (PFA) solutions. Brains were removed, post‐fixed in 4% paraformaldehyde overnight at 4°C, and cryoprotected in 30% sucrose for 2 days. Brains were frozen in Optimal Cutting Temperature compound (OCT) in dry ice and stored at −80°C. Brain slices were cut 16 μm thick throughout the hypothalamic area. Slides were dried at room temperature for 6 h and then stored at −20°C. After incubation at room temperature for 20 min, the slides were rehydrated in PBS for 10 min. The slices were then permeabilized for 10 min in 0.1% Triton X‐100 in PBS and blocked for 1 h with 3% BSA and/or mouse on M.O.M.® Mouse blocking solution on Mouse Blocking Reagent Vector Laboratories. The slides were incubated with primary antibody (dilutions: 1:200 to 1:800) overnight at 4°C, afterward they were washed three times with PBS for 10 min each time and incubated with the appropriate secondary conjugated antibody (dilutions: 1:500 to 1:800). After 1 h at room temperature, the slices were washed three times with PBS. VECTASHIELD Antifade Mounting Medium with DAPI mounting buffer was used to finalize the slides. Staining was visualized with a Zeiss Axiophot microscope or with an Olympus FV1000MPE multi‐photon microscope or a Nikon Eclipse Ti2 series confocal microscope, all equipped with dedicated software. Two‐three technical replicates/brain.
2.5.2. Free‐floating
Brain slices were cut at 35 μm and washed 3 times with PBS for 5 min. They were blocked in PBS + 0.3% Triton X‐100 and 3% Donkey Serum for 1 h and incubated in PBS + 0.3% Triton X‐100 and 1% Donkey Serum with the primary antibody (dilution: 1:500) overnight at 4°C. Then the slices were washed 3 times for 10 min and incubated with the respective secondary antibody in PBS + 0.3% Triton X‐100 and 1% Donkey Serum (dilutions: 1:500 or 1:1800) for 2 h at room temperature. Slices were mounted on microscope slides and the cover slide was placed with VECTASHIELD Antifade Mounting Medium with DAPI mounting buffer.
2.5.3. RNAscope in situ hybridization
Mice were either not treated, injected with Leptin (L) (2 g/kg mouse weight), or injected with Vehicle (V). Organs were washed with 0.9% NaCl and then perfused with 4% PFA. Brains were extracted and left in 4% PFA for 24 h. Brains were then dehydrated in 10%, 20%, 30% sequential sucrose solutions. Finally, they were stored in OCT at −80°C until cryostat sectioning. Slides with a single/double 10 μm brain slice were then used the following day for in situ hybridization following the manufacturer's instructions (ACD, a Biotechne Brand, Minneapolis, MN). The KCNB1 probes target a 20 bp region in the C‐terminus of the channel (the stop signal in the NULL is in the exon coding for the S4 segment). The slices were observed and imaged with a Leica TCS SP8 tauSTED 3X (SP8 tauSTED 3X) super‐resolution microscope. Analysis was performed with FIJI software.
2.6. Primary neuronal cultures
The detailed procedure used to obtain pure cultures of selected hypothalamic neuronal sub‐populations is described in Ref. [11]. Briefly, the brains of E13‐E14 embryos were extracted and the hypothalamus was isolated. The hypothalamus was cut into 4–6 pieces and a single‐cell suspension was obtained by enzymatic digestion. A specific population of hypothalamic neuronal cells was isolated based on specific surface markers using the Miltenyi Biotech MACS technology. Thus, cells were linked to magnetic beads via cell‐surface antigen antibodies. The suspension was then passed through a magnetic column allowing the isolation of the targeted neurons. In this study, leptin receptor‐expressing cells were isolated. Cells were grown for seven days in vitro (DIV7) on ready‐to‐use poly‐d‐lysine‐coated glass in 24‐well plates. Cultures were washed 3 times for 5 min with PBS, fixed using a 50% methanol, 50% acetone solution for 20 min on ice. Cells were washed 3 times with PBS for 10 min each time and blocked for 1 h at room temperature with 3% BSA in PBS. Cells were incubated overnight at 4°C with the primary antibodies (dilutions: 1:800–1:500) and then washed three times with PBS, followed by application of the appropriate secondary conjugated antibodies (dilutions: 1:800–1:500). After 1 h at room temperature, the cells were washed three times with PBS. All slides were mounted in VECTASHIELD Antifade Mounting Medium with DAPI mounting buffer and stored at 4°C. Staining was visualized with a Zeiss Axiophot microscope or with an Olympus FV1000MPE multi‐photon microscope or a Nikon CREST X‐Light LFOV confocal microscope, all equipped with dedicated software.
2.7. Antibody validation
The antibodies used in this study have all been well reported in the literature. Since the KCNB1 and the LepR proteins are the two most critical proteins in this study we further validated the specificity of their antibodies by immunofluorescence and Western blot analysis (Figure 1, Figure S1).
FIGURE 1.
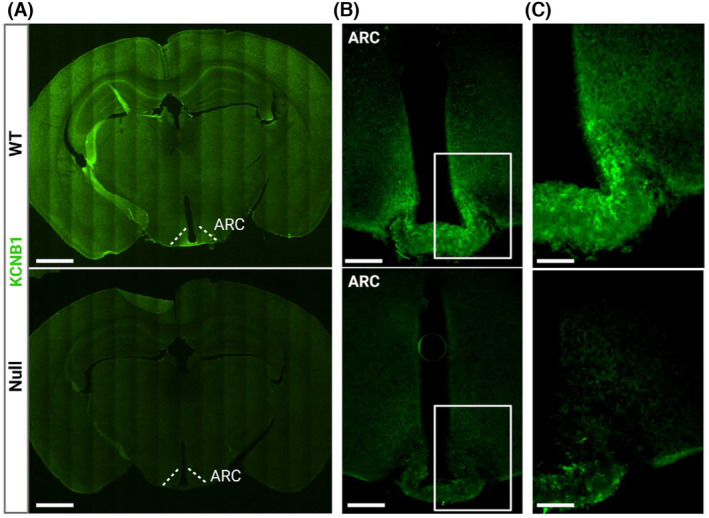
KCNB1 is expressed in the hypothalamus. (A) Representative confocal tiles images of brain slices of the indicated genotypes stained with KCNB1 antibody (green). The ARH area is indicated. Scale bar 1 mm. (B) Representative confocal images of the mediobasal area of the hypothalamus of the indicated genotypes stained with KCNB1 antibody. Scale bar 200 μm. (C) Magnifications of the ARH areas boxed in (B). Scale bar 100 μm.
2.8. Statistical analysis
Quantitative data are presented as mean ± standard error of the mean (SEM). The estimated sample size, N, needed to detect a meaningful difference between groups was calculated using power analysis with alpha = 0.05 and power = 0.8. 12 Normality and log‐normality tests (D′ Agostino and Pierson) were calculated for normal distribution. The level of significance, assumed at the 95% confidence limit or greater (p < .05), was calculated using GraphPad Prism with one of the following: unpaired or paired Student's t‐test; one‐way ANOVA with Sidak and Tukey's post hoc test; one‐way repeated measures ANOVA; non‐parametric one‐way ANOVA Kruskal‐Wallis with post hoc test; one‐tail correlation and regression analysis with 95% confidence.
3. RESULTS
3.1. KCNB1 is broadly expressed in the hypothalamus
To gain insight into the role of KCNB1 in the brain, we performed immunofluorescence (IF) analysis in slices cut from the brains of wild‐type (WT) mice and homozygous Kcnb1 NULL KI mice, called NULL, that we characterized previously. 8 The Kcnb1 NULL allele bears a nonsense (STOP) mutation in the exon coding for the S4 transmembrane domain of the KCNB1 protein that gives rise to a truncated, not functional protein (~37 kDa vs. ~110 kDa), that is quickly degraded. In three experiments, along with the cortex and hippocampus, where the presence of KCNB1 is well established, significant immunoreactivity to KCNB1 was detected in the hypothalamus (Figure 1A), prominently in the Arcuate nucleus (ARH, Figure 1B,C). KCNB1 was not detected in NULL sections, as expected (Figure 1A–C). Western blot/densitometry (WBD) analysis confirmed the presence/absence of KCNB1 in the hypothalami of WT/NULL mice (Figure S1).
3.2. KCNB1 influences the transcriptomic profile of the hypothalamus
Given the cellular diversity of the hypothalamus, to better focus the role of KCNB1 we analyzed the transcriptomic profile of WT and NULL hypothalami. Out of 46 078 genes sampled, 5% mRNA transcripts were significantly upregulated, and 3.4% transcripts were significantly downregulated in NULL compared to WT (cutoff log2 change −0.5 to +0.5 and .05 adjusted p value) (Figure 2A,B). This suggests that KCNB1 strongly influences hypothalamic function post‐transcriptionally. The transcripts to be mostly dysregulated were those implicated in ion transport and the regulation of membrane potential, as expected, and metabolic and oxidative pathways, consistent with the important role that the hypothalamus plays for metabolism and energy homeostasis and the well‐established role of KCNB1 as oxidative stress sensor (Figure 2C). 8 , 10 , 13 , 14 Accordingly, QIAGEN Ingenuity Pathway Analysis (QIAGEN IPA) predicted metabolic (log/p‐value = 11.5) and cellular growth (log/p‐value = 7.8) pathways to be among the ones most altered 15 (Figure 2D).
