

Abstract
T-cell large granular lymphocyte leukemia (T-LGLL), which is associated with autoimmune diseases, has been described. However, T-LGLL with B-cell dyscrasias presenting as autoimmune hemolytic anemia has been less frequently reported. Here, we report a rare case of combined papillary thyroid cancer (PTC), discuss the possible relationship between autoimmune hemolytic anemia (AIHA) and TLGLL, and review the literature. The patient presented with pancytopenia and systemic jaundice. Fortunately, the patient responded well to hormone and immunosuppressive (methotrexate) therapy. However, this insight is based on one rare case, and the pathogenesis of this disease requires further clinical research for clarification.
Keywords: T-LGLL, AIHA, IL-6, PTC, MGUS
Introduction
T-cell large granular lymphocytic leukemia (T-LGLL) is an indolent lymphoproliferative disorder caused by cytotoxic T-cells and natural killer (NK) cells. This clinically rare disease is characterized by the clonal expansion of large granular lymphocytes [1,2]. The initial clinical manifestations are mainly neutropenia including recurrent oral ulcers, fever secondary to bacterial infection, splenomegaly in 20-50% of cases, and, less commonly, lymphadenopathy [3]. T-LGLL is often associated with immune disorders, including autoimmune hemolytic anemia (AIHA), immune thrombocytopenia, and rheumatoid arthritis (RA), which often coexist with secondary tumors [3]. STAT3 and STAT5B mutations are the most common mutations in T-LGLL and NK chronic lymphoproliferative disorders [4]. In most cases, the immunophenotype is CD3+, TCRαβ+, CD4-, CD5dim, CD8+, CD16+, CD27-, CD28-, CD45R0-, CD45RA+, and CD57+ [5]. AIHA is characterized by hemolysis, which is the breakdown of red blood cells, mediated by autoantibodies and/or complement. Activated macrophages, tumor lymphocytes, and cytokines are also involved in the process [6].
We report a case of T-cell large granular lymphocytic leukemia (T-LGLL) with autoimmune hemolytic anemia (AIHA), B-cell clones, and papillary thyroid carcinoma (PTC). We provided some new insights into the diagnosis and etiology of the disease and analyzed the literature retrospectively.
Case presentation
A 56-year-old woman was treated at a local hospital for shortness of breath after activity with nausea and vomiting for 10 months prior to admission. Her condition gradually worsened with jaundice and fever over the previous half month. In April 2023, she was admitted to the Department of Hematology at Hebei Provincial People’s Hospital; her vital signs were normal, and her main symptoms were severe anemia and generalized jaundice.
The laboratory examination results upon admission are presented in Table 1. Serum immunofixation electrophoresis was positive, and a dyscrasia M-protein precipitation band was visible on electrophoresis of the IgG kappa light chain type. Computed tomography (CT) of the abdominopelvic region suggested splenomegaly. Peripheral blood and bone marrow smears revealed an increased ratio of lymphoid lineage cells and LGL (Figure 1). The bone marrow biopsy and immunostaining revealed positive staining as presented in Figure 2. Positron emission tomography-CT (PET-CT) revealed enlargement of the liver and spleen (Figure 3). Additionally, PET-CT indicated a high metabolic low-density nodule in the middle of the left thyroid gland. Combined with the results of B-mode ultrasound and pathological examination, the lesion was diagnosed as PTC (Figure 4). Flow cytometry (FC) of peripheral blood illustrated that the clonal T-cell large granular lymphocytes (T-LGLs) expressed CD3, CD57, CD8, CD45RA, and CD16. These cells additionally exhibited partial CD57 expression and low CD5 and CD2 expression, and they were negative for CD4 and CD56. Monoclonal B-cells expressed CD5dim, whereas they were negative for CD10 (Figure 5). The TCR rearrangements were TCRαβ+, and TCRγΔ+.
Table 1.
