

Abstract
In this study, we propose a practical approach for producing a heterobimetallic Ni(II)–Ce(III) diimine complex from an extended salen-type ligand (H2L) to serve as an electrocatalyst for CO2 reduction and demonstrate an outstanding overall efficiency of 99.6% of the cerium–nickel complex and integrate it into applicable cell assemblies. We optimize not only the catalyst, but the operational conditions enabling successful CO2 electrolysis over extended periods at different current densities. A comparison of electrochemical behavior in H-cell and zero-gap cell electrolyzers suggests potential applications for industrial scale-up. In the H-cell electrolyzer configuration, the most elevated efficiency in CO production was achieved with a selectivity of 56.96% at −1.01 V vs RHE, while HCOO– formation exhibited a selectivity of 32.24% at −1.11 V vs RHE. The highest TON was determined to be 14657.0 for CO formation, followed by HCOO– with a TON of 927.8 at −1.11 V vs RHE. In the zero-gap electrolyzer configuration, the most efficient setup toward CO production was identified at a current density (CD) of 75 mA cm–2, a flow rate of 10 mL min–1, operating at 60 °C and utilizing a low KOH concentration of 0.1 M to yield a maximum faradaic efficiency (FECO) of 82.1% during 24 h of stable electrocatalysis.
Keywords: CO2 electrocatalysis, salen ligand, cerium nickel complex, heterobimetallic complex, zero-gap cell electrolyzer
1. Introduction
The reduction of CO2 is pivotal in attaining climate neutrality and mitigating energy challenges.1−7 The development of efficient catalysts emerges as a crucial determinant, facilitating pathways to expedite the conversion of CO2 into valuable, environmentally friendly resources through electroreduction.8 The transformation of CO2 via electroreduction into industrial chemicals and usable fuels stands as a promising solution to these challenges. Despite the comprehensive study of traditional metallic catalysts, their inadequate durability, scarcity, high cost and substantial overpotentials hinder their practical application in real-world scenarios.9,10 Hence, one of the key challenges is the development of catalysts that can efficiently convert CO2 into high-value products such as syngas, methane, or ethylene. This requires catalysts with high selectivity and activity, as well as stability under the harsh conditions of electrosynthesis.11 Among many catalysts for the CO2 reduction reaction (CO2RR), Fe-based and Cu-based assemblies are one of the primary catalysts capable of converting CO2 into multicarbon products. However, the long-term stability of these catalysts at high current densities is often unsatisfactory, which hampers commercially relevant CO2 electrolysis.12−16 The OER (Oxygen Evolution Reaction) is a critical process often occurring at the anode during CO2 electroreduction, which plays a key role in balancing charge and maintaining efficient overall cell operation.17
In recent years, there has been growing interest in heterobimetallic catalysts due to their unique properties and potential for enhancing the selectivity and activity of CO2 electroreduction.18−21 It has been proven that synergistic effects between two different metal centers in a heterobimetallic catalyst can lead to improved catalytic performance compared to single-metal catalysts.18−21 Here, we investigate the electrocatalytic performance of a heterobimetallic Ni–Ce complex for CO2 reduction. By leveraging the complementary properties of nickel and cerium, we developed a catalyst that exhibits high selectivity and activity for the conversion of CO2. In addition, the use of Schiff-base ligands with alkoxy groups in the cerium–nickel complex introduces bicompartmental ligands, facilitating coordination with various metal ions. Our heterobimetallic complex is applied for the first time as a catalyst for electrosynthesis from CO2 feedstock. The combination of Ni and Ce efficiently activates the O=C=O bonds, where cerium with its Lewis acidic property pivotally shuttles CO2/bicarbonate to the reactive Ni center, thus considerably enhancing the electroreduction process.22−26 Comparisons with bare ligand and nickel-ligand systems highlight the catalytic synergy in the nickel–cerium complex, emphasizing the indispensable role of cerium in optimal CO2 electrocatalysis. Through a combination of electrochemical and spectroscopic techniques, we gain insights into the CO2 reduction process on the Ce–Ni molecule and optimize its performance for practical CO2 electroreduction applications. Furthermore, we explore the stability and durability of the Ce–Ni complex under prolonged electrochemical conditions to assess its potential for industrial-scale CO2 conversion technology.27−29 Meticulous electrochemical characterizations in both H-cell and zero-gap cell configurations reveal the cerium–nickel compound’s ability to produce diverse carbon-based products, showcasing its versatility. These promising outcomes position the cerium–nickel complex as a valuable electrocatalyst with significant implications for industrial applications in CO2 electrocatalysis.
2. Experimental Section
2.1. Synthesis of the Heterobimetallic Cerium-Nickel Complex
The synthesis of all three compounds, namely the H2L salen ligand (L), NiL, and CeNiL, involved the application of facile protocols sourced from the literature (Scheme 1 and Supporting Information).30
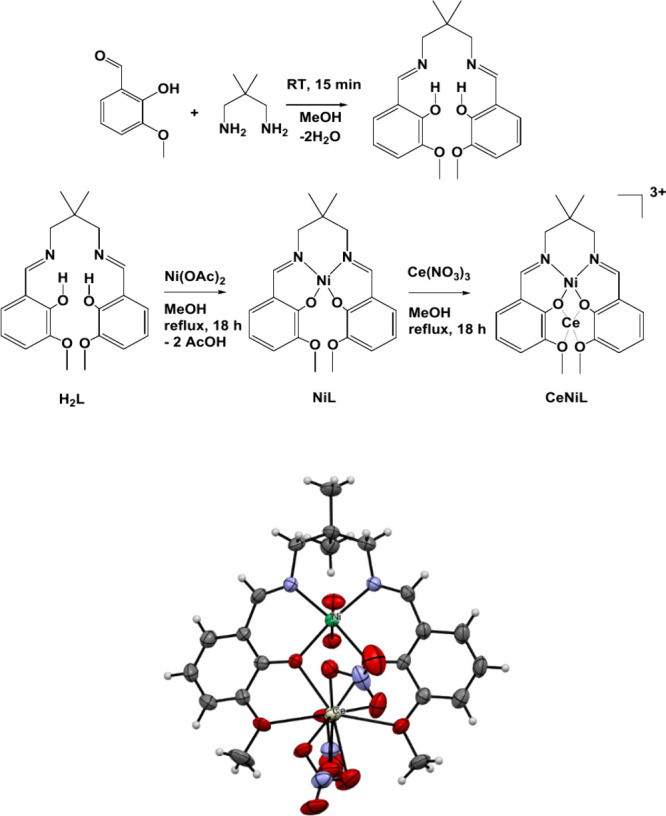
Scheme 1. (Top) One-Step Synthesis of the Salen-Type LigandH2L, Monometallic ComplexesNiL and the Heterobimetallic CongenerCeNiL, the NO3– Anions of CompoundCeNiL as well as the Crystal Water Molecules of the Applied Ni and Ce Salt Are Omitted in the Drawings for the Sake of Simplicity; (Bottom) Molecular Structure of DinuclearCeNiL as Determined by Single Crystal X-ray Diffraction Analyses, the Axial Ligands That Are Linked to the Ni Center Stem from the Crystal Water in the Applied Nickel Precursor, and the Thermal Ellipsoids Were Drawn up to the 50% Probability Level and the H Atoms Are Omitted for Clarity.
