

ABSTRACT
Stroke, a major neurological disorder and a leading cause of disability and death, often inflicts damage upon other organs, particularly the kidneys. While chronic kidney disease (CKD) has long been established as a significant risk factor for cerebrovascular disease, stroke can induce renal dysfunction, manifesting as acute kidney injury (AKI) or CKD. Mounting clinical and basic research evidence supports the existence of a bidirectional brain‐kidney crosstalk following stroke, implicating specific mechanisms and pathways in stroke‐related renal dysfunction. This review analyzes pertinent experimental studies, elucidating the underlying mechanisms of this cerebro‐renal interaction following stroke. Additionally, we summarize the current landscape of clinical research investigating brain‐kidney interplay and discuss potential challenges in the future. By enhancing our understanding of the scientific underpinnings of brain‐kidney crosstalk, this review paves the way for improved treatment strategies and outcomes for stroke patients. Recognizing the intricate interplay between the brain and kidneys after stroke holds profound clinical implications.
Keywords: AKI, brain–kidney interaction, CKD, renal dysfunction, stroke
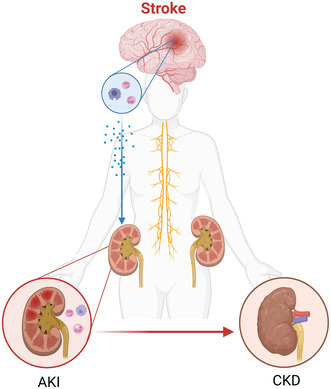
After a stroke, multiple pathways in the neurohumoral system are activated. At the same time, the blood–brain barrier is compromised, and pro‐inflammatory substances are released from the brain into the bloodstream, consequently activating the peripheral inflammatory immune response. As an acute injury, stroke triggers renal hemodynamic disturbances and an acute ischemic state in the kidney, thereby promoting the development of AKI or CKD.
Abbreviations
- ACR
albumin–creatinine ratio
- ACTH
adrenocorticotropic hormone
- ADH
antidiuretic hormone
- AIS
acute ischemic stroke
- AKI
acute kidney injury
- BBB
blood–brain barrier
- BP
blood pressure
- CBF
cerebral blood flow
- CE
cardioembolism
- CI‐AKI
contrast‐induced AKI
- CKD
chronic kidney disease
- CNS
central nervous system
- CRP
C‐reactive protein
- CTA
computed tomography angiography
- CysC
cystatin C
- eGFR
estimated glomerular filtration rate
- EMT
epithelial–mesenchymal transition
- END
neurological deterioration
- EPVS
enlarged perivascular spaces
- ESRD
end‐stage renal disease
- EVs
extracellular vesicles
- EVT
endovascular therapy
- GCs
glucocorticoids
- GFAP
glial fibrillary acidic protein
- GFR
glomerular filtration rate
- HK‐2
human kidney 2
- HT
hemorrhagic transformation
- HTPA axis
the hypothalamic–pituitary–adrenal axis
- ICH
intracerebral hemorrhage
- ICUs
intensive care units
- IGFBP7
insulin‐like growth factor‐binding protein 7
- IL‐18
interleukin‐18
- IL‐1β
interleukin‐1β
- ILs
interleukins
- IS
ischemic stroke
- JAMs
junctional adhesion molecules
- KDIGO
kidney disease: improving global outcomes
- KIM‐1
kidney injury molecule 1
- LAA
large‐artery atherosclerosis
- MiRs
microRNAs
- MMPs
matrix metalloproteinases
- MRI
magnetic resonance imaging
- mRS
modified Rankin scale
- mt‐Nd6
mitochondrially encoded NADH dehydrogenase 6
- NGAL
neutrophil gelatinase‐associated lipocalin
- NK
natural killer
- PA
pressure autoregulation
- RAAS
renin–angiotensin–aldosterone system
- RBF
renal blood flow
- RBMEC
rat‐brain microvascular endothelial cells
- ROS
reactive oxygen species
- RVR
renal vascular resistance
- SAH
subarachnoid hemorrhage
- SAO
small artery occlusion
- SCr
serum creatinine
- SNGFR
single nephron glomerular filtration rate
- SNS
sympathetic nervous system
- SOE
stroke of other determined etiology
- SUE
stroke of undetermined etiology
- T2D
type 2 diabetes mellitus
- TGF‐β
transforming growth factor beta
- TIMP‐2
tissue inhibitor of metalloproteinases‐2
- TJs
tight junction proteins
- TNFs
tumor necrosis factors
- TNF‐α
tumor necrosis factor alpha
- TOAST
the trial of org 10172 in acute stroke treatment
- Tregs
regulatory T cells
- UACR
urine albumin‐to‐creatinine ratio
- WML
white‐matter lesions
- ZO
zonula occludens
1. Introduction
Stroke, a leading cause of mortality and disability worldwide, initiates a cascade of physiological responses extending beyond the central nervous system (CNS) [1]. Among the systemic consequences of stroke, renal dysfunction—manifesting as AKI and CKD—has emerged as a critical yet often‐overlooked area of concern. These conditions, while diverse in their clinical presentation, both represent pathophysiological states of the kidney that occur as a response to the brain–renal crosstalk initiated by a stroke.
AKI, characterized by a rapid decline in renal function, often marks the initial phase of kidney impairment following a stroke [2, 3]. If unresolved, this acute renal injury can progressively transition into CKD, a long‐term condition characterized by persistent kidney damage and a gradual loss of renal function over time [4].
This review seeks to elucidate the complex mechanisms underpinning the relationship between stroke and renal impairment, focusing on inflammatory factor release, blood–brain barrier (BBB) impairment, immune cell response, and the role of extracellular vesicles (EVs) and microRNAs (MiRs). By exploring these intertwined pathways, we hope to shed light on the systemic nature of stroke and its impact on renal health.
As we delve into the intricate dynamics of neuro–renal interactions following stroke, we also underscore the therapeutic implications of these findings, which could pave the way for novel strategies to mitigate stroke‐induced renal damage. The aim of this review is to investigate the brain–kidney axis between renal injury after stroke to provide more opportunities for early detection and potential therapeutic directions for patients in the clinical setting. At the same time, we acknowledge the gaps in our current understanding, emphasizing the need for further research in this fascinating intersection of neurology and nephrology.
2. Stroke and Renal Dysfunction: Clinical Insights
The cerebro–renal interaction is an emerging research focus due to its substantial implications for patient outcomes and overall healthcare management. In this section, we delve into the clinical dimensions of post‐stroke renal dysfunction, starting with the epidemiological evidence that underscores the frequency and severity of this often‐overlooked consequence of stroke.
2.1. Epidemiology of Kidney Injury Post‐Stroke
Stroke, a neurologically devastating disease, can concurrently instigate peripheral organ pathologies, including kidney dysfunction. Understanding this cerebro‐renal relationship is imperative given the ubiquitous nature of AKI and CKD—clinical conditions that typically lead to alterations in glomerular filtration rate (GFR), urine output, and relevant biomarkers [5, 6, 7].
However, the incidence of AKI and CKD following stroke is a contentious subject, with reported morbidity rates exhibiting substantial variation across clinical studies [8, 9, 10]. Meta‐analyses conducted in 2018 reported an incidence rate of 9.61% and 11.6% respectively, for AKI following stroke [8, 10]. Consequences of stroke‐induced renal insufficiency often include severe neurological sequelae and poorer patient outcomes, underscoring the clinical relevance of studying the brain‐kidney interplay in the context of stroke. Figure 1 lists relevant clinical manifestations and abnormal examination findings observed in patients with post‐stroke kidney injury.
FIGURE 1.
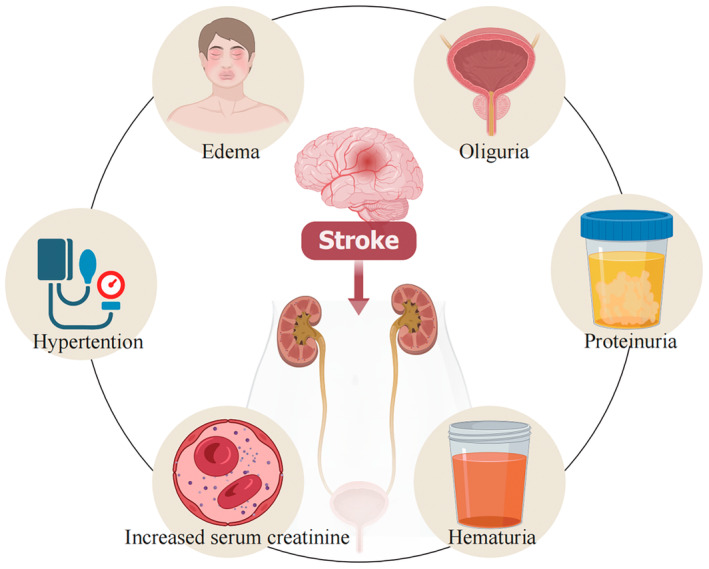
This chart lists relevant clinical manifestations and abnormal examination results that may be seen in patients with post‐stroke kidney injury.
2.2. Estimated Glomerular Filtration Rate (eGFR) as a Prognostic Indicator Post‐Stroke
Renal dysfunction, characterized by a persistent decline in renal function, can precipitate structural damage, severe clinical syndromes, accumulation of waste metabolites, and abnormal proteinuria discharge. In current clinical practice, the evaluation of renal function primarily relies on indicators like eGFR, albuminuria, and other related markers [11, 12, 13]. Progressive deterioration of these markers often heralds a poor prognosis in stroke patients [14, 15].
GFR is typically represented by surrogate markers such as serum creatinine (SCr) and urine output, serving as effective guides for clinical diagnosis. The eGFR is a critical diagnostic measure reflecting renal function. It quantifies the filtration capacity of the glomeruli, the kidneys' minute filtering units, assessing the volume of blood cleared of creatinine per minute. Frequently used to monitor and diagnose chronic kidney disease, eGFR considers variables like blood creatinine levels, age, gender, and body size [16]. Among these, creatinine‐based eGFR has amassed substantial clinical evidence, establishing itself as a reliable predictor of adverse outcomes post‐stroke, including recurrent stroke, cardiovascular events, stroke‐related disability, re‐hospitalization, and all‐cause mortality [17, 18, 19, 20].
