

Abstract
RNA-dependent protein kinase (PKR) may have a positive regulatory role in controlling tumor growth and progression in hepatocellular carcinoma (HCC). However, the downstream substrates and the molecular mechanism of PKR in the growth and progression of HCC have not been clarified. In this study, mass spectrometry analysis was performed with immunoprecipitated samples, and 4.1R was identified as a protein that binds to PKR. In transfected COS7 cells, an immunoprecipitation experiment showed that 4.1R binds to wild-type PKR, but not to a kinase-deficient mutant PKR, suggesting that PKR binds to 4.1R in a kinase activity-dependent manner. In HCC cell lines, HuH7 and HepG2, the expression level of 4.1R protein was shown to be regulated by protein expression and activation of PKR. Interestingly, high expression of 4.1R, as well as PKR, is associated with a worse prognosis in HCC. PKR increased HCC cell growth in both anchorage-dependent and anchorage-independent manners, whereas 4.1R was involved in HCC cell growth only in an anchorage-independent manner, not in an anchorage-dependent manner. The rescue experiment indicated that increased anchorage-independent growth of HCC cells by PKR might be caused by 4.1R. In conclusion, PKR associates with 4.1R and promotes anchorage-independent growth of HCC. The PKR-4.1R axis might be a new therapeutic target in HCC.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-75142-5.
Keywords: eIF-2 kinase, Erythrocyte membrane band 4.1 protein, Liver neoplasms, Cell line, Prognosis
Subject terms: Cancer, Molecular biology, Gastroenterology, Oncology
Introduction
Hepatocellular carcinoma (HCC) is the fourth most common tumor worldwide1. About 840,000 new cases of liver cancer are diagnosed each year, and 780,000 people die from this disease2.
Transarterial chemoembolization (TACE) or resection is recommended for patients with intermediate-stage HCC3. For advanced tumors, systemic drugs such as sorafenib, lenvatinib, regorafenib, cabozantinib, and ramucirumab are used. Even with the above treatments, a median survival of 10–16 months is expected in advanced-stage HCC4,5. Therefore, it would be useful to elucidate the molecular mechanisms to search for new treatment methods.
Double-stranded RNA-dependent protein kinase (PKR), also known as eukaryotic initiation factor 2-α kinase 2 (EIF2AK2), is a serine-threonine protein kinase that is expressed throughout the body6. Initially, PKR was described as an anti-viral protein of the innate immune system induced by interferon7. In previous research, it was shown that PKR modulates several signal transduction pathways, such as the mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT)8,9, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways10. It was also reported that PKR could function as both an oncogene and as a tumor suppressor gene in a tissue-dependent manner11,12. As examples, survival was significantly shorter with high-grade PKR expression than with low-grade expression in small-sized peripheral lung cancer13, and there were much higher PKR activity levels in human breast cancer cell lines compared to non-transformed mammary epithelial cell lines14. Conversely, much lower proliferative activity was seen in tumor cells with high PKR expression in thyroid carcinoma15.
In HCC, upregulation of PKR was seen in tumor tissues compared with the surrounding tissues16,17. In hepatitis C virus (HCV)-related HCC cell lines, c-Fos and c-Jun activities were upregulated by PKR to increase HCC cell proliferation18. However, the molecules that function upstream or downstream of PKR are largely unknown. In this study, to screen molecules that bind PKR, mass spectrometry (MS) analysis was performed with immunoprecipitated protein samples after Flag-PKR expression plasmid was transfected into HEK293T cells. This resulted in 4.1R being identified as a downstream molecule binding with PKR.
4.1R (gene name EPB41) is a member of the protein 4.1 family that was originally discovered as a membrane protein in human red blood cells19. Four paralogues have been identified in the protein 4.1 family in vertebrates, and all four are relatively ubiquitously expressed. The proteins are now named 4.1R, 4.1N, 4.1G, and 4.1B20. Protein 4.1 is a scaffold protein. Studies in erythrocytes indicate that protein 4.1 forms a complex with spectrin and actin, interacts with transmembrane proteins, and participates in cell morphology21.
There is evidence for a connection between protein 4.1 and cancer; 4.1B interacts with the tumor suppressor cell adhesion molecule 1 (CADM1), and loss of 4.1B is linked to malignant potential in non-small cell lung cancer cells, breast cancer, and renal cancer22–26. However, the relationship between 4.1R and HCC has rarely been reported, and there are many unknowns. In this study, the aim was to analyze the role of 4.1R and the importance of the interaction between PKR and 4.1R in HCC.
Materials and methods
Cell culture
The human HCC cell lines HuH7 and HepG2, HEK293T cells, and the monkey kidney cell line COS-7 (Japanese Collection of Research Bioresources, Osaka, Japan) were grown and maintained in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific). Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air, and the culture medium was changed three times per week.
Western blotting (WB)
RIPA buffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5% v/v NP-40, and 1% v/v SDS) was added to cells for protein extraction. Next, 15–20 µg of protein were separated on 4%-12% Bis–Tris Gels (Thermo Fisher Scientific) and transferred to an Immun-Blot PVDF Membrane for Protein Blotting (BIO-RAD, Hercules, CA, USA) and then blocked with 1% non-fat milk in Tris-buffered saline-Tween 20 (TBS-T) for 1 h at room temperature. The blocked membrane was then incubated with primary antibodies overnight at 4 °C. All antibodies used are listed in Supplementary Table 1. Secondary antibodies purchased from Cytiva (Washington, DC, USA) were used. Bands were labeled using the ECL Prime Kit (Cytiva) and visualized with ImageQuant LAS 4000 (Cytiva). The density of the bands was quantified by normalization to β-actin using Image J Software (National Institutes of Health, Bethesda, MD, USA).
Immunoprecipitation (IP)
COS7 cells were transfected with Flag or 6Myc-PKR and Flag or 6Myc-4.1R using Lipofectamine LTX reagent (Thermo Fisher Scientific). After 24 h, cells were lysed in the above lysis buffer. Lysates were centrifuged at 15,000 g at 4 °C for 10 min, and supernatants were pre-cleared with the antibody against Flag or Myc at 4 °C for 1 h and then incubated with the Protein G Sepharose (Cytiva) at 4 °C for 1 h. Sepharose was washed four times with lysis buffer, suspended in a sample buffer, boiled at 95 °C for 5 min, and then incubated on ice. Samples were then subjected to polyacrylamide gel containing SDS‐PAGE followed by WB.
