

Abstract
Precancerous lesions provide insight into tumor development as well as prognostication, since distinguishing high-risk from benign disease will stratify clinical management. In a recent issue of The Journal of Pathology, Ghosh et al performed comprehensive genomic characterization of the precancerous lesion leukoplakia, comparing RNA and DNA with peripheral blood, normal mucosa, and squamous cell carcinoma (SCC) of the gingivobuccal region of the oral cavity from the same 28 individuals. The data paint a picture of increasing mutation and early caspase-8 inactivation on the background of inflammation with decreasing immune surveillance in the progression from benign leukoplakia to SCC. This research points to an opportunity for disease intercept at the premalignant niche prior to the development of malignancy.
Keywords: head and neck squamous cell carcinoma, leukoplakia, CASP8
Many cancers evolve through a natural history of progression, beginning with benign precancerous lesions that present a clinical conundrum, since only a small proportion will progress to malignancy. In the oral cavity, leukoplakia (oral leukoplakia or OLK) is a white thickening of the mucosa, which progresses to overt cancer in ~9% of individuals and our understanding of this process has, until recently, been limited [1]. Previous studies have identified immunological dysregulation, increasing mutation burden as well as specific changes to signaling pathways and epidermal growth factor receptor (EGFR) amplification with similarity as well as differences compared with squamous cell carcinoma (SCC), the predominant cancer of the oral cavity [2-4].
In a recent issue of The Journal of Pathology, Ghosh et al studied OLK arising in the gingivobuccal region of the oral cavity from 28 individuals all of whom reported tobacco use (the majority of whom (25/28) used chewing tobacco). By comparing DNA and RNA profiles with concurrent SCC and adjacent normal skin in the same individual, the authors describe a sequence of events beginning with mutation induction followed by immune dysregulation, further mutation, and eventual tumor formation [5]. A causal relationship was identified on the basis of mutations shared between SCC and OLK, which were only present in those SCC-OLK pairs in close proximity (all 22 pairs in close proximity shared mutations, while 0 of 6 pairs separated by the greatest distance did not). A statistically significant relationship between distance and percent of mutations shared and mutation burden followed a trajectory from normal mucosa, to OLK without SCC (n = 11), to OLK with SCC, and then SCC (having the highest burden of mutation). Of note CASP8, a gene previously identified to be heavily mutated in cancers with a strong inflammatory component [6], was the driver-gene most frequently shared, and mathematical modeling based on allele frequency suggested that this mutation occurred early, even earlier than TP53, pointing to a gatekeeper event in chewing tobacco-associated SCC [5]. In addition, increased expression and mutational activity of the endogenous apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC) deaminases (associated with tissue damage-driven SCC [7] and SCC of the head and neck [8]) were found to be concomitant with increased mutation burden in both SCC and OLK [5]. Interestingly, signatures of mutation associated with APOBEC activity were found in three OLK lesions, suggesting that aberrant APOBEC activity can be triggered in precancer. Since efforts to characterize normal tissues have yet to reveal significant APOBEC activity [9], these data hint that this mode of mutagenesis may be active before the onset of overt carcinogenesis. One caveat here is that the number of mutations identified were low and more in-depth characterization using whole-genome sequencing would provide definitive answers to the question of timing for APOBEC mutagenesis in the oral cavity [8]. With respect to the timing of tumor progression, molecular modeling based on mutation data estimated the average time taken for progression of OLK to SCC to be around 7 years, with a substantial number of patients having an average time of around 3 years and others more than 18 years. However, no patient-reported data were collected in this respect (the time OLK was present prior to SCC diagnosis), which would have been helpful for confirmation of this calculated timeline.
In agreement with earlier studies [1,4,10], analysis of immune cell complement identified strong decreases in CD8+ T-cells and strong increases in macrophages, mast cells, and CD4+ regulatory T-cells in SCC compared with leukoplakia [5]. Taken together with the mutation analysis, these data paint a picture of increasing mutation, inflammation, and early CASP8 inactivation on the background of decreasing immune surveillance in the progression from OLK to SCC (Figure 1).