2.2. Materials and Reagents
Heterobimetallic cerium–nickel complex electrocatalyst inks were prepared by combining 2 mg of the electrocatalyst with 2 mg carbon black in 2 mL of methanol. To enhance the adhesion of the ink, 20 μL of a 5 wt % Nafion 117 solution (Sigma-Aldrich) was incorporated as a binder. The resulting catalytic inks underwent sonication for 30 min and were subsequently sprayed onto a 1 × 1 cm surface of carbon paper (TGP-H-60, Thermo scientific) for H-cell characterization, whereas for zero-gap cell measurements, 8 cm2 gas diffusion layers (GDE, CeTech) were applied (W1S1011-365 μm) and fully dried under vacuum overnight.
2.3. Characterization Methods
The catalytic loading was determined by weighing before and after spray-coating as 0.2 mg cm–2. Note that for XPS characterization the GDLs were prepared similarly only without the addition of Nafion in order to avoid suppression of other elements intensity by mainly carbon and oxygen. The electrochemical properties of the electrocatalysts were systematically examined through both homogeneous and heterogeneous approaches. Homogeneous electro-characterization was specifically employed to assess catalyst responsiveness to CO2 and involved cyclic voltammetry measurements. Glassy carbon served as the working electrode in 10 mL of acetonitrile with 0.1 M TBAPF6 as the supporting electrolyte, employing a scan rate of 30 mV s–1. Conversely, heterogeneous measurements were conducted to elucidate real electrochemical phenomena that could be encountered in scaled up applications. Here, linear sweep voltammetry (LSV) was performed in 0.1 M CsHCO3 aqueous electrolyte, as H2 formation was lowest in the case of CsHCO3. Initially, Ar gas (99.99%) was purged for 15 min through the CsHCO3 solution to remove air. The experiments were then carried out in 0.1 M CsHCO3 solution saturated with gaseous CO2 (99.99%) approximately for 1 h at a flow rate of 10 mL min–1 until the pH of the saturated solution reached 6.8. Cyclic voltammetry (CV) measurements under heterogeneous conditions were conducted to study the electrocatalytic effciency toward CO2 reduction reaction (e-CO2RR) in an aqueous electrolyte solution. The nickel–cerium–ligand assembly (CeNiL) was physisorbed on carbon paper as supportive electrode with an effective loading of 0.2 mg cm–2 and tested in a three-electrode configuration with CeNiL as working, Ag/AgCl/1 M KCl as reference and platinum wire as counter electrode. Cyclic voltammetry measurements were performed under Ar (red) and CO2 (blue) in 0.1 M CsHCO3 at pH 6.8 electrolyte solution. All heterogeneous electrochemical measurements were carried out in an H-type cell, where compartments were separated by a Nafion membrane, unless otherwise noted. The reference electrode was Ag/AgCl (with saturated KCl as the filling solution), and a Pt wire served as the counter electrode. Before measurements, the electrolyte solution (0.1 M CsHCO3) was preferentially purged with CO2 for 1 h at a flow rate of 50 mL min–1 and then bubbled continuously with CO2 at 10 mL min–1 during the test. Potentiostatic chronoamperometry (CA) in an H-type cell was conducted to measure the consumed electrons during electrosynthesis in coulomb by integration of the current over time. Throughout the electrolysis, CO2 gas was introduced into the cathodic compartment at a flow rate of 10 mL min–1 to maintain a CO2-saturated environment. The voltage on the working electrode was incrementally adjusted, ranging from −0.61 to −1.31 V vs RHE, and held steady for 1 h with stirring at each potential to record the corresponding chronoamperometric curve. The electrochemical active surface area (ECSA, cm2) was calculated by double-layer capacitance CDL, which was measured by conducting CV within a 100 mV window centered at 0.78 V vs RHE. All potentials were eventually transformed to the reversible hydrogen electrode reference through the following relationship:
| 1 |
The different current densities (ic, mA cm–2) were plotted as a function of scan rate (v, mV s–1) with a slope equal to the CDL (μF cm–2). The ECSA can be obtained by comparing the correlation CDL (μF) to a smooth planar surface (CREF, μF cm–2) which was often assumed to be 40 μF cm–2 following these equations:
| 2 |
| 3 |
A comprehensive structural characterization was conducted using NMR, FTIR, UV–vis, and XPS techniques, verifying the proposed catalytic structure (Figures S1–S12). NiL and CeNiL were deposited on carbon paper through drop casting using a methanol mixture and underwent X-ray photoelectron spectroscopy (XPS) analysis both before (see Figures S3 and S4) and after the electroreduction (refer to Figures S5–S7). The XPS survey scans encompassed the corresponding Ni 2p, Ce 3d, N 1s, O 1s binding energy regions (see Figures S3–S7). In the Ni 2p3/2 region, the main peak at 856.3 eV is situated at a typical nickel(II) position, while the primary peaks for N 1s and O 1s are at 399.9 and 532.55 eV, respectively.11,18 The XPS measurements further revealed the presence of cerium mainly as Ce3+ and to a minor extent as Ce4+.19−21 The Ni 2p region partially overlaps with the one from Ce 3d, adding complexity to the analysis. However, the XPS scans demonstrate that the catalyst remains stable throughout the course of electrocatalysis (Figures S6 and S7). All zero-gap cell experiments related to CO2 electroreduction were conducted using an electrochemical configuration as illustrated in Figure S39. The cathode gas diffusion electrode (GDE), prepared with a catalyst (geometric active area of 1 mg cm–2), was separated from the anode by an anion exchange membrane (PiperION A40-HCO3). The membrane was conditioned overnight in 1 M KOH before being washed with Milli-Q water before electrolysis. The anode employed a Ti fleece with a loading of 1 mg cm–2 IrO2. A liquid electrolyte (0.1 M KOH) was introduced into the anolyte chamber on each side of the anion exchange membrane. Gaseous CO2 was fed into the cell behind the cathode GDE and diffused into the catalyst layer. Utilizing a temperature-controlled humidifier, the relative humidification of the CO2 gas was adjusted based on the applied current density. For each CO2 reduction experiment, fresh electrolyte was prepared, and it was circulated through the electrochemical cell using peristaltic pumps at a rate of 10 mL min–1. An automatic mass flow controller maintained the flow of the input CO2 (99.99%) at 100 sccm throughout each experiment.