A multicentric, randomized controlled trial studying acute intracerebral hemorrhage (ICH) patients from 21 countries demonstrated that lower eGFR values correlated with a higher risk of death or major disability at 90 days. However, there are also issues of generalizability arising from the clinical trial population [19]. According to the kidney disease: improving global outcomes (KDIGO) CKD Work Group, a decreased GFR is defined as GFR < 60 mL/min per 1.73 m2 (GFR categories G3a–G5) [6]. This renal dysfunction is associated with a higher incidence of new cardiovascular events, stroke‐related disability, recurrent stroke, and an increasing rate of hospital readmissions, thereby intensifying both short‐term and long‐term mortality [15, 21].
Interestingly, a number of studies suggest that, akin to low eGFR, a high eGFR could also indicate a poor prognosis in stroke patients, even after adjusting for confounding factors such as age, sex, and risk factors [7, 22, 23]. However, a prospective cohort study by Luo observed a differential association: higher eGFR was related to increased all‐cause mortality, while lower eGFR correlated with higher all‐cause mortality, stroke recurrence, and disability [23]. The strengths of this study include its large sample size of stroke patients from China. The limitation is the potential selection bias, as patients with missing baseline serum creatinine or lost to follow‐up within one year were not included in the study.
Notably, high eGFR might not reflect accurate renal function under conditions of substantial muscle mass loss, such as old age, amputations, paralysis, and chronic muscle disease [24]. Elevated eGFR can also manifest in early stages of diabetic nephropathy, and conditions like youth, type 2 diabetes mellitus (T2D), and heavy alcohol consumption are recognized predisposing factors for high eGFR. In contrast, low eGFR, but not high eGFR, is highly relevant to cerebral small‐vessel occlusive stroke in elderly patients [22]. Thus, patient comorbidities and other influencing factors could contribute to the observed eGFR changes post‐stroke. While renal insufficiency is frequently linked to poor stroke prognosis, current evidence is insufficient to support the utility of renal dysfunction in stroke risk stratification [25].
2.3. The Implications and Progression of AKI Following Stroke
As per the 2013 KDIGO Clinical Practice Guidelines, AKI is characterized as an abrupt deterioration in renal function, as indicated by an increased SCr (≥ 1.5 times the baseline value within 7 days or ≥ 26.5 μmol/L within 48 h), or diminished urine output (< 0.5 mL/kg/h for 6 h) [5, 26]. AKI is a clinical syndrome with a diverse etiology, resulting from direct renal damage and acute impairment of kidney function, and can be categorized into different stages based on SCr or urine output. Recent evidence posits cystatin C, interleukin‐18 (IL‐18), glutathione‐S‐transferase, insulin‐like growth factor‐binding protein 7 (IGFBP7), and tissue inhibitor of metalloproteinases‐2 (TIMP‐2) as potential biomarkers for early diagnosis of AKI via plasma or urine tests [13]. Current epidemiological evidence suggests that even transient, mild AKI can significantly impact the clinical outcomes of stroke, increasing disability and mortality rates [27, 28, 29].
Indicators related to renal function can directly signify the progression of AKI. A study in 2015 revealed a correlation between higher albumin–creatinine ratio (ACR) and lower eGFR with an elevated risk of AKI in patients with comorbidities like diabetes and hypertension [30]. In line with extant clinical studies, post‐stroke proteinuria has been associated with AKI, mirroring the relationship observed with eGFR. Retrospective analysis indicates that proteinuria exacerbates secondary AKI and increases one‐year mortality in stroke patients undergoing cerebral angiography. However, due to the low proportion of participants with proteinuria, this research was unable to analyze the dose‐effect relationship between proteinuria and outcomes [31]. After controlling for baseline comorbidities, it has been identified that baseline renal function is a potent independent predictor of short‐term survival post‐acute stroke and a risk factor for subsequent AKI [2]. Biomarkers indirectly linked with stroke have also been associated with AKI and poorer stroke prognosis [32]. Furthermore, a glycaemia‐based nomogram shows potential in predicting AKI in patients with ischemic stroke (IS) post‐endovascular therapy (post‐EVT) [33].
The medications or treatments administered to stroke patients post‐admission may only have minimal correlation with the incidence and severity of AKI [9, 19, 34]. Nevertheless, AKI might predominantly be triggered by the stroke itself, rather than the associated angiography and endovascular treatment post‐stroke. Studies suggest that neither thrombolytic therapy nor other related treatments for IS exacerbate AKI progression [34]. A clinical trial encompassing data from various specialized intensive care units (ICUs) from academic medical centers found a lower incidence of AKI, regardless of patients undergoing computed tomography angiography (CTA) or endovascular treatment [9]. Another study in 2016 illustrated that early intensive antihypertensive treatment for ICH positively impacted the incidence of 90‐day mortality or severe disability, irrespective of whether patients had a reduction in eGFR or not [19]. However, as renal insufficiency progresses, therapeutic efficacy diminishes, and it is significantly linked to adverse outcomes [9, 19, 35]. In a multivariate logistic regression analysis, researchers discovered that patients with albuminuria had a five‐fold higher risk of developing contrast‐induced AKI (CI‐AKI) compared to stroke patients without albuminuria [31]. In summary, the early detection and identification of AKI during the initial stages of stroke progression can serve as a crucial guide for improving the poor prognosis of stroke and mitigating the mortality associated with renal injury.
2.4. The Implications and Progression of CKD Following a Stroke
According to the KDIGO clinical practice guidelines, CKD is characterized by abnormalities of renal structure or function that persist for over 3 months, with a diagnostic threshold set at a GFR of less than 60 mL/min per 1.73 m2 and an ACR of 30 mg/g or higher [6]. CKD is a prevalent clinical condition with a higher incidence among high‐risk groups including the elderly, diabetics, and hypertensives. Notably, sensitive biomarkers of CKD do not entirely align with those of AKI. For instance, neutrophil gelatinase‐associated lipocalin (NGAL) and urinary kidney injury molecule 1 (KIM‐1), markers of renal tubular injury, are elevated in the context of CKD, but not AKI [11].
The brain and kidney share similar hemodynamic characteristics, including effective dynamic autoregulation capabilities. CKD and stroke can reciprocally influence each other's onset and progression by affecting hemodynamics and causing vascular structure disruptions. While many researchers are currently focusing on stroke incidence in CKD patients following vascular disease onset, the primary objective of this article is to explore the development of CKD in scenarios where stroke is the precipitating and leading cause, even long after the stroke occurrence.
Stroke patients often present with hypertension due to increased pressure, which further diminishes renal blood supply, exceeding the compensatory capacity of the kidneys. This can result in renal dysfunction manifestations such as reduced eGFR, proteinuria, and oliguria. Albuminuria, a marker of glomerular integrity, has been recognized in numerous studies as a potent clinical indicator for assessing the risk of adverse outcomes in patients with IS, particularly in predicting early neurological deterioration (END) and white‐matter lesions (WML) [31, 36, 37, 38].
The development of CKD can also forecast an unfavorable prognosis in IS patients, including recurrent stroke risks and heightened mortality [39, 40, 41]. In a multicenter cohort study involving patients with acute ischemic stroke (AIS) undergoing thrombolysis, CKD‐associated renal impairment was found to reduce thrombolysis efficacy without raising hemorrhagic complications, differing from AKI. This effect may primarily be attributed to the decrease in fibrinolysis and variance in infarct size [39]. Post‐stroke CKD signals an increase in localized brain damage, disturbances in the body's water and electrolyte balance, and in severe cases, end‐stage organ failure such as uremia.
3. Impact of Various Stroke Subtypes on Renal Function
Stroke is a complex disorder encompassing several subtypes, primarily including IS and intracerebral hemorrhage. In the trial of Org 10,172 in acute stroke treatment (TOAST) classification, IS is further categorized into five subtypes: large‐artery atherosclerosis (LAA), cardioembolism (CE), small artery occlusion (SAO), stroke of other determined etiology (SOE), and stroke of undetermined etiology (SUE) [42]. Patients may initially present with one stroke subtype and subsequently develop other subtypes. For instance, patients with IS may develop cerebral hemorrhage secondary to the natural progression of the disease or due to inappropriate treatment.
Each stroke subtype demonstrates unique clinical characteristics, with varied incidence and severity of accompanying renal injury. It has been reported that patients with ICH or aneurysmal subarachnoid hemorrhage (SAH) exhibit enhanced renal clearance. This elevated renal clearance can lead to subtherapeutic concentrations of renally eliminated drugs, resulting in decreased drug efficacy and potentially, medical complications [43].
Table 1 provides a summarization of renal dysfunction manifestations and related adverse prognosis associated with different stroke subtypes. However, it is crucial to note that current studies regarding each stroke subtype are not exhaustive. Thus, this table offers a partial snapshot of specific renal impairment manifestations and related prognostic outcomes for the various subtypes of stroke.
TABLE 1.
