

Abstract
Background
Tumors harboring two or more PIK3CA short variant (SV) (“multi-hit”) mutations have been linked to improved outcomes with anti-PIK3CA-targeted therapies in breast cancer. The landscape and clinical implications of multi-hit PIK3CA alterations in clinically advanced prostate cancer (CAPC) remains elusive.
Objective
To evaluate the genomic landscape of single-hit and multi-hit PIK3CA genomic alterations in CAPC.
Patients and Methods
The Foundation Medicine FoundationCore database was used to identify 19,978 CAPC tumors that underwent hybrid capture-based comprehensive genomic profiling to evaluate all classes of genomic alterations (GA) and determine tumor mutational burden (TMB), microsatellite instability (MSI), genomic ancestry, single-base substitution mutational signatures, and homologous recombination deficiency signature (HRDsig). Tumor cell PD-L1 expression was determined by IHC (Dako 22C3).
Results
18,741 (93.8%) tumors were PIK3CA wild type (WT), 1155 (5.8%) featured single PIK3CA SV, and 82 (0.4%) featured multi-hit PIK3CA SVs. Single-hit (6.6 versus 3.8; p < 0.0001) and multi-hit (12.8 versus 3.8; p < 0.0001) featured more driver GA per tumor than PIK3CA WT CAPC, as well as higher prevalence of MMR mutational signature, MSI high status, and TMB levels versus PIK3CA WT (p < 0.0001). Other differences in GA included higher frequencies of GA in BRCA2 in multi-hit versus WT (18.3% versus 8.5%; p = 0.0191), ATM in multi-hit versus WT (13.4% versus 5.6%; p = 0.02) and PTEN in single-hit versus WT (40.2% versus 30.1%; p < 0.0001). Homologous recombination deficiency signatures were higher in PIK3CA WT versus single-hit (11.2% versus 7.6%; p = 0.0002). There were no significant differences in PD-L1 expression among the three groups.
Conclusions
Identification of multi-hit PIK3CA GA in CAPC highlights a potentially unique phenotype that may be associated with response to anti-PIK3CA targeted therapy and checkpoint inhibition, supporting relevant clinical trial designs.
Key Points
| Activation of the PI3K pathway in prostate cancer can predispose the patient toward more aggressive and castration-resistant growth. |
| We found that PIK3CA-mutated clinically advanced prostate cancer (CAPC) occurs less frequently in patients of African genomic ancestry and has a higher frequency of MSI-H, high TMB, and COSMIC mutational signatures associated with mismatch repair deficiency versus wild-type PIK3CA CAPC. |
| Single-hit and multi-hit PIK3CA alterations may be a potential biomarker for PIK3CA and PD-L1 inhibition. |
Background
Prostate cancer is the most common malignancy among men in the USA. The 5-year survival rate of men with localized prostate cancer nears 100%, though this significantly decreases to about 30% in men with non-regional metastatic disease [1]. Treatment of metastatic castration-sensitive prostate cancer (mCSPC) includes androgen-deprivation therapy (ADT) plus a second-generation anti-androgen (AA) or docetaxel, or, most recently, triple therapy; however, nearly all patients progress to metastatic castration-resistant prostate cancer (mCRPC) within 2–3 years, at which point systemic therapy options are typically limited in efficacy [2–4].
The National Comprehensive Cancer Network (NCCN) guidelines recommend genetic and biomarker testing in patients with regional or distant metastatic prostate cancer, focusing predominantly on germline mutations, DNA damage response genes, microsatellite instability, mismatch repair deficiency, and tumor mutational burden [2]. While there have been advances in molecular biomarkers in prostate cancer, there is still a major need for accurate and predictive biomarkers to help guide treatment decisions [5].