Cell proliferation
Cell viability was quantified by an MTT assay (Promega, Madison, WI, USA). After 24 h of siRNA or plasmid transfection in 6-well plates, cells were resuspended, and then 2 × 103 cells were seeded in 96-well plates. At each time point, cells were treated with MTT reagent and incubated for 60 min. Absorbance at 492 nm was recorded.
Colony formation assay
A suspension of 1.5 × 104 − 3 × 104 cells in a culture medium containing 0.3% Agar (Fuji Film Wako Chemicals) was overlaid onto the precast 0.6% agar in a well of a 6-well culture plate. The cells were incubated at 37 °C for a period of two weeks. Using a light microscope, each well was photomicrographed evenly, and the average of the colony area in a square of 0.49 mm2 was determined. Each well was then stabilized with 4% paraformaldehyde and stained with 0.005% (v/w) crystal violet (Fuji Film Wako Chemicals) solution in 10% ethanol, and the entire well was photographed.
Mass spectrometry (MS)
For MS analysis, duplicate samples of HEK293T cells were transfected with three plasmids (Flag-PKR wild, Flag-PKR K296R, and pcDNA3 as control). The cells were lysed with lysis buffer (20 mM HEPES–KOH [pH 7.4], 150 mM NaCl, 1% Triton-X100, and protease inhibitor cocktail). After the cell debris was removed by centrifugation at 15,000 rpm for 10 min at 4 °C, the resulting supernatant was immunoprecipitated using Dynabeads protein (Thermo Fisher Scientific) by anti-Flag M2 antibody (Sigma-Aldrich, St. Louis, MO, USA). The beads were washed with wash buffers (10 mM HEPES–KOH [pH 7.5], 150 nM NaCL, and 0.1% Triton-X100). The beads were then incubated at 100 °C for 5 min in elution buffer (elution buffer: 10 mM HEPES KOH [pH 7.5], 150 mM NaCl, 0.05% Triton X100, 0.5 mg/mL Flag peptide). The eluted proteins were precipitated with 100% (w/v) trichloroacetic acid and washed with acetone. Precipitates produced by this process were re-dissolved in digestion buffer, consisting of 100 mM ammonium bicarbonate, 0.05% decyl β-D-glucopyranoside, and 7 M guanidine hydrochloride. This was followed by treatment with lysyl endopeptidase (Fuji Film Wako Chemicals, Osaka, Japan) at 37 °C for 3 h, further digestion with Trypsin TPCK (Sigma-Aldrich) at 37 °C for 12 h, and then centrifugation at 15,000 rpm for 1 min. Liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis was then performed on the resulting supernatant, and MS analysis was performed twice for each sample. An EasynLC1200 system (Thermo Scientific) and Q Exactive HF-X (Thermo Scientific) were used for the LC–MS/MS analyses. The analytical column for LC was a C18 silica resin-packed capillary column (diameter 10 μM and length 12 cm; Nikkyo Technos, Tokyo, Japan). Using solvents A (0.1% formic acid) and B (0.1% formic acid/80% acetonitrile), peptide separation was performed with a flow rate of 300 nL/min and a gradient of B from 0–40% for 80 min. The Data-Dependent Acquisition (DDA) mode was used for the MS and MS/MS measurements. The mass resolution for MS was 60,000, and that for MS/MS was 15,000. The m/z measurement range for MS was 380–1,500, and that for MS/MS was 200–2,000. The AGC target and maximum IT for MS were set to 3e6 and 60 ms, respectively, and for MS/MS to 1e5 and 45 ms, respectively. On the MS/MS scan, 20 precursor ions were selected per MS scan with an exclusion time of 12 s. Proteome Discoverer 2.2 software (Thermo Fisher Scientific) was used for the MS/MS data set analyses. The SEQUEST algorithm with Swiss-Prot (Human: 20,386 entries) as the protein database was used for peptide identification, with tolerances of 10 ppm for precursor ions and 0.02 Da for fragment ions. The digestion enzyme used was trypsin, allowing up to two missed cleavages, with oxidation (M), carbamidomethylation (C), and protein N-terminal acetylation added as modifications. A label-free quantification method using precursor ions was used to quantify proteins.
RNA extraction, cDNA synthesis, and real-time RT-PCR
Total RNA was extracted with TRIzol reagent (Thermo Fischer Scientific). Extracted RNA was reverse-transcribed using TaqMan Reverse Transcription Reagents (Thermo Fisher Scientific). The qRT-PCR assays were performed in a real-time PCR system (LightCycler 480 Instrument II, Roche Diagnostics Inc., Basel, Switzerland), and mRNA expression was assessed by the comparative cycle threshold (Ct) method (2−ΔCt). The PKR, 4.1R, and GAPDH primer and probe sets were obtained from Thermo Fisher Scientific (ID nos. Hs00169345, Hs01057085, and Hs0275899, respectively).
Chemicals
The PKR inhibitor, oxindole/imidazole compound (C16), was purchased from Merck (Darmstadt, Germany) and solubilized in dimethylsulfoxide (DMSO). The final concentrations of the PKR inhibitor in the in vitro experiments are described in the corresponding sections that follow.
RNA interfering
The small interfering RNAs (siRNAs) targeting PKR and 4.1R were obtained from Horizon Discovery (Cambridge, UK), and the transfection of cells was performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) following the manufacturer’s protocol. The sequences of siRNA are presented in Supplementary Table 2.
Expression vector
The cDNAs of wild PKR, point mutant K296R of PKR, and 4.1R were constructed by a PCR-based approach. Flag or 6Myc-PKR and Flag or 6Myc-4.1R were subcloned into pcDNA3 vector using EcoRI and XhoI restriction sites. Each plasmid (1 μg/mL-2.5 μg/mL) was transfected using Lipofectamine LTX (Thermo Fisher Scientific). For siRNA and plasmid co-transfection, the plasmid was transfected 24 h after transfection of siRNA.