Figure 1.
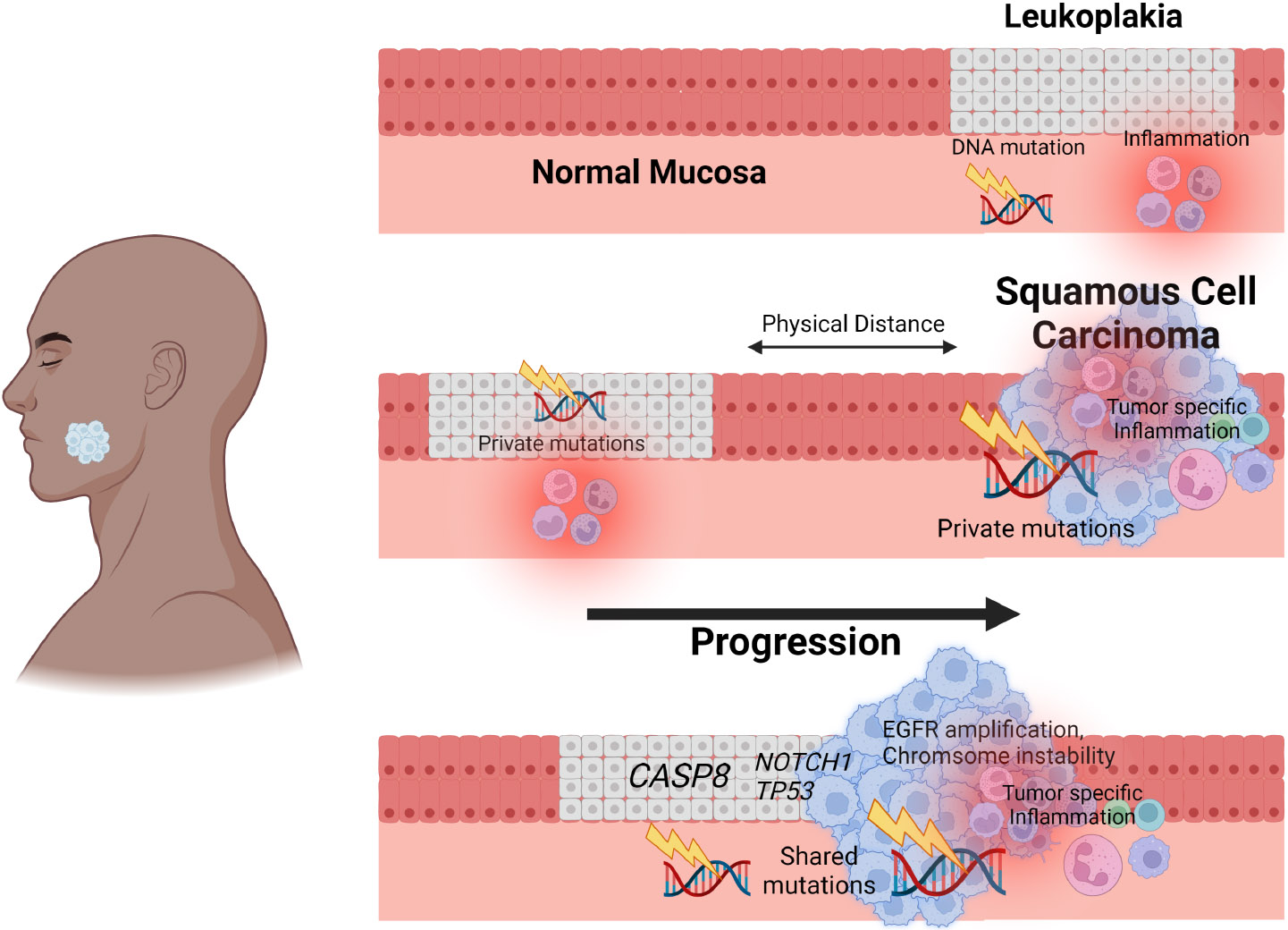
Integrative analysis identifies signatures of gingivobuccal oral cancer initiation and progression. Analysis of precancerous leukoplakia and matched squamous cell carcinoma (SCC) from the same individuals identifies that leukoplakia-SCC pairs in close proximity share molecular characteristics and highlights a sequence of events beginning with CASP8 mutation and culminating in immune dysregulation that leads to tumor initiation and progression.
