

Abstract
Cancers eventually kill hosts even when infiltrated by cancer-specific T cells. We examined whether cancer-specific T cell receptors of CD4+ T cells (CD4TCRs) from tumor-bearing hosts can be exploited for adoptive TCR therapy. We focused on CD4TCRs targeting an autochthonous mutant neoantigen that is only presented by stroma surrounding the MHC class-II negative cancer cells. The 11 most common tetramer-sorted CD4TCRs were tested using TCR-engineered CD4+ T cells. Three TCRs were characterized by convergent recombination for which multiple T cell clonotypes differed in their nucleotide sequences but encoded identical TCR α- and β-chains. These preferentially selected TCRs destroyed tumors equally well and halted progression through reprogramming of the tumor stroma. TCRs represented by single T cell clonotypes were similarly effective only if they shared CDR elements with preferentially selected TCRs in both α- and β-chains. Selecting candidate TCRs based on these characteristics can help identify TCRs that are potentially therapeutically effective.
One-sentence summary:
Selecting candidate TCRs based on convergent recombination can help identify TCRs with potential therapeutic efficacy.
INTRODUCTION:
Somatic mutations cause cancer and therefore are found in all types of malignancies (1). Many of these mutations represent non-synonymous single nucleotide variants (nsSNVs) in tumor DNA, absent from the germline genome. These nsSNVs cause single amino acid substitutions, are the basis of individually distinct (“unique”) tumor-specific antigens (2, 3) and are targetable by adoptive transfer of mutation-specific T cells (2). These mutation-encoded, tumor-specific antigens are now usually referred to as “neoantigens”. Findings from patients treated with immune checkpoint inhibitors (ICI) or adoptively transferred tumor infiltrating lymphocytes (TIL) support the notion that these neoantigens are effective T cell targets in humans (4). Unfortunately, such immunotherapies achieve long-term survival only in a fraction of patients with certain types of cancers, and relapse remains common (5, 6). ICI and TIL therapies seem to rely on converting the endogenous tumor-specific T cells into an active, tumor-killing state. However, reactivated T cells may return to an inactive state once re-exposed to the cancer cells and the tumor microenvironment (7, 8).
An alternative approach, now also being applied in humans (9, 10), is to isolate T cell receptors (TCRs) from neoantigen-specific CD8+ T cells (CD8TCRs) and express these CD8TCRs in healthy T cells from peripheral blood for adoptive transfer. Indeed, neoantigen-specific CD8TCR-engineered T cells can eliminate large, established solid tumors in mice after adoptive T cell transfer (11, 12). However, the targeted neoantigen needs to be artificially overexpressed for eradication whereas cancer cells expressing the unmanipulated autochthonous neoantigen regularly escape immune-mediated destruction (11).
Some clinical data using neoantigen-specific TILs suggest the potential use of CD4+ T cells in immunotherapy (13–15). TIL populations usually consist of various effector cells, and it has been unclear from clinical studies whether one or multiple TCRs of tumor infiltrating CD4+ T cells (CD4TCRs) are sufficient for effective immunotherapy. We have shown recently that a single CD4TCR expressed by engineered CD4+ T cells can destroy established tumors when targeting an unmanipulated autochthonous neoantigen (16). However, it remains unclear how to predict therapeutic efficacy of CD4TCRs from a polyclonal T cell response of a tumor-bearing host (mouse and human), and how effective CD4TCRs mediate tumor destruction when used in adoptive therapy settings.
Here, we use the autochthonous and syngeneic UV-induced cancer cell model 6132A (17) to explore the selection of therapeutically effective neoantigen-specific CD4TCRs isolated from tumors and/or peripheral blood of cancer-bearing mice. Among the 11 most frequently occurring CD4TCRs, three TCRs were each made by multiple T cell clonotypes (18) differing in their α- and β-chain nucleotide sequences but encoding identical amino acid sequences, which is also referred to as convergent recombination (19). Adoptive transfer of T cells engineered with either one of these preferentially selected TCRs (20) resulted in destruction of aggressively growing tumors and the reprogramming of its stroma. This effect was dependent solely on stroma recognition, and not on direct cancer cell targeting. The other eight TCRs, represented by single T cell clonotypes were only effective therapeutically when they shared CDR elements with at least one preferentially selected TCR in both chains. Thus, our study identifies characteristics of neoantigen-specific CD4TCRs that can help to predict therapeutic efficacy against progressing solid tumors.
RESULTS:
Hosts with progressing tumors respond with multiple CD4+ T cell receptors to an immunodominant neoantigen.
6132A cancer cells harbor an immunodominant L47H mutation in the ribosomal protein L9 (mL9) which is presented on the MHC class II haplotype I-Ek (2). To understand whether tumor-bearing hosts generate a response to this unmanipulated autochthonous neoantigen, we used a mL9-I-Ek-tetramer to analyze tumors and spleens from normal syngeneic mice bearing 6132A tumors that had grown for over 2 weeks (Fig. 1A). Indeed, 6132A tumors were infiltrated with mL9-I-Ek-tetramer-binding CD4+ T cells (median frequency 1.4%) (Fig. 1B and Fig. S1). Single cell TCR sequencing of mL9-tetramer-binding CD4+ T cells showed the relative frequencies of TCRs (Fig. 1C). On average, we obtained 162 T cells from a tumor sample harboring 45 different TCRs and 202 T cells from a spleen sample harboring 55 different TCRs (Table S1). Our aim was to determine which of these CD4TCRs could be utilized for adoptive transfer of TCR-engineered CD4+ T cells, and if any, by which mechanisms these TCRs mediate anti-tumor activity.
Fig. 1. Convergent recombination of different T cell clonotypes encoding identical, preferentially selected TCRs against the mutant neoantigen mL9.
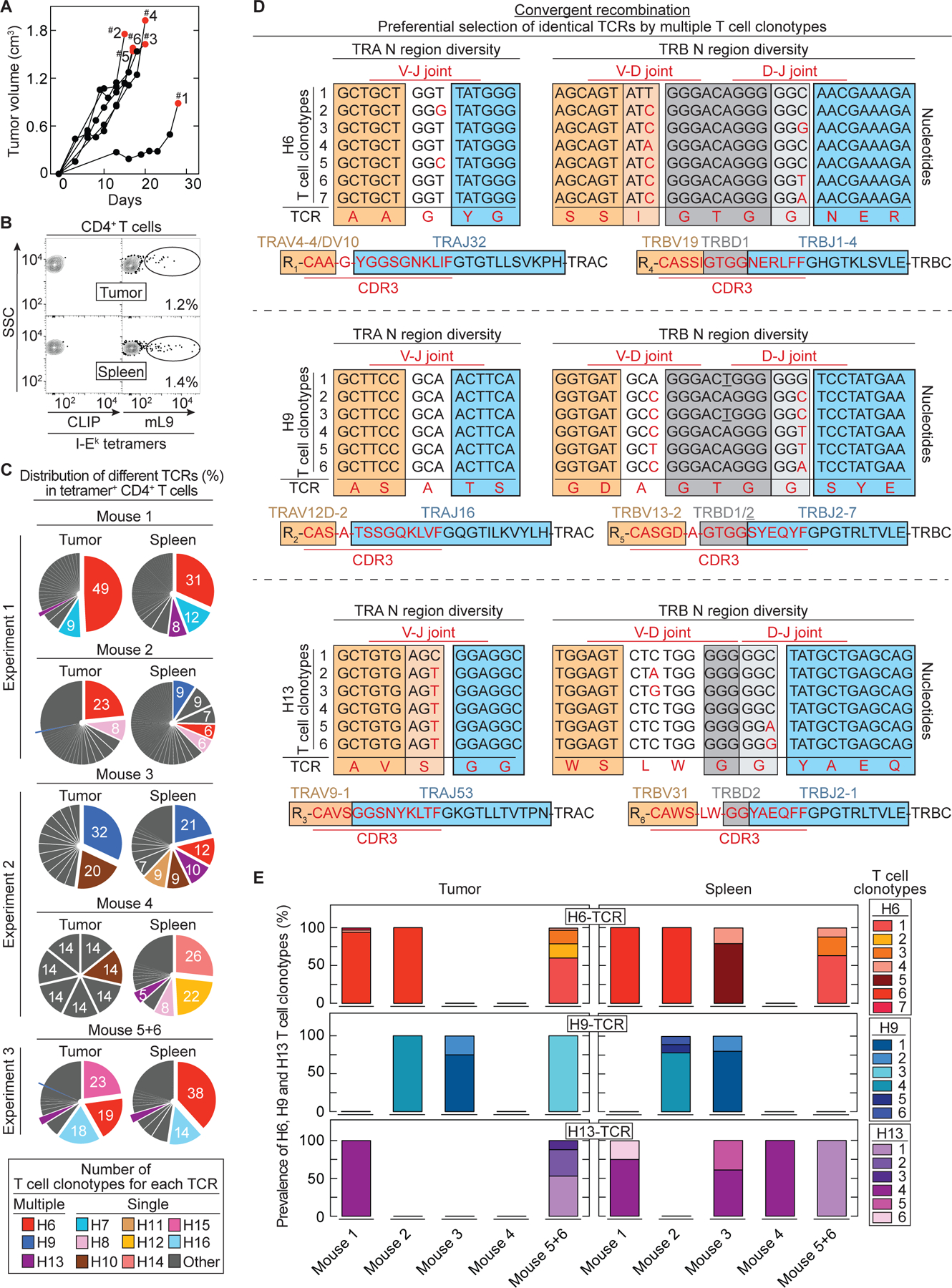
(A) 6132A tumor fragments were injected s.c. into C3H/HeN mice. Six mice are shown which developed tumors after fragment injection (55%, 11/20 injected C3H/HeN mice) and were used for TCR analysis. Results were compiled from three independent experiments. Red dots indicate day of T cell analysis. (B) An example is shown of T cells isolated from spleen and tumor sorted for live, CD3+, CD4+ and mL9-tetramer+ specificity. Percentages of mL9-tetramer positive T cells are indicated. CLIP-tetramer staining was used as negative control. (C) Frequencies of paired TCR CDR3 amino acid sequences in mL9-teramer sorted CD4+ T cells obtained from tumor and spleen of the six analyzed mice. (D) Identification of different T cell clonotypes encoding an identical TCR based on N nucleotide sequence diversity in the TRA and TRB V(D)J joints. This was determined for the TCRs H6 (upper TCR), H9 (middle TCR) and H13 (bottom TCR). (E) Frequency of the different T cell clonotypes encoding an identical TCR (either H6, H9 or H13) among the analyzed mice.