Confocal image acquisition
All images were obtained using an upright laser scanning microscope (A1R, Nikon) with a 25 × water immersion objective lens (Apo LWD 25 × NA 1.10). Image stacks comprising more than 70 optical sections with 2-μm Z-steps were acquired from the surface, corresponding to an area of 502 × 502 μm2 (1024 × 1024 pixel2, 0.49 μm pixel− 1). Before observation, Hoechst 33,342 staining (1 μg/ml, 30 min, room temperature) was performed. An excitation laser at 403 nm was applied, and fluorescence signals were detected at wavelengths of 425–475 nm via GaAsP-type PMT. All images were filtered using Nikon NIS-Elements ver. 5.20 software. For image stacks, “median filter (3 × 3)” was applied to the reconstructed 3D image.
UALCAN
UALCAN (http://ualcan.path.uab.edu/) is a comprehensive web portal for analyzing cancer OMICS data27. In this study, expression of 4.1R was analyzed in UALCAN based on sample types of HCC tissues and normal liver tissues.
The Kaplan–Meier Plotter
The Kaplan–Meier Plotter (https://kmplot.com/analysis/) is a web server for the discovery and validation of survival biomarkers28. The survival-analysis tool in the Kaplan–Meier Plotter was used to analyze the prognostic significance of 4.1R and PKR mRNA. Survival analysis was performed using the Kaplan–Meier method, and hazard ratios (HRs) with 95% confidence intervals (CIs) and a log-rank P-value were calculated.
Statistical analysis
All statistical analyses were performed using Microsoft Excel (Microsoft, Redmond, WA, USA). Student’s t-test was performed to compare between two groups, and one-way ANOVA was performed for comparisons among multiple groups. All values are shown as means ± standard deviations (SDs). P values were categorized as follows: *P < 0.05; **P < 0.01; and ***P < 0.001.
Results
PKR interacts with 4.1R in a kinase activity-dependent manner
To determine the binding molecules of PKR, LC–MS/MS analysis was performed. Briefly, HEK293T cells were transfected with Flag-PKR plasmid, Flag-PKRK296R plasmid, a mutant deficient in kinase activity29, or control plasmid, followed by LC–MS/MS analysis four times using the immunoprecipitated samples by anti-Flag antibody. The data that could be reproduced four times were analyzed. Of the 105 proteins that were precipitated with wild-type PKR, 13 proteins were not detected to bind with kinase-dead PKR, and they are shown in Table 1. Of the PKR-binding proteins identified, 4.1R was the protein of interest. 4.1R is required to efficiently focus mitotic spindle poles, and knockdown of 4.1R causes defective metaphase and multi-nuclei, suggesting that 4.1R plays an important role in cell cycle progression30. In addition, we previously reported that 4.1R was required for the oncogenic role of CADM1 in small-cell lung cancer (SCLC) 31. Then, among the 13 proteins, 4.1R was selected. The interaction between 4.1R and wild-type PKR was confirmed by IP using two distinct epitope-tagged proteins in transfected COS7 cells (Fig. 1A, B). Moreover, PKR-V5 protein was co-localized with endogenous 4.1R protein on the cell membrane in HEK293FT cells (Supplementary Fig. 1).
Table 1.
Candidate proteins with which PKR interacts in a kinase activity-dependent manner.
| Protein name | Accession number in UniProt* |
|---|---|
| 14–3-3 protein gamma | P61981 |
| 14–3-3 protein beta/alpha | P31946 |
| 14–3-3 protein eta | Q04917 |
| Casein kinase II subunit alpha’ | P19784 |
| Casein kinase II subunit beta | P67870 |
| Protein 4.1 | P11171 |
| Casein kinase II subunit alpha | P68400 |
| Zinc finger CCCH domain-containing protein 14 | Q6PJT7 |
| Trifunctional enzyme subunit alpha, mitochondrial | P40939 |
| DBIRD complex subunit ZNF326 | Q5BKZ1 |
| Isoform 2 of chromatin target of PRMT1 protein | Q9Y3Y2 |
| U6 snRNA-associated Sm-like protein LSm2 | Q9Y333 |
| Apoptotic chromatin condensation inducer in the nucleus | Q9UKV3 |
Fig. 1.

PKR interacts with 4.1R depending on its kinase activity. (A) COS7 cells were transfected with the indicated plasmids, followed by IP and WB. The top panel shows the co-precipitation of PKR with 4.1R by IP with anti-Flag antibody and WB with anti-Myc antibody. The second panel shows the expression of 4.1R by IP with anti-Flag antibody and WB with anti-Flag antibody. The lower two panels show the expression level of each protein without IP. (B) COS7 cells were transfected with the indicated plasmids, followed by IP and WB. The top panel shows the co-precipitation of 4.1R with PKR by IP with anti-Flag antibody and WB with anti-Myc antibody. The second panel shows the expression of PKR by IP with anti-Flag antibody and WB with anti-Flag antibody. The lower two panels show the expression level of each protein without IP. (C) COS7 cells were transfected with the indicated plasmids, followed by IP and WB. The top panel shows the co-precipitation of wild type PKR or PKRK296R with 4.1R by IP with anti-Flag antibody and WB with anti-Myc antibody. The second panel shows the expression of 4.1R by IP with anti-Flag antibody and WB with anti-Flag antibody. The lower two panels show the expression level of each protein without IP.
Next, to determine whether the interaction of 4.1R with PKR depends on PKR activation, the binding of 4.1R to a kinase-deficient mutant PKRK296R was tested by IP. As shown in Fig. 1C, 4.1R failed to bind to PKRK296R in transfected COS7 cells (Fig. 1C), indicating that PKR interacts with 4.1R in a kinase activity-dependent manner, or, in other words, 4.1R is a downstream substrate of PKR. The interaction of endogenous 4.1R with PKR-V5 was demonstrated, but this interaction was not seen with PKRK296R (Supplementary Fig. 2).
Because 4.1R belongs to the 4.1 protein family including 4.1B, 4.1G, and 4.1N, the binding of the family members to PKR was then checked. Interestingly, 4.1B, 4.1G, and 4.1N failed to bind to PKR (Supplementary Fig. 3), suggesting that the 4.1R-PKR interaction is specific within the 4.1 protein family.
PKR affects the expression of 4.1R protein in HCC cell lines
To clarify the importance of 4.1R in HCC, the expression level of 4.1R was examined in HCC cell lines. WB was performed using three different HCC cell lines, HuH7 cells, HLE cells, and HepG2 cells, and abundant expression of 4.1R was found in all three cell lines at different levels (Fig. 2A).
Fig. 2.