Phosphatidylinositol 3-kinase (PI3K) activation is one of the initial signals in the mammalian target of rapamycin (mTOR) pathway, which has been strongly linked to prostate cancer progression and metastasis [6]. Prior studies have suggested a significant crosstalk with androgen receptor (AR) signaling, whereby PI3K inhibition is often associated with activation of AR-related genes and vice versa, signifying that PI3K axis may offer possible alternative therapeutic targets. Despite this, combination of pan-PI3K plus AR inhibitors have shown little efficacy [7–9]. However, multiple mutations in PIK3CA, an oncogene in the PI3K pathway, create an additive effect of single mutants and are hypersensitive to PI3K inhibition [10]. Targeting oncogenic mutations, PIK3CA has been relatively underexplored in prostate cancer, but has been more successful in other malignancies [11]. Our hypothesis was that PIK3CA could emerge as a drugagble target for men with refractory clinically advanced disease. Therefore, we sought to determine the genomic landscape and clinical implications of single- and multi-hit PIK3CA alterations in clinically advanced prostate cancer (CAPC).
Methods
The Foundation Medicine FoundationCore database was used to identify 19,978 CAPC that underwent hybrid capture-based tissue comprehensive genomic profiling (CGP). Approval for this study, including a waiver of informed consent, was obtained from the Western Institutional Review Board (protocol no. 20152817). Clinicopathological data confirming that all cases were clinically advanced and metastatic CAPC, including patient age, routine histology and immunohistochemical staining results, and confirmation of the diagnosis, were extracted from medical records and pathology reports. All patients with CAPC had developed either local progression or metastatic disease that had progressed at the time of CGP. The biopsy location of the specimen, such as whether it was from a primary or metastatic site, was determined by the accompanying pathology report for each case. The vast majority of patients had stage IV disease at the time the sequencing test was ordered by the treating physician. In addition, the vast majority of patients had been treated with hormone deprivation regimens, a subset with radiation treatments and an additional subset with systemic chemotherapy prior to the submission of a sample for CGP. CGP was performed on US Food and Drug Administration (FDA)-approved, hybridization-captured adaptor ligation–based libraries using DNA extracted from formalin-fixed paraffin-embedded tumor in a CLIA- and CAP-certified laboratory (FoundationOne®CDx, Foundation Medicine, Inc.). All samples forwarded for DNA extraction contained a minimum of 20% tumor nuclei. The samples were assayed for exons from at least 324 cancer-related genes, plus select introns from at least 31 genes frequently rearranged in cancer. All mutations included were pathogenic. Patient samples were sequenced and evaluated for genomic alterations including base substitutions, insertions, deletions, copy number alterations (amplifications and homozygous deletions), and gene fusions/rearrangements, as previously described [12, 13]. The bioinformatics processes used in this study included Bayesian algorithms to detect base substitutions, local assembly algorithms to detect short insertions and deletions, a comparison with process-matched normal control samples to detect gene copy number alterations, and an analysis of chimeric read pairs to identify gene fusions as previously described [12, 13]. Tumor mutational burden was determined on 0.8–1.1 Mb of sequenced region, as previously described [14, 15]. Assessment of microsatellite instability was performed from DNA sequencing using a fraction-based algorithm interrogating at least 1500 loci, as previously described [15]. All non-germline mononucleotide repeats with lengths ≥ 6 bp and with sufficient sequence coverage were evaluated. Single-base substitution signatures were determined based on deconvolution of the COSMIC mutational signatures v2 to yield coefficient weights representing the contributions of the signatures in each sample [16].
The genetic ancestry for each patient was predicted using a single-nucleotide polymorphism (SNP)-based approach. Briefly, the profiled SNPs that overlapped with those captured in phase 3 of the 1000 Genomes Project were projected down to five principal components, which were then used to train a random forest classifier to identify the following continental geographic ancestry groups: European, African, East Asian, South Asian, and admixed American [17]. PD-L1 expression was determined by immunohistochemistry (IHC) performed on FFPE tissue using the Dako 22C3 PD-L1 antibody, according to the manufacturer’s instructions (catalog number SK006). PD-L1 expression was stratified into three categories based on the fraction of stained tumor cells: negative (< 1%), low positive (1–49%), and high positive (≥ 50%). HRDsig was called using a machine learning-based algorithm, as previously described. Briefly, an extreme gradient boosting (XGB) machine learning model interpreted a broad set of genome-wide copy number and short variant features from segmented copy number profiles [18].