Summary of the different kidney impairments and relevant prognosis associated with various stroke subtypes.
| Stroke subtype | Kidney impairments | Relevant prognosis | References | Authors | Sample; mean age, year; follow‐up |
|---|---|---|---|---|---|
| Ischemic stroke |
|
|
[37] | Kanamaru et al. 2017 | The stroke center at Nippon Medical School Hospital {n = 294}; 73.6 ± 11.9 years; No mention |
|
[38] | Suda et al. 2017 | The stroke center at Nippon Medical School Hospital {n = 284}; 72 years; No mention | ||
|
|
[15] | Lee et al. 2013 | The VISP trial {n = 3673}; ≥ 44.6 years; 2 years | |
|
|
[7] | Luo et al. 2014 | ACROSS{n = 4836}; ≥ 41 years; 1 year | |
|
|
[14] | Husseinet al. 2017 | The GWTG‐Stroke program {n = 232,236}; ≥ 65 years; No mention | |
|
|
[21] | Husseinet al. 2018 | The GWTG‐Stroke program {n = 204,652}; ≥ 65 years; 1 year | |
|
[18] | Nugroho et al. 2018 | The Shiga Stroke Registry (SSR) {n = 2813}; ≥ 18 years; No mention | ||
| LAA |
|
|
[44] | Yeh et al. 2015 | The prospective Taiwan Stroke Registry {n = 8052}; ≥ 25.9 years; 6 months |
|
|
[45] | Liu et al. 2016 | The Chengdu Stroke Registry {n = 1645}; 62.9 years; No mention | |
| CE |
|
[46] |
Kudo et al. 2012 | Yamagata City Hospital, Saiseikan, Japan {n = 525}; 74.1 ± 12.1 years; 1 year | |
| SAO |
|
|
[36] | Umemura et al. 2014 | The stroke center in Chubu Rosai Hospital {n = 85}; 69.8 years; No mention |
|
|
[47] | Yang et al. 2017 | The Department of Neurology at Third Affiliated Hospital of Soochow University {n = 210}; 67.17 ± 12.35 years; No mention | |
| SOE | |||||
| SUE |
|
|
[48] | Chua et al. 2021 | Taiwan Stroke Registry {n = 3775}; 66.6 ± 14.5 years; 1 year |
| Hemorrhagic stroke | |||||
| ICH |
|
|
[35] | Fukuda‐Doi et al. 2021 | ATACH‐2 {n = 974}; ≥ 47 years; 90 days |
|
|
[40] | Ovbiagele et al. 2014 | The GWTG‐Stroke program {n = 5,113,059}; (Patient with CKD) 72.5 years; (Patient without CKD) 67.3 years; No mention | |
|
[49] | Snarska et al. 2016 | The Department of Neurology, Medical University Hospital Bialystok {n = 129}; 64.80 ± 15.76 years; No mention | ||
|
[46] | Kudo et al. 2012 | Yamagata City Hospital, Saiseikan, Japan {n = 525}; 74.1 ± 12.1 years; 1 year | ||
| SAH |
|
[43] | Morbitzer et al. 2019 | Neurosciences ICU in a tertiary care academic medical center {n = 50 + 30 = 80}; 57.2 ± 10.7 years; No mention | |
4. Is There a Link Between AKI and CKD Following Stroke?
AKI is a clinical condition marked by rapid deterioration of renal function, which is often accompanied by hypovolemia, sepsis, and nephrotoxic drugs. Various vasoconstrictive substances, including catecholamines, are released due to the activation of the sympathetic system following an acute stroke, leading to a systemic redistribution of blood flow. This prioritizes blood supply to the heart and brain, but significantly diminishes renal blood flow, thereby inducing a state of renal ischemia.
On the other hand, CKD, a chronic or progressive renal disorder, is characterized by inflammation‐induced myofibroblast transformation, proliferation, capillary disintegration, and thinning [4]. The stroke often elevates plasma levels of interleukin‐1 beta (IL‐1β). When a stroke occurs, transforming growth factor beta (TGF‐β) overexpression can activate downstream Smad signaling, contributing to renal fibrosis [50, 51, 52]. Additionally, stroke often results in upregulated expression of C‐reactive protein (CRP) and reactive oxygen species (ROS), which mediate renal tissue fibrosis through a TGF‐β1‐dependent mechanism, indirectly upregulating the downstream Smad3 signaling pathway [53].
Epidemiological studies highlight the complex interplay between AKI and CKD in stroke patients. Patients who suffer from AKI are at a higher risk of developing CKD, but differentiating the individual contributions of AKI and shared risk factors such as diabetes, hypertension, proteinuria, and coronary artery disease to CKD is a challenge [54]. AKI‐induced changes such as myofibroblast transformation, proliferation, fibrosis, and vascular disintegration are often implicated in the development of CKD [3, 43]. Figure 2 illustrates the potential pathological processes of AKI and CKD following a stroke. Moreover, AKI's indirect contribution to CKD progression, by influencing hemodynamic disturbances and renal load in stroke patients, is noteworthy.
FIGURE 2.
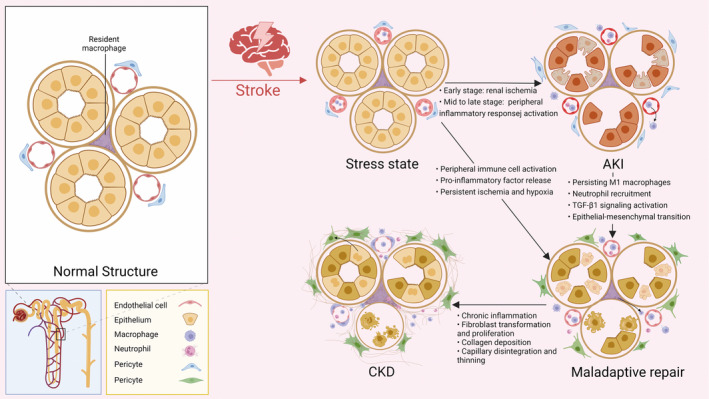
Schematic of the pathologic process of AKI and CKD after stroke. This diagram illustrates the changes that occur in renal tubular tissue following a stroke, leading to AKI and CKD. Initially, the kidneys experience ischemia and hypoxia due to high vascular resistance, which can result in acute tubular cell necrosis and endothelial cell damage, causing AKI. In the mid to late phase, brain macrophages are activated and released into the blood, further causing activation of peripheral immune cells (e.g., renal resident macrophages) as well as neutrophil recruitment. Large numbers of leukocytes infiltrate the kidney, M1 phenotype macrophages are continuously expressed, and maladaptive repair of the renal tubules occurs. Stimulated by pro‐inflammatory and other injury factors, pericytes separate from the endothelium, resulting in capillary thinning. Additionally, epithelial or endothelial cells may undergo epithelial‐mesenchymal transition (EMT), transforming into mesenchymal cells through TGF‐β1 signal pathway activation and overexpression of MiRs. Pericytes proliferate and differentiate to produce myofibroblasts, which promote collagen deposition in the kidney and cause renal fibrosis, ultimately leading to and the development of CKD.
In conclusion, stroke‐associated AKI doesn't directly lead to CKD. AKI is more as an acute pathophysiology of the kidneys which is related to the progression of peripheral neurological and circulatory conditions following stroke. Instead, it is related to maladaptive repair of abnormal or necrotic renal tubules due to repeated stimulation, caused by underlying diseases such as high vascular resistance, coronary artery disease, and diabetes mellitus [4].
5. Mechanisms Underlying Renal Impairment Post‐Stroke: Hemodynamic and Neuroendocrine Pathways
The kidneys and brain sustain homeostasis through a complex interplay via neuroendocrine pathways [55]. A stroke, however, may disturb this balance by overstimulating the CNS and sympathetic nervous system (SNS) and impairing the BBB. This allows inflammatory mediators and immune‐related substances from the brain to enter the bloodstream, inciting systemic circulatory inflammatory immune responses that indirectly affect the kidney. Stroke‐related renal failure is believed to arise from the combined effects of these neuroendocrine and inflammatory‐immune pathways, along with other pathophysiological mechanisms [56, 57, 58]. Figure 3 depicts the potential mechanisms and pathways underlying renal dysfunction following a stroke. This section explores these mechanisms in‐depth.
FIGURE 3.
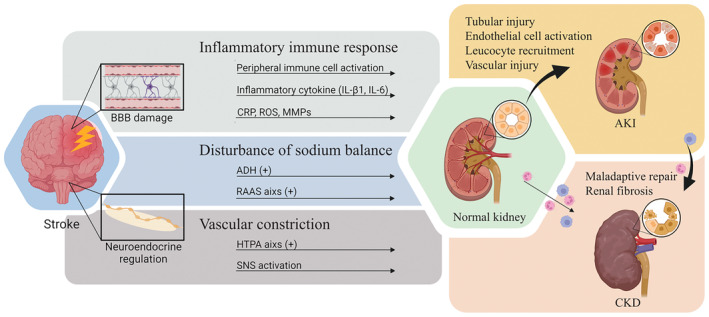
Schematic overview of the mechanisms and pathways involved in renal impairment after stroke. Renal impairment after stroke involves two major pathways: The inflammatory immune pathway and the neurohumoral pathway. In the inflammatory immune pathway, the BBB is compromised after stroke, leading to the release of pro‐inflammatory substances from the brain into the blood and the activation of peripheral inflammatory immune responses. The neurohumoral pathway involves the activation of the RAAS axis, the HTPA axis and the SNS. Multiple vasoconstrictor substances and related hormones act on receptor targets, resulting in hemodynamic disturbances and an acute ischemic state in the kidney. Multiple mechanisms promote the development of AKI or CKD in the kidney, while stroke as an acute injury promotes the progression of AKI to CKD.
5.1. Hemodynamic Mechanisms and Autoregulation
Despite serving different roles, the brain and kidneys share a key feature: robust pressure autoregulation (PA), which maintains stable blood flow despite variations in perfusion or blood pressure (BP) levels [59, 60]. Cerebral vasculature maintains constant flow even when arterial BP varies between 50 and 150 mmHg, while renal autoregulation maintains a steady renal blood flow with BP ranges of 80–180 mmHg [61]. Factors such as age, gender, comorbidities, anesthetic drugs, and vasoactive agents can influence these autoregulatory boundaries [62].
Post‐stroke cerebral autoregulation impairments can lead to hypoperfusion, altered microvascular pressure, BBB integrity loss, and microbleeds [63, 64]. A retrospective study of dynamic nomogram for predicting acute kidney injury in patients with acute ischemic stroke has indicated that compromised cerebral autoregulation significantly correlates with renal dysfunction during the acute phase (< 6 h) of AIS. But single‐center population selection bias and the constant revision of treatment protocols may lead to decreased predictive performance [64]. This disturbance in cerebral blood flow post‐stroke indirectly contributes to renal hemodynamic alterations.