Convergent recombination by multiple T cell clonotypes indicates TCRs preferentially selected by hosts with autochthonous or transplanted progressive tumors.
We analyzed the 11 most common TCRs found in tumors and spleens of six mice (Fig. 1C). The two most frequent TCRs (H6 and H9) from mouse #1, #2, #3 and #5+6 were found in tumor and spleen tissue. Interestingly, the amino acid sequences of the CDR3s of the H6- and H9-TCR were generated by multiple different T cell clonotypes as determined by different N-nucleotides between the V(D)J joints (Fig. 1D). Seven different T cell clonotypes in at least four different mice encoded the H6-TCR while six different T cell clonotypes in at least three different mice encoded the H9-TCR (Fig. 1E). This convergent recombination by multiple T cell clonotypes encoding identical TCRs is in agreement with preferential selection. Compellingly, even though mouse #4 had only very few mL9-I-Ek-tetramer-binding T cells, we still detected in the spleen a less frequent TCR (H13) that was again characterized by recombinational convergence of multiple T cell clonotypes. Six different T cell clonotypes found in at least four different mice encoded TCR H13 (Fig. 1D and 1E). Furthermore, H13 was also among the TCR-response detected in the spleen of the 6132 mouse that developed the original 6132A tumor, indicating that convergent recombination was not restricted to mice with transplanted tumors (Fig. S2).
Convergent recombination defined therapeutically effective TCRs.
All three preferentially selected TCRs were cloned into retroviral vectors and transduced into splenic T cells from C3H CD8-/- mice. TCR-engineered CD4+ T cells were adoptively transferred into C3H Rag-/- mice bearing large and established solid 6132A tumors (Fig. 2A). Strikingly, TCR H6 destroyed tumors within 10 days after transfer, (Fig. 2B). Transfer of αmL26-TCR T cells, a CD4TCR T cell targeting the irrelevant mutant ribosomal protein L26 (21) (found in another UV-induced C3H tumor, 6139B) had no effects since 6132A tumors progressed similarly to untreated controls (Fig. 2B). TCRs H9 and H13 had similar therapeutic efficacies as H6 (Fig. 2C), even though 5 out of 8 mice treated with H13 relapsed after 50 days.
Fig. 2. Therapeutically effective TCRs cause tumor destruction followed by long-term growth arrest and can be predicted by CDR elements of preferentially selected TCRs.

(A) Outline of adoptive transfer using TCR-engineered T cells. (B – C) Spleens from C3H CD8-/- mice were used as CD4+ T cell source for TCR engineering. C3H Rag-/- mice bearing 6132A tumors were treated with TCR-engineered CD4+ T cells 21 to 25 days after cancer cell injection as indicated by the arrow head. Total number of mice (n) is indicated. (B) Average tumor sizes were 0.558 cm3 ± 0.122 cm³ standard deviation at day of treatment. Data are summarized from three independent experiments. (left) Treatment was performed with H6-T cells (n = 6). (middle) Mice treated with αmL26-T cells, which are specific against an irrelevant antigen (n = 4) have the same outcome as (right) untreated mice (n = 4). (C) Average tumor sizes were 0.378 cm3 ± 0.156 cm³ standard deviation at day of treatment. Data are summarized from two independent experiments. Treatment with different TCR-engineered CD4+ T cells is indicated from left to right, top to bottom: H7 (n = 4), H8 (n = 3), H9 (n = 5), H10 (n = 4), H11 (n = 6), H12 (n = 5), H13 (n = 8), H14 (n = 6), H15 (n = 6) and H16 (n = 6). (D) The 11 TCRs fell into three groups based on therapeutic failure or efficacy (defined by >25% shrinkage of tumor volume) and whether being generated by multiple or single clonotypes. Color coding indicates whether CDR elements were shared in TRA and/or TRB with preferentially selected TCRs. (E – F) TCR-treatment Group 1: H6, H9, H13 (total n = 16). Group 2: H11, H12, H14, H15, H16 (total n = 29). Group 3: H7, H8 and H10 (total n = 11). (E) The three groups were compared in a survival analysis (*p ≤ 0.5, ***p ≤ 0.001 significance, n.s. – not significant). Log-rank test was used to determine significance indicated in black while significance indicated in red used the Wilcoxon test. (F) Probability of relapse at day 40 or 80 after start of T cell transfer among the three TCR-treatment groups. **p ≤ 0.01 and ***p ≤ 0.001 significance determined using a two-tailed Fischer’s exact test (n.s. – not significant).
TCRs from single clonotypes can also be therapeutically effective when sharing CDR elements in both TRA and TRB with TCRs identified by convergent recombination.
We also cloned all eight TCRs represented by single T cell clonotypes (Fig. S3) into retroviral vectors and used again TCR-transduced CD4+ T cells for adoptive transfer into C3H Rag-/- mice bearing large, established solid 6132A tumors. Interestingly, only H12 was as effective as H6 and H9 while H11, H14, H15 and H16 also destroyed tumors, but the majority of mice relapsed more rapidly. TCRs H7, H8 and H10 had almost no anti-tumor effects (Fig. 2C).
When further evaluating the amino acid sequence of TCRs made by single T cell clonotypes, we discovered that some TCRs share CDR elements with preferentially selected TCRs (Fig. 2D): the V(D)J-elements of H11 and H14 are almost identical to H6 except for a single amino acid difference in the α-chains and a single amino acid difference in the β-chains precisely at the site of V(D)J rearrangement. TCR H12 uses the identical β-chain of H6 and an α-chain in which the V region from H6 was recombined with the J region of H9. TCR H15 is almost identical to H13 except for two amino acid differences in the α-chain, precisely at the site of V(D)J rearrangement. TCR H16 uses the same α-chain V gene as H6 combined with a different J gene and the same β-chain V gene as H13 combined with a different J gene. Notably, the therapeutically failing TCR H7 shared the identical β-chain with H6 but had a unique α-chain. TCRs H8 and H10 had completely unique V(D)J-usages and thus completely different amino acid sequences in their respective α- and β-chains.