In the clinic, treatment of head and neck cancer with immune checkpoint inhibition (ICI), such as nivolumab and pembrolizumab (antiPD-1), have demonstrated efficacy in a subpopulation of patients but limited or no clinical response in others. This resistance to ICI has been related to a number of mechanisms, including tumor cell adaption with malfunction of the antigen-presenting machinery, altered T-cell function, defective dendritic cells, alterations of metabolism, and upregulation of additional checkpoints (i.e. TIM3, TIGIT, LAG3) [10]. In this context, the correlation of oral tumor progression associated with depletion of CD8+ T cells begs the questions of what comes first, or is there a mechanism of action that connects these observations? That Ghosh et al found a gradient of immune cell complement transformation from normal to OLK and then on to malignancy indicates a synchronous evolution of genetic alteration. As carcinogens are resulting in genetic alterations, the simultaneous mechanical and chemical irritation of mucosa sets up a protumor inflammatory infiltrate in the premalignant niche. This combination is in essence a synergy that allows the party to rage without the parent home.
In summary, the study by Ghosh et al identifies both genetic and immune environmental characteristics in the premalignant niche that are drivers for the development of head and neck cancer. The multi-omics characterization helps determine the transformation and progression of head and neck squamous cell carcinoma. The identification of early events in CASP8, followed by pathogenic NOTCH1, HRAS and TP53 mutation, increased APOBEC activity, EGFR amplification, chromosome arm level instability, and finally alterations of the immune microenvironment all allow for a potential disease intercept opportunity at the point of premalignant lesion identification.
Footnotes
Invited commentary for Ghosh et al. Integrative analysis of genomic and transcriptomic data of normal, tumour, and co-occurring leukoplakia tissue triads drawn from patients with gingivobuccal oral cancer identifies signatures of tumour initiation and progression. J Pathol 2022; 257:593–606.
No conflicts of interest were declared.
References
- 1.Farah CS. Molecular, genomic and mutational landscape of oral leukoplakia. Oral Dis 2021; 27: 803–812. [DOI] [PubMed] [Google Scholar]
- 2.Foy JP, Bertolus C, Ortiz-Cuaran S, et al. Immunological and classical subtypes of oral premalignant lesions. Oncoimmunology 2018; 7: e1496880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farah CS, Jessri M, Bennett NC, et al. Exome sequencing of oral leukoplakia and oral squamous cell carcinoma implicates DNA damage repair gene defects in malignant transformation. Oral Oncol 2019; 96: 42–50. [DOI] [PubMed] [Google Scholar]
- 4.Das D, Maitra A, Panda CK, et al. Genes and pathways monotonically dysregulated during progression from normal through leukoplakia to gingivo-buccal oral cancer. NPJ Genom Med 2021; 6: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh A, Das C, Ghose S, et al. Integrative analysis of genomic and transcriptomic data of normal, tumour, and co-occurring leukoplakia tissue triads drawn from patients with gingivobuccal oral cancer identifies signatures of tumour initiation and progression. J Pathol 2022; 257: 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rooney MS, Shukla SA, Wu CJ, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015; 160: 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho RJ, Alexandrov LB, den Breems NY, et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci Transl Med 2018; 10: eaas9668. [DOI] [PubMed] [Google Scholar]
- 8.Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature 2020; 578: 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martincorena I, Fowler JC, Wabik A, et al. Somatic mutant clones colonize the human esophagus with age. Science 2018; 362: 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cramer JD, Burtness B, Le QT, et al. The changing therapeutic landscape of head and neck cancer. Nat Rev Clin Oncol 2019; 16: 669–683. [DOI] [PubMed] [Google Scholar]