3. Results and Discussion
3.1. Homogeneous and Heterogenous Electrocatalysis Experiments of CeNiL Complex
The electrochemical characteristics of 1 mM CeNiL in acetonitrile were explored through CV measurements, in which glassy carbon served as the working electrode with 0.1 M TBAPF6 as the supporting electrolyte, and a scan rate of 30 mV s–1. As depicted in Figure 1a (red curve) and b, distinct one-electron redox peaks appeared at −1.0 and −1.62 V vs NHE, representing ligand and metal-centered electroreduction creating a ligand radical anion and Ni1+, as determined by in situ spectroelectrochemistry measurements (vide infra). The CV of the CeNiL on carbon paper was further investigated under Ar (Figure 1c, red curve) and CO2 (Figure 1c, blue curve) in aqueous conditions at pH = 6.8. In both cases, a marked increase in catalytic current was observed at a potential ranging from −1.62 to −1.75 V vs RHE, also corresponding to the reduction reaction of residual Ce4+ to Ce3+ (Figure 1c).31 Here the Lewis acid character of cerium facilitates the coordination of CO2/bicarbonate at these potentials and enhances the electroreduction of CO2 through the interplay with Ni, providing proton coupled electron transfers (PCET).
Figure 1.

(a) Comparison of cyclic voltammograms of CeNiL dissolved in acetonitrile under argon and CO2 containing 0.1 M TBAPF6 as supporting electrolyte with glassy carbon as working, platinum wire as counter and nonaqueous pseudo-Ag/AgCl as reference electrode with a scan rate of 30 mV s–1. (b) CV curves of CeNiL dissolved in acetonitrile under CO2 containing 0.1 M TBAPF6 as supporting electrolyte. (c) CV curves at a scan rate of 30 mV s–1CeNiL/CB WEs with Ar and CO2 saturated 0.1 M CsHCO3 obtained in a H-cell. (d) Cyclic voltammograms of CeNiL/CB catalyst at different sweep rates of 10–90 mV s–1 from 0.70 to 0.87 V vs RHE in 0.1 M CsHCO3. Insert: a linear plot of capacitive current versus scan rate. (e) Bode plot recorded via electrochemical impedance spectroscopy in the frequency range of 1 × 10–1 Hz to 1 × 105 Hz with a perturbation amplitude of 10 mV. (f) Cell current vs time plot at different half-cell potentials (V vs RHE).
The first metal-centered oxidation at E1/2 = 0.625 V and Ep = 0.63 V vs NHE involves ring-centered oxidation where Ce3+ is oxidized to a Ce4+. In the cyclic voltammogram (Figure S21), “curve crossing” is observed, where the current on the return scan surpasses that on the forward scan. This phenomenon results from the increasing concentration of the active catalyst due to in situ generation, leading to higher catalytic current as the cyclic voltammetry progresses.22,23,27,32−34 Linear sweep voltammetry (LSV, Figure S16) was conducted in a 0.1 M CsHCO3 aqueous electrolyte, with CsHCO3 chosen for its amending effect on the electroreduction of CO2 (e-CO2R).41 In the CO2 saturated aqueous 0.1 M CsHCO3 medium, the cell current observed can stem not only from the electroreduction of CO2 but also from H2 gas generation, making it challenging to distinguish solely from LSV whether the observed peaks can be attributed to e-CO2RR or the hydrogen evolution reaction (HER). In our study, a pair of sharp and broad peaks were observed at Ecat = −1.02 V and Ecat = −1.62 V vs NHE for complex CeNiL in CO2-saturated media (Figure S16), indicating e-CO2RR and HER, respectively. The higher current density observed in the CO2-saturated environment compared to the Ar environment yet affirmed the assignment to the CO2 reduction reaction under those conditions.
The variation in total current density and scan rates was also recorded as CVs in CO2-saturated media (Figure 1b,d), showing an increase in current density with higher scan rates. Due to the slow nature of e-CO2RR, the peak was not observed at −1.02 V vs RHE beyond a certain scan rate (>60 mV s–1). A substantial increase in current density was observed upon CO2 saturation, accompanied by a broad wave in the linear sweep voltammetry beginning at −1.62 V vs NHE. This observation once again suggests that the immobilized CeNiL on carbon paper catalyzes the reduction of CO2.29,30,35−40
As the reduction processes involve electron and proton transfers, electrochemical impedance spectroscopy (EIS) was employed to evaluate the charge-transfer resistance of the Ni(II)–Ce(III) diimine complex for carbon dioxide reduction electrolysis. This method was employed capturing the impedance spectrum within a frequency range of 105–0.01 Hz, with a perturbation amplitude of 10 mV (Figure 1e). Initially, two platinum electrodes were employed in a single-cell configuration with the 0.1 M CsHCO3 aqueous electrolyte, serving as a control experiment to ascertain the electrolyte resistance. Subsequently, the setup was transitioned to an H-cell configuration with a Nafion membrane, enabling the determination and subtraction of the membrane resistance from the electrolyte resistance. Further experiments involved replacing one platinum electrode with a carbon paper electrode as the working electrode. Lastly, the carbon paper, coated with CeNiL, served as the working electrode for the complete electrochemical cell evaluation through EIS (Figure 1e). The resulting fitted and calculated impedance data, as well as resistance values for each cell component (electrolyte solution, membrane, carrier electrode) in the carbon dioxide reduction cell system are summarized in Table 1. The detailed characterization based on EIS revealed negligible losses in the applied electrochemical cells. Evidently, CeNiL demonstrated the lowest charge transfer resistance, indicating enhanced electrocatalytic kinetics (Figure 1e). Controlled potential electrolysis (CPE) was then performed at various potentials, ranging from −0.8 to −1.3 V vs RHE, over 1 h, resulting in current densities ranging from 6.5 to 21.45 mA cm–2 (see Figure 1f).