5.2. Neuroendocrine Pathway Over‐Activation
5.2.1. The Hypothalamic–Pituitary–Adrenal (HPA) Axis
The HPA axis is a servocontrol system involving direct actions and feedback interactions [65]. Post‐stroke, increased blood cortisol levels are observed, correlating with injury severity and location [66, 67, 68, 69]. The primary cortisol hormones—glucocorticoids (GCs), mineralocorticoids, and sex hormones—can impact renal function following a stroke. Specifically, GCs can affect glomerular function, leading to a short‐term increase in GFR and proteinuria [70, 71]. High cortisol levels correspond with increased renal vascular resistance (RVR) and renal blood flow (RBF), affecting single nephron glomerular filtration rate (SNGFR) and consequently increasing systemic GFR [72].
5.2.2. The Renin‐Angiotensin‐Aldosterone System (RAAS)
The RAAS rapidly activates post‐stroke, resulting in the upregulation of RAAS‐related substances, with angiotensin II playing a crucial role [73]. High levels of aldosterone have been associated with poor stroke outcomes [74]. Additionally, angiotensin II is known to induce pro‐inflammatory factor IL‐6 production, while aldosterone stimulates ROS production, increasing endothelial cell permeability and promoting renal fibrosis [75, 76].
5.3. The Activation of SNS
AIS triggers a stress response, leading to SNS activation and increased release of catecholamines such as epinephrine and norepinephrine [77]. This provokes renal vasculature constriction, leading to pre‐renal insufficiency [78, 79], and potentially causing acute kidney injury due to prolonged renal ischemia and hypoxia [4]. Hence, the SNS plays a significant role in the early phase of stroke, despite its detrimental impact on renal hemodynamics.
In summary, post‐stroke renal impairment arises from complex, interwoven neuroendocrine and hemodynamic pathways. A comprehensive understanding of these mechanisms will provide valuable insight into effective therapeutic strategies to minimize renal damage following stroke.
6. Mechanisms of Renal Impairment Post‐Stroke: Inflammatory Mediators and Immune Response
The breakdown of the BBB, neuroendocrine activation, and the systemic dispersion of brain‐derived inflammatory‐immune substances can instigate a peripheral sterile inflammatory response [80, 81]. Stroke‐induced sterile inflammation in the brain triggers a multifaceted immune response, involving an array of immune cells, including microglia/macrophages, neutrophils, astrocytes, and T cells [82]. These cells orchestrate a large‐scale release of inflammatory mediators such as interleukins (ILs), tumor necrosis factors (TNFs), and chemokines [57, 83]. These processes lead to the widespread distribution of inflammatory factors and immune cells, with the kidneys being particularly vulnerable to their effects. Prolonged exposure to inflammatory stimuli can provoke a series of renal structural changes, including myofibroblast accumulation and collagen deposition, culminating in renal fibrosis [84, 85].
Inflammatory pathways stimulate renal fibrosis after stroke. Therefore, inhibiting inflammation becomes an effective therapeutic strategy to alleviate renal disease after stroke. Such a strategy underscores the necessity of understanding the cascading effects of stroke‐induced inflammation on kidney health and function.
6.1. BBB Impairment in Post‐Stroke Renal Injury
The BBB is a critical biochemical barrier that sustains CNS homeostasis. It controls the passage of essential nutrients and metabolic waste products via the expression of a variety of ion transport proteins and channels [80, 86]. The BBB is composed of tight junctions between brain microvascular endothelial cells that regulate paracellular diffusion between adjacent endothelial cells. The transmembrane tight junction proteins (TJs) encompass claudins, occludin, tricellulin, junctional adhesion molecules (JAMs), and accessory proteins such as zonula occludens (ZO) proteins [87].
BBB damage is a cardinal pathological characteristic of stroke that typically commences early (usually within 6 h post‐stroke) and persists [88]. The increased permeability of the BBB during a stroke permits easier entry of solutes into the circulation, contributing to the development of vasogenic edema and hemorrhagic transformation (HT) [89, 90]. Initial BBB damage in AIS primarily stems from hypoxia and nutrient deprivation brought about by persistent post‐stroke hypoperfusion. This stressor stimulates an increase in the expression of matrix metalloproteinases (MMPs) and ROS in endothelial cells and infiltrating neutrophils, leading to the aberrant expression of linker proteins. Subsequent neuroinflammation contributes to irreversible BBB damage [80, 81].
BBB damage is a necessary precursor to secondary renal damage following stroke. The early translocation of pro‐inflammatory factors, immune‐related substances, and upstream regulatory signals of pathways from the brain injury site into the circulation via the BBB may be a critical step in the evolution of AKI. Further, functional BBB deficiency and the upregulated expression of hormones, such as antidiuretic hormone (ADH) and aldosterone, give rise to systemic circulating ion disturbances that exacerbate renal insufficiency. A deeper understanding of BBB damage mechanisms and the potential for transporting various types of regulatory factors into the circulation to reach the kidney will aid researchers in further exploring the signal pathways and mechanisms implicated in renal damage at different stages of stroke. Consequently, safeguarding the BBB and reducing its permeability during stroke onset might represent a potential therapeutic strategy to mitigate renal impairment post‐stroke.
6.2. Interplay of Immune Cells in Stroke‐Induced Brain and Kidney Injury
Stroke elicits a complex immune response that involves a broad array of immune cells. Among these, monocytes/macrophages, neutrophils, natural killer (NK) cells, and T cells are notable for their substantial impact on both stroke‐induced brain injury and subsequent peripheral organ damage, particularly in the kidneys.
6.2.1. Monocytes/Macrophages
The response to IS is rapidly initiated by the activation of resident immune cells in the CNS, the monocytes/macrophages [91]. Within hours of the onset of IS, numbers of microglia in the border zone of the ischemic area increases rapidly which are induced to release a plethora of cytotoxic components, contributing to an inflammatory environment that exacerbates neuronal damage [92]. This inflammatory milieu is not confined to the CNS but permeates the systemic circulation, as these activated monocytes/macrophages translocate into peripheral tissues. In the kidney, their ongoing pro‐inflammatory secretions contribute to renal dysfunction, fostering the development of fibrosis, a chronic kidney condition characterized by an excess accumulation of fibrous connective tissue. Therefore, regulation of monocyte/macrophage activation and response post‐stroke can potentially offer a therapeutic avenue to mitigate deleterious effects on both brain and kidney function.
6.2.2. Neutrophils
Neutrophils, the most abundant white blood cells, are another critical player in the immune response to stroke. Following a stroke, neutrophils are quickly mobilized to the brain, where they intensify the inflammatory response through the release of pro‐inflammatory cytokines, chemokines, and ROS [93]. Beyond their damaging role in the CNS, these activated neutrophils migrate into the systemic circulation, instigating widespread inflammation that impacts peripheral organs, including the kidneys. In the renal tissue, neutrophils potentiate injury by releasing harmful substances and amplifying the inflammatory response, underscoring the necessity of strategies to moderate neutrophil activation to alleviate stroke‐induced renal damage.
6.2.3. NK Cell
NK cells, the immune system's initial defenders against infections and diseases, are activated and recruited to the brain following stroke. Their contribution amplifies the inflammatory environment in the brain, leading to more extensive damage [94]. Systemically, the stroke‐induced activation of NK cells disrupts immune homeostasis, which potentially heightens the susceptibility of peripheral organs, such as the kidneys, to inflammatory insult.
6.2.4. T Cell
T cells play a nuanced role in stroke. They can perpetuate the post‐stroke inflammatory response, contributing to exacerbated brain damage. Nevertheless, certain subtypes, specifically regulatory T cells (Tregs), can restrain the immune response and limit tissue damage [95]. Systemically, these activated T cells traverse the body and localize in various organs, including the kidneys, where their impact diverges based on their subtype. Effector T cells can provoke inflammation and instigate AKI, while Tregs may mitigate renal injury through suppression of the inflammatory response.
In summary, understanding the complex interplay of these immune cells in stroke‐induced brain and subsequent renal damage is paramount. This knowledge could pave the way for the development of therapeutic interventions targeting these cells to alleviate both the primary brain injury and secondary renal damage ensuing stroke.
6.3. Systemic Inflammatory Response and Oxidative Stress Post‐Stroke: The Role of pro‐Inflammatory Factors
Stroke instigates a series of biochemical responses, prominently featuring the production and release of pro‐inflammatory factors and oxidative stress products from microglia, neutrophils, and endothelial cells in ischemic brain tissue. Subsequent to the disruption of the BBB, these cells expel cytokines (e.g., ILs, tumor necrosis factor alpha (TNF‐α), chemokines), and brain‐derived antigens (e.g., enolase, S100b, and glial fibrillary acidic protein (GFAP)) into the bloodstream [83, 96]. These bioactive substances can stimulate both distant organs and the peripheral immune system. Evidence of brain‐derived antigens and specific antibodies can be detected in the tonsils and lymph nodes of stroke patients [97, 98].
In the context of stroke, particularly as observed in animal models, systemic immune responses are noticeably altered. Post‐stroke, an observable increase in the presence of various peripheral immune cells, including lymphocytes, monocytes, and neutrophils, is seen in the circulatory system. This increase is often paired with anatomical changes in immune organs, such as the reduction of spleen volume. These peripheral immune cell responses underscore the systemic nature of immune activation following a stroke [90].
The activation of these immune cells results in the production of an array of pro‐inflammatory factors and oxidative stress products. The subsequent release of these substances into the bloodstream triggers a systemic inflammatory immune response, contributing to secondary injury. The implications of these immune responses post‐stroke suggest a significant interplay between the CNS and the peripheral immune system. Understanding this interaction is crucial in exploring the mechanisms behind the secondary effects of stroke on peripheral organs and potentially designing therapeutic interventions to mitigate these effects. Several substances integral to the inflammatory immune response post‐stroke are discussed in detail below.
6.3.1. ILs
ILs can be broadly categorized into anti‐inflammatory and pro‐inflammatory classes. Notably, IL‐6 demonstrates variable roles in different phases of stroke—acute, subacute, and long‐term [99]. Serum concentrations of IL‐6 are significantly elevated after stroke onset [100]. IL‐6 exhibits pleiotropic effects, playing a pro‐inflammatory role in the development of AKI in the short‐term, and contributing to renal fibrosis in the long term by promoting the upregulation of multiple fibrotic genes in the kidney, including α2‐procollagen, TGF‐β, and plasminogen activator inhibitor‐1 [101, 102, 103].