Categorical prevalence comparisons made between groups were evaluated using Fisher’s exact testing with alpha set to 0.05. Two-sided p values were calculated for each comparison and false discovery rate (FDR) was adjusted using the Benjamini–Hochberg procedure. Statistical significance was defined as FDR-corrected p ≤ 0.05. Statistics, computation, and plotting were carried out using Python 2.7.18 (Python Software Foundation) and R 4.2.3 (R Foundation for Statistical Computing).
Results
The clinical and genomic features of the 19,978 cases of CAPC are shown in Table 1. A total of 18,741 (93.8%) of the CAPC were PIK3CA wild type (WT), 1155 (5.8%) featured a single PIK3CA SV mutation, and 82 (0.4%) featured more than one (“multi-hit”) PIK3CA SV mutations (Fig. 1). The median age of patients with CAPC with PIK3CA WT (68; IQR 61–74), single hit PIK3CA SV (70; IQR 62–76), and multi-hit PIK3CA SV (69; IQR 63–73) appeared similar. When compared with PIK3CA WT CAPC, both the single-hit (6.6 versus 3.8; p < 0.0001) and multi-hit (12.8 versus 3.8; p < 0.0001) featured more driver GA per tumor (Fig. 2A–C). At 14.0%, African genetic ancestry was more frequent in PIK3CA WT CAPC than in single-hit (10.4%; p = 0.0010) and multi-hit (10.2%; not significant) cases. The frequencies of East Asian, South Asian, and admixed American ancestry ranged from less than 1% to 6% and were similar in all groups in this study.
Table 1.
Clinical and genomic features of PIK3CA mutational landscape in clinically advanced prostate cancer
| PIK3CA mutations | WT | Single hit | Multi-hit | WT versus single-hit p value† |
WT versus multi-hit p value† |
Single-hit versus multi-hit p value† |
|---|---|---|---|---|---|---|
| Cases | 18,741 | 1155 | 82 | – | – | – |
| Median age (range) years | 68 (34–89+) | 70 (40–89+) | 69 (44–89+) | < 0.0001 | NS | NS |
| GA/tumor | 3.8 | 6.6 | 12.8 | < 0.0001 | < 0.0001 | < 0.0001 |
| Genomic ancestry | ||||||
| AFR | 14.0% | 10.4% | 10.3% | 0.0010 | NS | NS |
| EUR | 76.9% | 81.8% | 83.3% | 0.0002 | NS | NS |
| Pathogenic genomic alterations | ||||||
| APC | 8.2% | 14.1% | 24.4% | < 0.0001 | < 0.0001 | 0.0757 |
| AR | 14.9% | 15.2% | 12.2% | NS | NS | NS |
| ATM | 5.6% | 6.8% | 13.4% | NS | 0.0238 | NS |
| BRCA1 | 1.2% | 1.7% | 4.9% | NS | NS | NS |
| BRCA2 | 8.5% | 8.9% | 18.3% | NS | 0.0191 | NS |
| CCND1 | 3.7% | 1.8% | 2.4% | 0.0011 | NS | NS |
| CDK12 | 5.6% | 3.6% | 3.7% | 0.0092 | NS | NS |
| CDKN2A | 2.5% | 5.1% | 6.1% | < 0.0001 | NS | NS |
| CTNNB1 | 5.0% | 11.8% | 22.0% | < 0.0001 | < 0.0001 | NS |
| PTEN | 30.1% | 40.2% | 32.9% | < 0.0001 | NS | NS |
| RB1 | 5.3% | 6.8% | 11.0% | NS | NS | NS |
| SPOP | 9.8% | 7.4% | 8.5% | 0.012 | NS | NS |
| TMPRSS2 | 31.5% | 37.1% | 23.2% | 0.0003 | NS | NS |
| TP53 | 39.6% | 43.0% | 39.0% | 0.0485 | NS | NS |
| Microsatellite instability and TMB | ||||||
| MSI high | 2.5% | 12.4% | 35.4% | < 0.0001 | < 0.0001 | < 0.0001 |
| TMB ≥ 10 | 3.9% | 16.0% | 50.0% | < 0.0001 | < 0.0001 | < 0.0001 |
| TMB ≥ 20 | 2.2% | 13.4% | 42.7% | < 0.0001 | < 0.0001 | < 0.0001 |
| Homologous recombination deficiency (HRDsig) | ||||||
| HRDsig positive | 11.2% | 7.6% | 5.1% | 0.0002 | NS | NS |
| COSMIC trinucleotide signature | ||||||
| MMR | 3.2% | 12.9% | 35.5% | < 0.0001 | < 0.0001 | < 0.0001 |
| POLE | 0.0% | 0.4% | 2.4% | 0.0002 | 0.0008 | NS |
| PD-L1 IHC | ||||||
| PD-L1 low positive | 11.5% | 12.4% | 8% | NS | NS | NS |
| PD-L1 high positive | 0.9% | 1.0% | 0% | NS | NS | NS |
Fig. 1.