The expression level of 4.1R protein is regulated by protein expression and activation levels of PKR in HCC cell lines. (A) Three HCC cell lines, HuH7, HLE, and HepG2 cells, were seeded in a 6-well, flat-bottomed plate, and cell lysates from three independent wells of each cell line were loaded, followed by WB with anti-4.1R antibody. Overexpression (B) and knockdown (C) of PKR and C16 treatment (D) affected the expression level of 4.1R protein. (B) HuH7 cells were transfected with or without Flag-PKR, followed by WB with anti-PKR antibody (top panel), anti-4.1R antibody (middle panel), and anti-actin antibody (bottom panel). The quantified PKR:actin ratio (center panel) and the 4.1R at 130 kDa:actin ratio (right panel) of the WB are shown. (C) HuH7 cells were transfected with control siRNA and two PKR siRNAs, followed by WB with anti-PKR antibody (top panel), anti-4.1R antibody (middle panel), and anti-actin antibody (bottom panel). The quantified PKR:actin ratio (center panel) and the 4.1R at 130 kDa:actin ratio (right panel) of WB are shown. (D) HuH7 cells were treated with DMSO or 1, 2, or 3 μM of C16 for 24 h, respectively, followed by WB with anti-phospho-PKR antibody (top panel), anti-PKR antibody (second panel), anti-4.1R antibody (third panel), and anti-actin antibody (bottom panel). The quantified PKR:actin ratio (center panel) and the 4.1R at 130 kDa: actin ratio (right panel) of WB are shown. The values in the figure represent fold change to those of control. Means ± SD values of three independent experiments are shown (*p < 0.05, **p < 0.01, ***p < 0.001 compared to control groups by the two-sided Student’s t-test or Tukey’s test).
Next, to determine the functional relationship between 4.1R and PKR, the effect of PKR on the protein expression of 4.1R was tested. As shown in Fig. 2B, overexpression of PKR increased the expression of 4.1R protein in HuH7 cells (Fig. 2B). Consistent with the result of PKR overexpression, knockdown of PKR by siRNA decreased the expression of 4.1R protein in HuH7 cells (Fig. 2C). On the other hand, knockdown of 4.1R by siRNA failed to affect the expression of PKR protein (Supplementary Fig. 4). In addition, knockdown (Supplementary Fig. 5A), as well as overexpression (Supplementary Fig. 5B), of PKR did not affect the mRNA of 4.1R in HuH7 and HepG2 cells on qRT-PCR. Moreover, the expression level of 4.1R protein was downregulated by C16, an inhibitor of phosphorylation of PKR32, in a dose-dependent manner, according to the phosphorylation levels of PKR (Fig. 2D). These results indicated that the expression level of 4.1R protein is regulated by protein expression and activation levels of PKR.
Expression of PKR and 4.1R associated with a poor prognosis of HCC patients
To clarify the importance of 4.1R in HCC prognosis, the expression levels of 4.1R were compared between HCC tissues and normal liver tissues using the available online database, UALCAN. As shown in Fig. 3A, the expression levels of 4.1R were significantly higher in HCC tissues (n = 371) than in normal liver tissues (n = 50) (p = 1.62E-12) (Fig. 3A). Accordingly, immunostaining for the surgically removed tissues in HCC patients for which both tumor and nontumor tissue were preserved was performed. In the case with HCC tissue that stained strongly for 4.1R, the HCC tissue stained more strongly than nontumor tissue surrounding the tumor (Supplementary Fig. 6A). Moreover, in the same case, the HCC tissue also stained more strongly for PKR than nontumor tissue, as for 4.1R (Supplementary Fig. 6B).
Fig. 3.
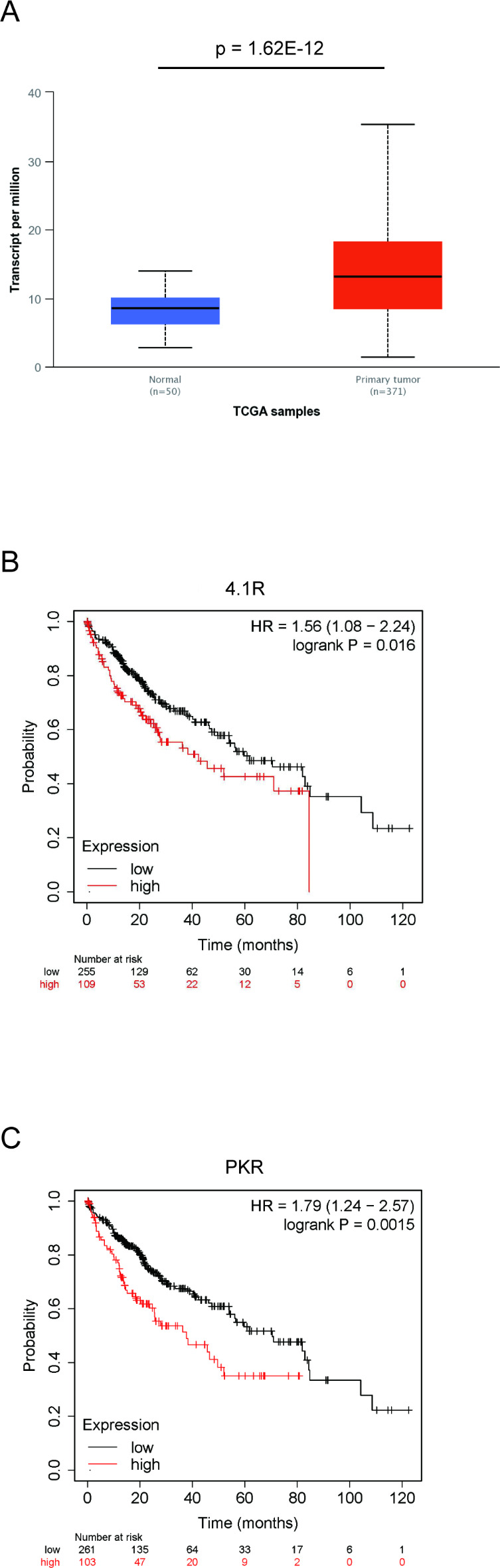
High expression of 4.1R in HCC patients leads to a poor prognosis. (A) 4.1R levels in HCC tissue and normal adjacent tissue were analyzed by the UALCAN database (p = 1.62E-12 compared to the normal group by Student’s t-test). (B, C) Survival rates based on the expression level of 4.1R (B) and PKR (C) were analyzed by the Kaplan–Meier Plotter database (p = 0.016, 0.0015, respectively, compared to the low expression group by the log-rank test).