Based on therapeutic success and representation by either multiple or single clonotypes, the 11 TCRs fell into three groups (Fig. 2E). Group 1 encompasses the preferentially selected TCRs (H6, H9 and H13). Each of these TCRs was therapeutically effective but has its own unique amino acid sequence characterized by convergent recombination of multiple clonotypes. Group 2 (H11, H12, H14, H15 and H16) is composed of TCRs that are also therapeutically effective but derived from single T cell clonotypes. Remarkably, all TCRs in group 2 share CDR elements in both chains with the preferentially selected TCRs (group 1). Group 3 TCRs (H7, H8 and H10) are also derived from single clonotypes but fail therapeutically. These group 3 TCRs lack CDR elements of group 1 TCRs in either one or both chains. Interestingly, therapeutic success was similar between mice treated with TCRs from group 1 and 2 (Fig. 2E), except that relapse occurred earlier with TCRs from group 2 (Fig. 2F). By contrast, mice treated with TCRs from group 3 had no significant survival improvements compared to mice treated with the control TCR αmL26 (Fig. 2E).
These results using H6 (preferentially selected, therapeutically effective), H10 (single clonotype, lacking elements, therapeutically failing), H12 (single clonotype, with shared elements, therapeutically effective) and the control TCR αmL26 were confirmed in another UV-induced tumor model 4102 (17) which was engineered to express the mL9 neoantigen (Fig. S4).
Strikingly, we found that the well-studied model antigen moth cytochrome c (MCC) also induces a CD4TCR response that consists of TCRs characterized by convergent recombination and TCRs from single clonotypes with shared elements (22) (Fig. S5). When introducing MCC into 6132A and 4102, mice treated with TCRs characterized by convergent recombination (group 1) showed again a significant (p = 0.005) therapeutic advantage over mice treated with either TCRs from single clonotypes with shared elements (group 2) or mice treated with the control TCR αmL26 (p = 0.0001, Fig. S6). Thus, in two distinct tumor systems targeting autochthonous neoantigen or model antigen, convergent recombination identified TCRs that mediated superior anti-tumor immunity upon adoptive transfer.
CD4TCRs cause destruction of tumor vessels but not of preexistent vasculature.
To determine how neoantigen-specific CD4TCRs caused tumor shrinkage, we used the tumor window technology and longitudinal confocal microscopy (23) to follow the cellular and vascular events that occurred in the first three weeks after CD4TCR T cell transfer. Window frames were implanted into a dorsal skinfold of mice. A circular hole of 1 cm diameter was dissected from one side of the skin flap by removing the skin with its fascial plane while leaving intact the opposite skin layer with its fascial plane and associated vasculature. Cerulean-labeled 6132A cancer cells were then injected under the remaining fascia before covering the opening with a glass pane. During the following 14 to 16 days, tumors developed dorsal to the window. When mice were treated with H6-T cells, DiD-labelled red blood cells were also injected to visualize the blood flow. A custom-made precision holder was used for the window frames to be able to focus on the exact same positions of multiple different areas in the tumors and to revisit these areas on different days. Thus, we could examine longitudinally the progressive changes of vasculature, blood flow, and cancer cells in a defined area over time after treatment with H6-T cells (Fig. 3A). Macroscopic regression of the tumors began about 4 to 5 days after T cell transfer and correlated with the disappearance of the flow of the DiD-labelled red blood cells in the tortuous tumor vessels. Quantification of the images during the early/first phase of tumor shrinkage showed that the area covered by vessels regressed by about 50% comparing day 4 with day 6 after H6-T cell transfer while the area covered by cancer cells regressed on average by 70% (Fig. 3B). Flow cytometric analyses revealed an increase in dead endothelial cells in tumor tissue 6 – 8 days after transfer of H6-T cells (Fig. 3C). Furthermore, these tumor tissues had significantly (p = 0.02) higher IFN-γ and TNF values compared to the control tumors from mice that received no or mL26-specific T cells (Fig. 3D and 3E). During the later/second phase of tumor shrinkage, the windows appeared flooded with unstained particles and assumed a ground-glass appearance consistent with debris resulting from cellular destruction (Fig. 3A). Histology of tumor tissue taken at day 6 after T cell transfer verified large areas of destroyed vessels and dead cells mainly in the tumor center. By contrast, pre-existing vessels stayed intact in the surrounding normal tissue at the tumor margin where cancer cells survived and T cells accumulated (Fig. S7 and Fig. S8A). However, starting around day 10 after T cell transfer, the windows cleared, and patches of cancer cells became visible within regular non-tortuous thinner vasculature. This characterized the fully arrested stage in which the cancer cells remained long-term.
Fig. 3. Stroma recognition by CD4+ T cells is sufficient to cause tumor destruction followed by growth arrest.
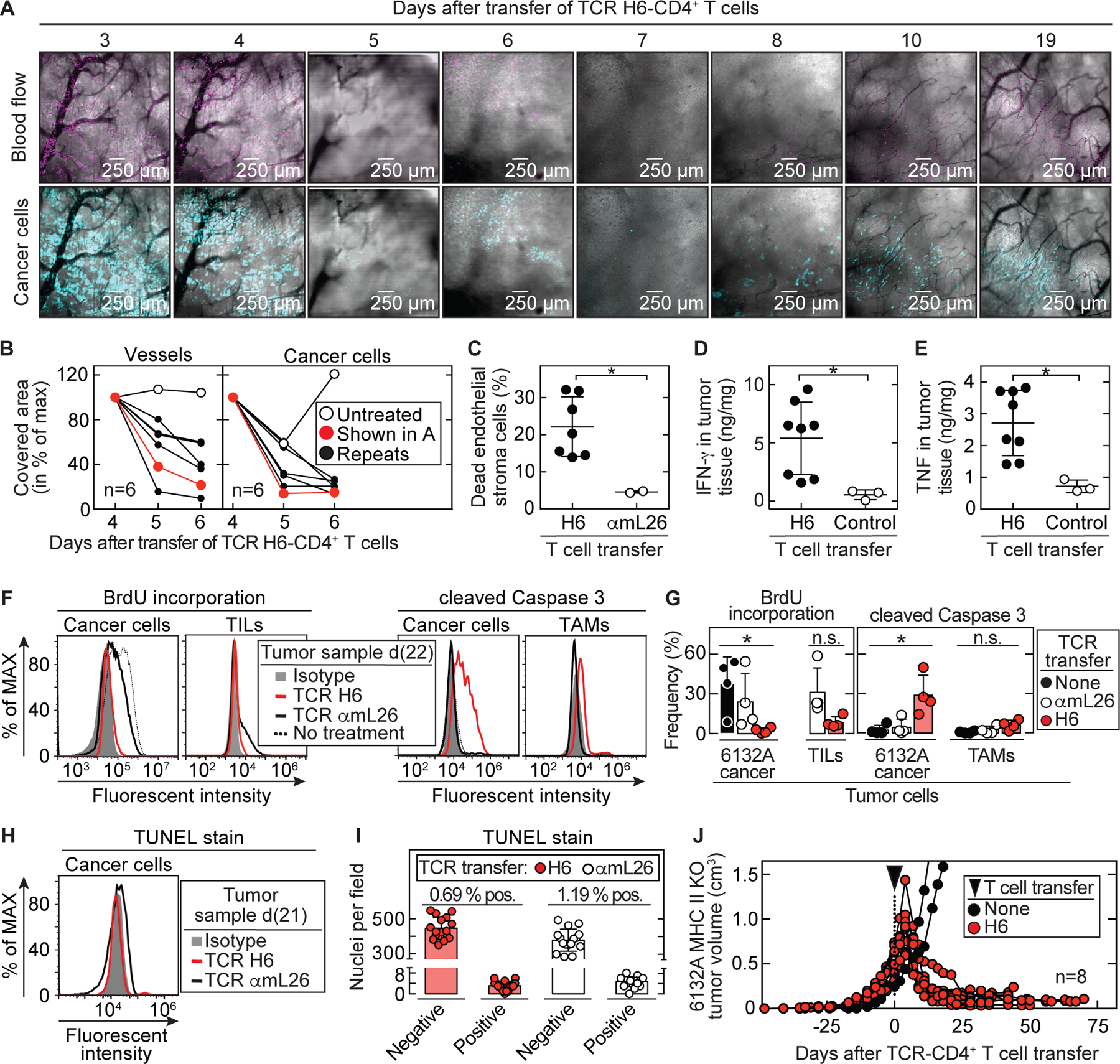
(A) Example of longitudinal microscopy in 6132A-cerulean tumor bearing C3H Rag-/- mice after transfer of H6-T cells. Tumor areas were randomly chosen before therapy and analyzed for (B) vessel and cancer cell reduction (total n = 6). DiD-labeled erythrocytes were used to visualize blood flow. Imaged area (in pixels) that was covered by vessels (black) or cancer cells (blue) from day 4 was set to 100%. Following days were assigned as percentage of maximum covered area. Indicated are an untreated control mouse (open circle) and the H6-treated mouse (red) shown in (A). Histology of tumor and vessel destruction on day 6 are shown in Fig. S7. (C – E) Tumor tissue was analyzed on day 6, 7 and 8 after therapy by flow cytometry. Control tumors received either no T cells (total n = 1) or αmL26-T cells (total n = 2) and were analyzed at day 8. Results are means ± SD from two independent experiments. Significance between groups was determined by a two-tailed Student’s t-test with *p ≤ 0.05. (C) Tumors were analyzed for dead endothelial cells (Sytox-positive, CD146 and CD31 double-positive cell