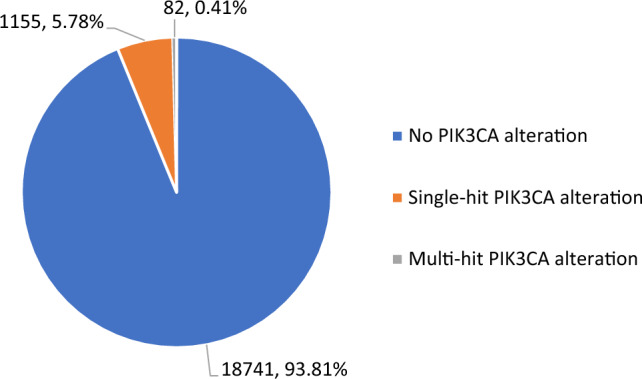
Frequency of PIK3CA alterations in clinically advanced prostate cancer
Fig. 2.

Long-tail plots of genomic alterations in CAPC based on PIK3CA mutation status. A: No PIK3CA SV mutations. B: One PIK3CA mutation only. C: Two or more PIK3CA mutations
Single-hit (12.9%; p < 0.0001) and multi-hit (35.4%; p < 0.0001) PIK3CA SV CAPC featured significantly higher prevalence of MMR mutational signatures than PIK3CA WT (3.2%). Single-hit (0.4%; p = 0.0002) and multi-hit (2.4% p = 0.0008) PIK3CA SV CAPC also featured significantly higher prevalence of POLE mutational signatures than PIK3CA WT (0.0%). MSI high status was significantly more common in both PIK3CA single-hit (12.4% versus 2.5%; p < 0.0001) and multi-hit (35.4% versus 2.5%; p < 0.0001) compared with PIK3CA WT. Median tumor mutational burden (TMB) was also higher in single-hit PIK3CA (2.5 mut/Mb; IQR 1.25–5.00) and multi-hit (7.5 mut/Mb; IQR 1.74–45.02) mutations compared with PIK3CA WT (1.7 mut/Mb; IQR 0.87–3.60). Noteworthy differences in GA of potential importance included significantly higher frequencies of GA in BRCA2 in multi-hit versus WT (18.3% versus 8.5%; p = 0.0191), ATM in multi-hit versus WT (13.4% versus 5.6%; p = 0.0238), and PTEN in single-hit versus WT (40.2% versus 30.1%; p < 0.0001), and lower frequencies of GA in CDK12 (3.6% versus 5.6%; p = 0.0092), and SPOP (7.4% versus 9.8%; p = 0.0122) in single-hit versus WT. Homologous recombination deficiency signatures were higher in the PIK3CA WT versus single-hit (11.2% versus 7.6%; p = 0.0002). There were no significant PD-L1 expression differences among the three groups. Examples of double hit PIK3CA mutations in CAPC are shown in Figs. 3 and 4. Finally, although a formal germline test was not performed on the patients included in this study, germline status was predicted from analysis of the sequencing data and no specific germline alterations were associated with the PIK3CA status of the prostate cancer samples of any of the prostate cancer groups.
Fig. 3.