Table 1. Cell Parameters Extracted via Electrochemical Impedance Measurements.
| WE | CE | Rsol/Ω | Rcarrier/Ω | R/CeNiΩ | RMe/Ω | CCeNi/F | CPE-T | CPE-P |
|---|---|---|---|---|---|---|---|---|
| Pt | Pt | 3.2 × 101 | 6.5 × 105 | 2.9 × 102 | 9.5 × 10–5 | 8.8 × 10–1 | ||
| GC | Pt | 3.2 × 101 | 5.7 × 105 | 2.9 × 102 | 5.4 × 10–5 | 8.9 × 10–1 | ||
| CeNi | Pt | 3.2 × 101 | 1.5 × 106 | 3.8 × 102 | 2.9 × 102 | 8.7 × 10–6 | 2.4 × 10–4 | 8.7 × 10–1 |
Subsequently, electrocatalysis experiments were performed in a H-cell electrolyzer system (Figure S25). Product analysis, conducted via 1H NMR spectroscopy and gas-chromatography (Nexis GC-2030), revealed formate as the predominant liquid product, with CO and H2 identified as the gaseous compounds (see Figure 2b–f).
Figure 2.

(a) Long-term stability test of CeNiL, cell current vs time at −1.11 V vs RHE and FE of formate. (b) Faradaic efficiencies of CeNiL/CB, NiL/CB, and L/CB catalyst for CO, H2, and HCOO– obtained during 1 h electrolysis at −1.0 V vs RHE. (c) Faradaic efficiencies of CeNiL/CB for CO, H2, and HCOO– obtained during 1 h electrolysis at each potential displayed. (d) Faradaic efficiencies of CeNiL/CB for CO, H2, and HCOO– obtained during 1 h electrolysis at each current density displayed. (e) 1H NMR spectra of the liquid products formed after CO2 reduction at −1.21 V vs RHE by CeNiL/CB catalyst modified carbon paper electrode in 0.1 M CO2-saturated CsHCO3 solution. Insert: Production of traces of MeOH and propane-1,2-diol. (f) GC-BID chromatogram of gaseous products formed after CO2 reduction at −1.01 V vs RHE for 1 h by CeNiL/CB catalyst modified carbon paper electrode in 0.1 M CO2-saturated CsHCO3 solution.
Over 24 h of CPE with CeNiL, formate was found to be the main liquid product with a faradaic efficiency (FE) of 32.24% at −1.1 V vs RHE (see Figure 2b–d). Furthermore, both the FE and current density remained relatively constant over 24 h of CPE (Figure 2e). CO exhibited faradaic efficiencies ranging from 4.31 to 56.96%, with product formation decreasing independently toward more negative potentials (see Figure 2c). At −1.1 V vs RHE, formate production was favored with a substantial increase in its FE (32.24%, Figure 2c). CO production was already observed at less negative potentials starting from −0.8 V (Figure 2c). Control measurements employing an unmodified salen ligand and nickel salen complex exhibited diminished product formation establishing the enhanced CO2 reduction capabilities of CeNiL (see Figure 2b–d). X-ray photoelectron spectroscopy (XPS) analysis of the coated carbon paper before and after the electrocatalysis reaction confirmed the stability of CeNiL throughout the CO2 reduction process (see Figures S3–S7). Methods for quantification of the respective gaseous and liquid compounds are described in Figures S28–S30. The faradaic efficiency of H2 production was 16.92% at Ecat = −0.91 V vs RHE, which increased to 85.93% at Ecat = −1.21 V vs RHE (see Figure 2c). The most elevated efficiency in CO production was achieved with a selectivity of 54.62% at −1.01 V vs RHE, while HCOO– formation exhibited a selectivity of 21.8% at −1.11 V vs RHE. The highest TON was determined to be 14656.98 for CO formation, followed by HCOO– with a TON of 927.75 at −1.11 V vs RHE. Notably, HCOO– and CO displayed distinct TON and TOF values (see SI for calculation), indicating that CeNiL exhibits greater activity in the generation of CO. The stability assessment of CeNiL was conducted through an extended chronoamperometry at −1.11 V. As depicted in Figure 2a, neither the current density nor the faradaic efficiency (FE) of formate exhibited a noticeable decline over a duration of 6 days of electrolysis, showcasing the stability of CeNiL. In a final observation, it is however suggested that the electrocatalyst-electrolyzer architecture could be even further optimized regarding its stability as the system’s FE experienced a minor drop from 32% (32.24% HCOO– at −1.11 vs RHE) to 29.2% after 200 h of reaction time. Furthermore, consistent results in both linear sweep voltammetry (Figures 2b and S14) and X-ray photoelectron spectroscopy (Figures S4–S7) before and after the prolonged reaction underscore excellent stability in activity. Poststability tests show that there were no significant changes in the valences of Ni and Ce, with Ni maintaining its Ni2+ state and Ce existing in the mixed states of Ce3+ and Ce4+ (vide supra, Figures S6 and S7). The ratio of Ce3+:Ce4+ yet showed a slight increase after the stability tests, attributable to the reduction process.
