

Abstract
The PTCD3 gene product (protein PTCD3 or MRPS39) forms the entry channel of the mitochondrial small ribosomal subunit and binds to single‐stranded mRNA. Here, we expand on the clinical manifestations of PTCD3 pathogenic variants by describing an early‐onset patient with Leigh‐like syndrome and two patients with milder form of disease, with combined oxidative phosphorylation deficiency. A 34‐year‐old male and his 33‐year‐old sister both have horizontal nystagmus, pronounced rough tremor, truncal ataxia, dysmetria, spasticity and hyperreflexia. The basal respiration rate decreased significantly for the male patient and his mother (p < 0.0001) compared to the controls. The whole genome sequencing analysis revealed two heterozygous variants in the PTCD3: c.1182T>A, p.(Tyr394Ter) and c.805C>T, p.(His269Tyr). Tyr394Ter variant ablates the C‐terminal half of the protein, including a significant portion of the central fold. In silico modelling for the variant His269Tyr shows that the inclusion of the slightly larger tyrosine sidechain is well tolerated, with no significant change in either the position or the movement of the surrounding area. The third case is a 9‐year‐old boy, who has a global developmental delay, central hypotonia, hyperreflexia and abnormal MRI. PTCD3 pathogenic variant c.538+4A>G was identified by whole exome sequencing. To test the variant's effect on splicing, an RT‐PCR experiment was performed, which revealed skipping of an out‐of‐frame exon 7.
Keywords: Leigh‐like syndrome, pentatricopeptide repeat, PTCD3
Synopsis.
Loss of function genetic variants in the PTCD3 gene are usually associated with severe progressive disease with neurodevelopmental problems and early death; here we describe also a new and a milder form of this disease—combined oxidative phosphorylation deficiency, which presents with optic atrophy, ataxia and neuropathy.
1. INTRODUCTION
Mitochondrial ribosomes or mitoribosomes, similar to cytoplasmic ribosomes, have a large 39S subunit (mt‐LSU) and a small 28S subunit (mt‐SSU). 1 The product of the pentatricopeptide repeat domain 3 (PTCD3) gene contributes to the function of the mitochondrial small ribosomal subunit. PTCD3 (or mS39) forms the entry channel of the mitoribosome and binds to single‐stranded mRNA. 2 The entire C‐terminal two‐thirds of PTCD3 consists of multiple pentatricopeptide repeats (PPR), which are necessary for interaction with RNA. PPR‐containing proteins are rare in mammalian cells; however, there is growing evidence of their, particularly PTCD3, involvement in mitochondrial metabolism. 3 PTCD3 is expressed in all tissues. 4
The PTCD3 knock‐out mutant of Drosophila is prenatally lethal, with defects in mitochondrial function and neuronal development. 5 Similarly, PTCD3 loss‐of‐function mice exhibit neurodevelopmental defects and early lethality. 6 Currently, there are three familial cases reported with Leigh or Leigh‐like syndrome (progressive early necrotising encephalopathy) due to loss‐of‐function variants of the PTCD3, which cause reduced expression of the PTCD3 protein and altered mitochondrial respiration. 7 , 8 The first described patient presented with early respiratory failure, progressive neurodegenerative disease and Leigh‐like brain magnetic resonance lesions. 7 Three additional patients from two families had developmental delay, seizures, respiratory insufficiency, optic nerve hypoplasia, abnormal brain magnetic resonance imaging and died in the first or second decade. 8
Here, we expand on the clinical descriptions of PTCD3 variants by presenting one early‐onset Leigh‐like syndrome patient and two combined oxidative phosphorylation deficiency patients, each cohort with unique PTCD3 pathogenic variants.