Laboratory findings before treatment
| Reference Range | |||
|---|---|---|---|
| CBC | WBC | 3.27*10^9/L↓ | (3.5-9.5) *10^9/L |
| NE | 1.5*10^9/L↓ | (1.8-6.3) *10^9/L | |
| LY | 1.55*10^9/L | (1.1-3.2) *10^9/L | |
| EO | 0↓ | (0.02-0.52) *10^9/L | |
| RBC | 1.94*10^12/L↓ | (3.8-5.1) *10^9/L | |
| HB | 61 g/L↓ | (115-150) g/L | |
| PLT | 105*10^9/L↓ | (125-300) *10^9/L | |
| Coombs test | Direct Coombs test | Positive | Negative |
| Direct Coombs Cd3 test | Positive | Negative | |
| ANA | ANA | Negative | Negative |
| Anti-Ro52 | 33↑ | Negative ≤15 | |
| SPE | ALPHA1 | 6.5%↑ | (1.0-3.2)% |
| Biochemistry | T.P | 57.0 g/L↓ | (65-85) g/L |
| GLOB | 39.6 g/L↓ | 40-55 g/L | |
| ALP | 121.8 U/L | (50-135) U/L | |
| TBIL | 90.5 µmol/L↑ | (0-23) μmol/L | |
| IBIL | 74.4 μmol/L↑ | / | |
| AST | 77.3 U/L↑ | (13-35) U/L | |
| LDH | 1056.1 U/L↑ | (120-250) U/L |
Notes: ↑Denotes above the upper limit of the reference range, and ↓denotes below the lower limit of the reference range. Abbreviations: CBC, complete blood count; WBC, white blood cell; NE, neutrophil; LY, lymphocyte; EO, eosinophil; RBC, red blood cell; Hb, hemoglobin; PLT, plate; ANA, antinuclear antibody; SPE, serum protein electrophoresis; T.P, total protein; GLOB, globulin; ALP, alkaline phosphatase; TBIL, total bilirubin; IBIL, indirect bilirubin; AST, aspartate amino transferase; LDH, lactate dehydrogenase.
Figure 1.

Smears of peripheral blood and bone marrow cells. A: Peripheral blood smears. B: Bone marrow cell smear (magnification, ×1000). Blue arrow: abnormal lymphocytes.
Figure 2.

The immunostaining of bone marrow biopsy specimen. A: ×100. The H&E staining of bone marrow. Bone marrow hyperplasia was hyperactive (about 90%). B: ×400. The H&E stain of bone marrow. The proportion of erythroid system increased, the granulocyte system decreased, the megakaryocytes slightly increased, and lymphocytes and plasma cells were scattered. C: ×40. CD3 positive. D: ×40. CD20 positive.
Figure 3.

PET/CT finding. A: Diffuse bone marrow is hypermetabolism. B: Liver and spleen were enlarged but no metabolic abnormalities were observed.
Figure 4.

The image of thyroid gland B-mode ultrasound and immunostaining of thyroid gland biopsy specimen. A: The image of thyroid gland B-mode ultrasound. The left lobe of the thyroid gland shows a hypoechoic nodule with indistinct margins (highlighted by the red arrow). B: ×100. The H&E stain of thyroid gland. Hyperplasia and crowding with papillary structure.
Figure 5.

Flow cytometry of peripheral blood. A-G. The purple cell population is clonal T-LGL. The aberrant lymphocytes population expressed CD3, CD57, CD8, CD45RA, CD16; partially expressed CD57; low expressed CD5, CD2, negative CD4, CD56. H, I. The red cell population is monoclonal B-cells. The aberrant lymphocytes population expressed CD5dim; negative CD10.
Based on these clinical manifestations and laboratory findings, diagnoses of T-LGLL, AIHA with monoclonal gammopathy of undetermined significance (MGUS), and PTC were considered. The patient was administered moxifloxacin to fight infection, and treatments were additionally administered to improve anemia and increase white blood cell counts. After a clear diagnosis, methylprednisolone infusion was administered for 10 days to control hemolysis (80 mg/day), suspended red blood cells were infused to improve anemia, and liver and gastric protection was performed to support symptomatic treatment. AIHA responds to steroid therapy and transfusion support. After discharge, the patient was given oral methylprednisolone 60 mg/day, and the dose was gradually reduced. During the follow-up visit, oral methotrexate 5 mg/week was added, and the dose was gradually increased to 8 mg/week. The patient’s laboratory findings and clinical symptoms were significantly improved (Figure 6).
Figure 6.