populations) (total n = 7). (D) IFN-γ and (E) TNF concentrations in tumor tissue were determined (total n = 8). (F – I) 6132A-ECFP was used for injection into C3H Rag-/- mice. (F – G) Tumors were left untreated (total n = 4) or treated with either H6- (total n = 4) or αmL26-T cells (total n = 4). Mice were injected with BrdU twice a day for three consecutive days before tumor tissue was isolated at day 20 – 25 after T cell transfer. (F) A representative flow cytometry analysis is shown. (left) 6132A-ECFP cancer cells and TILs (CD3+, CD4+ and mL9-tetramer+) were analyzed by flow cytometry for frequency of BrdU incorporation. (right) 6132A-ECFP cancer cells and TAMs (CD11b+, F4/80+) were analyzed by flow cytometry for activation of cleaved caspase 3. (G) Significance between groups of 6132A cancer cells was determined by an ordinary one-way ANOVA with *p ≤ 0.05 (n.s. – not significant). Results are compiled from three independent experiments. (H – I) Tumors were treated either with H6- or αmL26-T cells. Tumor tissue was isolated at day 20 – 22 after T cell transfer (H) Life 6132A-ECFP cancer cells were analyzed by TUNEL-stain using flow cytometry. One representative flow cytometry analysis is shown out of two independent experiments. (I) DNA damage on formalin-fixed paraffin-embedded 6132A tumor slides was determined using TUNEL stain by immunohistochemistry. Eight fields were counted per slide. Shown is the total number of nuclei that were either stained negative or positive for TUNEL. The proportion (%) of TUNEL positive nuclei was slightly higher (p = 0.0017) in αmL26-treated control samples (1.19 ± 0.45 %) compared to H6-treated samples (0.69 ± 0.39 %). (J) C3H Rag-/- mice bearing 6132A MHC II KO tumors (red, total n = 8) were treated with H6-T cells 31 to 35 days after cancer cell injection, indicated by the arrow head. Spleens from C3H CD8-/- mice were used as CD4+ T cell source for TCR-engineering. Average tumor sizes were 0.530 cm3 ± 0.170 cm³ standard deviation at day of treatment. Data are summarized from two independent experiments. Shown are untreated tumors (black, total n = 2) as control.
CD4TCRs cause long-term tumor growth arrest.
Strikingly, after the bulk of the tumor mass had been destroyed and shrunken to small sizes, the remaining tumors persisted over the entire observation periods (≥75 days) (Fig. 2C). H6-T cells persisted in the peripheral blood for months (Fig. S8B), and there was no notable decrease in the intensity of CD4+ T cell infiltration even at the longest observation time point (124 days, Fig. S8C). To determine whether the stable size was the result of an equilibrium between cancer cell growth and death, we injected mice with BrdU for three consecutive days. We found that proliferation of 6132A cancer cells had ceased almost completely in the tumors remaining small after treatment with the H6-TCR (Fig. 3F, left). Surprisingly, a large fraction of 6132A cancer cells showed cleavage of caspase 3 as determined by flow cytometry (Fig. 3F, right). Both findings were exclusively dependent on using the mL9-specific H6-TCR (Fig. 3G). Since the tumor stayed at a small size even though cancer cells were non-proliferative and positive for cleaved caspase 3, we investigated whether cancer cells could be readapted in vitro. Indeed, when removing tumors from the H6-T cell-treated host, cancer cells started to grow in vitro 60 days later, and a stable cell line was recovered which was able to induce new tumors in vivo and also could be treated again with H6-T cells (Fig. S9). Since cleaved caspase 3 can also be associated with DNA instability (24), we compared DNA damage in 6132A tumors when arrested after H6-T cell treatment with actively growing after treatment with αmL26-T cells (Fig. 3H and 3I). Interestingly, no elevated DNA damage was detected in the arrested tumor using TUNEL-stain for either of the two situations. In addition, we determined whether the number of mutations had changed by performing whole-exome and RNA-sequencing (Table S2) of in vitro readapted 6132A cancer cells from untreated, H6- or αmL26-treated tumors. The expression of nsSNVs by these three cell lines was virtually indistinguishable, showing that growth arrest and its reversion in vitro did not lead to any notable acquisition of additional mutations.
Stromal recognition is sufficient for tumor destruction and long-term growth arrest.
6132A cancer cells lack expression of MHC class II (25) and are therefore representative for most human cancers. Thus, 6132A cancer cells are not recognized directly by H6-CD4+ T cells but H6-T cells recognize similarly well CD11b+ cells and F4/80+ cells isolated from the stroma of 6132A tumors (16) indicating that stromal dendritic cells and macrophages become CD4+ T cell targets by presenting neoantigen. To determine whether the effect of CD4+ T cells depended solely on stroma recognition, we genetically deleted the beta chain of the I-E MHC class II molecule in 6132A cancer cells. H6-T cells were similarly capable of permanently shrinking and halting tumor progression of both, parental and I-Ek-deficient 6132A cancer cells (Fig. 3J).
TCR efficacy in vivo was not reliably predicted by in vitro responses of TCR transduced T cells.
We aimed to understand whether in vitro characterizations of our CD4TCRs correlate with features of preferential selection and in vivo efficacy. We stimulated all 11 CD4TCR-engineered T cell populations in vitro with dilutions of either mutant or wild type L9 peptide presented by spleen cells from wild type C3H/HeN mice and compared IFN-γ secretion values (Fig. 4A). None of the 11 CD4TCRs recognized wild type L9 peptide. Within the group of preferentially selected TCRs, H6 was able to detect very low amounts of mL9 peptide (EC50: 0.1 nM) whereas H9 (EC50: 10 nM) and H13 (EC50: 1 nM) needed 100x or 10x more peptide for effective stimulation. The single clonotype TCRs H11, H12, H14, H15 and H16, which share elements with preferentially selected TCRs, all detected low mL9 peptide amounts (EC50: 0.5 nM) almost as well as H6 and were more sensitive than H9 and H13. Single clonotype TCRs lacking elements from preferentially selected TCRs showed either no IFN-γ response (H7, H8) or were only stimulated by very high amounts of mL9 peptides (H10, EC50: 1,000 nM).
Fig. 4. Analysis of TCR-engineered CD4+ T cells in vitro did not reliably predict therapeutic value in vivo.