2. MATERIALS AND METHODS (SUPPLEMENT 1 METHODS IN DATA S1)
2.1. Patients
2.1.1. Cases 1 and 2
A 34‐year‐old male (case 1) and his 33‐year‐old sister (case 2) both have horizontal nystagmus, pronounced rough tremor, truncal ataxia, dysmetria, spasticity and hyperreflexia. Both are legally blind, retaining only light perception, experiencing mild cognitive decline and being non‐ambulatory. They are from a non‐consanguineous family without a remarkable family history. Both patients were born from uncomplicated pregnancies and their early development was normal. Vision problems began at the age of 2 years, when the clinical diagnosis of horizontal nystagmus was established for case 1 with progression over the next 2 years leading to optical nerve atrophy. An instability and a progressive gait abnormality at the age of 6 years led to a neurology consult, where dysmetria, severe tremor and truncal ataxia were described. EEG, echocardiography and hearing tests did not reveal any pathology, but EMG/NCV showed motor polyneuropathy. Metabolic investigations of amino acids in blood and urine, and oligosaccharides in urine, as well as transferrin isofocusing were normal, except for a mild elevation of lactic acid 2.9–3.2 mmol/L (reference: 2.4 mmol/L). Urinary organic acid analysis were within reference range for both of them, except 3‐OH propionic acid in case 1 (169.98 mM/M creat; N 0–20) and in case 2 it was increased to 58.63 mM/M creat (N 0–20).
A slightly milder presentation of identical clinical symptoms affected his sister's health (case 2), and genetic testing was initiated on the hypothesis of mitochondrial pathology or hereditary ataxia for both. The results of mtDNA sequencing, Friedreich ataxia repeat expansion analysis, NGS ataxia gene panel (case 1), nucleotide repeat expansion analysis for all known types of spinocerebellar ataxia (case 1) and whole exome sequencing (case 2) did not reveal the cause of the disease. Both patients received education at a special school for the visually impaired.
Brain magnetic resonance imaging for case 1 was normal at age 20, however, due to the severe tremor, it was not possible to repeat it recently without sedation, which was refused by the patient and his family (Videos S1 and S2).
2.1.2. Case 3
The third case was identified using the GeneMatcher tool. 9 The patient is a 9‐year‐old boy from a full‐term uneventful pregnancy, born through spontaneous vaginal delivery, with a birth weight of 3 kg. His parents are double first cousins with no familial history of similar conditions. The parents have one healthy daughter, and the mother has experienced previously three spontaneous abortions. The patient has had a global developmental delay since birth with no seizures. At 9 years of age, he was able to sit and crawl, babble and make sounds. He has dysphagia with frequent episodes of choking and no sphincter control. The clinical evaluation revealed that weight and height were below the third percentile with normal head circumference. He has a myopathic face, bilateral squints and nystagmus, bushy eyebrows and a sialorrhea. He showed central hypotonia with peripheral hypertonia and hyperreflexia. No organomegaly was observed. Laboratory investigation revealed normal results of blood lactate, ammonia, creatine phosphokinase, cholesterol and electrolytes in variable ages (2, 6 and 7 years). Organic acids in urine by tandem MS were done several times and only showed border‐low free carnitine. Brain magnetic resonance imaging (MRI) at 6 years of age showed a bilateral symmetrical globus pallidus signal intensity with an elevated lactate peak. Repeated brain MRI at 8 years of age showed cystic changes, gliosis and volume loss involving bilateral posterior putamina with high signal intensity in T2 and FLAIR, low signal intensity in T1 and no associated susceptibility effect or diffusion restriction. The performed MR spectroscopy shows no other abnormal metabolites detected, except lactate peak. The brainstem, vermis and cerebellum appear unremarkable. These clinical manifestations align with a Leigh‐like syndrome.
3. RESULTS AND DISCUSSION
Two variants in the PTCD3 were identified via whole genome sequencing (Supplement 1 Methods in Data S1) in cases 1 and 2. The results were validated by Sanger sequencing, followed by family segregation analysis, which confirmed their trans position in the gene. The first variant NM_017952.6: c.1182T>A, p.(Tyr394Ter) hereinafter Tyr394Ter is localised in exon 15 (Figure 1A). This nonsense change has a high pathogenicity metascore and has not been previously reported in GnomAD. The CADD score of this variant is 36. 10 Semi‐automated ACMG classification attributes PVS1 and PM2 criteria, identifying it as likely pathogenic. 11 This pathogenic variant ablates the C‐terminal half of the protein (red; Figure 1B), including a significant portion of the central fold; therefore, proteins harbouring this variant cannot adopt a native fold and cannot be correctly incorporated into the mitochondrial ribosome. Western blot analysis of PTCD3 does not differ between patients and controls, concluding that the Tyr394Ter variant does not cause nonsense‐mediated decay (Supplement 1 in Data S1, Figure 1).