Hematoxylin and eosin-stained images of a double-hit PIK3CA mutated CAPC in a needle biopsy from a poorly differentiated Gleason 10 prostate cancer (upper left and upper right) in a 60-year-old man, which progressed to clinically advanced disease. On comprehensive genomic profiling, this tumor was MSI high with TMB of 30 mutations/Mb. Two PIK3CA missense mutations were detected: R38H (middle) and Y1021H (lower). There was a BRCA2 N1784fs*3 inactivating frameshift mutation. Other alterations included short variant mutations PTEN N323fs*2, APC D1636fs*14, ASXL1 G646fs*12, DICER1 G87fs*41, EP300 K277fs*6, JAK1 K860fs*16, KMT2C (MLL3) N2842fs*21, MSH6 F1088fs*5, MYST3 R864Q, PLCG2 R732C, and TP53 R248Q. In addition to indications for checkpoint inhibitors associated with MSI high and high TMB status, PARP inhibitor indications associated with the heterozygous BRCA2-inactivating mutation are also identified. The double-hit PIK3CA mutation also raises the possibility that this tumor may also be potentially sensitive to PIK3CA inhibitors, such as alpelisib
Fig. 4.

Hematoxylin-stained images of a biopsy of a recurrent high-grade prostate cancer presenting as a pelvic mass in a 75-year-old man (upper left and upper right). The tumor was positive for NKX3.1, PSA, and synaptophysin on IHC staining. Comprehensive genomic profiling revealed that this tumor was MSI high and had a TMB of 15 mutations/Mb. In addition to two PIK3CA mutations (I1058F and R88Q), his tumor featured a TMPRSS2-ERG rearrangement (middle panel) along with SV mutations in ASXL1 G646fs*12, AXIN1 R22*, BCORL1 P1681fs*20, CTNNB1 S45P, MSH3 K383fs*32, NOTCH2 R1931H, PTCH1 L517fs*25, RNF43 G659fs*41, TGFBR2 R528C TP53 R175H, and TP53 M243T. The copy number plot revealed losses in AR and PTEN. In addition to potential checkpoint inhibitor-based treatments, the activation of the mTOR pathway by both PIK3CA mutations and the PTEN loss raises the possibility of mTOR or PIK3CA inhibitors as potential therapy options
Discussion
With advances in tumor genomics and available therapies in prostate cancer, there has been a need for biomarker-driven treatments. For example, the recent approval of poly(ADP-ribose) polymerases (PARP) inhibitors in advanced prostate cancer offers hope for personalized therapy, particularly for those with mutations in DNA damage response genes [19, 20]. However, there continues to be a need for other reliable biomarkers and therapy targets.
Abnormalities in the PI3K pathway are detected in 70–100% of advanced prostate cancer cases [21]. Studies have demonstrated a reciprocal feedback mechanism between the AR and the PI3K/AKT pathways, whereby inhibition of one leads to activation of the other [22]. Therefore, it is thought that castration-resistant prostate cancer may develop resistance to antiandrogens through the PI3K/AKT pathway, particularly through a PI3KCA-activating mutation [23]. The rates of PI3KCA mutations in literature ranges between 5.5 and 11.5%, which is similar to our study that had a prevalence of 6.2% [24]. Multiple mutations, in PIK3CA, an oncogene in the PI3K pathway, have been shown to combine the effects of single mutants [10]. PIK3CA mutation has been associated with poor prostate cancer prognosis and, in conjunction with PTEN loss, accelerates castration-resistant cancer growth [25]. Consistent with prior reports, we identified molecular aberrations (single- and multi-hit PIK3CA) compared with wild-type PIK3CA that may correlate with more advanced prostate cancer at presentation [26].
Despite the initial optimism, the first studies featuring inhibition of the PI3K pathway did not achieve the expected success in the treatment of prostate cancer. Trials of pan-class I PI3K inhibitors given alone or in combination with abiraterone/enzalutamide were stopped early due to futility or failure to reverse resistance [7, 8]. Selective PI3Kβ and PI3Kδ inhibitors have shown little efficacy in prostate cancer, as well as other malignancies [27, 28]. However, in a phase 3, randomized clinical trial targeting the AKT pathway using ipatasertib, an AKT inhibitor, in combination with abiraterone, there was an improvement in radiographical progression-free survival among patients with metastatic CRPC with PTEN loss, suggesting there may be utility in targeting this pathway in select patients [29]. Additionally, specifically targeting PIK3CA using alpelisib in combination with the estrogen receptor degrader fulvestrant has been shown to improve progression-free survival in PIK3CA-mutated estrogen receptor positive breast cancer, which has been granted FDA approval [11]. Specific inhibition of PIK3CA may represent improved biologic targeting compared with other PI3K subunits [11]. Additionally, multi-hit PIK3CA mutations have been shown to be hypersensitive to PI3K inhibition in cells, thereby further highlighting the need to further assess its role as a biomarker [10]. Early trials have also demonstrated response to PIK3CA inhibition in other solid malignancies, such as head and neck, colorectal, and ovarian cancer [30]. To date, alpelisib remains the only selective PI3K inhibitor approved in solid tumors and given the proposed AR-resistance pathway through the PI3K pathway; further studies evaluating PIK3CA inhibition in selected patients may represent an alternative, or even synergistic, therapeutic strategy.