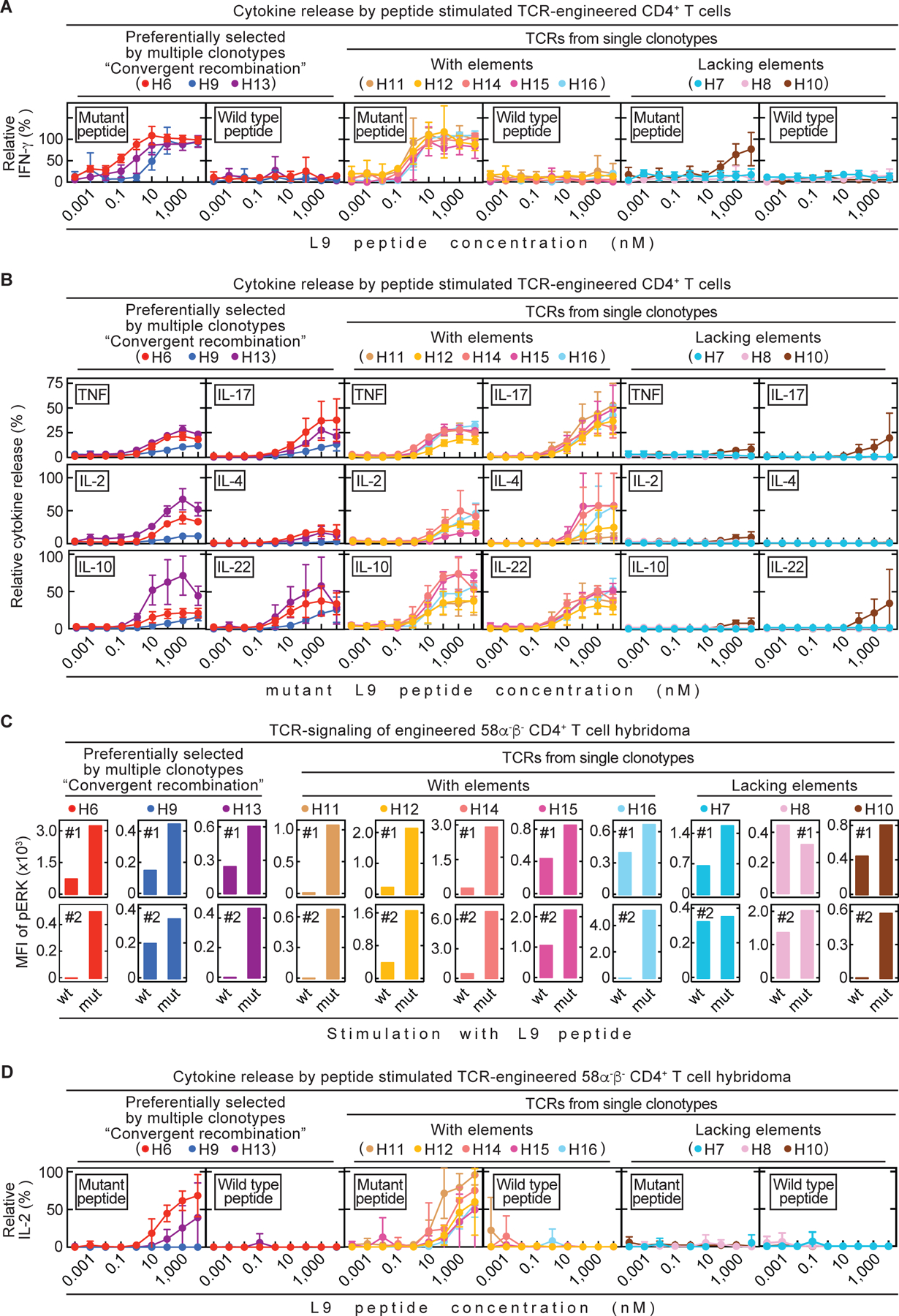
All 11 TCRs were tested in vitro. (A – B) Spleens from C3H CD8-/- mice were used as source for CD4+ T cells. TCR-engineered CD4+ T cells were co-cultured 24 h with C3H/HeN spleen cells and various mutant or wild type L9 peptide concentrations. Data are means ± standard deviation and compiled from two independent experiments. (A) Supernatants were analyzed for IFN-γ concentrations by ELISA. (B) Supernatants were analyzed for various cytokines by flow cytometry. (C – D) TCR-engineered 58α-β- CD4+ T cell hybridomas were used for co-cultures together with LK35 B cell hybridoma as antigen presenting cell (APC) of either mutant or wild type L9 peptide. (C) Phosphorylation of ERK1/2, as a measure of TCR-signaling, was determined by flow cytometry (Mean fluorescent intensity (MFI)). Life, TCR β-chain positive 58α-β- cells were analyzed. Shown are both (#1, #2) independently performed experiments. (D) Co-cultures were performed for 24 h using various mutant or wild type L9 peptide concentrations. Supernatants were analyzed for IL-2 by ELISA. Data are means ± standard deviation and compiled from two independent experiments.
The IFN-γ response in combination with other cytokines might more reliably predict the in vivo efficacy of our CD4TCRs. Therefore, we also determined cytokine values of TNF, IL-2, IL-4, IL-10, IL-17 and IL-22 after stimulation with different amounts of mL9 peptide (Fig. 4B). Again, T cells engineered with the TCRs H11, H12, H14, H15 and H16, which have shared elements, reliably produced as much TNF, IL-2, IL-17 and IL-22 as T cells engineered with the preferentially selected TCRs H6 and H13 while H9 always led to secretion of low cytokine values. However, there seems to be a difference in release of IL-4 and IL-10. The preferentially selected TCRs secreted almost no IL-4, which is in contrast to single clonotype TCRs with shared elements. The same seems to be true for IL-10. Interestingly, the preferentially selected TCR H13 also induces a stronger release of IL-10 and is associated with eventual relapse in vivo which is in contrast to H6 and H9. Furthermore, H11 and H12 as single clonotype TCRs with shared elements, also resulted in only low values of IL-4 and IL-10. Yet, tumors treated with H12 stay in long-term growth arrest while tumors treated with H11 relapse regularly within 25 days after T cell transfer. Additionally, TCRs H11, H14, H15 and H16 outperformed TCR H9 in the comprehensive cytokine analysis in vitro, yet in vivo, treatment with H11, H14, H15 and H16 was fraught with early relapse. The TCR-engineering of bulk T cells from the spleen can bias in vitro assays since the endogenous TCR may influence the strength of the response (26). Therefore, we made use of the TCR-negative 58α-β- CD4+ T cell hybridoma (27) to normalize the peptide L9 T cell response in vitro and generated 11 TCR-engineered 58α-β- CD4+ T cell lines (Fig. S10). We first determined the strength of TCR-signaling by phosphorylation of ERK, which is an indicator for T cell activation, (28) using flow cytometry (Fig. 4C). Preferentially selected TCRs (H6, H9 and H13) and single clonotype TCRs with shared elements (H11, H12, H14, H15 and H16) as well as the TCR H10 (lacking elements), all had increased mean fluorescent intensity (MFI) values of phosphorylated ERK when stimulated with mL9 peptide in comparison to wtL9 peptide. We did not observe a consistent increase in MFI of phosphorylated ERK in the TCRs H7 and H8. Additionally, we also analyzed the ability of the 11 TCR-engineered 58α-β- CD4+ T cell lines to secrete IL-2 (Fig. 4D). As before, none of the TCR-engineered 58α-β- cells recognized wild type L9. Yet again, the single clonotype TCRs H11, H12, H14, H15 and H16 demonstrated a strong IL-2 response and only the preferentially selected TCR H6 worked comparably well in vitro.
Since in vitro assays using peptide seem to be inconsistent for the understanding of in vivo efficacy of CD4TCRs, we investigated the cytokine response of TCR-engineered CD4+ T cells when stimulated with 6132A-tumor associated macrophages (TAMs) isolated from established tumors (TAMs, Fig. 5A). The preferentially selected TCRs H6 and H13 released high amounts of IFNγ, IL-2 and IL-10, and some degree of IL-17, while H9 only showed high release of IL-10, small secretion of IFNγ and IL-17, and almost no IL-2. Single clonotype TCRs with shared elements (H11, H12, H14, H15 and H16) responded similarly well to TAMs as H6 and H13. High secretion of IFNγ, IL-2 and IL-10 and some degree of IL-17 were detected. The single clonotype TCRs H7, H8 and H10 (lacking elements) failed to release any cytokines except for IL-10 at amounts that were comparable to the preferentially selected TCRs. Overall the cytokine response of TCR-transduced T cells to TAMs correlated as poorly as responses to peptide with in vivo efficacy.
Fig. 5. NO expression in 6132A tumor-associated macrophages is induced by T cells when transduced with therapeutically effective CD4TCRs.