FIGURE 1.
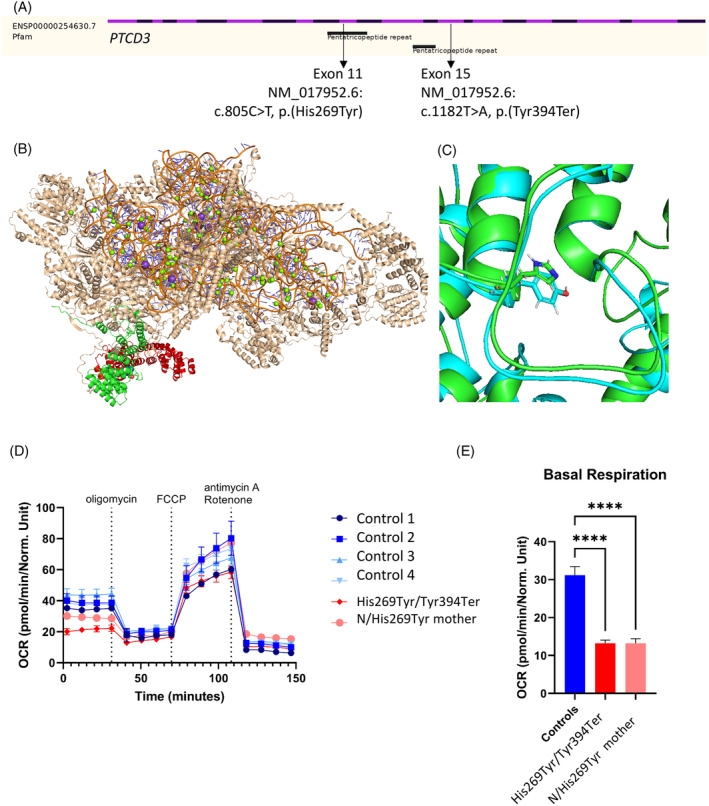
PTCD3 variants c.805C>T, p.(His269Tyr) and c.1182T>A, p.(Tyr394Ter) analysis. (A) PTCD3 gene structure with variant localisation in exon 11th and 15th. (B) Cryo‐EM structure of the mt‐SSU (PDB: 6GAZ) showing PTCD3 (green/red). Red denotes the part of the structure c‐terminal to the Tyr394Ter mutation. (C) MD of WT (green) and His269Tyr (blue) PTCD3 show similar backbone and sidechain placement. (D) Oxygen consumption rate (OCR) were measured with XF96 extracellular flux analyser (Seahorse, Agilent) in fibroblasts of two PTCD3 individuals and controls. OCR in pmol/min/Norm. Unit. The data show mean values ± SEM (number of replicates: patient = 8; control = 17). (E) The data show the mean values ± SEM of basal respiration (number of replicates: patient = 8; control = 17), ****p < 0.0001.