FIGURE 2.
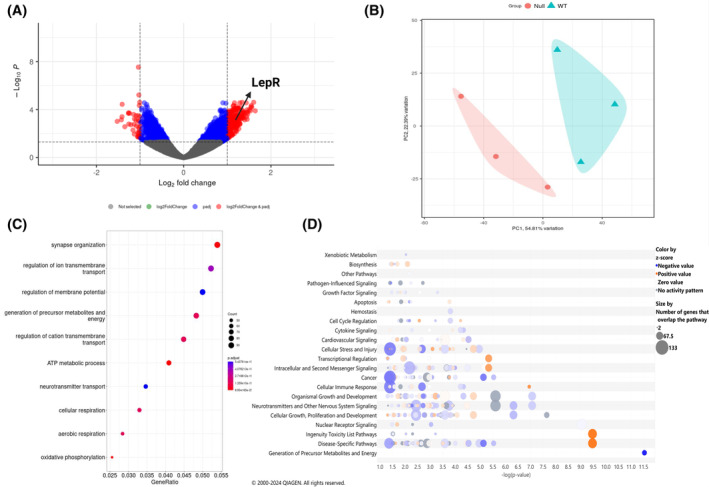
Transcriptome analysis reveals dysregulated leptin signaling in the NULL. (A) Volcano plot of NULL versus WT RNA transcripts. Significantly different transcripts (p ≤ .05) are indicated in the colored region (Blue and Red). Red‐colored transcripts are up‐regulated and down‐regulated with a cut‐off log2change of ±0.5. Up‐regulated red‐colored genes are shown on the right (log2change 0.5–2) and downregulated on the left (log2change −0.5 to −2). (B) Principal component analysis of NULL (pink cluster) versus WT (blue cluster). (C) Dotplot Gene Ontology over‐representation analysis showing down‐regulated cellular pathways in NULL versus WT. The color of the dot depends on the value of p‐adjust, and its size is determined by the count of differentially expressed genes (DEGs) related to the respective pathway in the analyzed set of DEGs (map color keys along with dot size ones are shown on the right). (D) Ingenuity Pathway Analysis bubble plot of predicted activated or inactive canonical pathways in NULL versus WT as per z‐score, which represents a measure of the predicted direction of the pathway activity. Each bubble represents an individual pathway that is included in the canonical pathway category (i.e. generation of precursor metabolites and energy, disease‐specific pathways, nuclear receptor signaling); the bubble size is directly proportional to the number of genes that overlap the pathway; bubble color represents z‐scores as per the legend (blue negatively activated, orange positively activated, gray no activity predicted). X‐axis indicates −log(p‐value). Y‐axis indicates categories of canonical pathways.
3.3. Metabolism is altered in NULL mice
Transcriptome analysis underscores significant changes in metabolic processes including leptin signaling (Log2FoldChange of the leptin receptor = 1.14, p < .000115, Figure 2A). As the hormone is a major mediator of feeding behavior and energy expenditure, we assessed basic metabolic parameters including body weight, average food consumption, circulating leptin, adipose tissue, glucose, and insulin levels in WT and NULL mice. To eliminate the influence of social behavior on food consumption we maintained the animals single‐caged. Mice were fed ad libitum with standard chow to avoid confounding effects associated with high‐fat diet, including rewiring of the ARH, increased activation of ARH neurons, and inflammation. 16 , 17 , 18 Growth (weight‐age relationships) and average chow consumed per day for WT and NULL mice are illustrated in Figure 3A,B. NULL mice grow normally and do not develop appreciable weight anomaly; however, the animals consume moderately but significantly less chow than WT. Accordingly, the levels of leptin are significantly lower in NULL animals compared to WT, at any developmental stage, from P4 pups to 5‐month‐old adults (Figure 3C–E. As blood sampling is technically challenging in pups, we measured their leptin mRNA levels in adipose tissue). To determine why a decrease in food intake does not lead to weight loss in the NULL, we examined the body mass composition of the animals. Twenty‐weeks‐old NULL mice had less fat than WT mice, collected from visceral pads (epididymal in males and periovarian in females), retroperitoneal pads, and mesenteric pads (Figure 3F,G). MRI analysis confirmed the decrease in body fat in the NULL (Figure 3H). The analysis further revealed an increase in lean mass (Figure 3I) and hydration (Figure 3J) in the NULL that compensated for the body fat decrease. In both genotypes, the amount of fat correlated with circulating leptin, a finding largely expected since adipose tissue produces the hormone (Pearson's correlation coefficients R WT = 0.96 and R NULL = 0.99 respectively, Figure 3K). However, while in the WT for one gram of fat there are 2.79 pg/mL leptin, in the NULL the fat/leptin ratio is 1.84 pg/mL, a significant ~40% decrease (p < .00013, regression analysis, Figure 3K). KCNB1 is expressed in islet of Langerhans beta‐cells. 19 Since insulin is a modulator of hypothalamic neurons, we assessed blood glucose and circulating insulin in WT and NULL animals under various feeding conditions. Glucose levels (collected as graphically illustrated in Figure 4D) are not significantly different in the two genotypes under normal conditions (ad libitum food, control, C), or fasting conditions in the absence/presence of leptin (control fasting, CF and leptin fasting, LF, Figure S2A). A glucose load elicited a rapid increase in blood glucose in both WT and NULL mice, which was metabolized fairly similarly in both genotypes (Figure S2B). Insulin levels taken under ad libitum conditions were also not different between WT and NULL mice (Figure S2C). In summary, these data reveal the existence of metabolic anomalies in the NULL animals. However, those mice maintain normal body weight along with insulin and glucose, suggesting they have developed long‐term compensatory mechanisms to adapt to their chronically altered leptin homeostasis.
FIGURE 3.
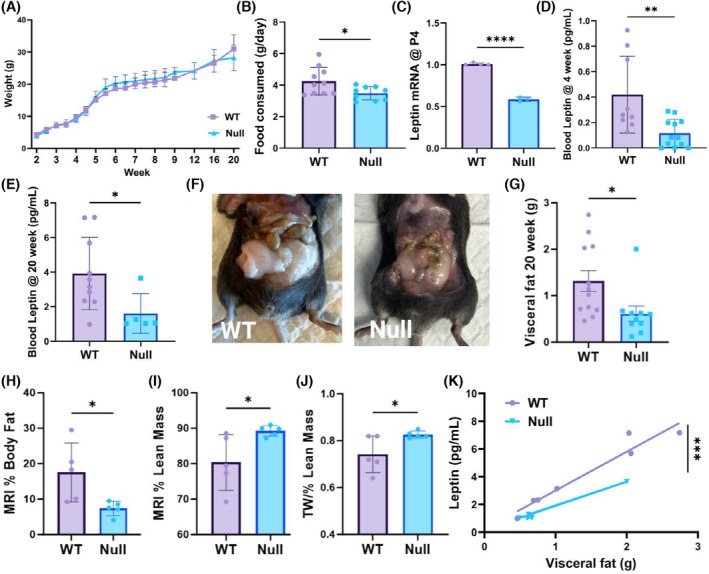
NULL mice present metabolic anomalies. (A) Weight‐age relationships for WT (squares N = 10) and NULL (triangles, N = 9). The animals were weighted twice/week. (B) Average chow consumed per day for WT and NULL mice. Chow measurements were taken daily during weeks 1–9. N = 10 WT and N = 9 NULL mice. (C) Leptin mRNA levels in WT and NULL P4 pups. N = 5 WT and N = 3 NULL mice. (D, E) Blood leptin in 4 and 20 weeks old WT and NULL mice. N = 5–14 animals. (F) Representative images of adipose tissue in 20 weeks old WT and NULL mice. (G) Visceral fat in 20 weeks old WT and NULL mice. N = 12 WT and N = 10 NULL mice. (H–J) MRI measurements of body fat (H); lean mass (I) and hydration (J) in WT and NULL mice. Fat and lean are expressed as percent over total body mass. Hydration is expressed as total water (TW), over lean. N = 5 mice/genotype. (K) Relationships between blood leptin and adipose tissue in WT and NULL mice. Data were linearly fit with slopes 2.79 (correlation coefficient, R = 0.96) and 1.84 (R = 0.99) pg/g⋅mL for WT and NULL respectively. p < .00013 (regression analysis). *p < .05, **p < .01, ***p < .001, ****p < .0001 (two‐tailed t‐test, correlation and regression analysis).
FIGURE 4.