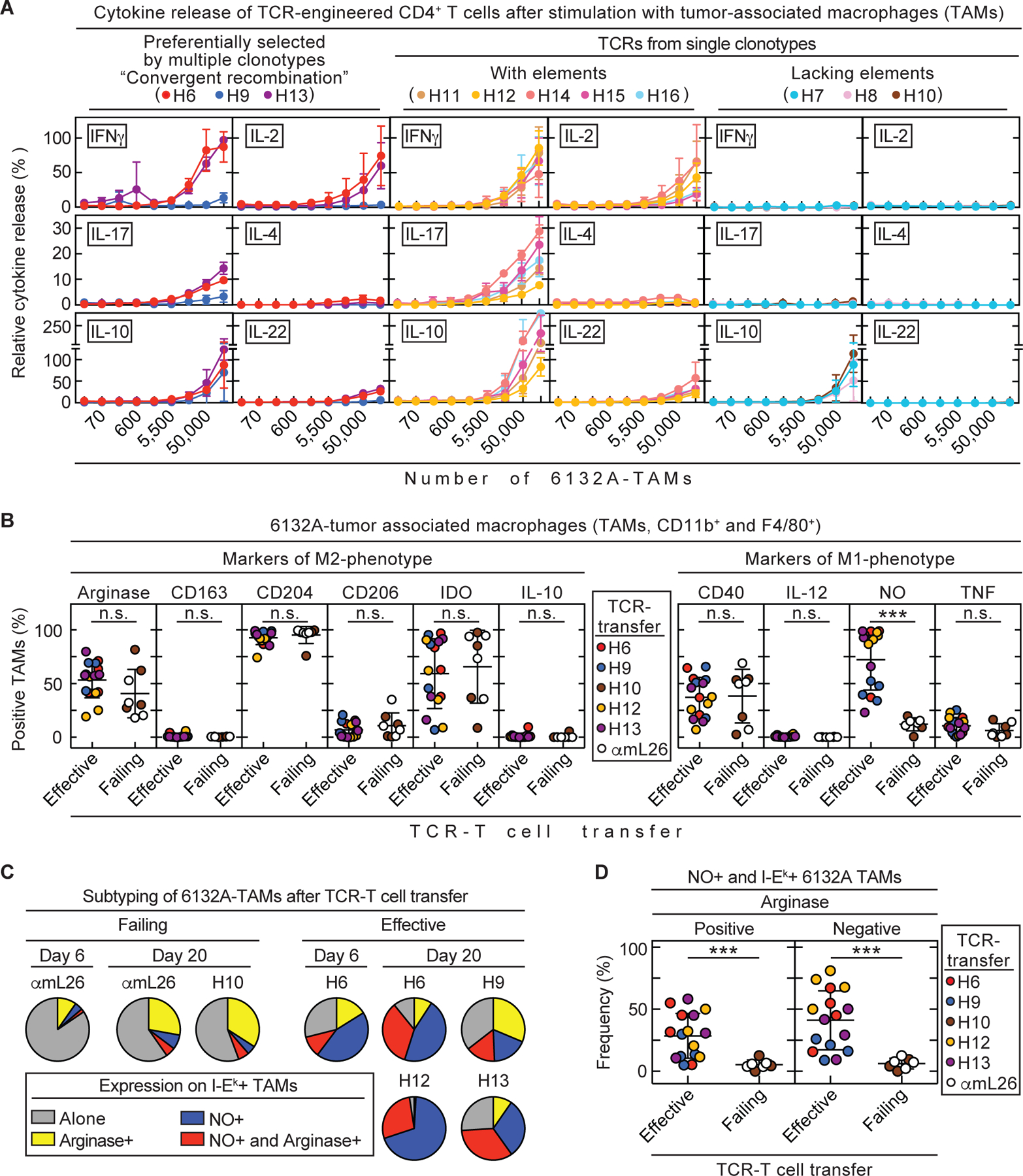
(A – D) Spleens from C3H CD8-/- mice were used as CD4+ T cell source for TCR-engineering. (A) TCR-engineered CD4+ T cells were co-cultured 24 h with 3-fold dilutions of tumor-associated macrophages (F4/80+ cells) isolated from 6132A tumors grown in C3H Rag-/- mice. Supernatants were analyzed for various cytokines by flow cytometry. Data are means ± standard deviation and compiled from two independent experiments. (B – D) C3H Rag-/- mice bearing 6132A tumors were treated with either H6- (n = 4), H9- (n = 4), H10- (n = 4), H12- (n = 4), H13- (n = 4) or αmL26- (n = 4) TCR-engineered T cells 21 to 23 days after cancer cell injection. Tumor tissue was isolated at day 20 – 22 after T cell transfer. Tumors were analyzed by flow cytometry for frequency of life CD11b+ and F4/80+ 6132A tumor-associated macrophages (TAMs) expressing M1-type (CD40, IL-12, NO and TNF) or M2-type (arginase, CD163, CD204, CD206, IDO and IL-10) markers. TCR-Treatment was divided into effective (H6, H9, H12 and H13, n = 16) and failing (H10 and αmL26, n = 8) therapy groups. Therapeutically effective TCRs are able to induce tumor shrinkage by more than >25% volume within 12 days after T cell transfer. All other TCRs are considered therapeutically failing which also includes the control TCR αmL26. Number (n) indicates the total number of tumors analyzed from independent mice. (B) Comparison of M1- and M2-type markers of TAMs from effective or failing TCR-T cell therapy. Significance between groups was determined by an unpaired, two-tailed Student’s t-test with ***p ≤ 0.001 (n.s. – not significant). Data are compiled from two independent experiments (C) MHC class II I-Ek positive TAMs were further analyzed by their frequency of expressing either arginase, NO, both or none. Data are compiled from three independent experiments (D) Frequency of NO and I-Ek expressing TAMs that were either positive or negative for arginase. Significance between groups was determined by an unpaired, two-tailed Student’s t-test with ***p ≤ 0.001. Data are compiled from two independent experiments.
CD4TCR efficacy correlates with ability to reprogram TAMs.
Since in vitro stimulation of TCR-transduced T cells unreliably predict in vivo efficacy of CD4TCRs, we focused on the interaction between stroma and CD4+ T cells required for tumor shrinkage and growth arrest. More than 80% of all CD11b+ cells in the 6132A microenvironment were F4/80+ TAMs (Fig. S11A). Thus, we examined which effect the different CD4TCRs might have on TAMs. Stromal recognition of TAMs by CD4TCRs was not associated with an increased death rate of TAMs since the number of non-viable TAMs did not differ significantly between untreated, H6- or αmL26-treated tumors (Fig. S11B). Two different TAM phenotypes, M1 and M2, have been described in the tumor microenvironment (29). In general, M2-TAMs promote tumor growth and are immunosuppressive while M1-TAMs are proinflammatory and tissue damaging (29). Therefore, we examined 6132A-TAMs for phenotypic changes in response to T cell transfer. Tumors were isolated at day 0, 6 and 20 after transfer of either H6- or αmL26-T cells, and TAMs were found to express the M2-type protein arginase (Fig. S12). Interestingly, we observed an increase of MHC class II I-Ek in almost all TAMs by day 20 after T cell transfer. However, this upregulation was not antigen-specific since it was similar after transfer of either H6- or αmL26-T cells (Fig. S12). Additionally, the fraction of TAMs expressing arginase also increased by day 20 but this again occurred independent of the antigen-specificity of the transferred T cells. Instead, H6-T cell transfer resulted in significant (p = 0.0001) induction of NO expression in almost all TAMs by day 20. This antigen-specific NO expression was absent when tumors were treated with αmL26 control T cells (Fig. S12). For a more comprehensive analysis on how phenotypic changes in TAMs predict the outcome of treatment, we analyzed TAMs for the expression of arginase, CD40, CD163, CD204, CD206, IDO, IL-10, IL-12, NO and TNF around 20 days after transfer of either therapeutically effective, preferentially selected TCRs H6, H9 and H13, or the single clonotype and therapeutically effective TCR H12, or the single clonotype but therapeutically failing TCR H10 using the αmL26-specific TCR as control (Fig. 5B). We only observed a significant (p = 0.0001) change in NO production of TAMs from 6132A tumors treated with therapeutically effective TCRs (72% of all TAMs are NO positive). Even the therapeutically effective TCR H9, which performed poorly in all of our in vitro stimulations, was able to induce NO production in TAMs while the therapeutically failing TCR H10, that also performed poorly in our in vitro stimulations, faltered. For further understanding of the TAM subpopulations, we analyzed the I-Ek-expressing TAM for their NO and arginase proportions. We found that TAMs from tumors treated with therapeutically effective TCRs consisted on average of 41% NO-producing TAMs while this cell population was minor (6%) in TAMs from mice treated with failing or control TCRs (Fig. 5C and 5D). This indicates that most TAMs present in an arrested tumor are of the M1-phenotype while TAMs found in growing tumors are mostly M2. However, TAMs that are double positive for NO and arginase were also detected in arrested tumors (28% versus 5% in growing tumors) showing that M2-TAMs are capable of producing NO without losing their M2-type identity. Thus, reprogramming of TAMs to produce NO correlated with therapeutically effective CD4TCRs.