In order to further probe the practical applicability of the investigated catalytic system, final measurements were conducted in a zero-gap cell electrolyzer. These assemblies are widely known to improve reaction efficiency by reducing the distance between the electrode and membrane, leading to lower resistance and better mass transfer, thus significantly enhancing achievable current densities.44 The here employed home-built zero-gap cell electrolyzer cell stack consisted of flow plates, sample and IrO2 electrodes, Teflon spacers, and a PiperION anion exchange membrane (40 μm) (Figure 3). In the pursuit of cost reduction for overall CO2 capture and conversion systems, attention is directed not only toward optimizing CO2 electrochemical reactors but also toward the capture and release of CO2 to the electrochemical cell. The previously considered inefficient KOH reduction, whose applicability was doubt, is now gaining attention as one of the most promising routes for developing an efficient integrated CO2 capture and conversion system involving the electrochemical reduction of CO2.45
Figure 3.
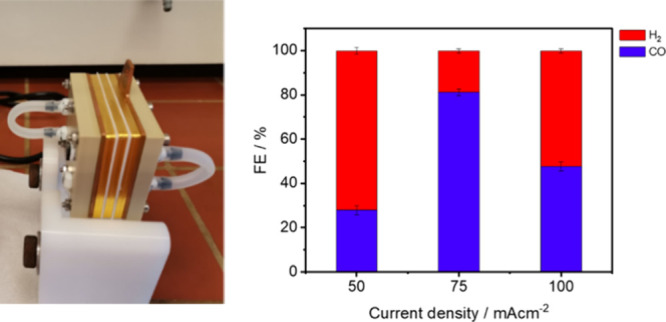
Performance of CeNiL/CB catalyst catalyst for the CO2 reduction to syngas in a home-built zero-gap electrolyzer cell stack at 50, 75, and 100 mA cm–2 at 60 °C after 24 h of electrolysis. All investigated GDEs possessed a catalytic loading of 0.5 mg cm–2 of active material.
Despite the promising findings in terms of cathodic and full cell electrical efficiency, the stability of the catalyst is crucial for commercial implementation. Commercial gas diffusion electrodes (GDEs) are known to suffer from stability issues, losing hydrophobicity and experiencing flooding over time. This behavior is exacerbated under pressure.42 For the investigated cerium nickel system the optimized operating parameters were determined to be a current density (CD) of 75 mA cm–2, a flow rate of 10 mL min–1, operating at 60 °C, and utilizing a low KOH concentration of 0.1M, yielding a maximum faradaic efficiency (FECO) of 82.1% (Figure 3). These findings offer promising insights toward the implementation of finely designed KOH electrolyzers for the integrated capture and conversion of CO2.
3.2. Mechanistic Considerations of the CO2 Electroreduction Reaction
During operando electrochemistry-UV–visible spectroscopy (EC-UV–vis) experiments conducted in both aqueous and dry acetonitrile, within the potential range of −0.5 to −1.7 V vs Ag/AgCl, significant spectral changes were observed. The absorption intensity below 250 and 280 nm (as shown in Figure 4a,b) decreased, indicating a ligand-centered reduction process (process 1). When the potential exceeded −0.8 V vs Ag/AgCl in aqueous acetonitrile or −1.3 V vs Ag/AgCl in dry acetonitrile, the UV–visible spectrum exhibited a notable alteration, with a bathochromic shift and a significant increase in absorption at 260 and 420 nm. This indicated the formation of a new spectroscopically active species, suggesting a nickel-centered reduction process (process 2) that leads to the generation of the corresponding Ni1+ complex. In aqueous acetonitrile under argon, the spectral changes were more pronounced compared to those under CO2, presumably due to the more favored formation of a nickel-hydride complex (Figure 4c). In order to substantiate these hypotheses, experiments were conducted utilizing KC8 as chemical reducing agent for CeNiL. The resulting UV–vis spectra (Figure S32) in dry and aqueous acetonitrile resembled the ones obtained by electrochemical reduction, thus convincing that identical catalyst species are generated. 1H NMR spectroscopy of the chemically reduced catalyst in nondried CD3CN revealed strong paramagnetism and an intense signal at approximately −6 ppm (Figure S33), which is in the reported range of hydride signals for Ni(I) complexes.43
Figure 4.
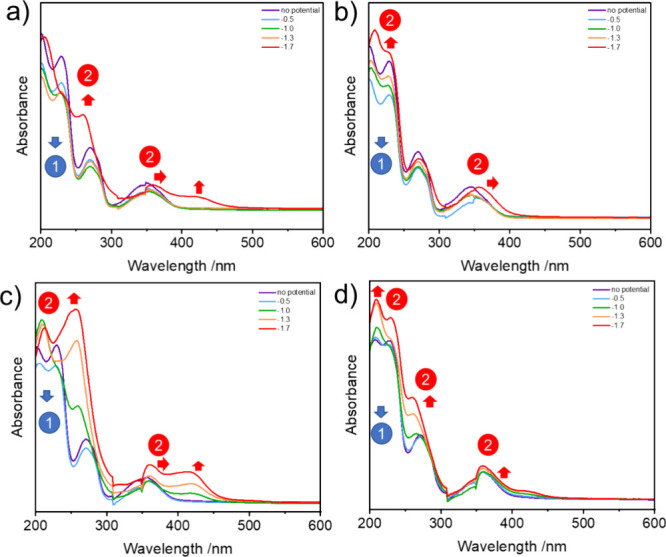
Spectroelectrochemistry of CeNiL with 0.2 M TBAPF6 in (a) ACN under Ar, (b) ACN under CO2, (c) ACN + 2% H2O under Ar, and (d) ACN + 2% H2O + CO2, at potentials from −0.5 to −1.7 V vs Ag/AgCl. Process 1: Ligand centered reduction; Process 2: Reduction of Ni2+ to Ni1+ and Ni-hydride formation. The kink at 350 nm stems from the lamp change of the employed instrument.