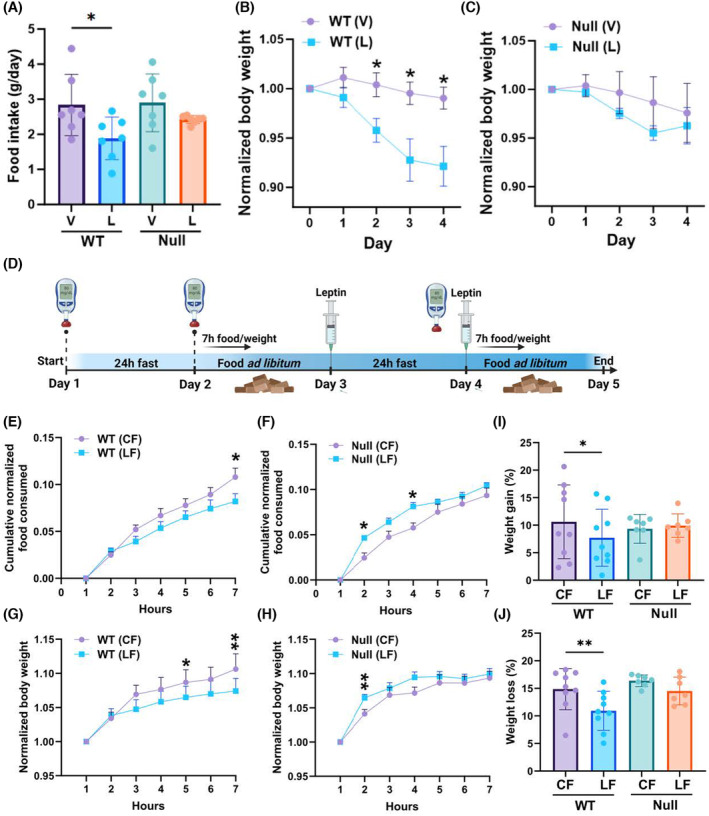
NULL mice are desensitized to leptin. (A) Food consumed per day by WT and NULL mice injected with vehicle, V (saline), or 3.0 g/kg body weight leptin, twice a day, from day 0 to day 4. (B) Body weight normalized to the weight of the animal at the beginning of the experiment (day 0) in control or leptin treatment for WT mice. (C) As in (B) for NULL mice. In (B) and (C) N = 7 mice/genotype/experimental condition. (D) Graphic illustration of the experimental protocol. Mice were fasted for 24 h (control fasting, CL) and then allowed food ad libitum for 24 h. The mice were then fasted again, but at the beginning and end of the fasting period, they were injected with 1.0 mg/kg body weight leptin (leptin fasting, LF). During the first seven hours after fasting, the food consumed by the animals and their weight was monitored hourly. Glucose level measurements were taken before the beginning of the experiment and at the end of the fasting periods. (E) Cumulative amount of food consumed after fasting during the first seven hours following CF (circles) or LF (squares) normalized to body weight (Equation 1) for WT mice. N = 9. (F) As in (E) for NULL mice. N = 7. (G) Weight recovered during the first seven hours following CF (circles) or LF (squares) normalized to initial body weight for WT mice. N = 9. (H) As in (G) for NULL mice. N = 7. (I) Weight gain expressed as percentage of the initial body weight (Equation 2) for WT or NULL animals following CF or LF. (J) Weight loss expressed as percentage of the initial body weight (Equation 2) for WT and NULL animals during CF or LF. *p < .05, **p < .01 (mixed‐effect model for paired values with Šídák's multiple comparisons test, two‐tailed paired t‐test).
3.4. NULL animals are desensitized to the anorexic stimulus of leptin
To further elucidate the mechanisms through which leptin regulates metabolism in the NULL in the short period, we subjected the animals to chronically elevated levels of leptin and determined how the hormone affected food consumption and body weight. Leptin induced a robust anorexic response in the WT animals leading to significant weight loss (Figure 4A,B). In contrast, the food consumed, and the weight loss did not significantly change the NULL animals subjected to leptin treatment compared to their control littermates (Figure 4A–C). In a second set of experiments, we subjected the mice to the protocol illustrated in Figure 4D, which was designed to assess the strength of the anorexic stimulus mediated by leptin, along with the effects of acute increase of leptin levels on metabolism. At the beginning of the experiment, the animals were fasted 24 h to offset satiety and induce strong appetite (CF). Then, mice were given ad libitum access to food for 24 h, an interval sufficient to recover normal weight. At the end of the recovery period, the animals were fasted again, but with one difference: they were injected intraperitoneally with 1.0 mg/kg body weight leptin at the beginning and at the end of the fasting cycle (LF). Food intake and body weight were monitored during the first seven hours post‐fasting. Blood glucose levels were measured before and at the end of the fasting period and were not different between WT and NULL mice (Figure S2A). Thus, the quantity of chow normalized to initial body weight that was progressively consumed by the animals (Equation 1) after CF or LF is compared in Figure 4E for WT and in Figure 4F for the NULL. In agreement with previous measurements (Figure 3B), NULL mice ate moderately less than WT mice following CF (normalized cumulative food consumed: WT = 0.11 ± 0.01 gr., N = 9; NULL 0.09 ± 0.008 gr., N = 7. Compare control curves in Figure 4E,F at hour 7). However, while acute leptin administration induced an expected and significant anorexic response in WT mice, it paradoxically increased appetite in the NULL. The different feeding behaviors of the WT and NULL were reflected in the weight they recovered after fasting (Figure 4G,H). Thus, WT mice consistently recovered less weight following LF compared to CF (Figure 4G). As a result, at the end of the seven hours period the weight gained by WT mice, expressed as a percentage of the initial body weight (Equation 2), was ~10% following CF but only ~7% following LF, a statistically significant difference (Figure 4I). Conversely, weight recovery in NULL mice was similar in CF or LF even though it was initially accelerated in the latter. As a result, at the end of the seven hours post‐fasting monitoring period the weight gained in percent by NULL mice was similar in both conditions (CF = ~9%; LF ~ 10%, Figure 4I). Likewise, the weight lost during fasting, expressed as a percentage of the initial body weight (Equation 2), which in first approximation gives a measure of energy expenditure, significantly decreased in WT mice from CF to LF, whereas it did not appreciably differ in NULL mice (Figure 4J). Overall, these data underscore multilevel desensitization of NULL animals to leptin, ranging from behavioral responses to anorexic stimuli to organismal metabolic processes, to energy consumption.
3.5. POMC biosynthesis is dysregulated in NULL
Since leptin regulates the production of POMC by hypothalamic neurons, we determined the levels of POMC in WT and NULL hypothalami by IF and WBD analysis at baseline or in response to acute increases in leptin levels. Representative images of POMC immunoreactivity in hypothalamic slices, along with quantifications, are illustrated in Figure 5A. Thus, immunoreactivity to POMC was leptin‐dependent in the WT. In contrast, in the NULL, POMC immunoreactivity was high at baseline and did not increase in the presence of leptin. Representative immunoblots of the amounts of POMC in the hypothalami of 4‐ and 24‐week‐old WT and NULL mice, are illustrated in Figure 5B. Two major differences can be immediately appreciated from the immunoblots (quantifications in Figure 5C,D). First, POMC levels are greater in NULL compared to WT at baseline (~5‐fold Figure 5C), and this difference more than doubles with age. Second, while acute leptin administration leads to an increase of POMC levels in WT mice, as expected (~3‐fold, Figure 5D) it does not significantly change POMC protein levels in the NULL, in agreement with the observed desensitization to leptin of this genotype. We conclude that KCNB1 is required for the endocrine function of POMC‐expressing neurons.
FIGURE 5.
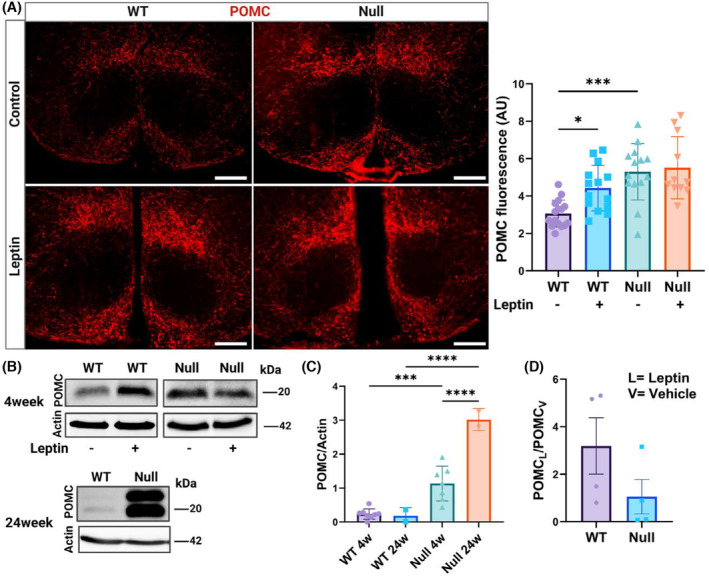
POMC homeostasis is dysregulated in NULL mice. (A) Representative Nomarski images of POMC immunofluorescence and quantifications in hypothalamic slices of WT or NULL in the absence/presence of leptin. Scale bars = 1 mm. N = 3 mice/genotype/experimental condition. (B) Representative Western visualizations of POMC protein in the hypothalami of 4 weeks old WT and NULL mice in the absence/presence of leptin and in the hypothalami of 24 weeks old WT and NULL mice in the absence of leptin. N = 2–8 mice/genotype/experimental condition. (C) Densitometric quantification of POMC protein in the hypothalami of 4 and 24 week‐old WT or NULL mice in the absence of leptin. (D) Densitometric ratio between POMC protein in the presence (POMCL) and absence (POMCV) of leptin in the hypothalami of 4 weeks old WT and NULL mice. Western blot loading controls were actin and Bradford colorimetric assay. Animals were injected with either vehicle or 1.0 mg/kg body weight leptin and sacrificed 90 min later. *p < .05, ***p < .001, ****p < .0001 (Kruskal‐Wallis test and post hoc; one‐way ANOVA and Šídák's multiple comparisons test).