6.3.2. CRP
CRP, an acute‐phase protein and immune mediator activated by complement, can permeate the BBB [104, 105]. Observations by Pecoits‐Filho et al. and Ladenvall et al. show elevated plasma CRP levels early in IS, and this condition also occurs after CKD and renal failure [52, 106].
CRP mediates the renal inflammatory response via a CD32‐NF‐κB‐dependent mechanism. Inflammatory signaling‐stimulated CRP notably upregulates pro‐inflammatory cytokines/chemokines (such as TNF‐α, IL‐1β), drives macrophage polarization toward the M1 phenotype, inhibits conversion to the M2 phenotype, and increases renal infiltration of F4/80+ macrophages through an NF‐κB‐dependent mechanism, triggering severe renal inflammation [107, 108, 109, 110]. Therefore, understanding these pro‐inflammatory factors and oxidative stress products is crucial in developing therapeutic strategies for stroke and its associated renal complications.
6.3.3. ROS
ROS broadly refer to oxygen‐derived free radicals and non‐free radicals possessing potent chemical reactivity [111]. In vitro, ROS instigate caspase‐3‐mediated damage to TJs, leading to increased permeability in rat brain microvascular endothelial cells (RBMEC) [112]. This phenomenon is associated with alterations in both the BBB and the renal filtration barrier.
As a continuation from the previous discussion on the role of pro‐inflammatory factors and oxidative stress products in stroke‐induced renal impairment, another critical aspect to consider is the contribution of EVs and MiRs in this pathological process. Their involvement is particularly noteworthy as they represent complex intercellular communication systems that influence disease progression and, hence, potential therapeutic targets. The specifics will be explained in the next section.
7. EVs and MiRs: Mediators of Post‐Stroke Renal Fibrosis and CKD
EVs and MiRs are hypothesized as crucial effectors in the progression of renal fibrosis and CKD following stroke [55]. EVs, lipid bilayer membrane structures, function as cellular messengers through the transport of various biomolecules like lipids, proteins, and nucleic acids. These biomolecules modulate the interaction between parental and recipient cells, subsequently influencing the recipient cell function [113]. In the context of fibrotic diseases, EVs serve as critical conduits of protofibrillar signaling, mediating the continuous deposition of diverse insoluble interstitial collagens [114].
MiRs are small, non‐coding RNA molecules with potent regulatory effects. Abnormal secretion or repression of numerous miRs has been linked to the progression of renal fibrosis. Stroke can induce alterations in miRs expression, with aberrant up inflammatory‐immune and downregulation of several miRs observed in the sera of young stroke patients [115].
MiR‐21, for example, facilitates renal fibrosis by enhancing TGF‐β signaling [116]. Additionally, in renal tubular epithelial cells from patients with renal fibrosis, an increased expression of miR‐34a is observed. MiR‐34a binds to and downregulates the expression of Klotho, an endogenous inhibitor of renal fibrosis. This overexpression of miR‐34a, induced by TGF‐β1, promotes the EMT in human kidney 2 (HK‐2) cells [117].
Several models of CKD, including those induced by obstruction, ischemia/reperfusion, mitochondrial miRs, and albumin overload, have demonstrated an upregulated expression of miR‐214. In vitro, miR‐214 disrupts mitochondrial oxidative phosphorylation in CKD by binding to specific targets, namely the mitochondrial genes mitochondrially encoded NADH dehydrogenase 6 (mt‐Nd6) and mt‐Nd4l [118]. Furthermore, miR‐184 and microRNA‐122‐5p have also been implicated in renal fibrosis [119, 120].
In conclusion, the regulation of EVs and aberrantly up‐ or down‐regulated miRs represent promising therapeutic targets for mitigating the progression of CKD following stroke.
8. Current Limitations and Future Research Directions
Our understanding of the interplay between stroke and renal impairment has substantially advanced over recent years, but several challenges and research opportunities remain, mainly centered around deciphering complex mechanisms, improving diagnostic tools, and developing effective therapies.
Firstly, we emphasize the potential benefit of early detection or prevention of brain‐kidney axis‐related complications, which may improve patient prognosis. We suggest that clinicians may include monitoring of kidney‐related markers such as blood creatinine and proteinuria as one of the important things in stroke care. The quest for improved diagnostic and prognostic tools also persists. Future research can focus on the identification of novel biomarkers that provide enhanced accuracy and reliability, potentially using high‐throughput omics technologies to discover novel disease markers.
Simultaneously, targeting the specific mechanisms of the neuroendocrine pathway and inflammatory response, we can investigate the relevance this may include interventions such as neuroprotective agents that simultaneously benefit renal function, or attenuating the systemic inflammatory response to inhibit the interaction between the two systems.
As well, while current studies have shed light on various immune cells, cytokines, and inflammatory mediators in driving stroke‐induced renal damage, gaps in our understanding persist. Specifically, more detailed explorations are needed on the exact mechanisms of fibrosis and extracellular matrix accumulation. Future research should leverage advanced techniques such as single‐cell RNA sequencing and proteomics to elucidate the role of different cell types and biomolecules involved in renal impairment post‐stroke.
Next, the intricate role of the autonomic nervous system, specifically the sympathetic and parasympathetic branches, and endocrine pathways in mediating the stroke's effects on kidney function, present another complex area of study. Although previous research has addressed these pathways, the dynamic and multifaceted nature of these interactions requires further examination, ideally through integrative methodologies, encompassing multi‐omics analyses, and state‐of‐the‐art imaging techniques.
The challenge of translating findings from animal models to human patients is another area where research can focus. The complexity of human disease demands a higher emphasis on translational studies, bridging the gap between animal models and clinical applications. This could involve the use of human‐derived organoid models, which can better replicate human physiological conditions.
9. Summary and Conclusion
In summary, the intricate relationship between stroke and renal impairment is an area of biomedical research that has gained considerable attention due to its profound implications for patient outcomes and management. The post‐stroke pathophysiological cascade involving immune cell activation, pro‐inflammatory factor release, oxidative stress, and the involvement of signaling molecules and EVs all contribute to renal impairment. This complex interplay underscores the multifaceted nature of stroke‐induced renal damage.
This review summarizes the existing clinical studies on post‐stroke kidney injury and integrates and screens the different literature for the same indicators and trends presented. Therefore, the review is mainly a selection of representative study results for presentation, which may be subject to potential biases such as selection bias and publication bias. Some of the literature with different trends from the mainstream findings were interpreted and analyzed in more detail, but some omissions cannot be ruled out.
Current research has significantly advanced our understanding of these interactions, identifying key mechanisms and highlighting the importance of the BBB and immune responses. However, numerous questions and challenges remain. These include the need for deeper mechanistic insights, advanced diagnostic and prognostic tools, translational studies to bridge the gap between animal models and human patients, and effective therapeutic interventions.
In conclusion, further research is necessary to unravel this complex interplay, leading to improved patient care strategies. By focusing on the highlighted challenges and utilizing innovative approaches, we can work toward a future with better management and therapeutic strategies for stroke patients at risk of renal impairment.
Ethics Statement
The authors have nothing to report.
Consent
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding: Research reported in this publication was supported by grant 82201429 (to P.H.) and grant 82471294 (to H.‐F.Z.) from the National Natural Science Foundation of China and grant 2024A1515012256 (to H.‐F.Z.) from the Natural Science Foundation of Guangdong Province, China and Research Support Plan for the Youth in the Department of Anesthesiology of Zhujiang Hospital.
The first two authors contributed equally to this work and should be considered co‐first authors.
Contributor Information
Hong‐Fei Zhang, Email: zhanghongfei@smu.edu.cn.
Pu Hong, Email: hppppp2018@smu.edu.cn.
Data Availability Statement
Authors confirm that all relevant data are included in the article.