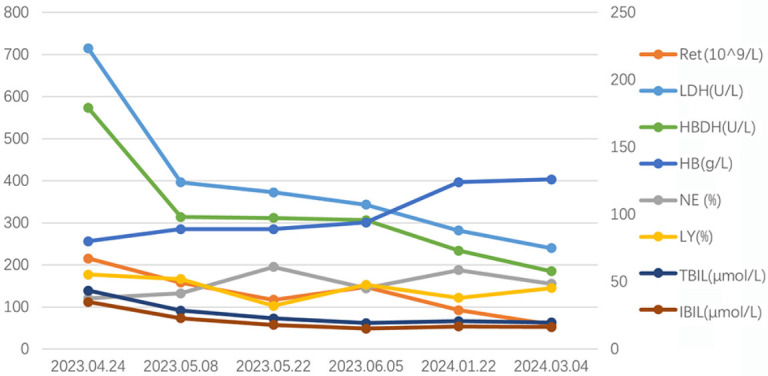
The changes of LDH, HBDH, TBIL, IBIL HB, Ret, NE, LY. The series of LDH, HBDH and Ret are drawn on the primary axis and the series of HB, NE, LY, TBIL and IBIL are drawn on the secondary axis. Abbreviations: LDH: lactate dehydrogenase; HBDH: hydroxybutyrate dehydrogenase; TBIL: total bilirubin; IBIL: indirect bilirubin; HB, hemoglobin; Ret: reticulocyte; NE: neutrophils; LY: lymphocyte.
Discussion
T-LGLL comprises a group of chronic lymphoproliferative disorders closely related to autoimmune diseases [1]. The first step in the diagnosis of T-LGLL is detecting an elevated number of circulating LGLs, as indicated by an LGL count of >2.0 × 109/L (normal: <0.3 × 109/L) [5-8]. However, the results of a peripheral blood smear, bone marrow biopsy, and immunophenotyping did not facilitate a simple diagnosis of T-LGLL in our case. No significant change in T- LGL counts was observed in the peripheral blood smear. However, Semenzato and others [8] found that an absolute LGL count of >2.0 × 109/L is not necessary for a diagnosis of T-LGLL, and T-LGLL can be diagnosed if these low-count LGL populations are clonal and the patient exhibits other clinical or hematological features such as RA or hematopoiesis [5,8]. Therefore, the diagnosis of T-LGLL was considered by combining the laboratory results and clinical manifestations of the patient with an observed increase in monoclonal B cell counts. Additionally, the patient had positive immunofixation electrophoresis with concomitant monoclonal gammaglobulinemia. This association of T-LGL with B-cell abnormalities was reported previously. The most common B-cell dyscrasias is MGUS, a disorder of unknown significance [9]. However, the IgG count was lower than normal in our patient, which has been rarely reported. Therefore, a more reasonable explanation in our view is the reduced immunoglobulin level in the background of T-LGLL, possibly attributable to the abnormal immune function of B- and T-cells. The abnormal function and number of B-cells make plasma cells unable to effectively mediate the humoral immune clearance of pathogens, leading to the continuous chronic antigen stimulation of T-cells involved in the immune response, and the clonal expansion of T-cells leads to T-LGLL [9].
The majority of patients with T-LGLL have autoimmune diseases. Before displaying obvious leukemia symptoms, some patients with T-LGLL can already have symptoms of autoimmune disease, suggesting that some autoimmune diseases are secondary and reflect an indolent course of leukemia [10]. The management of patients with AIHA requires appropriate methods to diagnose AIHA, a determination of whether AIHA is primary or secondary, and identification of the most effective treatment for specific patients [11]. Secondary causes mainly include lymphoproliferative disorders such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, and Waldenström macroglobulinemia; autoimmune diseases; infections; immune system disorders; and neoplasms [6,12,13]. T-LGLL is commonly associated with RA or other autoimmune diseases, but autoimmune hemolytic anemia is rarely reported. We reviewed the literature on AIHA with T-LGLL (Table 2) [14-18]. The findings highlighted the need for additional vigilance regarding the presence of an underlying lymphoid malignancy causing AIHA, and this case might have induced by T-LGLL.
Table 2.