Prostate cancer has classically been described as an “immunologically cold tumor,” and multiple prostate cancer trials evaluating programmed ligand-1 (PD-L1) inhibition in biomarker unselected patients have been unsuccessful [31–34]. However, pembrolizumab is FDA-approved for tumors with high microsatellite instability or “high” TMB (≥ 10 mutations per megabase of sequenced DNA) [35, 36]. Although MSI high and high TMB prostate cancer incidence are rare, in our cohort of multi-hit PIK3CA tumors, more than a third were MSI high and half had a TMB ≥ 10 mutations/Mb. We demonstrated a strong correlation between PIK3CA mutations and MSI high, and high TMB, as well as MMR and POLE single-base substitution mutational signatures. However, it is unclear if this is a causal relation and can help select patients for immunotherapy, or more likely just a correlation.
Our study has several limitations inherent to its design. It is retrospective and lacks data on clinical parameters, outcomes, and therapies used that may help better analyze and characterize the clinical relevance our molecular biomarker findings. Unfortunately, we do not know whether patients received prior cancer therapy or location of the biopsy specimen, whether it was obtained from the primary or metastatic site, which would have helped us better understand the evolution of the cancer and even provide some implications for treatment. Although the sample size for multi-hit PIK3CA mutations was low, thus limiting the generalizability, our study had a significant sample size of single-hit PIK3CA mutations, which showed a continued trend in the frequency of concurrent alterations in the multi-hit population. This favors the study of PIK3CA inhibitors and PD-1 and PD-L1 inhibitors in PIK3CA-altered prostate cancer regardless of the single-hit or multi-hit status. Additionally, our data is collected on the basis of a referral-based nature, which subjects it to selection bias of the clinically advanced cases; moreover, we could not account for unmeasured confounders. Despite several limitations, our study can generate relevant hypotheses for the selection and study of patients with prostate cancer, especially those with PIK3CA mutation and help inform clinical trial designs.
Conclusion
Activation of the PI3K pathway in prostate cancer can predispose toward more aggressive and castration-resistant growth. We found that PIK3CA-mutated CAPC occurs less frequently in patients of African genomic ancestry, has a higher frequency of MSI high, high TMB, and COSMIC mutational signatures associated with mismatch repair deficiency versus wild-type PIK3CA CAPC. Single-hit and multi-hit PIK3CA alterations may be a potential biomarker for PIK3CA and PD-L1 inhibition. Further studies are warranted to evaluate PIK3CA mutations as a biomarker for targeted therapies and immunotherapies in prostate cancer.
Declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Grivas: Consulting: Aadi Bioscience; AstraZeneca; Asieris Pharmaceuticals, Astellas Pharma, Bristol-Myers Squibb, Boston Gene, CG Oncology, Dyania Health, Lucence Health, Fresenius Kabi, G1 Therapeutics, Gilead, Guardant Health, ImmunityBio, Janssen, Merck KGaA, MSD, Pfizer, PureTech, Roche, SeaGen, Strata Oncology, Silverback Therapeutics. Research funding to institution: Acrivon Therapeutics; ALX Oncology, Bavarian Nordic; Bristol-Myers Squibb; Clovis Oncology; Debiopharm; Merck KGaA; Gilead; Pfizer; MSD; QED Therapeutics; GlaxoSmithKline; G1 Therapeutics; Mirati Therapeutics. Vasan: Consulting and advisory board activities from Magnet Biomedicine, Novartis, and Reactive Biosciences; grants from Gilead outside the submitted work; and a patent for US20210189503A1 pending to Memorial Sloan Kettering Cancer Center. Pavlick, Huang, Lin, Danziger, Sokol, Sivakumar, Graf, Ross: employed by Foundation Medicine, Inc. Basin, Miguel, Jacob, Goldberg, Spiess, Necchi, Kamat, Cheng, Basnet, and Bratslavsky declare that they have no conflicts of interest that might be relevant to the contents of this manuscript. Petros Grivas is an Editorial Board member of Targeted Oncology. Petros Grivas was not involved in the selection of peer reviewers for the manuscript nor any of the subsequent editorial decisions.