3.6. Canonical STAT3 signaling is impaired in the NULL
Binding of leptin to its receptor culminates in an anorexigenic stimulus mediated by the release of POMC. Therefore, to investigate the molecular basis for the abnormal production of POMC in the NULL we analyzed leptin signaling pathways, that are graphically illustrated in Figure 6A. To this end, we first assessed the amounts of the long isoform of the LepR in the hypothalami of WT and NULL by WBD. The analysis underscored moderate LepR overexpression in the latter (Figure 6B). This is consistent with the significant increase in its RNA levels observed with transcriptome analysis (Log2FoldChange = 1.14, p < .000115) and probably reflects a compensatory response to the low leptin levels circulating in the NULL animals. Typically, the LepR triggers POMC production via the Janus kinase 2, which activates, by phosphorylating Y705, the signal transducer and activator of transcription 3 (canonical JAK2/STAT3 pathway). Once activated, STAT3 is translocated into the nucleus where it acts as a transcription activator of POMC synthesis. 7 As expected, leptin caused STAT3 phosphorylation at Y705 in WT hypothalami (representative Western blot in Figure 6C and quantifications in Figure 6D). However, the hormone failed to trigger the phosphorylation of STAT3 in the NULL. STAT3 activation was further assessed in IF assays (Figure S3). In agreement with WBD analysis, in the WT hypothalamus, immunoreactivity to phosphorylated STAT3 at Y705 (pSTAT3) was leptin‐dependent as expected. In contrast, in NULL slices pSTAT3 signals were weaker compared to WT and did not change in the presence of leptin.
FIGURE 6.
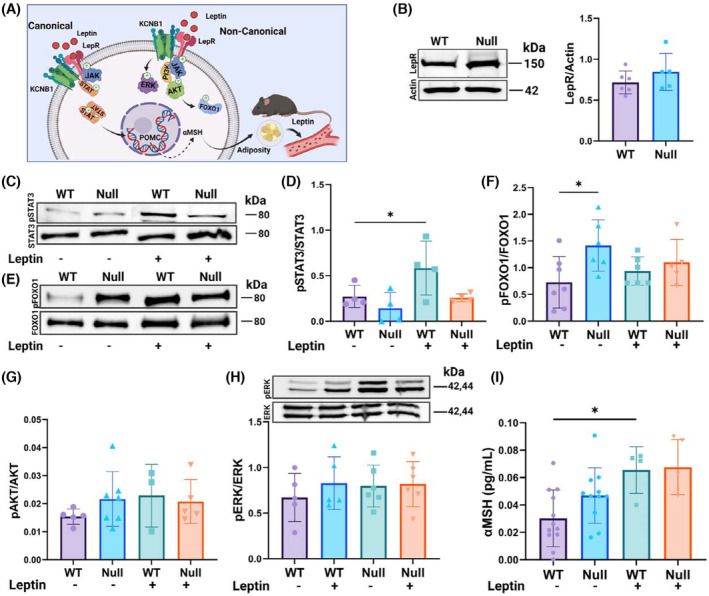
Canonical and non‐canonical leptin pathways are altered in NULL. (A) Graphical illustration of LepR signaling. Canonical (JAK2/STAT3) and non‐canonical PI3K/Akt/FOXO1 and ERK pathways are indicated. (B) Representative Western blot and densitometric quantification of the long isoform leptin receptor (LepR) in hypothalamic lysates of WT and NULL mice. (C) Representative Western blot of STAT3 phosphorylated at Y705 (pSTAT3) and total STAT3 protein (STAT3) in hypothalamic lysates of WT and NULL mice in absence/presence of leptin. (D) Densitometric quantifications of the fraction of pSTAT3/STAP calculated from Westerns as that in (C). (E) Representative Western blot of phosphorylated FOXO1 at S256 (pFOXO1) and total FOXO1 protein (FOXO1) in hypothalamic lysates of WT and NULL mice in the absence/presence of leptin. (F) Densitometric quantifications of pFOXO1/FOXO1 calculated from Westerns as those in (E). (G) ELISA quantifications of phosphorylated Akt at S473 (pAkt) over total Akt protein (pAkt/Akt) in hypothalamic lysates of WT and NULL mice in the absence/presence of leptin. (H) Representative Western blots of phosphorylated ERK1/2 at T202/T204 (pERK) and total ERK1/2 (ERK) protein in hypothalamic lysates of WT and NULL mice in the absence/presence of leptin and corresponding densitometric quantifications of the fraction of phosphorylated ERK1/2 over total ERK1/2 (pERK/ERK). (I) ELISA quantifications of αMSH in hypothalamic lysates of WT and NULL mice in the absence/presence of leptin. Western blot loading controls were actin and Bradford colorimetric assay. Animals were injected with either vehicle or 1.0 mg/kg body weight leptin and sacrificed 90 min later. *p < .05 (ANOVA with Tukey's post hoc).
3.7. Non‐canonical PI3K/Akt/FOXO1 and ERK signaling are upregulated in the NULL
Leptin can stimulate the production of POMC via non‐canonical pathways. One in particular, the insulin receptor substrate (IRS)–phosphatidylinositol 3‐OH kinase and protein kinase B (PI3K/Akt) pathway modulates suppression of feeding elicited by leptin administration but does not affect long‐term body weight regulation, which is normal in the NULL. 20 , 21 , 22 In this cascade, the binding of leptin to the LepR activates JAK2, which engages the Sar homology family member SH2B1. The latter recruits the insulin receptor, forming JAK2/SH2‐B/IRS complexes that mediate PI3K/Akt activation. Once Akt is released from the plasma membrane it phosphorylates Forkhead Box protein O1 (FOXO1), which by translocating out of the nucleus allows continuous POMC transcription. Representative Western blots of total and phosphorylated FOXO1 protein at S256 (FOXO1, pFOXO1) are illustrated in Figure 6E along with quantifications in Figure 6F. ELISA quantifications of total and phosphorylated Akt at S473 (Akt, pAkt) are shown in Figure 6G. Accordingly, the fractions of activated Akt (pAkt/Akt) and FOXO1 (pFOXO1/FOXO1) protein were greater in NULL compared to WT at baseline. In the presence of leptin, activated Akt and FOXO1 protein increased in WT, as expected. In contrast, the hormone was fairly ineffective in the NULL hypothalamus presumably because the pathway is constitutively activated. Another non‐canonical signaling pathway, the MAPK/ERK—Mitogen‐Activated Protein Kinase/Extracellular signal‐Regulated kinase (ERK1/2) also turned out to be chronically active in the NULL (Figure 6H). In summary, the suppression of STAT3 activity in the NULL is compensated by FOXO1 and ERK activation that causes constitutive POMC production. A similar conclusion is supported by QIAGEN IPA analysis of transcriptome data that predicts increased POMC production, and consequent anorexia, in the NULL (Figure S4). POMC is cleaved into five peptides including α‐melanocyte‐stimulating hormone (αMSH), which modulates neurons of the paraventricular nucleus of hypothalamus (PVH). To ascertain that the high levels of POMC in NULL are correctly processed, we measured αMSH protein, which resulted significantly increased in NULL compared to WT (ELISA, Figure 6I; IF, Figure S5). Together, these data indicate that KCNB1 is necessary for the signaling of the LepR. The absence of the channel results in the inactivation of the canonical STAT3 pathway and in the constitutive activation of non‐canonical pathways leading to POMC and one of its by‐products, αMSH, overproduction. As αMSH inhibits leptin release by the adipocytes, these results may explain why fat and circulating leptin are lower in the NULL in the presence of high levels of POMC. 23
3.8. KCNB1 and the LepR transcripts are expressed in POMC neurons
The observation that LepR signaling is dysregulated in hypothalamic neurons lacking KCNB1 may imply that the channel and the receptor operate in the same cells. The proopiomelanocortin neurons of the Arcuate nucleus (ARHPOMC) are likely candidate hosts, as these cells are the primary mediators of POMC homeostasis in the hypothalamus. To begin to test this, we carried out in situ mRNA hybridization using RNAscope. 24 Examples of hypothalamic slices hybridized with fluorescent probes against POMC, KCNB1, and the LepR are illustrated in Figure 7A. Both WT and NULL hypothalamic cells were positive to POMC, KCNB1, and the LepR, as expected. Notably, LepR transcripts are significantly increased in the NULL compared to WT, along with an increase of POMC transcript levels in agreement with biochemical data (Figure 7B,C). In contrast, the mRNA levels of KCNB1 are similar in the two genotypes (Figure 7D). Most importantly eye inspection reveals significant colocalization of POMC, KCNB1, and LepR mRNA transcripts, suggesting that the channel and the receptor are co‐expressed in POMC neurons where they may possibly form a complex (Figure 7E–H). Thus, analysis indicates that ~18% of KCNB1 mRNA transcripts over total transcripts are expressed in POMC+ cells and ~31% of LepR transcripts in POMC+ cells (KCNB1POMC/KCNB1TOT and LepRPOMC/LepRTOT, Figure 7I). This is expected, since two sub‐populations of POMC neurons one expressing the LepR and the other expressing the glucagon receptor have been identified. 4 Ninety‐eight percent of the cells expressing POMC transcripts, co‐express KCNB1 and LepR transcripts (POMC+_KCNB1+_LepR+, Figure 7I), in a ~1:1 ratio (LepRPOMC/KCNB1POMC, Figure 7I. The ratio is expressed in percent in the figure). Interestingly, in about 50% of the cases KCNB1 and LepR mRNA transcripts partially overlap (Figure 7H and OVERLAP KCNB1POMC_LepRPOMC in Figure 7I). The physical proximity of mRNA molecules may imply that the KCNB1 and the LepR protein are assembled and then trafficked together to the plasma membrane, a typical feature of interacting proteins. 25 These data are in agreement with the work of Yao and colleagues who constructed a mouse brain transcriptomic and cell type atlas. 26 Analysis of the atlas shows that KCNB1 mRNA is present in all hypothalamic neuron classes and is expressed in ~92% of ARH neurons (Figure S6). Likewise, POMC transcripts are maximally expressed in ARH neurons (~63%). Also, LepR transcripts are broadly expressed in ARH neurons where, in ~60% of the cases, they are colocalized with KCNB1 and POMC transcripts.