The resulting proposed electrocatalytic pathway 1 involves a ligand-centered reduction to form radical anions, which are catalytically active in producing HCOO– and particularly H2. In contrast, the proposed electrocatalytic pathway 2 involves a metal-hydride process, similarly leading to the production of H2, formic acid/formate, but additionally small amounts of CO, if the ligand is solely metalated with nickel (compare Figure 2b). These two pathways operate at different reduction potentials: pathway 1 occurs at lower reduction potentials (≤−1.0 V vs NHE), whereas pathway 2 favors H2/formate evolution through hydride formation at higher reduction potentials (≥−1.0 V vs NHE). The introduction of cerium into the catalytic framework, however, beneficially alternates its behavior (Figure 2b). The cerium ion as strong Lewis acid is capable of coordinating to HCO3–/CO2 and hence accelerates the HCO3– flux to the catalyst’s reactive center. Bringing the molecule of interest in spatial proximity to the reactive nickel hydride as well as lowering pH values (approximately 6.8) at the cathode surface shifting the HCO3–/CO2 equilibrium toward a higher CO2 concentration, it facilitates the hydrides interception by reacting with carbon dioxide instead of recombining with protons to form H2 gas. Considering the obtained faradaic efficiencies as well as the spectroscopic results from electrochemical and chemical reduction it is proposed that the synergistic effect of the complexed cerium ion enhances the kinetics of CO2 to CO conversion at low reduction potentials compared to the formation of H2 and formate.
4. Conclusions
In summary, this investigation focuses on employing a molecular, heterobimetallic cerium–nickel catalyst to electrochemically reduce CO2 to CO in H-cell and zero-gap cell setups. This process achieved maximum faradaic efficiencies of 54.6% for CO (FECO) and 21.8% for formate (FEHCOO–) in the H-cell setup with stable performance for 200 h. Electrocatalysis experiments in a zero-gap cell electrolyzer demonstrated a FECO of 82.1% at current densities of 75 mA cm–2.
Our study demonstrates that the explored CeNiL complex exhibits high selectivity for CO production from CO2 at low reduction potentials. This efficiency is enabled by the strongly Lewis acidic cerium ion in spatial proximity to the nickel center, substantially accelerating the CO2 to CO reduction process. Overall, this research presents a potent method for directly generating CO through electrocatalytic CO2 reduction using a readily available CeNiL-based molecular catalyst. Future investigations in our laboratory will explore the use of homobimetallic or heterobimetallic salen-type catalysts, which have the potential to exhibit superior activity toward electrocatalytic CO2 reduction, electrocatalytic N2 reduction and hydrogen evolution.
Acknowledgments
W.S. acknowledges the financial support of the Austrian Science Fund (FWF Standalone Projects P28167 “Heterogeneous catalysis for water oxidation and hydrogen evolution” and P32045 “Catalysts for biomass valorization”) and of the Austrian Research Promotion Agency FFG (“CO2Val”, FFG bridge project no. 883671). W.S. and D.K. also acknowledge the financial support by the LIT Project (LIT-2022-11-SEE-111, “e-COOL”) and by the “Klima- und Energiefonds” (the project is carried out within the framework of the “Energieforschungsprogramm 2022” (FFG project number: FO999903855)). W.S. extends appreciation for the insightful discussions on the characteristics of the CeNiL catalyst with Professor Christoph Topf from the Institute of Catalysis at JKU Linz.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsaem.4c02132.
The authors declare no competing financial interest.
Supplementary Material
References
- Coskun H.; Aljabour A.; de Luna P.; Farka D.; Greunz T.; Stifter D.; Kus M.; Zheng X.; Liu M.; Hassel A. W.; Schöfberger W.; Sargent E. H.; Sariciftci N. S.; Stadler P. Biofunctionalized conductive polymers enable efficient CO2 electroreduction. Sci. Adv. 2017, 3, 68. 10.1126/sciadv.1700686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J.; Qiu L.; Xin M.; He W.; Zhao W.; Dong J.; Xu G. Boosting Electrochemical CO2 Reduction on Copper-Based Metal-Organic Frameworks via Valence and Coordination Environment Modulation. Small 2024, 20, 2311060 10.1002/smll.202311060. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Qian J.; Zhang G.; Zhang B.; He Y. Electrochemical CO2-to-CO conversion: A comprehensive review of recent developments and emerging trends. Sep. Purif. Technol. 2024, 330, 125177 10.1016/j.seppur.2023.125177. [DOI] [Google Scholar]
- Lodh J.; Paul S.; Sun H.; Song L.; Schöfberger W.; Roy S. Electrochemical organic reactions: A tutorial review. Front. Chem. 2023, 10, 956502 10.3389/fchem.2022.956502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedić D.; Dorniak A.; Rinner U.; Schöfberger W. Recent Progress in (Photo-)-Electrochemical Conversion of CO2 With Metal Porphyrinoid-Systems. Front. Chem. 2021, 9, 685619 10.3389/fchem.2021.685619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari N.; Halder S.; Naskar S.; Ganguly S.; Pramanik K.; Yari F.; Dorniak A.; Schöfberger W.; Roy S. Coordinatively fluxional diazo-based organo-electrocatalyst for conversion of CO2 to C2 and C3 products. Mater.Today Catal. 2024, 5, 100049 10.1016/j.mtcata.2024.100049. [DOI] [Google Scholar]