The second is a missense variant NM_017952.6: c.805C>T, p.(His269Tyr) hereinafter His269Tyr, which is the first nucleotide in exon 11 and within the PPR domain. The allele frequency for this variant in the GnomAD database is 0.000004. 12 The histidine in this position is conserved in fish and mammals. The CADD score is 21.50. 10 A series of previously reported cryo‐EM structures 1 , 13 , 14 , 15 , 16 show that PTCD3 is located on the periphery of the large mitochondrial ribosome subunit and His269 is positioned towards the hydrophobic centre of the protein (Figure 1C). In silico modelling shows that the inclusion of the slightly larger tyrosine sidechain is well tolerated, with no significant change in either the position or the movement of the surrounding area. Therefore, it is unlikely that this pathogenic variant influences either the PTCD3 structure of or target binding. The rarity of the variant allowed us to use the PM2 ACMG classification and classify it as the variant of unknown significance. 11
The oxygen consumption rate (OCR) was measured with the XF96 extracellular flux analyser (Seahorse, Agilent) in fibroblasts of case 1 individual and his unaffected mother in comparison with controls (Figure 1D). The basal respiration rate decreased significantly for the case 1 patient and his mother (p < 0.0001) compared to controls (Figure 1D,E). OCR before the addition of oligomycin showed a reduction (~20 pmol/min/Norm. Unit) for the case 1 sample, compared to the mother and controls (~30–50 pmol/min/Norm. Unit). These results suggest that both PTCD3 variants Tyr394Ter and His269Tyr affect protein function, despite our modelling predictions. One of the identified variants Tyr394Ter meets the pathogenic criteria. In a patient this terminating variant is responsible for the significantly decreased basal respiration rate compared to controls. The similar results were observed in his unaffected mother, who is a carrier of the His269Tyr variant, concluding that both heterozygous unaffected and affected carriers exhibit a lower OCR. However, this lower rate is not enough to cause the disease, as there is no significant difference between the affected patient and his unaffected mother.
Furthermore, in silico, protein analysis confirms that the Tyr394Ter terminating variant prevents the binding of the PTCD3 protein to mt‐SSU. The His269Tyr variant and the non‐Leigh‐like clinical presentation add to the complexity of cases 1 and 2. The genetic change His269Tyr has been classified as a variant of uncertain significance. Additional information obtained by us – being in trans position with a pathogenic variant and fitting with family segregation studies allows us to reclassify it as likely pathogenic.
In case 3, WES revealed a homozygous variant of uncertain significance in intron 7 of the PTCD3 NM_017952.6: c.538+4A>G, which was confirmed by Sanger sequencing. This variant was also absent from the gnomAD database. 12 The CADD score is 21.30. 10 Splicing Prediction Pipeline algorithm predicted a high risk of alteration for the splice site (98.41%). 17 Segregation analysis showed that the parents are heterozygous carriers, and the non‐affected normal sister is homozygous for the normal variant. To test the variant's effect on the splicing, an RT‐PCR experiment was performed, which revealed the skipping of exon 7 (124 bp) (Figure 2).
FIGURE 2.
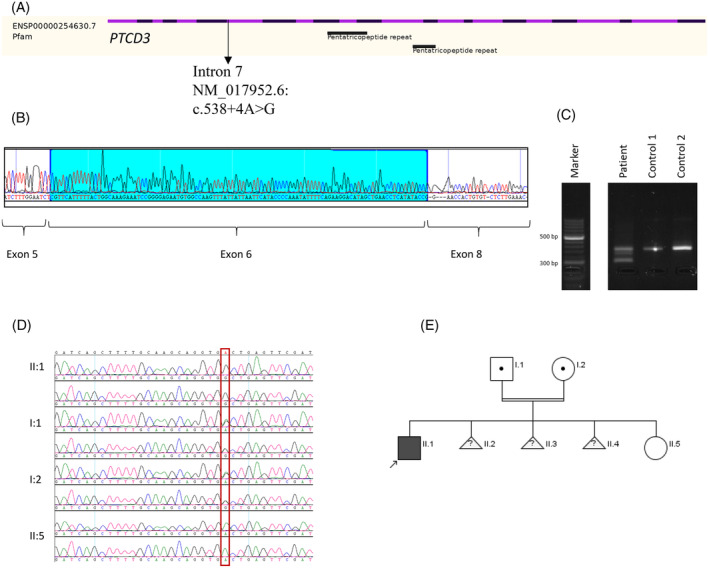
PTCD3 variant NM_017952.5:c.538+4A>G analysis. (A) PTCD3 gene structure with variant localisation in intron 7. (B) Band from agarose gel was sequenced using Sanger sequencing, which revealed skipping of Exon 7 (124 bp). (C) The band of the aberrant transcript was cut out using a sterile blade from 2% agarose gel. DNA extraction was performed using QIAquick Gel Extraction Kit (QIAGEN). (D) Familial segregation studies confirming homozygous c.538+4A>G variant in proband, heterozygous parents and homozygous normal unaffected sister. (E) Family pedigree.