DISCUSSION:
In this study, we show that selecting candidate TCRs based on convergent recombination can help identify TCRs that are therapeutically effective. Not only the TCRs made by multiple T cell clonotypes but also TCRs made by a single T cell clonotype had therapeutic value when they shared CDR elements in paired α- and β-chains with the TCRs characterized by convergent recombination. Identical TCRs encoded by different nucleotide sequences have been considered to be “preferentially selected” (20), because different T cell clonotypes expressing the same TCR developed independently multiple times in vivo. Thus, convergent recombination adds an important host-generated quality indicator for a “best-fit” TCR (30). Beneficial clinical outcome has been linked statistically to the increased occurrence of convergent recombination in bulk TCR β-chain sequencing only (31–33). However, presence of convergent recombination only in TCR β-chain sequences did not distinguish the therapeutically effective TCR H6 from the failing TCR H7. Therefore, predicting a therapeutic TCR depended on finding convergence in T cell clonotypes by paired α- and β-chain analyses.
Currently, the focus is on finding in vitro assays that can predict which TCR will be successful for immunotherapeutic interventions (34). Our in vitro analyses did not reliably predict the in vivo outcome. Both CD4TCRs H6 and H9 caused long-term growth arrest after tumor destruction although H6 responded well and H9 poorly to peptide or TAMs that present the tumor antigen. However, we do not know whether TCRs from single T cell clonotypes with strong responses to the mutant peptide or other types of in vitro activity could fail in vivo. Using mice that lacked endogenous T cells was essential to exclude the participation of such T cells (35, 36) and to evaluate stromal reprogramming as well as long-term outcome between preferentially selected TCRs and TCRs generated by single T cell clonotypes. However, this reductionist approach has also limitations because it does not examine how the efficiency of our T cell therapy might be affected by pre-conditioning regimens in immunocompetent mice (37).
Most human epithelial cancers do not express MHC class II and do not allow for direct recognition by CD4+ T cells, as observed in our tumor models (25) even though melanoma represents a notable exception (38–40). Nevertheless, adoptive transfer of CD4+ T cells has been shown to eradicate disseminated Friend virus-induced erythroleukemia, and these cancer cells were found to be MHC class II negative (41). A decrease in targeted lesions and growth control of the persistent cancer has also been achieved in patients after transfer of in vitro-expanded mutation-specific CD4+ TIL populations (13, 14). Loss of antigen or MHC are common causes of relapse after immune therapy with CD8+ T cells (42). In our model, relapse after CD4TCR therapy retained the targeted neoantigen (16). Although the CD4TCRs targeted the neoantigen only on stroma, spatial restriction in the tumor environment can favor the escape of antigen-loss variants (43). Therefore, another reason why we did not observe antigen loss variants might be that we targeted an antigen essential for cell survival and growth that is characterized by genetic loss of the wild-type allele (21). Loss of heterozygosity (LOH) of essential genes is increasingly being recognized as an underestimated potent class of cancer-specific targets (21, 44, 45) and can become a paradigm shift for cancer therapy (46). Previous reports showed destruction of tumor vessels followed by ischemic necrosis of large areas of solid tumors by effects of IFN-γ and/or TNF (47–50) which we also observe through antigen-specific release of IFN-γ and TNF by tumor-infiltrating CD4TCR-T cells. After tumor destruction, the surviving cancer cells persisted at tumor margins nourished by the pre-existent non-tumor vasculature that is resistant to IFN-γ and TNF (51). The tumor microenvironment is widely considered to be tumor-promoting (52), immuno-suppressive (53) and a barrier for effective CD8+ T cell therapy. Indeed, a part of stromal TAMs in untreated or control-TCR treated mice expressed arginase but only few expressed NO consistent with an immunosuppressive environment (54, 55). Changes in MHC II expression on TAMs in tumors treated with non-specific T cells were observed which is consistent with bystander infiltration and activation of non-specific T cells in cancer and viral diseases (56–58). Nonetheless, we observed antigen-specific reprogramming of M2-type TAMs together with appearance of M1-type TAMs. Therefore, our approach of identifying and utilizing CD4TCRs for adoptive T cell transfer gives evidence for the concept that the immunosuppressive, tumor-promoting microenvironment can be targeted and reprogrammed by tumor-infiltrating neoantigen-specific CD4+ T cells.
NO is known for its reversible cytostatic effect on cancer cells (59), and previous studies showed that CD4+ T cells producing IFN-γ and TNF signal TAMs to activate nitric oxide synthase (60, 61), thereby preventing the outgrowth of cancer cell inocula. This is in line with our observation that effective T cells secreting IFN-γ and TNF occur together with TAMs that produce NO. Histochemistry showed T cells densely infiltrating non-proliferating cancer cells forming a “stalemate” with T cells maintaining cancer cells in growth arrest without eradicating them. Thus, growth arrest was not due to an equilibrium of growth and death of cancer cells as in previous studies targeting tumor stroma with CD8+ T cells (62, 63). The reversibility we observed also appears to exclude growth arrest due to CD4+ T cell-induced senescence (64). Instead, we found that cleaved caspase 3 positive arrested cancer cells without damaged DNA could recover. This has been reported in other studies (65), is consistent with NO being an anti-apoptotic regulator of caspase 3 activity in vivo (66) and is now referred to as “anastasis” (67, 68). It also had been proposed that cleaved caspase 3 could cause genetic instability and might be involved in carcinogenesis (24, 65). However, one reason for accumulation of mutations are errors during DNA replication (69). Since the cancer cells in our model are growth arrested, no DNA replication occurs and thus the acquisition of new mutations is hindered. Indeed, we found no significant increase in mutations, which is consistent with lack of DNA damage, in cancer cells readapted after treatment.
Together, our study shows that the clonally diverse CD4+ T cell response in progressive cancers harbors some CD4TCRs that are of therapeutic value in adoptive therapy settings. We suggest that convergent recombination in paired TCR chains can be used to identify these therapeutically effective CD4TCRs and that this strategy can become applicable when treating human cancers.
Materials and Methods
Study design
Objective of the study was to determine therapeutically effective TCRs used for adoptive transfer of TCR-engineered CD4+ T cells against established solid tumors. Animal experiments were approved by The University of Chicago Institutional Animal Care and Use Committee (IACUC). Cancer cells were injected s.c. in the shaved back of mice. Tumor volumes were measured along 3 orthogonal axes, every 2 – 3 days and were calculated as (a x b x c) ÷ 2. Mice were treated around 3 weeks after cancer cell injection when tumors were established. The number of TCR+ T cells was calculated based on transduction rate (determined by TCR Vβ-stain, on average ~ 30%) on the day of treatment prior to T cell transfer. Per recipient, 2x106 TCR+ CD4+ T cells were injected i.p. Mice were randomized into different treatment groups on the day of adoptive T cell transfer. Mice were euthanized when tumor sizes reached more than 2 cm3 or mice appeared hunched and weak. Relapsing tumors were allowed to reach 1.5 cm3 before mice needed to be euthanized. TCRs were defined as being therapeutically effective when tumor volume shrunk by more than 25% within 12 days after T cell transfer, otherwise TCRs were defined as therapeutically failing. Therefore, the control αmL26 TCR was also included in the therapeutically failing TCR group. Experimental replicates are included in figure legends.
Mice
3 to 8 months old female and male mice were used in this study. Mice were bred and maintained in a specific pathogen-free barrier facility at The University of Chicago according to IACUC guidelines. C3H/HeN mice were obtained from Envigo (Huntingdon, Cambridgeshire, United Kingdom, RRID:MGI:2160972). C3H Rag2-/- (C3H.129S6-Rag2tm1Fwa) mice were obtained from Douglas Hanahan (University of California, San Francisco, CA, USA). C3H CD8-/- (C3H.129S2-Cd8atm1Mak) mice were generated in house by crossing C3H/HeN mice with C57BL/6 CD8-/- mice purchased from the Jackson Laboratory (B6.129S2-Cd8atm1Mak, RRID:MGI:3789587) and then backcrossed with C3H/HeN for 20 generations. Spleen of C3H CD8-/- mice were used as T cell sources for TCR-engineering.