- Gonglach S.; Paul S.; Haas M.; Pillwein F.; Sreejith S. S.; Barman S.; De R.; Müllegger S.; Gerschel P.; Apfel U.-P.; Coskun H.; Aljabour A.; Stadler P.; Schöfberger W.; Roy S. Molecular cobalt corrole complex for the heterogeneous electrocatalytic reduction of carbon dioxide. Nat. Commun. 2019, 10, 3864. 10.1038/s41467-019-11868-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellumbi K.; Krisch D.; Rettenmaier C.; Awada H.; Sun H.; Song L.; Sanden S. A.; Hoof L.; Messing L.; Puring K. J.; Siegmund D.; Roldan Cuenya B.; Schöfberger W.; Apfel U.-P. Pushing the Ag-loading of CO2 electrolyzers to the minimum via molecularly tuned environments. Cell Rep. Phys. Sci. 2023, 4, 101746 10.1016/j.xcrp.2023.101746. [DOI] [Google Scholar]
- Chen Q.; Wang X.; Zhou Y.; Tan Y.; Li H.; Fu J.; Chen M.; Liu Q.; Wang X.; Zhou Y.; Tan Y.; Li H.; Fu J.; Liu M. Electrocatalytic CO2 Reduction to C2+ Products in Flow Cells. Adv. Mater. 2024, 36, e2303902 10.1002/adma.202404734. [DOI] [PubMed] [Google Scholar]
- Shen J.; Wang D. How to select heterogeneous CO2 reduction electrocatalyst. Nano Res. Energy 2024, 3, e9120096 10.26599/NRE.2023.9120096. [DOI] [Google Scholar]
- Bose P.; Mukherjee C.; Golder A. K. A NiII complex of the tetradentate salen ligand H2LNH2 comprising an anchoring–NH2 group: synthesis, characterization and electrocatalytic CO2 reduction to alcohols. Inorg. Chem. Front. 2019, 6 (7), 1721–1728. 10.1039/C9QI00353C. [DOI] [Google Scholar]
- Wu Z.-Z.; Zhang X.-L.; Yang P.-P.; Niu Z.-Z.; Gao F.-Y.; Zhang Y.-C.; Chi L.-P.; Sun S.-P.; DuanMu J.-W.; Lu P.-G.; Li Y.-C.; Gao M.-R. Gerhardtite as a Precursor to an Efficient CO-to-Acetate Electroreduction Catalyst. J. Am. Chem. Soc. 2023, 145 (44), 24338–24348. 10.1021/jacs.3c09255. [DOI] [PubMed] [Google Scholar]
- DuanMu J.-W.; Wu Z.-Z.; Gao F.-Y.; Yang P.-P.; Niu Z.-Z.; Zhang Y.-C.; Chi L.-P.; Gao M.-R. Investigation and Mitigation of Carbon Deposition over Copper Catalyst during Electrochemical CO2 Reduction. Precis. Chem. 2024, 2 (4), 151–160. 10.1021/prechem.4c00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M. R.; Sun S. P.; Duanmu J. W.; Yu P. C.; Wang Y. H.; Yang P. P.; Gao F. Y.; Chi L. P.; Niu Z. Z.; Wu Z. Z.; Zhang X. L.; Zhang Y. C. Facet-switching of rate-determining step on copper in CO2-to-ethylene electroreduction. Proc. Natl. Acad. Sci. U. S. A. 2024, 121 (25), e2400546121 10.1073/pnas.2400546121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.; Wang N.; Li X.; Lei H.; Wang Y.; Guo H.; Jin X.; Zhang Q.; Peng X.; Zhang X.-P.; Zhang W.; Apfel U.-P.; Cao R. Bioinspired iron porphyrins with appended poly-pyridine/amine units for boosted electrocatalytic CO2 reduction reaction. eScience 2022, 2, 623–631. 10.1016/j.esci.2022.06.003. [DOI] [Google Scholar]
- Guo K.; Li X.; Lei H.; Guo H.; Jin X.; Zhang X.-P.; Zhang W.; Apfel U.-P; Cao R. Role-Specialized Division of Labor in CO2 Reduction with Doubly-Functionalized Iron Porphyrin Atropisomers. Angew. Chem., Int. Ed. 2022, 61, e202209602 10.1002/anie.202209602. [DOI] [PubMed] [Google Scholar]
- Gao X.; Liu X.; Yang S.; Zhang W.; Lin H.; Cao R. Black phosphorus incorporated cobalt oxide: Biomimetic channels for electrocatalytic water oxidation. Chin. J. Catal. 2022, 43 (4), 1123–1130. 10.1016/S1872-2067(21)63937-2. [DOI] [Google Scholar]
- Zheng T.; Jiang K.; Ta N.; Hu Y.; Zeng J.; Liu J.; Wang H. Large-Scale and Highly Selective CO2 Electrocatalytic Reduction on Nickel Single-Atom Catalyst. Joule 2019, 3 (1), 265–278. 10.1016/j.joule.2018.10.015. [DOI] [Google Scholar]
- Bogart J. A.; Lewis A. J.; Medling S. A.; Piro N. A.; Carroll P. J.; Booth C. H.; Schelter E. J. Homoleptic Cerium(III) and Cerium(IV) Nitroxide Complexes: Significant Stabilization of the 4+ Oxidation State. Inorg. Chem. 2013, 52 (19), 11600–11607. 10.1021/ic401974t. [DOI] [PubMed] [Google Scholar]
- Jacobs G.; Keogh R.; Davis B. Steam reforming of ethanol over Pt/ceria with co-fed hydrogen. J. Catal. 2007, 245 (2), 326–337. 10.1016/j.jcat.2006.10.018. [DOI] [Google Scholar]
- Jia Z.; Ning S.; Tong Y.; Chen X.; Hu H.; Liu L.; Ye J.; Wang D. Selective Photothermal Reduction of CO2 to CO over Ni-Nanoparticle/N-Doped CeO2 Nanocomposite Catalysts. ACS Appl. Nano Mater. 2021, 4 (10), 10485–10494. 10.1021/acsanm.1c01991. [DOI] [Google Scholar]
- Bogart J. A.; Lewis A. J.; Medling S. A.; Piro N. A.; Carroll P. J.; Booth C. H.; Schelter E. J. Homoleptic cerium(III) and cerium(IV) nitroxide complexes: significant stabilization of the 4+ oxidation state. Inorg. Chem. 2013, 52, 11600. 10.1021/ic401974t. [DOI] [PubMed] [Google Scholar]
- Bose P.; Mukherjee C.; Golder A. K. A NiII complex of the tetradentate salen ligand H2LNH2 comprising an anchoring–NH2 group: synthesis, characterization and electrocatalytic CO2 reduction to alcohols. Inorg. Chem. Front. 2019, 6, 1721. 10.1039/C9QI00353C. [DOI] [Google Scholar]
- Möller F.; Piontek S.; Miller R. G.; Apfel U.-P. Frontispiece: From Enzymes to Functional Materials—Towards Activation of Small Molecules. Chem.—Eur. J. 2018, 24, 147. 10.1002/chem.201880762. [DOI] [PubMed] [Google Scholar]