Genetic changes that affect splice sites are frequently not disease‐associated due to residual correct splicing events; many pathogenic variants suspected of affecting splice sites are classified as of uncertain significance or benign variants. However, PTCD3 seems to be particularly vulnerable to pathological splice site errors, in spite of pLI‐score of 2.487e−22 (ranked 18 512 most intolerant of LoF pathogenic variants out of 19 704 genes under study). 18 A Leigh‐like syndrome has previously been shown to be caused by altered PTCD3 splicing; two missense variants (c.1918C>G and c.902C>T) alter splicing in exons 12 and 23, respectively, in these patients. 8
In addition to confirming the alterations in the splicing machinery as one of the most important pathogenetic mechanisms of the PTCD3, we add the report of the homozygous intronic variant c.538+4A>G as a function‐altering pathogenic variant. This genetic change abolishes the consensus splice site and causes exon 7 skipping, which is essential for all protein‐coding transcripts in all tissues. 4 The clinical symptoms of case 3 meet the criteria of Leigh‐like syndrome: a progressive neurodegenerative disease with abnormal brain magnetic resonance imaging that affects the basal ganglia with an elevated lactic acid signal.
The phenotype of two patients (cases 1 and 2) presented here has been suggestive of mitochondrial pathology since their early age. The symptoms of both siblings come from multiple systems that resemble a combined oxidative phosphorylation deficiency. The heat map was generated with clinical features of previously and currently published patients, including cases 1 and 2 (Figure 3), and showed optic atrophy and nystagmus in six of seven patients. Defects in mitochondrial replication, RNA transcription and translation initiate early‐onset optic atrophy, as these cells are highly vulnerable to mitochondrial dysfunction. 19 Global developmental delay was described in all Leigh‐like patients, except our presented cases 1 and 2, who have only mild cognitive decline. Severely affected PTCD3 patients have spasticity, abnormal EEG, seizures, myoclonus, dystonia, dysphagia, polyneuropathy (Figure 3, cases 4, 5 and 7) and respiratory deficiency. This is a severe progressive disease, with neurodevelopmental problems and early death caused by loss of function genetic variants in PTCD3. 7 , 8 Cases 1 and 2 patients have ataxia, tremor, paraplegia, spasticity and polyneuropathy. We hypothesise that PTCD3 missense pathogenic variants lead to a milder form of combined oxidative phosphorylation deficiency, probably due the higher level of residual PTCD3.
FIGURE 3.
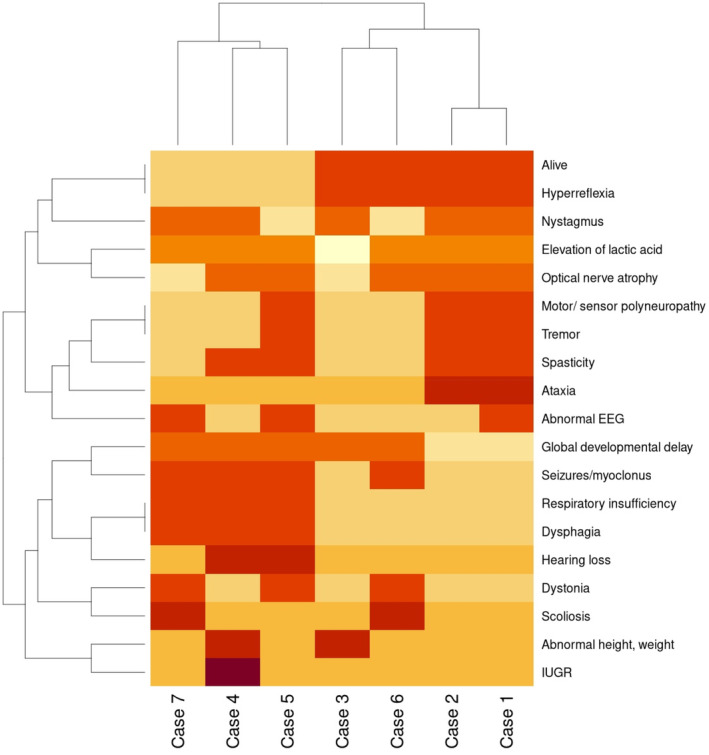
Heat map of clinical symptoms in PTCD3 patients. Cases 1, 2 and 3 described here in the current report. Case 4, patient with PTCD3 pathogenic variants c.415‐2A>G and c.1747_1748insCT (p.Phe583Serfs*3). 7 Case 5, patient 1 with PTCD3 pathogenic variants c.1453‐1G>C and c.1918C>G. 8 Case 6, patient 2.1 with PTCD3 pathogenic variants c.710del and c.902C>T. 8 Case 7, patient 2.2 with PTCD3 pathogenic variants c.710del and c.902C>T. 8