References
- 1. Murray C. J. L., Aravkin A. Y., Zheng P., et al., “Global Burden of 87 Risk Factors in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019,” Lancet 396, no. 10258 (2020): 1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Covic A., Schiller A., Mardare N. G., et al., “The Impact of Acute Kidney Injury on Short‐Term Survival in an Eastern European Population With Stroke,” Nephrology, Dialysis, Transplantation 23, no. 7 (2008): 2228–2234. [DOI] [PubMed] [Google Scholar]
- 3. Ferenbach D. A. and Bonventre J. V., “Mechanisms of Maladaptive Repair After AKI Leading to Accelerated Kidney Ageing and CKD,” Nature Reviews Nephrology 11, no. 5 (2015): 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Venkatachalam M. A., Weinberg J. M., Kriz W., and Bidani A. K., “Failed Tubule Recovery, AKI‐CKD Transition, and Kidney Disease Progression,” Journal of the American Society of Nephrology 26, no. 8 (2015): 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kellum J. A. and Lameire N., “Diagnosis, Evaluation, and Management of Acute Kidney Injury: A KDIGO Summary (Part 1),” Critical Care 17, no. 1 (2013): 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stevens P. E. and Levin A., “Evaluation and Management of Chronic Kidney Disease: Synopsis of the Kidney Disease: Improving Global Outcomes 2012 Clinical Practice Guideline,” Annals of Internal Medicine 158, no. 11 (2013): 825–830. [DOI] [PubMed] [Google Scholar]
- 7. Luo Y., Wang X. W., Wang Y. L., et al., “Association of Glomerular Filtration Rate With Outcomes of Acute Stroke in Type 2 Diabetic Patients: Results From the China National Stroke Registry,” Diabetes Care 37, no. 1 (2014): 173–179. [DOI] [PubMed] [Google Scholar]
- 8. Arnold J., Ng K. P., Sims D., Gill P., Cockwell P., and Ferro C., “Incidence and Impact on Outcomes of Acute Kidney Injury After a Stroke: A Systematic Review and Meta‐Analysis,” BMC Nephrology 19 (2018): 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qureshi A. I., Aslam H., Zafar W., et al., “Acute Kidney Injury in Acute Ischemic Stroke Patients in Clinical Trials,” Critical Care Medicine 48, no. 9 (2020): 1334–1339. [DOI] [PubMed] [Google Scholar]
- 10. Zorrilla‐Vaca A., Ziai W., Connolly E. S., Geocadin R., Thompson R., and Rivera‐Lara L., “Acute Kidney Injury Following Acute Ischemic Stroke and Intracerebral Hemorrhage: A Meta‐Analysis of Prevalence Rate and Mortality Risk,” Cerebrovascular Diseases 45, no. 1–2 (2018): 1–9. [DOI] [PubMed] [Google Scholar]
- 11. Koyner J. L., Coca S. G., Thiessen‐Philbrook H., et al., “Urine Biomarkers and Perioperative Acute Kidney Injury: The Impact of Preoperative Estimated GFR,” American Journal of Kidney Diseases 66, no. 6 (2015): 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Veldhuisen D. J., Ruilope L. M., Maisel A. S., and Damman K., “Biomarkers of Renal Injury and Function: Diagnostic, Prognostic and Therapeutic Implications in Heart Failure,” European Heart Journal 37, no. 33 (2016): 2577–2585. [DOI] [PubMed] [Google Scholar]
- 13. Wasung M. E., Chawla L. S., and Madero M., “Biomarkers of Renal Function, Which and When?,” Clinica Chimica Acta 438 (2015): 350–357. [DOI] [PubMed] [Google Scholar]
- 14. El Husseini N., Fonarow G. C., Smith E. E., et al., “Renal Dysfunction Is Associated With Poststroke Discharge Disposition and in‐Hospital Mortality: Findings From Get With the Guidelines‐Stroke,” Stroke 48, no. 2 (2017): 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee M., Markovic D., and Ovbiagele B., “Impact and Interaction of Low Estimated GFR and B Vitamin Therapy on Prognosis Among Ischemic Stroke Patients: The Vitamin Intervention for Stroke Prevention (VISP) Trial,” American Journal of Kidney Diseases 62, no. 1 (2013): 52–57. [DOI] [PubMed] [Google Scholar]
- 16. Inker L. A. and Titan S., “Measurement and Estimation of GFR for Use in Clinical Practice: Core Curriculum 2021,” American Journal of Kidney Diseases 78, no. 5 (2021): 736–749. [DOI] [PubMed] [Google Scholar]
- 17. Miwa K., Koga M., Nakai M., et al., “Etiology and Outcome of Ischemic Stroke in Patients With Renal Impairment Including Chronic Kidney Disease Japan Stroke Data Bank,” Neurology 98, no. 17 (2022): E1738–E1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nugroho A. W., Arima H., Miyazawa I., et al., “The Association Between Glomerular Filtration Rate Estimated on Admission and Acute Stroke Outcome: The Shiga Stroke Registry,” Journal of Atherosclerosis and Thrombosis 25, no. 7 (2018): 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng D. N., Sato S., Arima H., et al., “Estimated GFR and the Effect of Intensive Blood Pressure Lowering After Acute Intracerebral Hemorrhage,” American Journal of Kidney Diseases 68, no. 1 (2016): 94–102. [DOI] [PubMed] [Google Scholar]
- 20. He P. P., Li H., Zhang Z. X., et al., “Change in the Estimated Glomerular Filtration Rate Over Time and Risk of First Stroke in Hypertensive Patients,” Journal of Epidemiology 33, no. 3 (2023): 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. El Husseini N., Fonarow G. C., Smith E. E., et al., “Association of Kidney Function With 30‐Day and 1‐Year Poststroke Mortality and Hospital Readmission Get With the Guidelines‐Stroke,” Stroke 49, no. 12 (2018): 2896–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Putaala J., Haapaniemi E., Gordin D., et al., “Factors Associated With Impaired Kidney Function and Its Impact on Long‐Term Outcome in Young Ischemic Stroke,” Stroke 42, no. 9 (2011): 2459–2464. [DOI] [PubMed] [Google Scholar]
- 23. Luo Y., Wang X. W., Matsushita K., et al., “Associations Between Estimated Glomerular Filtration Rate and Stroke Outcomes in Diabetic Versus Nondiabetic Patients,” Stroke 45, no. 10 (2014): 2887–2893. [DOI] [PubMed] [Google Scholar]
- 24. Delanaye P., Schaeffner E., Ebert N., et al., “Normal Reference Values for Glomerular Filtration Rate: What do We Really Know?,” Nephrology, Dialysis, Transplantation 27, no. 7 (2012): 2664–2672. [DOI] [PubMed] [Google Scholar]
- 25. Ntaios G., Lip G. Y. H., Lambrou D., et al., “Renal Function and Risk Stratification of Patients With Embolic Stroke of Undetermined Source,” Stroke 49, no. 12 (2018): 2904–2909. [DOI] [PubMed] [Google Scholar]
- 26. Mok V., Nixon J., Hu J., and Ma D., “The Impact of Perioperative Acute Kidney Injury/Failure on Short and Long Surgical Outcomes,” Anesthesiology and Perioperative Science 1, no. 2 (2023): 9. [Google Scholar]
- 27. Hoste E. A. J., Clermont G., Kersten A., et al., “RIFLE Criteria for Acute Kidney Injury Are Associated With Hospital Mortality in Critically Ill Patients: A Cohort Analysis,” Critical Care 10, no. 3 (2006): R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pande S. D., Roy D., Khine A. A., et al., “Acute Kidney Injury Without Need for Dialysis, Incidence, Its Impact on Long‐Term Stroke Survival and Progression to Chronic Kidney Disease,” BMJ Open 12, no. 5 (2022): e050743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y. L., Wang D. X., Li H., et al., “Association of Acute Kidney Disease With the Prognosis of Ischemic Stroke in the Third China National Stroke Registry,” BMC Nephrology 23, no. 1 (2022): 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grams M. E., Sang Y. Y., Ballew S. H., et al., “A Meta‐Analysis of the Association of Estimated GFR, Albuminuria, Age, Race, and Sex With Acute Kidney Injury,” American Journal of Kidney Diseases 66, no. 4 (2015): 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tao Y. M., Dong W., Li Z. L., et al., “Proteinuria as an Independent Risk Factor for Contrast‐Induced Acute Kidney Injury and Mortality in Patients With Stroke Undergoing Cerebral Angiography,” Journal of Neurointerventional Surgery 9, no. 5 (2017): 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang F., Shi Z., Liu X. Y., et al., “The Role of the Monocyte‐To‐Lymphocyte Ratio in Acute Ischemic Stroke Patients With Acute Kidney Injury,” Mediators of Inflammation 2022 (2022): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu C. F., Li X. H., Xu Z. H., et al., “Construction of a Glycaemia‐Based Signature for Predicting Acute Kidney Injury in Ischaemic Stroke Patients After Endovascular Treatment,” Journal of Clinical Medicine 11, no. 13 (2022): 3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gadalean F., Simu M., Parv F., et al., “The Impact of Acute Kidney Injury on in‐Hospital Mortality in Acute Ischemic Stroke Patients Undergoing Intravenous Thrombolysis,” PLoS One 12, no. 10 (2017): e0185589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fukuda‐Doi M., Yamamoto H., Koga M., et al., “Impact of Renal Impairment on Intensive Blood‐Pressure‐Lowering Therapy and Outcomes in Intracerebral Hemorrhage Results From ATACH‐2,” Neurology 97, no. 9 (2021): E913–E921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Umemura T., Senda J., Fukami Y., et al., “Impact of Albuminuria on Early Neurological Deterioration and Lesion Volume Expansion in Lenticulostriate Small Infarcts,” Stroke 45, no. 2 (2014): 587–590. [DOI] [PubMed] [Google Scholar]
- 37. Kanamaru T., Suda S., Muraga K., et al., “Albuminuria Predicts Early Neurological Deterioration in Patients With Acute Ischemic Stroke,” Journal of the Neurological Sciences 372 (2017): 417–420. [DOI] [PubMed] [Google Scholar]
- 38. Suda S., Kanamaru T., Okubo S., et al., “Urinary Albumin‐To‐Creatinine Ratio Is Associated With White Matter Lesions Severity in First‐Ever Stroke Patients,” Journal of the Neurological Sciences 373 (2017): 258–262. [DOI] [PubMed] [Google Scholar]
- 39. Power A., Epstein D., Cohen D., et al., “Renal Impairment Reduces the Efficacy of Thrombolytic Therapy in Acute Ischemic Stroke,” Cerebrovascular Diseases 35, no. 1 (2013): 45–52. [DOI] [PubMed] [Google Scholar]