Next, the prognostic values of 4.1R and PKR were analyzed in HCC patients using the available online database, Kaplan–Meier plotter. Interestingly, high expression of 4.1R in HCC tissues was associated with a worse prognosis in HCC patients (n = 364, OS: HR = 1.56, 95% CI = 1.08–2.24, p = 0.016) (Fig. 3B). In addition to 4.1R, high expression of PKR in HCC tissues was also associated with a poor prognosis in HCC patients (n = 364, OS: HR = 1.79, 95% CI = 1.24–2.57, p = 0.0015) (Fig. 3C). These results indicated that high expressions of PKR and 4.1R in HCC are factors associated with a poor prognosis for these patients.
Silencing of PKR and 4.1R suppresses anchorage-independent proliferation of HCC cell lines
To examine the effect of 4.1R on cell proliferation in HCC cell lines, 4.1R was knocked down using siRNA in HuH7 cells and HepG2 cells. Transfection efficiency of two siRNAs against 4.1R was confirmed by WB in HuH7 cells (Supplementary Fig. 4) and HepG2 cells (Supplementary Fig. 7).
An MTT assay of cultured cells on plates was then performed to assess anchorage-dependent proliferation of HCC cells. Knockdown of 4.1R caused no significant change in anchorage-dependent proliferation of HuH7 cells (Fig. 4A, top panel) and HepG2 cells (Fig. 4A, bottom panel).
Fig. 4.

Suppression of anchorage-independent proliferation by silencing of PKR or 4.1R in HCC cell lines. (A) HuH7 cells (top panel) and HepG2 cells (bottom panel) on culture dishes were transfected with control siRNA or two 4.1R siRNAs, followed by MTT assay. The black line, blue line, and yellow line show the results for control, 4.1R siRNA1, and 4.1R siRNA2, respectively. (B) Huh7 and HepG2 cells were transfected with control siRNA or two 4.1R siRNAs, followed by colony formation assay. Three representative images of colony formation on soft agar and their quantified colony area with control siRNA or two 4.1R siRNAs, respectively, in HuH7 (B, top panels) and HepG2 cells (B, bottom panels) are shown. (C) HuH7 cells (top panel) and HepG2 cells (bottom panel) on culture dishes were transfected with control siRNA or two PKR siRNAs, followed by MTT assay. The black line, blue line, and yellow line show the results for control, PKR siRNA1, and PKR siRNA2, respectively. (D) Huh7 and HepG2 cells were transfected with control siRNA or two PKR siRNAs, followed by colony formation assay. Three representative images of colony formation on soft agar and their quantified colony area with control siRNA or two PKR siRNAs, respectively, in HuH7 (D, top panels) and HepG2 cells (D, bottom panels) are shown. (E) Huh7 and HepG2 cells were transfected with the indicated siRNA and plasmid, respectively, followed by colony formation assay. Three representative images of colony formation on soft agar and their quantified colony area after transfection of the indicated siRNA and plasmid, respectively, in HuH7 (B, top panels) and HepG2 cells (B, bottom panels) are shown. The values indicate the mean ± SD values of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control group by the two-sided Student’s t-test or Tukey’s test.
Next, a colony formation assay was performed to assess anchorage-independent proliferation of the HCC cell lines. The 3D reconstructed images of the nucleus of the colony were first confirmed using a confocal microscope (Supplementary Fig. 8). Colony area was then evaluated using light microscopy to quantify the colony formation of the HCC cell lines in soft agar. As shown in Fig. 4B, silencing of 4.1R decreased anchorage-independent proliferation of HuH7 cells (Fig. 4B, top panel) and HepG2 cells (Fig. 4B, bottom panel).
In contrast to 4.1R, the MTT assay showed that knockdown of PKR suppressed anchorage-dependent proliferation of HuH7 cells (Fig. 4C, top panel) and HepG2 cells (Fig. 4C, bottom panel), corresponding to our previous report18 in which siRNAs with different sequences were used in HuH7 cells.
As well as 4.1R inhibition, PKR silencing also decreased anchorage-independent proliferation of HuH7 cells (Fig. 4D, top panel) and HepG2 cells (Fig. 4D, bottom panel) in the colony formation assay, and the decrease of colony formation by PKR knockdown was rescued by 4.1R overexpression in HuH7 cells (Fig. 4E, top panel) and HepG2 cells (Fig. 4E, bottom panel).
These results demonstrated that PKR silencing inhibited HCC cell growth in both anchorage-dependent and anchorage-independent manners, whereas 4.1R knockdown inhibited HCC cell growth only in an anchorage-independent manner, but not in an anchorage-dependent manner. In addition, the rescue experiment showed that 4.1R acts as a downstream molecule of PKR, at least in HCC cell growth, in an anchorage-independent manner.
Overexpression of PKR and 4.1R enhances anchorage-independent proliferation of HCC cell lines
To confirm the results of the above loss-of-function approach, a gain-of-function approach using an overexpression experiment was performed in HCC cell lines. The transfection efficiency of 4.1R plasmid was confirmed by WB in HuH7 and HepG2 (Supplementary Fig. 9).
The MTT assay showed that the overexpression of 4.1R caused no significant change in the anchorage-dependent proliferation of HuH7 cells (Fig. 5A, top panel) and HepG2 cells (Fig. 5A, bottom panel), whereas the colony formation assay demonstrated that overexpression of 4.1R increased the anchorage-independent proliferation of HuH7 cells (Fig. 5B, top panel) and HepG2 cells (Fig. 5B, bottom panel).
Fig. 5.