FIGURE 7.
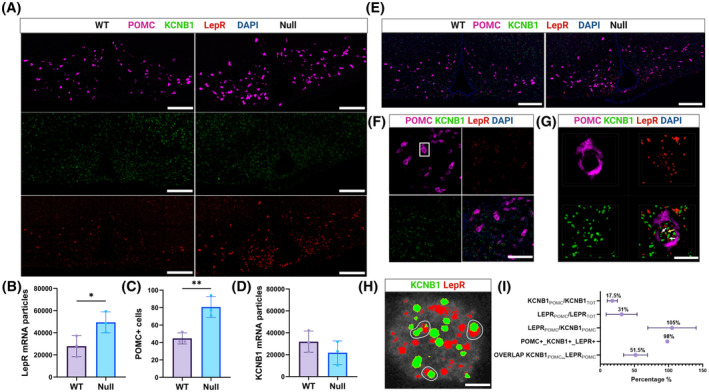
In situ hybridization reveals the presence of KCNB1 and LepR mRNA in POMC neurons. (A) Representative confocal images of brain slices showing the hypothalamus of WT and NULL hybridized with fluorescent probes for POMC (magenta), KCNB1 (green), and the LepR (red.) Scale bars 1 mm. (B) Mean number of LepR mRNA transcripts per image in WT and NULL slices. (C) Mean number of cells per image expressing POMC mRNA transcripts (POMC+) in WT and NULL slices. (D) Mean number of KCNB1 mRNA transcripts image in WT and NULL slices. (E) Overlap of the images in (A). Note the presence of many cells co‐expressing POMC, KCNB1, and LepR signals. Scale bars 1 mm. (F) Representative confocal images of an area of the ARH expressing POMC, KCNB1, and LepR mRNA transcripts and overlap (lower right image). The boxed area represents a single cell. Scale bars 30 μm. (G) Magnifications of the boxed area in (F), showing POMC, KCNB1, and LepR signals. The image in the lower right corner is a z‐stack of 18 images. Spots of colocalization of single KCNB1 and LepR mRNA molecules are indicated by arrows. Scale bars 10 μm. (H) Representative image of a single POMC+ cell showing individual KCNB1 and LepR mRNA molecules. Partial overlap of KCNB1 and LepR signals is indicated. (I) Percentage of KCNB1 and LepR mRNA transcripts present in POMC+ cells; percentage of POMC+ cells simultaneously expressing KCNB1 and LepR mRNA transcripts; mean ratio of single LepR and KCNB1 transcripts per POMC+ cell expressed in percent and percentage of KCNB1‐LepR mRNA transcripts partial overlaps per POMC+ cell. N = 119 cells, 3 biological replicates, and 8 technical replicates. *p < .05, **p < .01 (two‐tailed Student's t‐test).
3.9. KCNB1 is expressed in the POMC neurons of the arcuate nucleus
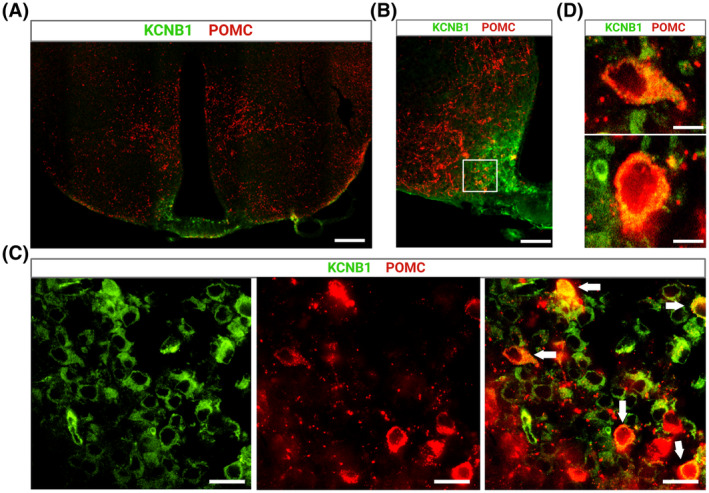
Representative images of hypothalamic slices co‐stained with KCNB1 (green) and POMC (red) antibodies are shown in Figure 8. Thus, in three experiments, POMC and KCNB1 immunoreactivity were distributed across the hypothalamus (Figure 8A,B). Importantly, KCNB1 immunoreactivity was prominent in the ARH. Within the ARH area, the number of KCNB1 positive (+) neurons was greater than the number of POMC+ neurons by a ~2:1 ratio (representative images in Figure 8C). However, the majority of POMC+ cells in the ARH co‐expressed KCNB1 (Figure 8C,D). We conclude that KCNB1 is expressed in ACRPOMC neurons.
FIGURE 8.
KCNB1 is expressed in ARHPOMC neurons. (A) Representative confocal image of a WT hypothalamic slice stained with KCNB1 (green) and POMC (red) antibody. Scale bar 1 mm. (B) Magnification of the ARH area in (A). Scale bar 200 μm. (C) Magnifications of the boxed area in (B) showing individual cells expressing KCNB1 (green), POMC (red), and overlap. Cells co‐expressing both proteins are indicated with arrows in the overlap image on the right. Scale bar 25 μm. (D) Representative magnifications of two cells indicated with arrows in (C), co‐expressing KCNB1 and POMC. Note the granular expression of KCNB1, reflecting cluster organization. Scale bar 10 μm.
3.10. KCNB1 is expressed in the AgRP neurons of the arcuate nucleus
Representative images of KCNB1 and AgRP colocalization in ARH neurons along with WBD analysis of AgRP levels in WT and NULL are illustrated in Figure S7. KCNB1 appears to be expressed in ARHAgRP neurons, in agreement with a previous study reporting the discovery of a leptin‐sensitive K+ current attributed to the channel and to its relative, KCNB2 (Figure S7A–C). 27 However, the lack of the channel only marginally affected AgRP biosynthesis at baseline, and for this reason, the role of KCNB1 in ARHAgRP cells was not investigated further (Figure S7D–F).