Analysis of AIHA complicated with TLGLL
| First author | Diagnosis | Surface marker | Primary AIHA/secondary AIHA | Treatment | Year |
|---|---|---|---|---|---|
| Qin [14] | PRCA, AIHA with TLGLL | CD2+, CD4-, CD5+, CD7+, CD8+, CD56+/-, CD57+, TCRα/β | NOT SURE | Methylprednisolone, CsA | (2016) |
| Muñoz [15] | Evans syndrome, RA with TLGLL | CD3+, CD4+, CD5+, CD7+, CD8 | AIHA secondary to TLGLL | gammaglobulin, corticosteroid therapy, and rituximab | (2021) |
| Alfano [16] | kidney transplantation, AIHA with TLGLL | —— | AIHA secondary to TLGLL | corticosteroids and immunosuppressive therapy | (2020) |
| Seibert [17] | Thymoma, PRCA, AIHA with TLGLL | CD3+, CD8+, CD2+, CD7+, CD57+, CD5-, CD56-, CD25- | AIHA secondary to thymoma | Corticosteroids, octreotide, MTX and methylprednisolone | (2022) |
| Kitchen [18] | HIGM syndrome, AIHA, intermittent lymphadenopathy with TLGLL | —— | NOT SURE | MTX | (2008) |
Notes: PRCA, pure red cell aplasia; RA, rheumatoid arthritis; HIGM, hyper-IgM; CsA, Cyclosporin A; MTX, Methotrexate.
We attempted to identify a possible link between these two diseases. The inflammatory response plays a major role in cancer development, including tumor initiation, promotion, progression, and metastasis. Cytokines are considered important mediators linking inflammation and cancer [19]. Friedman and others [20] found that the levels of cytokines (especially soluble interleukin-2 receptor, tumor necrosis factor-α, IL-6, and IL-8) in the culture supernatant of T-LGLL cells were significantly higher than those in healthy controls. IL-6 expression in this patient was high according to bone marrow biopsy (Figure 7). In addition, Zaninon and others [21] found significant increases in IL-6 and Il-10 expression in controls in a study of 123 patients diagnosed with AIHA. Therefore, the increased expression of IL-6 in patients might play a major role in the occurrence and development of PTC, T-LGLL, and AIHA.
Figure 7.

The immunostaining of cytokines. A: H&E staining of bone marrow IL-6. B: H&E staining of bone marrow IL-6. (magnification, ×400).
Abnormal LGL cells can cause neutropenia in patients. Therefore, it is necessary to be aware of repeated infection. Additionally, T-LGLL is an inert disease. The standard treatment of LGLL is immunosuppressive therapy, but this treatment strategy is mainly based on a small retrospective series of studies [5]. First-line therapy relies on the use of a single immunosuppressant, such as methotrexate, cyclophosphamide, or cyclosporine A. Since the first study on the efficacy of methotrexate, the drug has been considered to be the best first-line treatment, especially for patients with neutropenia. Oral cyclophosphamide has been given priority for the treatment of patients with major anemia [22]. Concerning the current case, the patient responded to immunosuppressive therapy. At present, the patient’s condition is stable, and follow-up has continued on outpatient basis.
In conclusion, our experience is limited to only one patient, and further studies are needed to clarify the relationship between T-LGL and B-cell abnormalities and autoimmune diseases as well as the underlying pathological mechanisms.
Acknowledgements
We thank Dr. Joe Barber Jr from Liwen Bianji (Edanz) (https://www.liwenbianji.cn) for editing the language of a draft of this manuscript.
Written informed consent for this case report was obtained from the patient.
Disclosure of conflict of interest
None.