Increased anchorage-independent proliferation by overexpression of PKR or 4.1R in HCC cell lines. (A) HuH7 cells (top panel) and HepG2 cells (bottom panel) were transfected with or without Flag-4.1R, followed by MTT assays on culture dishes. The blue line and black line show the results with and without Flag-4.1R, respectively. (B) Huh7 and HepG2 cells were transfected with or without Flag-4.1R, followed by colony formation assay. Two representative images of colony formation on soft agar and their quantified colony area with or without Flag-4.1R in HuH7 (B, top panels) and HepG2 cells (B, bottom panels) are shown. (C) HuH7 cells (top panel) and HepG2 cells (bottom panel) were transfected with or without Flag-PKR, followed by MTT assay on culture dish. The blue line and black line show the results with and without Flag-PKR, respectively. (D) Huh7 and HepG2 cells were transfected with or without Flag-PKR, followed by colony formation assay. Two representative images of colony formation on soft agar and their quantified colony area with or without Flag-PKR in HuH7 (D, top panels) and HepG2 cells (D, bottom panels) are shown. (E) Huh7 and HepG2 cells were transfected with the indicated plasmid and siRNA, respectively, followed by colony formation assay. Three representative images of colony formation on soft agar and their quantified colony area after transfection of indicated siRNA and plasmid, respectively, in HuH7 cells (top panels) and HepG2 cells (bottom panels) are shown. The values indicate the mean ± SD values of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control group by the two-sided Student’s t-test or Tukey’s test.
On the other hand, the MTT assay showed that PKR overexpression increased anchorage-dependent proliferation of HuH7 cells (Fig. 5C, top panel) and HepG2 cells (Fig. 5C, bottom panel), corresponding to our previous report18 in which a different plasmid was used in HuH7 cells. In the colony formation assay, PKR overexpression also increased anchorage-independent proliferation of HuH7 cells (Fig. 5D, top panel) and HepG2 cells (Fig. 5D, bottom panel). Moreover, the PKR-induced colony formation was cancelled by 4.1R knockdown (Fig. 5E).
These results indicated that PKR induces both anchorage-dependent and anchorage-independent growth of HCC cell lines, but 4.1R was only involved in anchorage-independent growth of HCC cell lines as a downstream target of PKR.
Discussion
PKR is a ubiquitously expressed enzyme that initiates an immune response to RNA virus infection. In RNA virus-infected cells, PKR recognizes its dsRNA, forms a dimer, and then undergoes autophosphorylation. This activates PKR, which phosphorylates eIF2α at serine 51, which is known to have antiviral activity6. HCV causes persistent infection of hepatocytes, and it is known that PKR is also mobilized during this infection to suppress HCV replication33. Besides dsRNA, many stimuli activate PKR; these stimuli include cytokines such as interferon, tumor necrosis factor alpha (TNF-α), and interleukin 1 (IL-1) 34–36, as well as some cellular stressors including arsenite, thapsigargin, and hydrogen peroxide37,38. Through its role in several signaling pathways, PKR is involved in the regulation of inflammation and immune responses.
Furthermore, several reports have suggested that, in liver cancer, PKR may have a positive regulatory role in controlling tumor growth and progression17,39. We have previously examined PKR phosphorylation in pairs of malignant and surrounding non-malignant tissues from patients with HCV-associated HCC and have shown that PKR protein levels are consistently increased in HCV-associated HCC tissues16.
Regarding the molecular mechanism of PKR in HCC progression, we previously reported that PKR upregulated c-Fos and c-Jun activities through activation of ERK1/2 and JNK, respectively, subsequently increasing HCC cell proliferation. Moreover, the coordinated upregulation of c-Fos and c-Jun signaling was confirmed in human HCC specimens18. Moreover, C16, which is a PKR inhibitor, suppressed proliferation of HCC cells in a dose-dependent manner both in vitro and in vivo. In addition, C16 also decreased angiogenesis in HCC tissue in the xenograft model due to downregulation of angiogenesis-related growth factors, showing the effects of PKR on the microenvironment of tumors40. However, until now, the downstream substrates that bind to PKR, other than eIF2α in normal cells, have not been clarified. In the present study, 4.1R was found to be a downstream substrate interacting with PKR, and the oncogenic role of this PKR-4.1R axis in HCC cells was clarified.
To date, 4.1R has been reported to bind to the cytoskeleton, spectrin or actin, and to act as hubs for membrane protein organization. In humans, the phenotype of 4.1R mutation was reported to be hereditary elliptocytosis41. As with human 4.1R, 4.1R-knockout mice are very anemic, but they do not have a substantially reduced mortality42,43. Various phenotypes of 4.1R-knockout mice have also been reported in cells or tissues other than red blood cells, for example bradycardia, long QT44, disorganization of gastric glands45, and learning and memory defects41. 4.1B, which is one member of the protein 4.1 family, is identified to have tumor suppressive functions in lung, breast, ovarian, and prostate cancers22–25,46,47. Similarly, the homolog 4.1N has also been reported as a tumor suppressor molecule in ovarian cancer, breast cancer, and non-small cell lung cancer (NSCLC) 48–50. 4.1R has also been reported as a tumor suppressor in meningiomas and colon cancer51,52. However, 4.1R has been reported to have a tumor-promoting function in small-cell lung cancer (SCLC) 31. Thus, the role of 4.1R may differ depending on the malignant disease. In HCC, using the available online database, as shown in Fig. 3A, the expression levels of 4.1R were significantly higher in HCC tissues than in normal liver tissues. However, Yang et al. reported that 4.1R expression was significantly decreased in HCC tissue specimens, especially in portal vein metastasis or intrahepatic metastasis, compared to normal tissues53. However, this report used Taiwanese databases and analyzed mainly cases of HBV-related HCC. Thus, the role of 4.1R in HCC may differ depending on the background or context.
In the present study, modulating the expression level of 4.1R did not affect anchorage-dependent growth on culture dishes, but it did affect anchorage-independent growth in soft agar in HCC cell lines. This result suggests that 4.1R, a PKR binding protein, would enhance the malignant features of HCC by promoting anchorage-independent growth of HCC cells. This function is similar to the results in SCLC reported by Funaki et al. Namely, they identified that 4.1R is associated with anchorage-independent proliferation; 4.1R binds CADM1 at the 4.1 protein-binding motif, and the 4.1R and CADM1 complex is responsible for the promotion of colony formation; moreover, knockdown of 4.1R suppressed the colony formation enhanced by CADM1, suggesting that the formation of CADM1-4.1R would have a tumor-promoting function in SCLC31.