3.11. KCNB1 and the LepR proteins colocalize in arcuate neurons
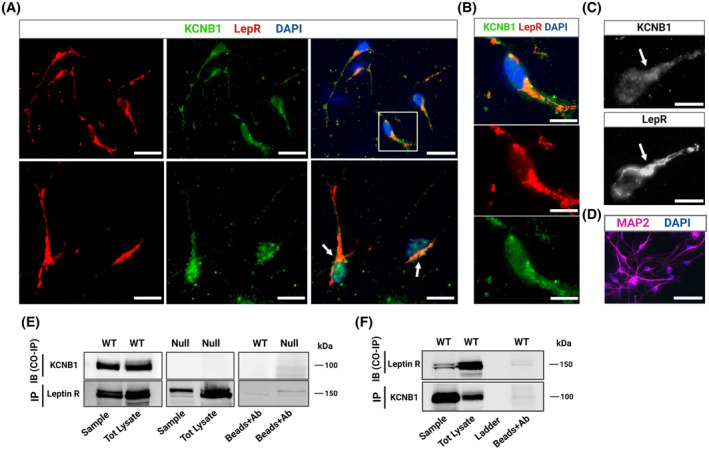
Representative confocal images of hypothalamic slices co‐stained with KCNB1 (green) and LepR (red) antibodies are shown in Figure S8A. Thus, overlap of KCNB1 and LepR signals can be observed, primarily within the ARH area (Figure S8B). To obtain a more detailed picture of the colocalization pattern of KCNB1 and the LepR at the level of the single cell, we employed a magnetic‐activated cell sorting (MACS) protocol, recently developed in house that allowed us to isolate neurons expressing the LepR from the hypothalami of E14‐E14 embryos and grow them in culture. 11 Images of primary hypothalamic neurons co‐stained for KCNB1 (green), and the LepR (red) are illustrated in Figure 9A. Colocalization in an exemplar neuron is illustrated in Figure 9B. Thus, in 2 experiments, the LepR was expressed in ~95 ± 5% of the neurons attesting the high selectivity of the method. The KCNB1 channel exhibits discrete distribution, a hallmark of this protein (Figure 9B). 28 Most importantly, KCNB1 and LepR immunofluorescence significantly overlap, primarily in the soma and in the proximal dendrite, consistent with the known expression pattern of the channel in neurons. 29 Manders analysis of twenty neurons (representative images are shown in Figure 9C) gave values of Pearson's correlation coefficient, R, ranging from a minimum of 0.74 to a maximum of 0.84 (R median = 0.77), and Manders's M1 coefficient in the 0.943–0.995 range (M1 median = 0.983) and Manders's M2 coefficient in the 0.872–0.979 range (M2 median = 0.954). 30 Microtubule‐associated protein 2 (Map2) was used as a marker for the neuronal purity of the cultures (Figure 9D). Overall, in the cells co‐expressing KCNB1 and the LepR overlap was significant, implying that a considerable number of KCNB1 channels may be associated with the LepR.
FIGURE 9.
KCNB1 forms stable complexes with the LepR. (A) Representative images of primary hypothalamic neurons stained for LepR (red) and KCNB1 (green) and overlap. In the composite image, DAPI is in blue color. Scale bars 20 μm. (B) Magnifications of the boxed area in (A) illustrating KCNB1‐LepR overlap. Colocalization occurred primarily in the soma and in the proximal dendrite. Typical clustered distribution of KCNB1 is also demonstrated. Scale bars 10 μm. (C) Representative example of colocalization analysis in a single neuron indicated with an arrowhead in (A), performed using Coloc 2 software (ImageJ). Top: 16‐bit image of KCNB1 green channel. Bottom: 16‐bit image of the LepR red channel. Arrows indicate maximal colocalization areas (hillock). Scale bars 50 μm. (D) Representative confocal images of primary LepR+ hypothalamic neurons stained with Map2. DAPI blue color. Scale bars 50 μm. (E) Representative co‐IPs of KCNB1 with the long isoform of the leptin receptor. LepR protein was immunoprecipitated from hypothalamic WT or NULL lysates and immunoblotted with KCNB1 antibody. Controls: Total lysates and beads conjugated to mouse IgG antibody. (F) In the representative immunoblots, KCNB1 protein was immunoprecipitated from hypothalamic WT lysates and immunoblotted with LepR antibody. Controls: Total lysates and beads conjugated to mouse GFP antibody. Experiments were carried out on the same antibody‐stripped membranes.
3.12. KCNB1 forms complexes with the LepR in hypothalamic neurons
We next performed coimmunoprecipitations (co‐IPs) to determine whether KCNB1 and the LepR physically interact. Representative examples of nine co‐IP experiments each performed on the same antibody‐stripped membranes, are illustrated in Figure 9E,F. Immunoprecipitating the LepR from WT hypothalamic lysates pulled down the KCNB1 channel (Figure 9E) and vice versa the channel pulled down the LepR (Figure 9F). As expected, the LepR immunoprecipitated from NULL hypothalamic lysates but did not pull down KCNB1 protein (Figure 9E). Additional controls performed with beads conjugated to an unspecific antibody (anti‐GFP) are illustrated in the right lanes of the blots. We conclude that KCNB1 and the LepR form stable complexes in hypothalamic cells.
3.13. c‐Fos expression is higher in ARH neurons of NULL
The nuclear protooncogene c‐Fos is an indirect marker of the electrical activity of many regions of the brain including the hypothalamus, as its expression increases when neurons fire action potentials. 31 , 32 Therefore, to determine whether KCNB1 plays a role in the excitability of arcuate neurons, we quantified c‐Fos mRNA transcripts in the hypothalami of WT and NULL mice by in situ hybridization using RNAscope. Thus, c‐Fos, POMC, and KCNB1 transcripts were co‐expressed in ARH cells, in agreement with IF data (Figure 10A–C. Since the channel is broadly expressed in the ARH, KCNB1 signals are not shown in Figure 10A), and previous studies. 26 Further, the amounts of POMC transcripts are significantly increased in the NULL compared to WT, in agreement with biochemical assessment of POMC protein (Figure 10D). Notably, c‐Fos transcription is significantly augmented in NULL neurons co‐expressing POMC and KCNB1 mRNA, compared to WT neurons (Figure 10E). Upon binding leptin, ARHPOMC neurons increase firing, which leads to c‐Fos production. In fact, acute leptin administration was associated with strengthened c‐Fos mRNA synthesis in WT arcuate neurons co‐expressing POMC and KCNB1 mRNA (Figure 10D,E). In contrast, c‐Fos mRNA levels did not vary in NULL neurons, presumably because the cells are constitutively active. We next assessed c‐Fos protein by WBD (Figure 10F,G) and IF (Figure S9). Thus, the amounts of c‐Fos protein quantified by WBD were greater in NULL compared to WT at baseline (representative WBs in Figure 10F and quantifications in Figure 10G). In the presence of leptin, they increased in WT, but remained high and stable in NULL. Likewise, c‐Fos immunoreactivity was significantly augmented in NULL arcuate slices compared to WT at baseline (Figure S9A and magnifications in Figure S9B. Quantifications in Figure S9C. All the slices used for c‐Fos analysis were cut from the same brains used for POMC analysis, Figure 5), whereas leptin administration increased immunoreactivity to c‐Fos in WT slices while leaving immunofluorescence unaffected in NULL slices. We conclude that KCNB1 is implicated in the modulation of the electrical activity of arcuate neurons.
FIGURE 10.
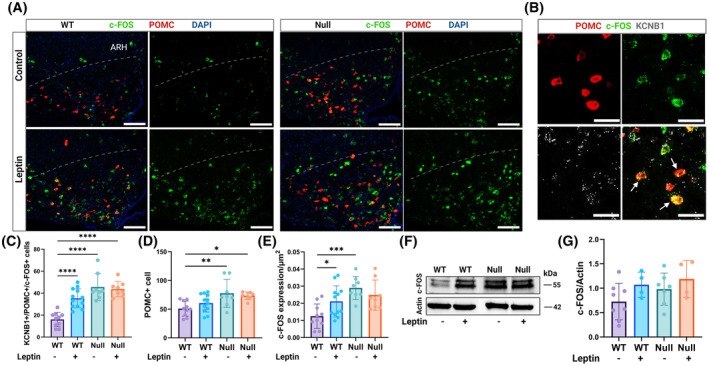
c‐Fos transcription is constitutively elevated in NULL arcuate neurons. (A) Representative confocal images of WT and NULL brain slices in the absence/presence of leptin, showing the ARH area under the dotted curved line, hybridized with fluorescent probes for c‐Fos (green), POMC (red), and DAPI (blue). Scale bars 100 μm. (B) Representative examples of ARH cells co‐expressing c‐Fos (green), POMC (red), and KCNB1 (dark gray) transcripts (arrows). Scale bars 20 μm. (C) Quantifications of cells positive for c‐Fos, POMC, and KCNB1 mRNA transcripts per image in WT or NULL cells in the absence/presence of leptin. N = 3 mice/genotype/experimental condition. N = 2445 cells. (D) Quantifications of cells expressing POMC mRNA transcripts per image in WT or NULL in the absence/presence of leptin. (E) Quantifications of the density of c‐Fos mRNA (number of transcripts per μm2) in WT or NULL in the absence/presence of leptin. (F) Representative Western blots of c‐Fos protein in WT and NULL hypothalamic lysates in the absence/presence of leptin. N = 4–8 mice/genotype/experimental condition. (G) Densitometric quantifications of c‐Fos protein normalized to actin in Western blots as shown in (F). Western blot loading controls were actin and Bradford colorimetric assay. Animals were injected with either vehicle or 1.0 mg/kg body weight leptin and sacrificed 90 min later. *p < .05, **p < .01, ***p < .001, ****p < .0001 (ANOVA with Tukey's post hoc).