- 40. Ovbiagele B., Schwamm L. H., Smith E. E., et al., “Hospitalized Hemorrhagic Stroke Patients With Renal Insufficiency: Clinical Characteristics, Care Patterns, and Outcomes,” Journal of Stroke and Cerebrovascular Diseases 23, no. 9 (2014): 2265–2273. [DOI] [PubMed] [Google Scholar]
- 41. Castro P., Azevedo E., Rocha I., Sorond F., and Serrador J. M., “Chronic Kidney Disease and Poor Outcomes in Ischemic Stroke: Is Impaired Cerebral Autoregulation the Missing Link?,” BMC Neurology 18 (2018): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. H. P. Adams, Jr. , Bendixen B. H., Kappelle L. J., et al., “Classification of Subtype of Acute Ischemic Stroke. Definitions for Use in a Multicenter Clinical Trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment,” Stroke 24, no. 1 (1993): 35–41. [DOI] [PubMed] [Google Scholar]
- 43. Morbitzer K. A., Jordan J. D., Dehne K. A., Durr E. A., Olm‐Shipman C. M., and Rhoney D. H., “Enhanced Renal Clearance in Patients With Hemorrhagic Stroke,” Critical Care Medicine 47, no. 6 (2019): 800–808. [DOI] [PubMed] [Google Scholar]
- 44. Yeh S.‐J., Jeng J.‐S., Tang S.‐C., et al., “Low Estimated Glomerular Filtration Rate Is Associated With Poor Outcomes in Patients Who Suffered a Large Artery Atherosclerosis Stroke,” Atherosclerosis 239, no. 2 (2015): 328–334. [DOI] [PubMed] [Google Scholar]
- 45. Liu B., Wang D., Hao Z., et al., “Reduction in Estimated Glomerular Filtration Rate (eGFR) Results in an Increased Risk of Spontaneous Hemorrhagic Transformation in Patients With Large‐Artery Atherosclerosis Stroke,” Current Neurovascular Research 13, no. 1 (2016): 75–81. [DOI] [PubMed] [Google Scholar]
- 46. Kudo K., Konta T., Degawa N., et al., “Relationship Between Kidney Damage and Stroke Types in Japanese Patients,” Clinical and Experimental Nephrology 16, no. 4 (2012): 564–569. [DOI] [PubMed] [Google Scholar]
- 47. Yang S., Cai J., Lu R., Wu J., Zhang M., and Zhou X., “Association Between Serum Cystatin C Level and Total Magnetic Resonance Imaging Burden of Cerebral Small Vessel Disease in Patients With Acute Lacunar Stroke,” Journal of Stroke and Cerebrovascular Diseases 26, no. 1 (2017): 186–191. [DOI] [PubMed] [Google Scholar]
- 48. Chua S.‐K., Hsieh F.‐I., Hu C.‐J., et al., “Prognostic Impact of Renal Dysfunction on Embolic Stroke of Undetermined Source‐Role Beyond CHA(2)DS(2)‐VASc Score: Results From Taiwan Stroke Registry,” European Journal of Neurology 28, no. 4 (2021): 1253–1264. [DOI] [PubMed] [Google Scholar]
- 49. Snarska K., Kapica‐Topczewska K., Bachorzewska‐Gajewska H., and Malyszko J., “Renal Function Predicts Outcomes in Patients With Ischaemic Stroke and Haemorrhagic Stroke,” Kidney & Blood Pressure Research 41, no. 4 (2016): 424–433. [DOI] [PubMed] [Google Scholar]
- 50. Ka S. M., Yeh Y. C., Huang X. R., et al., “Kidney‐Targeting Smad7 Gene Transfer Inhibits Renal TGF‐Beta/MAD Homologue (SMAD) and Nuclear Factor Kappa B (NF‐Kappa B) Signalling Pathways, and Improves Diabetic Nephropathy in Mice,” Diabetologia 55, no. 2 (2012): 509–519. [DOI] [PubMed] [Google Scholar]
- 51. Wu W., Wang X., Yu X., and Lan H.‐Y., “Smad3 Signatures in Renal Inflammation and Fibrosis,” International Journal of Biological Sciences 18, no. 7 (2022): 2795–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pecoits‐Filho R., Heimburger O., Barany P., et al., “Associations Between Circulating Inflammatory Markers and Residual Renal Function in CRF Patients,” American Journal of Kidney Diseases: The Official Journal of the National Kidney Foundation 41, no. 6 (2003): 1212–1218. [DOI] [PubMed] [Google Scholar]
- 53. You Y.‐K., Wu W.‐F., Huang X.‐R., et al., “Deletion of Smad3 Protects Against C‐Reactive Protein‐Induced Renal Fibrosis and Inflammation in Obstructive Nephropathy,” International Journal of Biological Sciences 17, no. 14 (2021): 3911–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rifkin D. E., Coca S. G., and Kalantar‐Zade K., “Does AKI Truly Lead to CKD?,” Journal of the American Society of Nephrology 23, no. 6 (2012): 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao Q., Yan T., Chopp M., Venkat P., and Chen J., “Brain‐Kidney Interaction: Renal Dysfunction Following Ischemic Stroke,” Journal of Cerebral Blood Flow and Metabolism 40, no. 2 (2020): 246–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mabuchi T., Kitagawa K., Ohtsuki T., et al., “Contribution of Microglia/Macrophages to Expansion of Infarction and Response of Oligodendrocytes After Focal Cerebral Ischemia in Rats,” Stroke 31, no. 7 (2000): 1735–1743. [DOI] [PubMed] [Google Scholar]
- 57. Petrovic‐Djergovic D., Goonewardena S. N., and Pinsky D. J., “Inflammatory Disequilibrium in Stroke,” Circulation Research 119, no. 1 (2016): 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Olsson T., Marklund N., Gustafson Y., and Nasman B., “Abnormalities at Different Levels of the Hypothalamic‐Pituitary‐Adrenocortical Axis Early After Stroke,” Stroke 23, no. 11 (1992): 1573–1576. [DOI] [PubMed] [Google Scholar]
- 59. Johnson P. C., “Review of Previous Studies and Current Theories of Autoregulation,” Circulation Research 15, no. Suppl (1964): 2–9. [PubMed] [Google Scholar]
- 60. Johnson P. C., “Autoregulation of Blood Flow,” Circulation Research 59, no. 5 (1986): 483–495. [DOI] [PubMed] [Google Scholar]
- 61. Lassen N. A., “Cerebral Blood Flow and Oxygen Consumption in Man,” Physiological Reviews 39, no. 2 (1959): 183–238. [DOI] [PubMed] [Google Scholar]
- 62. Meng L., Wang Y., Zhang L., and McDonagh D. L., “Heterogeneity and Variability in Pressure Autoregulation of Organ Blood Flow: Lessons Learned Over 100+Years,” Critical Care Medicine 47, no. 3 (2019): 436–448. [DOI] [PubMed] [Google Scholar]
- 63. Claassen J. A. H. R., Thijssen D. H. J., Panerai R. B., and Faraci F. M., “Regulation of Cerebral Blood Flowin Humans: Physiology and Clinical Implications of Autoregulation,” Physiological Reviews 101, no. 4 (2021): 1487–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhu G. G., Fu Z. X., Jin T. A., et al., “Dynamic Nomogram for Predicting Acute Kidney Injury in Patients With Acute Ischemic Stroke: A Retrospective Study,” Frontiers in Neurology 13 (2022): 987684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keller‐Wood M., “Hypothalamic‐Pituitary‐Adrenal Axis‐Feedback Control,” Comprehensive Physiology 5, no. 3 (2015): 1161–1182. [DOI] [PubMed] [Google Scholar]
- 66. Feibel J. H., Hardy P. M., Campbell R. G., Goldstein M. N., and Joynt R. J., “Prognostic Value of the Stress Response Following Stroke,” Journal of the American Medical Association 238, no. 13 (1977): 1374–1376. [PubMed] [Google Scholar]
- 67. Olsson T., Astrom M., Eriksson S., and Forssell A., “Hypercortisolism Revealed by the Dexamethasone Suppression Test in Patients [Corrected] With Acute Ischemic Stroke,” Stroke 20, no. 12 (1989): 1685–1690. [DOI] [PubMed] [Google Scholar]
- 68. Olsson T., “Urinary Free Cortisol Excretion Shortly After Ischaemic Stroke,” Journal of Internal Medicine 228, no. 2 (1990): 177–181. [DOI] [PubMed] [Google Scholar]
- 69. Hunter R., Ivy J. R., and Bailey M. A., “Glucocorticoids and Renal Na+ Transport: Implications for Hypertension and Salt Sensitivity,” Journal of Physiology (London) 592, no. 8 (2014): 1731–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Davis J. O. and Howell D. S., “Comparative Effect of ACTH, Cortisone and DCA on Renal Function, Electrolyte Excretion and Water Exchange in Normal Dogs,” Endocrinology 52, no. 3 (1953): 245–255. [DOI] [PubMed] [Google Scholar]
- 71. Connell J. M., Whitworth J. A., Davies D. L., Lever A. F., Richards A. M., and Fraser R., “Effects of ACTH and Cortisol Administration on Blood Pressure, Electrolyte Metabolism, Atrial Natriuretic Peptide and Renal Function in Normal Man,” Journal of Hypertension 5, no. 4 (1987): 425–433. [PubMed] [Google Scholar]
- 72. Smets P., Meyer E., Maddens B., and Daminet S., “Cushing's Syndrome, Glucocorticoids and the Kidney,” General and Comparative Endocrinology 169, no. 1 (2010): 1–10. [DOI] [PubMed] [Google Scholar]
- 73. Back C., Thiesen K. L., Skovgaard K., et al., “RAAS and Stress Markers in Acute Ischemic Stroke: Preliminary Findings,” Acta Neurologica Scandinavica 131, no. 2 (2015): 132–139. [DOI] [PubMed] [Google Scholar]
- 74. Dinh Q. N., Arumugam T. V., Young M. J., Drummond G. R., Sobey C. G., and Chrissobolis S., “Aldosterone and the Mineralocorticoid Receptor in the Cerebral Circulation and Stroke,” Experimental & Translational Stroke Medicine 4, no. 1 (2012): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brown N. J., “Contribution of Aldosterone to Cardiovascular and Renal Inflammation and Fibrosis,” Nature Reviews Nephrology 9, no. 8 (2013): 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ruiz‐Ortega M. and Egido J., “Angiotensin II Modulates Cell Growth‐Related Events and Synthesis of Matrix Proteins in Renal Interstitial Fibroblasts,” Kidney International 52, no. 6 (1997): 1497–1510. [DOI] [PubMed] [Google Scholar]
- 77. Meyer J. S., Stoica E., Pascu I., Shimazu K., and Hartmann A., “Catecholamine Concentrations in CSF and Plasma of Patients With Cerebral Infarction and Haemorrhage,” Brain: A Journal of Neurology 96, no. 2 (1973): 277–288. [DOI] [PubMed] [Google Scholar]
- 78. Fujii T., Kurata H., Takaoka M., et al., “The Role of Renal Sympathetic Nervous System in the Pathogenesis of Ischemic Acute Renal Failure,” European Journal of Pharmacology 481, no. 2–3 (2003): 241–248. [DOI] [PubMed] [Google Scholar]