Ethics approval
Western Institutional Review Board (Protocol No. 20152817).
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Availability of data and materials
Available in repository.
Author contributions
Michael F. Basin: analysis and interpretation of data, manuscript writing; Carla M. Miguel: analysis and interpretation of data, manuscript writing; Joseph M. Jacob: analysis and interpretation of data, critical manuscript review; Hanan Goldberg: critical manuscript review; Petros Grivas: critical manuscript review; Philippe E. Spiess: critical manuscript review; Andrea Necchi: critical manuscript review; Ashish M. Kamat: critical manuscript review; Dean C. Pavlick: acquisition, analysis, critical manuscript review; Richard S.P. Huang: critical manuscript review, Douglas I. Lin: critical manuscript review; Natalie Danziger: acquisition, analysis, critical manuscript review; Ethan S. Sokol: critical manuscript review; Smruthy Sivakumar: critical manuscript review; Ryon Graf: critical manuscript review; Liang Cheng: critical manuscript review; Neil Vasan: critical manuscript review; Jeffrey Ross: conception and design of work, analysis, interpretation, critical manuscript review; Alina Basnet: critical manuscript review; Gennady Bratslavsky: conception and design of work, analysis, interpretation, critical manuscript review.
References
- 1.Prostate Cancer—Cancer Stat Facts. https://seer.cancer.gov/statfacts/html/prost.html.
- 2.Schaeffer EM, Srinivas S, Adra N, An Y, Barocas D, et al. NCCN Guidelines version 1.2023 Prostate Cancer. J Natl Compr Canc Netw. 2022;20(12):1288–98. [DOI] [PubMed] [Google Scholar]
- 3.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6(2):76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamada S, Iguchi T, Kato M, Asakawa J, Kita K, Yasuda S, et al. Time to progression to castration-resistant prostate cancer after commencing combined androgen blockade for advanced hormone-sensitive prostate cancer. Oncotarget. 2018;9(97):36966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alarcón-Zendejas AP, Scavuzzo A, Jiménez-Ríos MA, Álvarez-Gómez RM, Montiel-Manríquez R, Castro-Hernández C, et al. The promising role of new molecular biomarkers in prostate cancer: from coding and non-coding genes to artificial intelligence approaches. Prostate Cancer Prostatic Dis. 2022;25(3):431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer. 2017;81:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotte SJ, Chi KN, Joshua AM, Tu D, Macfarlane RJ, Gregg RW, et al. A Phase II Study of PX-866 in patients with recurrent or metastatic castration-resistant prostate cancer: Canadian Cancer Trials Group Study IND205. Clin Genitourin Cancer. 2019;17(3):201-208.e1. [DOI] [PubMed] [Google Scholar]
- 9.Massard C, Chi KN, Castellano D, de Bono J, Gravis G, Dirix L, et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur J Cancer. 2017;76:36–44. [DOI] [PubMed] [Google Scholar]
- 10.Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science. 2019;366(6466):714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–40. [DOI] [PubMed] [Google Scholar]
- 12.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milbury CA, Creeden J, Yip WK, Smith DL, Pattani V, Maxwell K, et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PLoS One. 2022;17(3):e0264138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and Pan-Tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn. 2019;21(6):1053–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connelly CF, Carrot-Zhang J, Stephens PJ, Frampton GM. Somatic genome alterations in cancer as compared to inferred patient ancestry. Cancer Res. 2018;78(13_Supplement):1227. [Google Scholar]
- 18.Moore JA, Chen KT, Madison R, Newberg JY, Fleischmann Z, Wang S, et al. Pan-cancer analysis of copy-number features identifies recurrent signatures and a homologous recombination deficiency biomarker to predict poly (ADP-Ribose) polymerase inhibitor response. JCO Precis Oncol. 2023:7:e2300093. [DOI] [PubMed] [Google Scholar]