4. DISCUSSION
This study was undertaken to answer important questions concerning the role of Kv channels in the brain. Here, we report that Kv channel KCNB1 is expressed in the neurons of the hypothalamus where it forms macromolecular complexes with the LepR in a sub‐set of these, including the ARHPOMC cells. Transcriptome analysis underscores dysregulated leptin signaling in the NULL. Accordingly, NULL mice have less fat and circulating leptin and are desensitized to the anorexic stimuli of the hormone. The lack of KCNB1 is associated with robust overproduction of POMC, via the non‐canonical PI3K/Akt/FOXO1 and ERK pathways, while the canonical STAT3 signaling is inhibited. In turn, more αMSH is produced, which stops the synthesis of leptin by the adipocytes. In this regard, it is known that disruption of PI3K/Akt signaling in ARHPOMC neurons promotes acute suppression of feeding elicited by leptin administration but does not affect long‐term body weight regulation. 22 This is consistent with our findings that NULL animals retain normal weight. The absence of the channel is associated with increased synthesis of proto‐oncogene c‐Fos protein an indirect marker of neuronal activity. Most importantly, c‐Fos, levels do not change in the NULL in response to leptin. The use of c‐Fos to assess KCNB1 regulation of neuronal excitability is a limitation of this study, dictated in part by the cellular diversity of the hypothalamus that makes electrophysiological recordings challenging. Nonetheless, decreased repolarizing K+ current due to the lack of the KCNB1 channel is the most likely explanation for the high c‐Fos levels detected in the NULL cells, but alternative, non‐exclusive mechanisms are conceivable. For example, changes in secretion/signaling of other regulating factors such as serotonin, glucagon‐like peptide 1 receptor or neuropeptide Y could cause increased c‐Fos expression. Likewise, alterations in other channels, for example K(ATP) whose trafficking is leptin‐dependent in pancreatic beta‐cells, or TRPC5, which is also leptin‐dependent, could further promote hyperexcitability. 33 , 34 , 35 , 36 With the current knowledge, it cannot be ruled out that KCNB1 modulates leptin signaling through its conducting function, for example by causing fluctuations in the resting membrane potential. However, since the channel and the LepR form a stable complex, it is more likely that KCNB1 regulates the function of the LepR through physical interactions, and vice versa. These considerations lead us to propose a working model, graphically illustrated in Figure 11, that predicts that the coupling of the KCNB1 channel and the LepR in a single macromolecular complex allows certain hypothalamic neurons like the ARHPOMC cells to synergistically combine electrical and endocrine functions. Hence, when KCNB1 is missing, the function of the LepR is impaired causing metabolic dysfunction. While this model awaits validation, we notice that it is in agreement with previous studies that showed that in HEK cells co‐transfected with KCNB1 and the LepR, leptin shifted the voltage dependence of the channel toward hyperpolarizing voltages and may also explain previous results indicating that leptin failed to induce ARHPOMC depolarization and increased firing frequency when PI3K/Akt signaling was inhibited. 22 , 27 Considering that in certain circumstances, for example under conditions of cellular oxidative stress, KCNB1 can sequester Akt, the possibility exists that the channel might impinge on POMC production in a LepR‐independent manner via directly regulating Akt. 10 It is also worth noticing that KCNB1 mRNA transcripts have been detected in adipose tissue (The Human Protein Atlas Database). 37 This raises the possibility that if KCNB1 protein is functional in adipocytes, its lack could affect leptin secretion and/or LepR signaling in the NULL. Furthermore, while dysfunctional ARCPOMC neurons are likely involved in the metabolic anomalies of the NULL, it is also probable that other cells contribute to the overall phenotype. Moreover, KCNB1 is widely expressed in the Central Nervous System, and thus, the possibility exists that its absence in neurons that interact with hypothalamic neurons may have indirect, unknown effects, on hypothalamic function. The answer to this and other questions will require further studies, including a conditional knockout mouse, and were beyond the scope of this work.
FIGURE 11.
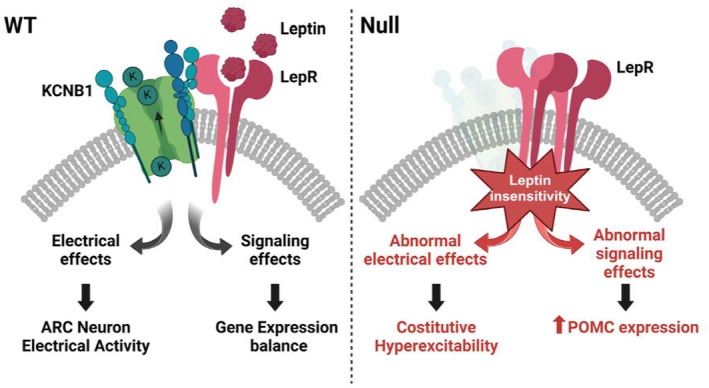
Proposed model of the function of KCNB1‐LepR complexes in ARHPOMC neurons. In WT cells, the conducting functions of the channel modulate neuron excitability. In addition, the allosteric changes associated with the opening/closing of KCNB1 are translated to the LepR modulating its function. When the channel is missing, like in NULL neurons, in addition to abnormal depolarization, the function of the LepR is impaired resulting in dysregulated POMC production.
Leptin modulates insulin release from pancreatic beta‐cells, which express KCNB1. 19 There, the channel conducts a repolarizing K+ current which has been proposed to inhibit insulin secretion in the presence of high glucose in vitro 38 (although recent evidence may suggest that KCNB1 modulates insulin secretion independently of its conducting function). 39 We did not detect significant changes in insulin homeostasis in the NULL animals, but this may be due to the different strains and diet used. The experiments in beta‐cells were carried out using Kcnb1 Knock Out (KO) mice, that are subject to significant K+ current remodeling. Jacobson and colleagues showed that in the islets of Langerhans of Kcnb1 KO, action potential repolarization and glucose‐induced insulin release were rescued by the activity of small conductance calcium‐activated K+ channels (SK), which appeared to be otherwise silent in WT islets. 40 Our findings align with previous reports that showed that inhibition of KCNB1 channels with Guangxitoxin‐1E or synthetic blockers had no effect on glucose tolerance in WT mice and that compared with WT mice, Kcnb1 KO mice had only mild hypoglycemia and largely normal responses to a glucose load. 41 , 42 However, mild metabolic abnormalities that would normally be amplified under hypercaloric regimens may have gone undetected in the mice used in this study that were fed with normal chow.
KCNB1 forms complexes with integrins named IKCs, that regulate the migration of excitatory glutamatergic neurons during corticogenesis. 8 Accordingly, Knock In mice harboring a missense (Kcnb1 R312H ) mutation found in children with developmental and epileptic encephalopathy (DEE), an umbrella term to describe pathologies in which severe developmental delay coexists with epilepsy, 43 , 44 exhibit neocortical malformations and defective pyramidal neuron morphology that are due to impairment of the signaling machinery of the IKC. 8 Since integrins are broadly expressed in the brain, we asked whether they were also present in KCNB1‐LepR complexes. We sampled potential interactions with integrin‐β5 which interacts with KCNB1 in the neocortex and was detected in the hypothalamus and integrin‐β1, which is perhaps the most abundant β‐integrin in the brain. Both coimmunoprecipitated with the LepR and KCNB1 (Figure S10). Integrin‐β1 gave the strongest signal suggesting that this is the predominant integrin interacting with the complex. The investigation of potential interactions of KCNB1‐LepR complexes with integrins in the hypothalamus was beyond the scope of this study and will be the matter of future investigations. However, a potentially significant implication of these findings is that Kv channels such as KCNB1 appear to give rise to multipronged macromolecular complexes that couple electrical activity with signaling functions. Furthermore, since the NULL mouse provides a model of DEE, as several nonsense mutations in the human KCNB1 gene have been found in DEE children, these findings may unveil the existence of genetic links between epileptic and metabolic disorders, paving the way to future investigations into the pathogenic mechanisms common to these diseases. 45
In conclusion, these results evidence the broad role played by KCNB1 in the brain, that appears to be implicated in the mechanisms underlying neurodevelopment, learning and memory, and metabolism. As obesity has become a global health concern, this study may provide insight into the molecular basis for this condition as well as a novel pharmacological target.
AUTHOR CONTRIBUTIONS
Elena Forzisi‐Kathera‐Ibarra performed literature search, research, and data analysis. Chanmee Jo performed Western blotting, Leonard Castillo performed behavioral analysis, Christian Roser performed immunofluorescence, Anika Gaur performed immunofluorescence and Western blotting, Prachi Lad performed immunofluorescence and Western blotting, Alessandro Bortolami analyzed data, Stefania Dutto performed behavioral analysis, Ping‐Yue Pan performed confocal analysis, Harini Sampath and Matthew Selby helped with the MRI analysis, Srinidi Venkateswaran developed software to count cells and Federico Sesti directed the study and wrote the manuscript.
DISCLOSURES
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Data S1..
ACKNOWLEDGMENTS
We thank Drs. Mark Rossi and Monahan for insightful discussions and help with the RNAscope. This work was supported by a NIA grant (R01AG060919) and an NSF grant (2030348) to FS.