- 79. Lopez‐Novoa J. M., Rodriguez‐Pena A. B., Ortiz A., Martinez‐Salgado C., and Lopez Hernandez F. J., “Etiopathology of Chronic Tubular, Glomerular and Renovascular Nephropathies: Clinical Implications,” Journal of Translational Medicine 9 (2011): 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jiang X., Andjelkovic A. V., Zhu L., et al., “Blood‐Brain Barrier Dysfunction and Recovery After Ischemic Stroke,” Progress in Neurobiology 163 (2018): 144–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nian K., Harding I. C., Herman I. M., and Ebong E. E., “Blood‐Brain Barrier Damage in Ischemic Stroke and Its Regulation by Endothelial Mechanotransduction,” Frontiers in Physiology 11 (2020): 605398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xiong X. X., Gu L. J., Zhang H. F., Xu B. H., Zhu S. M., and Zhao H., “The Protective Effects of T Cell Deficiency Against Brain Injury Are Ischemic Model‐Dependent in Rats,” Neurochemistry International 62, no. 3 (2013): 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bourhy L., Mazeraud A., Bozza F. A., Turc G., Lledo P.‐M., and Sharshar T., “Neuro‐Inflammatory Response and Brain‐Peripheral Crosstalk in Sepsis and Stroke,” Frontiers in Immunology 13 (2022): 834649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. LeBleu V. S., Taduri G., O'Connell J., et al., “Origin and Function of Myofibroblasts in Kidney Fibrosis,” Nature Medicine 19, no. 8 (2013): 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li L., Fu H., and Liu Y., “The Fibrogenic Niche in Kidney Fibrosis: Components and Mechanisms,” Nature Reviews Nephrology 18, no. 9 (2022): 545–557. [DOI] [PubMed] [Google Scholar]
- 86. Neuwelt E. A., Bauer B., Fahlke C., et al., “Engaging Neuroscience to Advance Translational Research in Brain Barrier Biology,” Nature Reviews Neuroscience 12, no. 3 (2011): 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chow B. W. and Gu C., “The Molecular Constituents of the Blood‐Brain Barrier,” Trends in Neurosciences 38, no. 10 (2015): 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. DiNapoli V. A., Huber J. D., Houser K., Li X., and Rosen C. L., “Early Disruptions of the Blood‐Brain Barrier May Contribute to Exacerbated Neuronal Damage and Prolonged Functional Recovery Following Stroke in Aged Rats,” Neurobiology of Aging 29, no. 5 (2008): 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Turner R. J. and Sharp F. R., “Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke,” Frontiers in Cellular Neuroscience 10 (2016): 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Seifert H. A., Hall A. A., Chapman C. B., Collier L. A., Willing A. E., and Pennypacker K. R., “A Transient Decrease in Spleen Size Following Stroke Corresponds to Splenocyte Release Into Systemic Circulation,” Journal of Neuroimmune Pharmacology 7, no. 4 (2012): 1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Guelke E., Gelderblom M., and Magnus T., “Danger Signals in Stroke and Their Role on Microglia Activation After Ischemia,” Therapeutic Advances in Neurological Disorders 11 (2018): 1756286418774254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nowicka D., Rogozinska K., Aleksy M., Witte O. W., and Skangiel‐Kramska J., “Spatiotemporal Dynamics of Astroglial and Microglial Responses After Photothrombotic Stroke in the Rat Brain,” Acta Neurobiologiae Experimentalis 68, no. 2 (2008): 155–168. [DOI] [PubMed] [Google Scholar]
- 93. Berry M. and Clatworthy M. R., “Immunotherapy for Acute Kidney Injury,” Immunotherapy 4, no. 3 (2012): 323–334. [DOI] [PubMed] [Google Scholar]
- 94. Gan Y., Liu Q., Wu W., et al., “Ischemic Neurons Recruit Natural Killer Cells That Accelerate Brain Infarction,” Proceedings of the National Academy of Sciences of the United States of America 111, no. 7 (2014): 2704–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yilmaz G., Arumugam T. V., Stokes K. Y., and Granger D. N., “Role of T Lymphocytes and Interferon‐γ in Ischemic Stroke,” Circulation 113, no. 17 (2006): 2105–2112. [DOI] [PubMed] [Google Scholar]
- 96. Whiteley W., Jackson C., Lewis S., et al., “Inflammatory Markers and Poor Outcome After Stroke: A Prospective Cohort Study and Systematic Review of Interleukin‐6,” PLoS Medicine 6, no. 9 (2009): e1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Planas A. M., Gomez‐Choco M., Urra X., Gorina R., Caballero M., and Chamorro A., “Brain‐Derived Antigens in Lymphoid Tissue of Patients with Acute Stroke,” Journal of Immunology 188, no. 5 (2012): 2156–2163. [DOI] [PubMed] [Google Scholar]
- 98. Choi Y. H., Laaker C., Hsu M., Cismaru P., Sandor M., and Fabry Z., “Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke,” International Journal of Molecular Sciences 22, no. 17 (2021): 9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wu F., Liu Z., Zhou L., et al., “Systemic Immune Responses After Ischemic Stroke: From the Center to the Periphery,” Frontiers in Immunology 13 (2022): 911661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rose‐John S., “Interleukin‐6 Family Cytokines,” Cold Spring Harbor Perspectives in Biology 10, no. 2 (2018): a028415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Grassl C., Luckow B., Schlondorff D., and Dendorfer U., “Transcriptional Regulation of the Interleukin‐6 Gene in Mesangial Cells,” Journal of the American Society of Nephrology: JASN 10, no. 7 (1999): 1466–1477. [DOI] [PubMed] [Google Scholar]
- 102. Zhang W., Wang W., Yu H., et al., “Interleukin 6 Underlies Angiotensin II‐Induced Hypertension and Chronic Renal Damage,” Hypertension 59, no. 1 (2012): 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nechemia‐Arbely Y., Barkan D., Pizov G., et al., “IL‐6/IL‐6R Axis Plays a Critical Role in Acute Kidney Injury,” Journal of the American Society of Nephrology 19, no. 6 (2008): 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Puthucheary Z., Tadie J.‐M., and Patel J. J., “C‐Reactive Protein in Immunometabolism: Spared From ‘Paying the Piper’,” Intensive Care Medicine 48, no. 1 (2022): 103–105. [DOI] [PubMed] [Google Scholar]
- 105. Hsuchou H., Kastin A. J., Mishra P. K., and Pan W., “C‐Reactive Protein Increases BBB Permeability: Implications for Obesity and Neuroinflammation,” Cellular Physiology and Biochemistry 30, no. 5 (2012): 1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ladenvall C., Jood K., Blomstrand C., Nilsson S., Jern C., and Ladenvall P., “Serum C‐Reactive Protein Concentration and Genotype in Relation to Ischemic Stroke Subtype,” Stroke 37, no. 8 (2006): 2018–2023. [DOI] [PubMed] [Google Scholar]
- 107. Devaraj S. and Jialal I., “C‐Reactive Protein Polarizes Human Macrophages to an M1 Phenotype and Inhibits Transformation to the M2 Phenotype,” Arteriosclerosis, Thrombosis, and Vascular Biology 31, no. 6 (2011): 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Li Z., Chung A. C. K., Zhou L., et al., “C‐Reactive Protein Promotes Acute Renal Inflammation and Fibrosis in Unilateral Ureteral Obstructive Nephropathy in Mice,” Laboratory Investigation 91, no. 6 (2011): 837–851. [DOI] [PubMed] [Google Scholar]
- 109. Liu F., Chen H. Y., Huang X. R., et al., “C‐Reactive Protein Promotes Diabetic Kidney Disease in a Mouse Model of Type 1 Diabetes,” Diabetologia 54, no. 10 (2011): 2713–2723. [DOI] [PubMed] [Google Scholar]
- 110. You Y.‐K., Huang X.‐R., Chen H.‐Y., Lyu X.‐F., Liu H.‐F., and Lan H. Y., “C‐Reactive Protein Promotes Diabetic Kidney Disease in Db/Db Mice via the CD32b‐Smad3‐mTOR Signaling Pathway,” Scientific Reports 6 (2016): 26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lennicke C. and Cocheme H. M., “Redox Metabolism: ROS as Specific Molecular Regulators of Cell Signaling and Function,” Molecular Cell 81, no. 18 (2021): 3691–3707. [DOI] [PubMed] [Google Scholar]
- 112. Alluri H., Stagg H. W., Wilson R. L., et al., “Reactive Oxygen Species‐Caspase‐3 Relationship in Mediating Blood‐Brain Barrier Endothelial Cell Hyperpermeability Following Oxygen‐Glucose Deprivation and Reoxygenation,” Microcirculation 21, no. 2 (2014): 187–195. [DOI] [PubMed] [Google Scholar]
- 113. Liu T., Zhang Q., Zhang J., et al., “EVmiRNA: A Database of miRNA Profiling in Extracellular Vesicles,” Nucleic Acids Research 47, no. D1 (2019): D89–D93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Brigstock D. R., “Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics,” Cells 10, no. 7 (2021): 1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tan K. S., Armugam A., Sepramaniam S., et al., “Expression Profile of MicroRNAs in Young Stroke Patients,” PLoS One 4, no. 11 (2009): e7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Patel V. and Noureddine L., “MicroRNAs and Fibrosis,” Current Opinion in Nephrology and Hypertension 21, no. 4 (2012): 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liu Y., Bi X., Xiong J., et al., “MicroRNA‐34a Promotes Renal Fibrosis by Downregulation of Klotho in Tubular Epithelial Cells,” Molecular Therapy 27, no. 5 (2019): 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bai M., Chen H., Ding D., et al., “MicroRNA‐214 Promotes Chronic Kidney Disease by Disrupting Mitochondrial Oxidative Phosphorylation,” Kidney International 95, no. 6 (2019): 1389–1404. [DOI] [PubMed] [Google Scholar]
- 119. Zanchi C., Macconi D., Trionfini P., et al., “MicroRNA‐184 Is a Downstream Effector of Albuminuria Driving Renal Fibrosis in Rats With Diabetic Nephropathy,” Diabetologia 60, no. 6 (2017): 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Liu Y., Dong Z.‐J., Song J.‐W., et al., “MicroRNA‐122‐5p Promotes Renal Fibrosis and Injury in Spontaneously Hypertensive Rats by Targeting FOXO3,” Experimental Cell Research 411, no. 2 (2022): 113017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Authors confirm that all relevant data are included in the article.