LRPPRC is another gene from PPR repeat family and bears similarity to the PTCD3. Pathogenic variants in LRPPRC are responsible for causing the French‐Canadian variant of Leigh syndrome. Due to the founder pathogenic variant NM_133259.4: c.1061C>T p.(Ala354Val), the French‐Canadian Leigh syndrome is common in the Saguenay‐Lac‐Saint‐Jean region of Quebec province in Canada. It represents a distinct phenotype characterised by severe acute metabolic and/or neurological crises resulting in a mortality rate that exceeds 80%. However, survivors beyond the age of 13 years have not encountered further metabolic or neurological crises. Additionally, these individuals exhibit symptoms such as truncal ataxia, mild intention tremors and developmental as well as language delays. 20 This group of patients with variants in LRPPRC presents a similar phenotype as our described cases 1 and 2.
PTCD3 is only peripheral to the small ribosomal subunit. However, PTCD3 binds to ribosomal initiation factors, 21 regulates mitochondrial translation 22 and its ablation causes metabolic defects. This is likely the molecular aetiology of PTCD3‐linked neurologic and metabolic diseases, including those presented here.
4. CONCLUSIONS
Novel PTCD3 loss‐of‐function intronic variant c.538+4A>G was described. This genetic change abolishes the consensus splice site and causes exon 7 skipping, which is essential for all protein‐coding transcripts in all tissues.
We hypothesise that pathogenic missense variants of PTCD3 cause a milder form of oxidative phosphorylation deficiency accompanied by symptoms of optic atrophy, ataxia, spasticity, paralysis and polyneuropathy.
AUTHOR CONTRIBUTIONS
BL and II designed and conceptualised the study. EF, ZK and IM performed clinical analysis of the patient. SP, JS and PZ performed bioinformatics, variant analysis and data analysis. II, NK and HA did molecular biology studies. JAM and MTA did respiratory chain analysis. NTW did in silico modelling. BL, EF, ZK, IM, JS, NTW and II drafted the manuscript. All authors were involved with revising the manuscript.
FUNDING INFORMATION
This work was supported by European Regional Development Fund [No. 1.1.1.1/18/A/096 ‘The determination of rare inherited diseases' causative mechanisms using whole genome sequencing approach’] and King Salman Center for Disability Research [Research Group No# KSRG‐2022‐105 and KSCDR‐RAC: 2180 004, NK]. SP was supported by the Estonian Research Council grant [PSG774]. This research was funded by U.S. National Science Foundation [MCB‐2024182 to NTW] and the U.S. National Institutes of Health [R15GM148890 to NTW].
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ETHICS STATEMENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Individuals affected with rare unidentified inherited diseases have been recruited in the Genome Database of the Latvian population (Riga, Latvia) in the framework of the ERDF research project ‘The determination of the causative mechanisms of rare inherited diseases using the whole genome sequencing approach’. The approval of the Latvian Central Committee of Medical Ethics (Protocol No. 2019‐3, Chapter 7, from 30.05.2019), covers all consent and data handling‐related issues for genetic research of the patients involved. Regarding the Saudi patient and his family members, the determination and consenting were carried out under a project (RAC#2120022), which was approved by the institutional review board of the King Faisal Specialist and Research Centre.
INFORMED CONSENT
Additional informed consent was obtained from all patients for which identifying information is included in this article.
ANIMAL RIGHTS
This article does not contain any studies with animal subjects performed by the any of the authors.
Supporting information
Data S1. Supporting information.
Video S1. Patient, case 1.