Forzisi‐Kathera‐Ibarra E, Jo C, Castillo L, et al. KCNB1‐Leptin receptor complexes couple electric and endocrine function in the melanocortin neurons of the hypothalamus. The FASEB Journal. 2024;38:e70111. doi: 10.1096/fj.202401931R
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are available at Dryad: https://doi.org/10.5061/dryad.7m0cfxq1g. The dataset will be published upon acceptance of the manuscript. A private, temporary URL can be accessed in the meantime upon request. Transcriptome data are available at the GEO repository of the National Institutes of Health and will be available with accession code: GSE246918 upon acceptance of the manuscript. Reviewers can access the data by using the temporary token ivqdgoouvdybbup.
REFERENCES
- 1. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8(5):571‐578. [DOI] [PubMed] [Google Scholar]
- 2. Clarke IJ. Hypothalamus as an endocrine organ. Compr Physiol. 2015;5(1):217‐253. [DOI] [PubMed] [Google Scholar]
- 3. Toda C, Santoro A, Kim JD, Diano S. POMC neurons: from birth to death. Annu Rev Physiol. 2017;79:209‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biglari N, Gaziano I, Schumacher J, et al. Functionally distinct POMC‐expressing neuron subpopulations in hypothalamus revealed by intersectional targeting. Nat Neurosci. 2021;24(7):913‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andermann ML, Lowell BB. Toward a wiring diagram understanding of appetite control. Neuron. 2017;95(4):757‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klok MD, Jakobsdottir S, Drent ML. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: a review. Obes Rev. 2007;8(1):21‐34. [DOI] [PubMed] [Google Scholar]
- 7. Kwon O, Kim KW, Kim MS. Leptin signalling pathways in hypothalamic neurons. Cell Mol Life Sci. 2016;73(7):1457‐1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bortolami A, Yu W, Forzisi E, et al. Integrin‐KCNB1 potassium channel complexes regulate neocortical neuronal development and are implicated in epilepsy. Cell Death Differ. 2022;30:687‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Forzisi E, Sesti F. Non‐conducting functions of ion channels: the case of integrin‐ion channel complexes. Channels. 2022;16(1):185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forzisi E, Yu W, Rajwade P, Sesti F. Antagonistic roles of Ras‐MAPK and Akt signaling in integrin‐K+ channel complex‐mediated cellular apoptosis. FASEB J. 2022;36(5):e22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elena KIF, Roser C, Gaur A, Sesti F. Isolation of targeted hypothalamic neurons for studies of hormonal, metabolic, and electrical regulation. J Vis Exp. 2023;198:e65674. [DOI] [PubMed] [Google Scholar]
- 12. Noordzij M, Tripepi G, Dekker FW, Zoccali C, Tanck MW, Jager KJ. Sample size calculations: basic principles and common pitfalls. Nephrol Dial Transplant. 2010;25(5):1388‐1393. [DOI] [PubMed] [Google Scholar]
- 13. Yu W, Shin MR, Sesti F. Complexes formed with integrin‐alpha5 and KCNB1 potassium channel wild type or epilepsy‐susceptibility variants modulate cellular plasticity via Ras and Akt signaling. FASEB J. 2019;33(12):14680‐14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei JF, Wei L, Zhou X, et al. Formation of Kv2.1‐FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J Cell Physiol. 2008;217(2):544‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30(4):523‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benani A, Hryhorczuk C, Gouazé A, et al. Food intake adaptation to dietary fat involves PSA‐dependent rewiring of the arcuate melanocortin system in mice. J Neurosci. 2012;32(35):11970‐11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rossi MA, Basiri ML, McHenry JA, et al. Obesity remodels activity and transcriptional state of a lateral hypothalamic brake on feeding. Science. 2019;364(6447):1271‐1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cansell C, Stobbe K, Sanchez C, et al. Dietary fat exacerbates postprandial hypothalamic inflammation involving glial fibrillary acidic protein‐positive cells and microglia in male mice. Glia. 2021;69(1):42‐60. [DOI] [PubMed] [Google Scholar]
- 19. Roe MW, Worley JF, Mittal AA, et al. Expression and function of pancreatic beta‐cell delayed rectifier K+ channels. Role in stimulus‐secretion coupling. J Biol Chem. 1996;271(50):32241‐32246. [DOI] [PubMed] [Google Scholar]
- 20. Duan C, Li M, Rui L. SH2‐B promotes insulin receptor substrate 1 (IRS1)‐ and IRS2‐mediated activation of the phosphatidylinositol 3‐kinase pathway in response to leptin. J Biol Chem. 2004;279(42):43684‐43691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115(4):951‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hill JW, Williams KW, Ye C, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118(5):1796‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoggard N, Hunter L, Duncan JS, Rayner DV. Regulation of adipose tissue leptin secretion by alpha‐melanocyte‐stimulating hormone and agouti‐related protein: further evidence of an interaction between leptin and the melanocortin signalling system. J Mol Endocrinol. 2004;32(1):145‐153. [DOI] [PubMed] [Google Scholar]
- 24. Wang F, Flanagan J, Su N, et al. RNAscope: a novel in situ RNA analysis platform for formalin‐fixed, paraffin‐embedded tissues. J Mol Diagn. 2012;14(1):22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bianchi L, Kwok SM, Driscoll M, Sesti F. A potassium channel‐MiRP complex controls neurosensory function in Caenorhabditis elegans . J Biol Chem. 2003;278(14):12415‐12424. [DOI] [PubMed] [Google Scholar]
- 26. Yao Z, van Velthoven CTJ, Kunst M, et al. A high‐resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature. 2023;624(7991):317‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baver SB, Hope K, Guyot S, Bjørbaek C, Kaczorowski C, O'Connell KMS. Leptin modulates the intrinsic excitability of AgRP/NPY neurons in the arcuate nucleus of the hypothalamus. J Neurosci. 2014;34(16):5486‐5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hwang PM, Fotuhi M, Bredt DS, Cunningham AM, Snyder SH. Contrasting immunohistochemical localizations in rat brain of two novel K+ channels of the Shab subfamily. J Neurosci. 1993;13(4):1569‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Du J, Tao‐Cheng JH, Zerfas P, McBain CJ. The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience. 1998;84(1):37‐48. [DOI] [PubMed] [Google Scholar]
- 30. Manders EMM, Verbeek FJ, Aten JA. Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc. 1993;169(3):375‐382. [DOI] [PubMed] [Google Scholar]
- 31. Dragunow M, Faull R. The use of c‐fos as a metabolic marker in neuronal pathway tracing. J Neurosci Methods. 1989;29(3):261‐265. [DOI] [PubMed] [Google Scholar]
- 32. Elmquist JK, Ahima RS, Maratos‐Flier E, Flier JS, Saper CB. Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology. 1997;138(2):839‐842. [DOI] [PubMed] [Google Scholar]
- 33. Ibrahim N, Bosch MA, Smart JL, et al. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144(4):1331‐1340. [DOI] [PubMed] [Google Scholar]
- 34. Zhan C. POMC neurons: feeding, energy metabolism, and beyond. Adv Exp Med Biol. 2018;1090:17‐29. [DOI] [PubMed] [Google Scholar]
- 35. Qiu J, Wagner EJ, Rønnekleiv OK, Kelly MJ. Insulin and leptin excite anorexigenic pro‐opiomelanocortin neurones via activation of TRPC5 channels. J Neuroendocrinol. 2018;30(2):e12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu Y, Shyng SL, Chen PC. Concerted trafficking regulation of Kv2.1 and KATP channels by leptin in pancreatic β‐cells. J Biol Chem. 2015;290(50):29676‐29690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tissue Expression of KCNB1—Summary—The Human Protein Atlas. Accessed June 20, 2022. https://www.proteinatlas.org/ENSG00000158445‐KCNB1/tissue [Google Scholar]
- 38. MacDonald PE, Ha XF, Wang J, et al. Members of the Kv1 and Kv2 voltage‐dependent K(+) channel families regulate insulin secretion. Mol Endocrinol. 2001;15(8):1423‐1435. [DOI] [PubMed] [Google Scholar]
- 39. Dai XQ, Manning Fox JE, Chikvashvili D, et al. The voltage‐dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia. 2012;55(6):1709‐1720. [DOI] [PubMed] [Google Scholar]
- 40. Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium‐activated and voltage‐gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. J Physiol. 2010;588(Pt 18):3525‐3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammälä CE, Philipson LH. Kv2.1 ablation alters glucose‐induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6(3):229‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li XN, Herrington J, Petrov A, et al. The role of voltage‐gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. J Pharmacol Exp Ther. 2013;344(2):407‐416. [DOI] [PubMed] [Google Scholar]
- 43. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15(3):304‐316. [DOI] [PubMed] [Google Scholar]
- 45. de Kovel CGF, Syrbe S, Brilstra EH, et al. Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. 2017;74(10):1228‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1..
Data Availability Statement
All data generated or analyzed during this study are available at Dryad: https://doi.org/10.5061/dryad.7m0cfxq1g. The dataset will be published upon acceptance of the manuscript. A private, temporary URL can be accessed in the meantime upon request. Transcriptome data are available at the GEO repository of the National Institutes of Health and will be available with accession code: GSE246918 upon acceptance of the manuscript. Reviewers can access the data by using the temporary token ivqdgoouvdybbup.