- Su X.; McCardle K. M.; Panetier J. A.; Jurss J. W. Electrocatalytic CO2 reduction with nickel complexes supported by tunable bipyridyl-N-heterocyclic carbene donors: understanding redox-active macrocycles. Chem. Commun. 2018, 54, 3351. 10.1039/C8CC00266E. [DOI] [PubMed] [Google Scholar]
- Zheng T.; Jiang K.; Ta N.; Hu Y.; Zeng J.; Liu J.; Wang V. Large-Scale and Highly Selective CO2 Electrocatalytic Reduction on Nickel Single-Atom Catalyst. Joule 2019, 3, 265. 10.1016/j.joule.2018.10.015. [DOI] [Google Scholar]
- Darvasiová D.; Šoral M.; Puškárová I.; Dvoranová D.; Vénosová B.; Bučinský L.; Zalibera M.; Dujnič V.; Dobrov A.; Schwalbe M.; Arion V. B.; Rapta P. Spectroelectrochemical, photochemical and theoretical study of octaazamacrocyclic nickel(II) complexes exhibiting unusual solvent-dependent deprotonation of methylene group. Electrochim. Acta 2019, 326, 135006 10.1016/j.electacta.2019.135006. [DOI] [Google Scholar]
- Nwabara U. O.; Heer M. P.; Cofell E. R.; Verma S.; Negro E.; Kenis P. J. A. Towards accelerated durability testing protocols for CO2 electrolysis. J. Mater. Chem. A 2020, 8, 22557. 10.1039/D0TA08695A. [DOI] [Google Scholar]
- Wang W.; Chu W.; Wang N.; Yang W.; Jiang C. Mesoporous nickel catalyst supported on multi-walled carbon nanotubes for carbon dioxide methanation. Int. J. Hydrogen Energy 2016, 41, 967. 10.1016/j.ijhydene.2015.11.133. [DOI] [Google Scholar]
- Michalke J.; Faust K.; Bögl T.; Bartling S.; Rockstroh N.; Topf C. Mild and Efficient Heterogeneous Hydrogenation of Nitroarenes Facilitated by a Pyrolytically Activated Dinuclear Ni(II)-Ce(III) Diimine Complex. Int. J. Mol. Sci. 2022, 23, 8742. 10.3390/ijms23158742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K.; Sato H.; Kaneeda M.; Kondo J. N. Synthesis and analysis of CO2 adsorbents based on cerium oxide. J. CO2 Utilization 2014, 8, 34. 10.1016/j.jcou.2014.10.001. [DOI] [Google Scholar]
- Jacobs G.; Keogh R.; Davis B. Steam reforming of ethanol over Pt/ceria with co-fed hydrogen. J. Catal. 2007, 245, 326. 10.1016/j.jcat.2006.10.018. [DOI] [Google Scholar]
- Jia Z.; Ning S.; Tong Y.; Chen X.; Hu H.; Liu L.; Ye J.; Wang D. Selective Photothermal Reduction of CO2 to CO over Ni-Nanoparticle/N-Doped CeO2 Nanocomposite Catalysts. ACS Appl. Nano Mater. 2021, 4, 10485. 10.1021/acsanm.1c01991. [DOI] [Google Scholar]
- Gao J.; Zhang H.; Guo X.; Luo J.; Zakeeruddin S. M.; Ren D.; Grätzel M. Selective C–C Coupling in Carbon Dioxide Electroreduction via Efficient Spillover of Intermediates As Supported by Operando Raman Spectroscopy. J. Am. Chem. Soc. 2019, 141, 18704. 10.1021/jacs.9b07415. [DOI] [PubMed] [Google Scholar]
- Devasia D.; Wilson A. J.; Heo J.; Mohan V.; Jain P. K. A rich catalog of C–C bonded species formed in CO2 reduction on a plasmonic photocatalyst. Nat. Commun. 2021, 12, 2612. 10.1038/s41467-021-22868-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh C.-T.; Burdyny T.; Kibria M. G.; Seifitokaldani A.; Gabardo C. M.; García de Arquer F. P.; Kiani A.; Edwards J. P.; De Luna P.; Bushuyev O. S.; Zou C.; Quintero-Bermudez R.; Pang Y.; Sinton D.; Sargent E. H. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. 10.1126/science.aas9100. [DOI] [PubMed] [Google Scholar]
- Gabardo C. M.; O’Brien C. P.; Edwards J. P.; McCallum C.; Xu Y.; Dinh C.-T.; Li J.; Sargent E. H.; Sinton D. Continuous Carbon Dioxide Electroreduction to Concentrated Multi-carbon Products Using a Membrane Electrode Assembly. Joule 2019, 3, 2777. 10.1016/j.joule.2019.07.021. [DOI] [Google Scholar]
- Merino-Garcia I.; Albo J.; Irabien A. Tailoring gas-phase CO2 electroreduction selectivity to hydrocarbons at Cu nanoparticles. Nanotechnology 2018, 29, 14001. 10.1088/1361-6528/aa994e. [DOI] [PubMed] [Google Scholar]
- Wang X.; Fu Y.; Tranca D.; Jiang K.; Zhu J.; Zhang J.; Han S.; Ke C.; Lu C.; Zhuang X. Regulating the Spin State of Nickel in Molecular Catalysts for Boosting Carbon Dioxide Reduction. ACS Appl. Energy Mater. 2021, 4, 2891. 10.1021/acsaem.1c00269. [DOI] [Google Scholar]
- Yun H.; Kim J.; Choi W.; Han M. H.; Park J. H.; Oh H.; Da Won H.; Kwak K.; Hwang Y. J. Understanding morphological degradation of Ag nanoparticle during electrochemical CO2 reduction reaction by identical location observation. Electrochim. Acta 2021, 371, 137795 10.1016/j.electacta.2021.137795. [DOI] [Google Scholar]
- Lum Y.; Yue B.; Lobaccaro P.; Bell A. T.; Ager Joel W. Optimizing C–C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction. J. Phys. Chem. C 2017, 121 (26), 14191–14203. 10.1021/acs.jpcc.7b03673. [DOI] [Google Scholar]
- Hernandez-Aldave S.; Andreoli E. Fundamentals of Gas Diffusion Electrodes and Electrolysers for Carbon Dioxide Utilisation: Challenges and Opportunities. Catalysts 2020, 10, 713. 10.3390/catal10060713. [DOI] [Google Scholar]
- Barton B. E.; Rauchfuss T. B. J. Hydride-Containing Models for the Active Site of the Nickel–Iron Hydrogenases. J. Am. Chem. Soc. 2010, 132 (42), 14877–14885. 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkort J. W.; Rajaei H. Voltage losses in zero-gap alkaline water electrolysis. J. Power Sources 2021, 497, 229864 10.1016/j.jpowsour.2021.229864. [DOI] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu Xin; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E. L.; Chan K.; Hahn Ch.; No̷rskov Jens K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119 (12), 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.