Anchorage-independent growth is a hallmark of carcinogenesis. After detachment from the ECM, normal cells are prevented from anchorage-independent growth by various forms of cell death, including anoikis, autophagy, entosis, and cell cycle arrest. Exposure to multiple stresses, such as loss of growth stimuli from the ECM, altered mechanical forces, cytoskeletal re-organization, reduced nutrient uptake, and increased reactive oxygen species (ROS) generation, leads to cell death. However, transformed cells acquire signaling pathways that regulate mechanical transduction, cytoskeleton re-organization, and metabolism, inhibiting cell death54. The soft agar colony formation assay is a well-established method for characterizing this capability in vitro55. Since cancer metastasis is a process in which cancer cells detach from the primary site, enter the vasculature or lymphatic vessels, localize, and reproduce at remote sites, anchorage-independent growth of cancer cells in vitro is a key aspect of the tumor phenotype, particularly with respect to metastatic potential54,56. In HCC, overcoming anoikis has also been reported to be associated with metastasis and a poor prognosis57. Then, PKR-4.1R axis might be associated with metastasis rather than carcinogenesis through anchorage-independent growth in HCC.
Although the localization of PKR and 4.1R was not confirmed in the present study, it is possible that PKR and 4.1R also form a complex and co-localize at the plasma membrane, since 4.1R generally functions to cross-link membrane proteins with the cytoskeleton, and CADM1–4.1R has been shown to co-localize at the plasma membrane in SCLC. 4.1R also has a FERM (four-point-one, ezrin, radixin, and moesin)-adjacent region (FA region) that is phosphorylated by protein kinase A (PKA) and protein kinase C (PKC). Activated PKR may regulate the mechanical properties of the 4.1R at the membrane by phosphorylating the FA region58. The above mechanisms may have allowed cells to re-organize the cytoskeleton and transmit signals from membrane proteins and acquire anchorage-independent proliferation. Considering that PKR is activated by viral infection, endoplasmic reticulum stress, and mechanical stress, such regulation of 4.1R by PKR may be an inherent cellular stress response of the organism.
In our previous study, PKR promoted anchorage-dependent proliferation through the MAPK signaling pathway18, whereas PKR also promotes anchorage-independent proliferation although its interaction with 4.1R (Fig. 6). In HCC, PKR might work in a multifunctional mode mediated by each different molecule. Therapies against the above processes or signaling pathways hold the potential to prevent or cure cancer metastasis. Since the results of the present study showed that 4.1R and PKR contributed to colony formation, future work will need to analyze whether the interaction of PKR and 4.1R affects cancer invasion and metastasis.
Fig. 6.

Schematic depicting the mechanism by which PKR enhances anchorage-independent proliferation via 4.1R and anchorage-dependent proliferation via mitogen-activated protein kinase (MAPK).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was partially funded by JSPS KAKENHI (No. JP21K08008) to YH, and supported partly by a Grant from International Joint Usage/Research Center, the Institute of Medical Science, the University of Tokyo. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data, and in writing the manuscript.
Author contributions
YO designed and performed all the experiments, analyzed the data, and wrote the manuscript; TW designed the experiments and revised the manuscript. TI (Inoue), YI (Ito), and TS provided advice and conducted the molecular experiments. YK, and RK assisted in part of the experiments. YI (Imai), YN, MK, OY, YT, MH, and MA provided technical advice on the molecular experiments and data analysis. TI (Imamura), YM, and YH supervised the experiments and critically revised the manuscript.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Villanueva, A. Hepatocellular carcinoma. N Engl. J. Med.380, 1450–1462 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Tada, T. et al. Role of hepatic resection in patients with intermediate-stage hepatocellular carcinoma: A multicenter study from Japan. Cancer Sci.108, 1414–1420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marrero, J. A. et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology68, 723–750 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Llovet, J. M. et al. Trial design and endpoints in hepatocellular carcinoma: AASLD consensus conference. Hepatology73, 158–191 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Samuel, C. E. Mechanism of interferon action: Phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. USA76, 600–604 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meurs, E. et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell62, 379–390 (1990). [DOI] [PubMed] [Google Scholar]
- 8.Takada, Y., Ichikawa, H., Pataer, A., Swisher, S. & Aggarwal, B. B. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene26, 1201–1212 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Wong, A. H. et al. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J.16, 1291–1304 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnet, M. C., Weil, R., Dam, E., Hovanessian, A. G. & Meurs, E. F. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol. Cell Biol.20, 4532–4542 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barber, G. N., Jagus, R., Meurs, E. F., Hovanessian, A. G. & Katze, M. G. Molecular mechanisms responsible for malignant transformation by regulatory and catalytic domain variants of the interferon-induced enzyme RNA-dependent protein kinase. J Biol Chem.270, 17423–17428 (1995). [DOI] [PubMed] [Google Scholar]
- 12.Yoon, C. H., Lee, E. S., Lim, D. S. & Bae, Y. S. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc Natl Acad Sci USA106, 7852–7857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roh, M. S. et al. Expression of double-stranded RNA-activated protein kinase in small-size peripheral adenocarcinoma of the lung. Pathol Int.55, 688–693 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Kim, S. H., Forman, A. P., Mathews, M. B. & Gunnery, S. Human breast cancer cells contain elevated levels and activity of the protein kinase PKR. Oncogene19, 3086–3094 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Terada, T., Maeta, H., Endo, K. & Ohta, T. Protein expression of double-stranded RNA-activated protein kinase in thyroid carcinomas: Correlations with histologic types, pathologic parameters, and Ki-67 labeling. Hum Pathol.31, 817–821 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Hiasa, Y. et al. Protein kinase R is increased and is functional in hepatitis C virus-related hepatocellular carcinoma. Am. J. Gastroenterol.98, 2528–2534 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Wang, X. et al. Double stranded RNA-dependent protein kinase promotes the tumorigenic phenotype in HepG2 hepatocellular carcinoma cells by activating STAT3. Oncol Lett.8, 2762–2768 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe, T. et al. Protein kinase R modulates c-Fos and c-Jun signaling to promote proliferation of hepatocellular carcinoma with hepatitis C virus infection. PLoS ONE.8, e67750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu, J., Fischman, D. A. & Steck, T. L. Selective solubilization of proteins and phospholipids from red blood cell membranes by nonionic detergents. J. Supramol. Struct.1, 233–248 (1973). [DOI] [PubMed] [Google Scholar]