Video S2. Patient, case 2.
ACKNOWLEDGEMENTS
Our thanks go to our patients. The family kindly agreed this case to be presented and video available for all physicians with the aim to supporting science. The authors thank KFMC Research Centre for partial support [IRF 019‐052].
[Correction added on 2 July 2024, after first online publication: The acknowledgements section has been updated.]
Lace B, Faqeih E, Kaya N, et al. The phenotypic spectrum of PTCD3 deficiency. JIMD Reports. 2024;65(5):297‐304. doi: 10.1002/jmd2.12424
Communicating Editor: Ron A Wevers
DATA AVAILABILITY STATEMENT
All data are available upon reasonable request.
REFERENCES
- 1. Amunts A, Brown A, Toots J, Scheres SHW, Ramakrishnan VR. The structure of the human mitochondrial ribosome. Science. 2015;348:95‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang G, Li H, Zhang H. Abnormal expression of mitochondrial ribosomal proteins and their encoding genes with cell apoptosis and diseases. Int J Mol Sci. 2020;21:8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manna S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie. 2015;113:93‐99. [DOI] [PubMed] [Google Scholar]
- 4. The data used for the analyses described in this manuscript were obtained from: dbGaP accession number phs000424.vN.pN. Accessed September 29, 2023. https://www.gtexportal.org/home/
- 5. Imura E, Enya S, Niwa R. A Drosophila melanogaster ortholog of pentatricopeptide repeat domain 3 (PTCD3) is essential for development. MicroPubl Biol. 2023;2023. doi: 10.17912/micropub.biology.000999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Groza T, Gomez FL, Mashhadi HH, et al. The international mouse phenotyping consortium: comprehensive knockout phenotyping underpinning the study of human disease. Nucleic Acids Res. 2023;51:D1038‐D1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borna NN, Kishita Y, Kohda M, et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics. 2019;20:9‐25. [DOI] [PubMed] [Google Scholar]
- 8. Muñoz‐Pujol G, Ortigoza‐Escobar JD, Paredes‐Fuentes AJ, et al. Leigh syndrome is the main clinical characteristic of PTCD3 deficiency. Brain Pathol. 2023;33:e13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD‐splice—improving genome‐wide variant effect prediction using deep learning‐derived splice scores. Genome Med. 2021;13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aibara S, Singh V, Modelska A, Amunts A. Structural basis of mitochondrial translation. elife. 2020;9:e58362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma H, Anand B. Ribosome assembly defects subvert initiation Factor3 mediated scrutiny of bona fide start signal. Nucleic Acids Res. 2019;47:11368‐11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kummer E, Leibundgut M, Rackham O, et al. Unique features of mammalian mitochondrial translation initiation revealed by cryo‐EM. Nature. 2018;560:263‐267. [DOI] [PubMed] [Google Scholar]
- 16. Itoh Y, Khawaja A, Laptev I, et al. Mechanism of mitoribosomal small subunit biogenesis and preinitiation. Nature. 2022;606:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leman R, Parfait B, Vidaud D, et al. SPiP: splicing prediction pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing. Hum Mutat. 2022;43:2308‐2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wiggs JL. Mitochondrial genetics and optic neuropathy. Annu Rev Vis Sci. 2015;1:97‐124. [DOI] [PubMed] [Google Scholar]
- 20. Debray F‐G, Morin C, Janvier A, et al. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J Med Genet. 2011;48:183‐189. [DOI] [PubMed] [Google Scholar]
- 21. Haque ME, Koc H, Cimen H, Koc EC, Spremulli LL. Contacts between mammalian mitochondrial translational initiation factor 3 and ribosomal proteins in the small subunit. Biochim Biophys Acta, Proteins Proteomics. 2011;1814:1779‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies SMK, Rackham O, Shearwood AMJ, et al. Pentatricopeptide repeat domain protein 3 associates with the mitochondrial small ribosomal subunit and regulates translation. FEBS Lett. 2009;583:1853‐1858. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information.
Video S1. Patient, case 1.
Video S2. Patient, case 2.
Data Availability Statement
All data are available upon reasonable request.