- 19.Fizazi K, Piulats JM, Reaume MN, Ostler P, McDermott R, Gingerich JR, et al. Rucaparib or physician’s choice in metastatic prostate cancer. N Engl J Med. 2023;388(8):719–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091–102. [DOI] [PubMed] [Google Scholar]
- 21.Cham J, Venkateswaran AR, Bhangoo M. Targeting the PI3K-AKT-mTOR pathway in castration resistant prostate cancer: a review article. Clin Genitourin Cancer. 2021;19(6):563.e1-563.e7. [DOI] [PubMed] [Google Scholar]
- 22.Tortorella E, Giantulli S, Sciarra A, Silvestri I. AR and PI3K/AKT in prostate cancer: a tale of two interconnected pathways. Int J Mol Sci. 2023;24(3):2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herberts C, Murtha AJ, Fu S, Wang G, Schönlau E, Xue H, et al. Activating AKT1 and PIK3CA mutations in metastatic castration-resistant prostate cancer. Eur Urol. 2020;78(6):834–44. [DOI] [PubMed] [Google Scholar]
- 24.Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 2020;21(12):1–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson HB, Li J, Meniel VS, Fennell CM, Waring P, Montgomery KG, et al. Identification of Pik3ca mutation as a genetic driver of prostate cancer that cooperates with pten loss to accelerate progression and castration-resistant growth. Cancer Discov. 2018;8(6):764–79. [DOI] [PubMed] [Google Scholar]
- 26.Choudhury AD, Higano CS, de Bono JS, Cook N, Rathkopf DE, Wisinski KB, et al. A phase I study investigating AZD8186, a potent and selective inhibitor of PI3Kβ/δ, in patients with advanced solid tumors. Clin Cancer Res. 2022;28(11):2257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarker D, Dawson NA, Aparicio AM, Dorff TB, Pantuck AJ, Vaishampayan UN, et al. A phase I, open-label, dose-finding study of GSK2636771, a PI3Kβ inhibitor, administered with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2021;27(19):5248–57. [DOI] [PubMed] [Google Scholar]
- 28.Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-kinase α-selective inhibition with Alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36(13):1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021;398(10295):131–42. [DOI] [PubMed] [Google Scholar]
- 30.Bou-Dargham MJ, Sha L, Sang QXA, Sang QXA, Zhang J. Immune landscape of human prostate cancer: immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer. 2020;20(1):572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graff JN, Liang LW, Kim J, Stenzl A. KEYNOTE-641: a Phase III study of pembrolizumab plus enzalutamide for metastatic castration-resistant prostate cancer. Future Oncol. 2021;17(23):3017–26. [DOI] [PubMed] [Google Scholar]
- 32.Antonarakis ES, Park SH, Goh JC, Shin SJ, Lee JL, Mehra N, et al. Pembrolizumab plus olaparib for patients with previously treated and biomarker-unselected metastatic castration-resistant prostate cancer: the randomized, open-label, phase III KEYLYNK-010 trial. J Clin Oncol. 2023;41(22):3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrylak DP, Ratta R, Gafanov R, Facchini G, Piulats JM, Kramer G, et al. KEYNOTE-921: phase III study of pembrolizumab plus docetaxel for metastatic castration-resistant prostate cancer. Future Oncol. 2021;17(25):3291–9. [DOI] [PubMed] [Google Scholar]
- 34.Barata P, Agarwal N, Nussenzveig R, Gerendash B, Jaeger E, Hatton W, et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J Immunother Cancer. 2020;8(2):e001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristescu R, Aurora-Garg D, Albright A, Xu L, Liu XQ, Loboda A, et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J Immunother Cancer. 2022;10:3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graf RP, Fisher V, Weberpals J, Gjoerup O, Tierno MB, Huang RSP, et al. Comparative effectiveness of immune checkpoint inhibitors vs chemotherapy by tumor mutational burden in metastatic castration-resistant prostate cancer. JAMA Netw Open. 2022;5(3):e225394–e225394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Available in repository.