- 20.Peters, L. L. et al. Four paralogous protein 4.1 genes map to distinct chromosomes in mouse and human. Genomics54, 348–350 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Discher, D., Parra, M., Conboy, J. G. & Mohandas, N. Mechanochemistry of the alternatively spliced spectrin-actin binding domain in membrane skeletal protein 4.1. J. Biol. Chem.268, 7186–7195 (1993). [PubMed] [Google Scholar]
- 22.Tran, Y. K. et al. A novel member of the NF2/ERM/4.1 superfamily with growth suppressing properties in lung cancer. Cancer Res.59, 35–43 (1999). [PubMed] [Google Scholar]
- 23.Zhang, Y. et al. Loss of expression of the differentially expressed in adenocarcinoma of the lung (DAL-1) protein is associated with metastasis of non-small cell lung carcinoma cells. Tumour Biol.33, 1915–1925 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Yageta, M. et al. Direct association of TSLC1 and DAL-1, two distinct tumor suppressor proteins in lung cancer. Cancer Res.62, 5129–5133 (2002). [PubMed] [Google Scholar]
- 25.Takahashi, Y. et al. Aberrant expression of tumor suppressors CADM1 and 4.1B in invasive lesions of primary breast cancer. Breast Canc.19, 242–252 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Nagata, M. et al. Aberrations of a cell adhesion molecule CADM4 in renal clear cell carcinoma. Int. J. Canc.130, 1329–1337 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Chandrashekar, D. S. et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia19, 649–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy, Á., Lánczky, A., Menyhárt, O. & Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep.8, 9227–9227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katze, M. G. et al. Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol. Cell Biol.11, 5497–5505 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang, S. H. et al. Mitotic regulation of protein 41R involves phosphorylation by cdc2 kinase. Mol. Biol. Cell.16, 117–127 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funaki, T. et al. CADM1 promotes malignant features of small-cell lung cancer by recruiting 4.1R to the plasma membrane. Biochem. Biophys. Res. Commun.534, 172–178 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Jammi, N. V., Whitby, L. R. & Beal, P. A. Small molecule inhibitors of the RNA-dependent protein kinase. Biochem. Biophys. Res. Commun.308, 50–57 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Tokumoto, Y. et al. Hepatitis C virus expression and interferon antiviral action is dependent on PKR expression. J. Med. Virol.79, 1120–1127 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Yeung, M. C., Liu, J. & Lau, A. S. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc. Natl. Acad. Sci. USA93, 12451–12455 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goh, K. C., deVeer, M. J. & Williams, B. R. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J.19, 4292–4297 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe, T., Imamura, T. & Hiasa, Y. Roles of protein kinase R in cancer: Potential as a therapeutic target. Cancer Sci.109, 919–925 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito, T., Yang, M. & May, W. S. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem.274, 15427–15432 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Ruvolo, P. P., Gao, F., Blalock, W. L., Deng, X. & May, W. S. Ceramide regulates protein synthesis by a novel mechanism involving the cellular PKR activator RAX. J Biol Chem.276, 11754–11758 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Shimada, A. et al. Aberrant expression of double-stranded RNA-dependent protein kinase in hepatocytes of chronic hepatitis and differentiated hepatocellular carcinoma. Cancer Res.58, 4434–4438 (1998). [PubMed] [Google Scholar]
- 40.Watanabe, T. et al. Therapeutic effects of the PKR inhibitor C16 suppressing tumor proliferation and angiogenesis in hepatocellular carcinoma in vitro and in vivo. Sci. Rep.10, 5133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gallagher, P. G. Hereditary elliptocytosis: spectrin and protein 4.1R. Semin Hematol.41, 142–164 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Shi, Z. T. et al. Protein 4.1R-deficient mice are viable but have erythroid membrane skeleton abnormalities. J. Clin. Invest.103, 331–340 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi, Z. T. et al. Protein 4.1-R knockout mice exhibit mechanically unstable erythrocytes and unexpected neurological defects. Blood90, 1168–1168 (1997).9242549 [Google Scholar]
- 44.Stagg, M. A. et al. Cytoskeletal protein 4.1R affects repolarization and regulates calcium handling in the heart. Circ. Res.103, 855–863 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Yang, S., Guo, X., Debnath, G., Mohandas, N. & An, X. Protein 4.1R links E-cadherin/beta-catenin complex to the cytoskeleton through its direct interaction with beta-catenin and modulates adherens junction integrity. Biochim. Biophys. Acta.1788, 1458–1465 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong, S. Y. et al. Protein 4.1B suppresses prostate cancer progression and metastasis. Proc. Natl. Acad. Sci. USA104, 12784–12789 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dafou, D. et al. Microcell-mediated chromosome transfer identifies EPB41L3 as a functional suppressor of epithelial ovarian cancers. Neoplasia12, 579–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xi, C. et al. Defective expression of Protein 4.1N is correlated to tumor progression, aggressive behaviors and chemotherapy resistance in epithelial ovarian cancer. Gynecol. Oncol.131, 764–771 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Ji, Z. et al. The membrane-cytoskeletal protein 4.1N is involved in the process of cell adhesion, migration and invasion of breast cancer cells. Exp Ther Med.4, 736–740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang, Z. et al. Correction: Protein 41N acts as a potential tumor suppressor linking PP1 to JNK-c-Jun pathway regulation in NSCLC. Oncotarget10, 6285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robb, V. A. et al. Identification of a third Protein 4.1 tumor suppressor, Protein 4.1R, in meningioma pathogenesis. Neurobiol. Dis.13, 191–202 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Lu, Y. et al. The protein 4.1R downregulates VEGFA in M2 macrophages to inhibit colon cancer metastasis. Exp. Cell Res.409, 112896 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Yang, X. et al. Integrative functional genomics implicates EPB41 dysregulation in hepatocellular carcinoma risk. Am. J. Hum. Genet.4, 275–286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng, Z., Wang, H., Liu, J., Deng, Y. & Zhang, N. Comprehensive understanding of anchorage-independent survival and its implication in cancer metastasis. Cell Death Dis.12, 629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borowicz, S. et al. The soft agar colony formation assay. J. Vis. Exp.92, e51998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strilic, B. & Offermanns, S. Intravascular survival and extravasation of tumor cells. Cancer Cell.32, 282–293 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Shimokawa, M. et al. Modulation of Nqo1 activity intercepts anoikis resistance and reduces metastatic potential of hepatocellular carcinoma. Cancer Sci.111, 1228–1240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baines, A. J. A FERM-adjacent (FA) region defines a subset of the 4.1 superfamily and is a potential regulator of FERM domain function. BMC Geno.7, 85 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.