References
- 1.Barilà G, Calabretto G, Teramo A, Vicenzetto C, Gasparini VR, Semenzato G, Zambello R. T cell large granular lymphocyte leukemia and chronic NK lymphocytosis. Best Pract Res Clin Haematol. 2019;32:207–216. doi: 10.1016/j.beha.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Loughran TP Jr, Kadin ME, Starkebaum G, Abkowitz JL, Clark EA, Disteche C, Lum LG, Slichter SJ. Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med. 1985;102:169–175. doi: 10.7326/0003-4819-102-2-169. [DOI] [PubMed] [Google Scholar]
- 3.Mohan SR, Maciejewski JP. Diagnosis and therapy of neutropenia in large granular lymphocyte leukemia. Curr Opin Hematol. 2009;16:27–34. doi: 10.1097/MOH.0b013e32831c8407. [DOI] [PubMed] [Google Scholar]
- 4.Muñoz-García N, Jara-Acevedo M, Caldas C, Bárcena P, López A, Puig N, Alcoceba M, Fernández P, Villamor N, Flores-Montero JA, Gómez K, Lemes MA, Hernández JC, Álvarez-Twose I, Guerra JL, González M, Orfao A, Almeida J. STAT3 and STAT5B mutations in T/NK-cell chronic lymphoproliferative disorders of large granular lymphocytes (LGL): association with disease features. Cancers (Basel) 2020;12:3508. doi: 10.3390/cancers12123508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129:1082–1094. doi: 10.1182/blood-2016-08-692590. [DOI] [PubMed] [Google Scholar]
- 6.Michalak SS, Olewicz-Gawlik A, Rupa-Matysek J, Wolny-Rokicka E, Nowakowska E, Gil L. Autoimmune hemolytic anemia: current knowledge and perspectives. Immun Ageing. 2020;17:38. doi: 10.1186/s12979-020-00208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loughran TP Jr. Clonal diseases of large granular lymphocytes. Blood. 1993;82:1–14. [PubMed] [Google Scholar]
- 8.Semenzato G, Zambello R, Starkebaum G, Oshimi K, Loughran TP Jr. The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood. 1997;89:256–260. [PubMed] [Google Scholar]
- 9.Viny AD, Lichtin A, Pohlman B, Loughran T, Maciejewski J. Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma. 2008;49:932–938. doi: 10.1080/10428190801932635. [DOI] [PubMed] [Google Scholar]
- 10.Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk. 2012;12:400–405. doi: 10.1016/j.clml.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971–2979. doi: 10.1182/blood-2016-11-693689. [DOI] [PubMed] [Google Scholar]
- 12.Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258–271. doi: 10.1002/ajh.10062. [DOI] [PubMed] [Google Scholar]
- 13.Crowther M, Chan YL, Garbett IK, Lim W, Vickers MA, Crowther MA. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036–4040. doi: 10.1182/blood-2011-05-347708. [DOI] [PubMed] [Google Scholar]
- 14.Qin X, Yu Y, Yan S, Wang R, Liu X, Chen C. Pure red cell aplasia and autoimmune hemolytic anemia sequentially occurring in a patient with large granular T-lymphocytic leukemia. Intern Med. 2016;55:1491–1496. doi: 10.2169/internalmedicine.55.5252. [DOI] [PubMed] [Google Scholar]
- 15.Muñoz AM, Orozco Niño JA, Seehaus CM, Giménez Conca AD, Chuliber FA, Lo Giudice LF, Bendek GE. Evans syndrome and rheumatoid arthritis as autoimmune manifestations of large granular T-cell leukemia. Medicina (B Aires) 2021;81:1060–1064. [PubMed] [Google Scholar]
- 16.Alfano G, Ferrari A, Fontana F, Damiano F, Solazzo A, Mori G, Cappelli G. Hemolytic anemia as presentation of T-cell large granular lymphocytic leukemia after kidney transplantation: a case report. Transplant Proc. 2020;52:1617–1618. doi: 10.1016/j.transproceed.2020.02.183. [DOI] [PubMed] [Google Scholar]
- 17.Seibert T, Loehrer PJ, O’Brien ARW. Thymoma with triple threat: pure red cell aplasia, autoimmune hemolytic anemia, and T-cell large granular lymphocytic leukemia. J Hematol. 2022;11:223–232. doi: 10.14740/jh1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitchen BJ, Boxer LA. Large granular lymphocyte leukemia (LGL) in a child with hyper IgM syndrome and autoimmune hemolytic anemia. Pediatr Blood Cancer. 2008;50:142–145. doi: 10.1002/pbc.20902. [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26:54–74. doi: 10.1016/j.smim.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Lumachi F, Basso SM, Orlando R. Cytokines, thyroid diseases and thyroid cancer. Cytokine. 2010;50:229–233. doi: 10.1016/j.cyto.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Zaninoni A, Fattizzo B, Pettine L, Vercellati C, Marcello AP, Barcellini W. Cytokine polymorphisms in patients with autoimmune hemolytic anemia. Front Immunol. 2023;14:1221582. doi: 10.3389/fimmu.2023.1221582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program. 2012;2012:652–659. doi: 10.1182/asheducation-2012.1.652. [DOI] [PubMed] [Google Scholar]