Cell lines
6132A and 4102 cancer cell lines originated from UV-treated C3H/HeN mice and were generated in our laboratory together with heart-lung fibroblasts as autologous normal tissue controls for each cancer cell line (17). The original primary tumors were minced and fragments were used to establish uncloned primary cultures of 6132A and 4102 cancer cells. These primary tumor cell cultures were only minimally expanded, and used for cell culture experiments and tumor induction in vivo. The 6132A-ECFP was generated by using retroviral transduction with the pMFG-ECFP vector as described before (23). 6132A-Cerulean was descripted before (70). Knockout of the H2-Eb1 gene results into I-E beta chain loss and therefore loss of MHC class II expression. The 6132A-H2-Eb1 knockout cell line was generated using CRISPR-Cas9. Single guide (sg) RNAs targeting exon 1 of the murine C3H H2-Eb1 gene were designed using the sg RNA design tool from the Broad Institute. The corresponding sense and antisense DNA oligomers (IDT, Coralville, IA, USA) were compared to other publications that also targeted H2-Eb1 to generate murine MHC class II knockout cancer cell lines. The DNA oligomers were annealed and cloned over an BbsI side into PX458. The sg RNA 5’ – AGGAGACACGAGAGTCAGAG – 3’ was successfully used to generate 6132A-H2-Eb1-/- cancer cells which were verified by Sanger-sequencing to have an indel and frameshift in exon 1. The 25mer of moth cytochrome c (MCC) and mL9 was cloned into the retroviral vector pMP71 (pMP71-mL9-P2A-eGFP and pMP71-MCC25-P2A-eGFP) and used to generate 6132A-MCC-GFP as well as 4102-MCC-GFP and 4102-mL9-GFP cell lines. Phoenix-ampho cells were transfected by calcium phosphate precipitation. Repeated rounds of transduction of either 6132A or 4102 with viral supernatants followed by fluorescence-based cell sorting (FACS Aria II, BD Biosciences, San Jose, CA, USA) derived highly GFP fluorescent cell lines which were cloned for homogeneous expression. The B cell hybridoma LK35 (71) was provided by Dr. Andrea Sant from the University of Rochester and maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine and 0.1 mM non-essential amino acids and cultured at 10% CO2 in a 37 °C dry incubator. Cancer cells were maintained in DMEM supplemented with 5% FBS (Gemini Bio-Products) and 2 mM L-glutamine (Life Technologies, Carlsbad, CA, USA) and cultured at 10% CO2 in a 37 °C dry incubator. Plat-E packaging cells (72) used for TCR gene transfer and Phoenix ampho used for gene transfer of neoantigens were maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 1µg/mL Puromycin and 1mg/mL Blasticidin (Invivogen, San Diego, CA, USA) and cultured at 5% CO2 in a 37 °C dry incubator. The 58α-β- CD4+ T cell hybridoma was provided by Dr. David Kranz from the University of Illinois Urbana-Champaign (27) and its TCR-engineered variants were maintained in RPMI 1640 (Corning, Corning, NY, USA) 10% FBS (Gemini, Sacramento, CA, USA), 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/mL penicillin, 100 µg/mL streptomycin (all purchased from Life Technologies, Carlsbad, CA, USA), 50 µM 2-mercaptoethanol (Thermo Fisher Scientific, Waltham, MA, USA) and 50 µg/mL gentamicin (VWR, Radnor, PA, USA) and cultured at 5% CO2 in a 37 °C dry incubator. Before use, cancer cell lines were authenticated by sequencing and/or co-culture with antigen-specific T cells and by morphology. All cell lines were shortly passaged after thawing of the initial frozen stock to generate master cell banks. Working batches were passaged no longer than 4 weeks.
Cell sorting, single cell sequencing and isolation of TCR genes
After harvesting tumor and spleen tissue between day 18 and 28 after injection of cancer cell fragments, single cell suspensions were prepared and stained for Sytox Blue (Helix NP Blue, Biolegend, San Diego, CA, USA, life/dead stain), CD3, CD4 and tetramer respectively, before viable tetramer binding CD4+ T cells were sorted (FACSAriaII, BD Bioscience, Franklin Lakes, NJ, USA). Samples from different mice were stained with TotalSeq™-C Hashtag antibodies #4, #6 and #8 (Biolegend, San Diego, CA, USA) and combined prior to sorting. The 10X Genomics (10X Genomics, Pleasanton, CA, USA) Chromium controller and the single cell 5’ dual index platform was used to generate TCR-libraries following manufacture protocol. Next generation sequencing was performed at the University of Chicago Genomics facility using NovaSeq 6000 (Illumina, San Diego, CA, USA). TotalSeq™-C Hashtag antibodies were used to demultiplex the different mice. Therefore, only 4 data points are shown in Fig. S1B. These data points are from tetramer sorts of mouse #1, #2, #3 + #4 and #5 + #6. Unfortunately, the TotalSeq™-C Hashtag procedure failed for mouse #5 and #6. Therefore, the two mice could not be separated. Obtained TCR sequences were codon optimized (GeneArt, Thermo Fisher Scientific, Waltham, MA, USA) and integrated into the pMP71 vector using NotI and EcoRI flanked restriction sides as described (73). The control TCR αmL26 was isolated from a T cell clone specific for the H96Y mutation in the ribosomal protein L26 which was identified in 6139B cancer cells and has been characterized before (16, 21). Additionally, the TCR H6 isolated from tumor-bearing mice is identical to a TCR identified in immunized mice (2, 16). MCC-specific TCR sequences 5c.c7, AND, M2.3 and M4.3 have been described before (22) and were also codon optimized and integrated into the pMP71 vector.
TCR-engineering of primary CD4+ T cells
TCR-engineering was conducted as previously described (74). A separate retroviral vector was generated for each TCR: pMP71-H6, -H7, -H8, -H9, H-10, H-11, -H12, -H13, -H14, -H15, -H16, -5c.c7, -AND, -M2.3, -M4.3 or -αmL26. Potential mispairing of transduced TCRs (75) was prevented by using a P2A element in TCR-vector designs. Plat-E packaging cells were transfected with pMP71-H6, -H7, -H8, -H9, H-10, H-11, -H12, -H13, -H14, -H15, -H16, -5c.c7, -AND, -M2.3, -M4.3 or -αmL26 by calcium phosphate precipitation. 42 h after transfection, virus supernatant was removed and filtrated through a 0.45 µm syringe filter (VWR, Radnor, PA, USA). Spleens were isolated and erythrocytes were lysed for 3 min with 0.017 M TRIS, 0.14 M ammonium chloride (both Sigma-Aldrich, St. Louis, MO, USA). Cells were cultured in complete medium containing Roswell Park Memorial Institute medium (RPMI 1640, Corning, Corning, NY, USA) 10 % FBS (Gemini, Sacramento, CA, USA), 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/mL penicillin, 100 µg/mL streptomycin (all purchased from Life Technologies, Carlsbad, CA, USA), 50 µM 2-mercaptoethanol (Thermo Fisher Scientific, Waltham, MA, USA), 50 µg/mL gentamicin (VWR, Radnor, PA, USA) and were supplemented with 40 U/mL recombinant human IL-2 (Peprotech, Rocky Hill, NJ, USA). The cell suspension was transferred into a 24-well plate (Greiner Bio-One, Kremsmuenster, Austria) coated with 1.4 µg/mL αCD3 (University of Chicago, Frank W. Fitch Monoclonal Antibody Facility, Clone 145–2C11.1) and 0.2 µg/mL αCD28 (Clone 37.51, Biolegend, San Diego, CA, USA) at a concentration of 3x106 cells/mL. On the subsequent day, 0.5 mL of corresponding virus supernatant containing 8 µg/mL protamine sulfate (Sigma-Aldrich, St. Louis, MO, USA) was added per well and cells were spinoculated (800 x g, 90 min, 32 °C). Overnight, a 12-well plate (Greiner Bio-One, Kremsmuenster, Austria) was coated with RetroNectin [12.5 µg/mL (TaKaRa)] and centrifuged with 1.5 mL virus supernatant (3000 x g, 90 min, 4 °C) on the next day. The virus supernatants were removed and 5x106 of CD4+ T cells in complete medium containing 40 U/mL IL-2 were transferred to the virus coated 12-well plate and followed by spinoculation (800 x g, 90 min, 32° C). Transduction rate was confirmed by flow cytometry using NovoCyte Quanteon (Agilent, Santa Clara, CA, USA) and T cells were used 3 days after transduction for adoptive transfer. For in vitro analyses, TCR-engineered CD4+ T cells were maintained in complete medium with 40 U/mL IL-2 and used after 4 days.
TCR-engineering of the 58α-β- CD4+ T cell hybridoma
Plat-E packaging cells were transfected with pMP71-H6, -H7, -H8, -H9, H-10, H-11, -H12, -H13, -H14, -H15 and -H16 by calcium phosphate precipitation. 42 h after transfection, virus supernatant was removed and filtrated through a 0.45 µm syringe filter (VWR, Radnor, PA, USA). A 24-well plate was coated overnight with 500 µL per well RetroNectin [12.5 µg/mL (TaKaRa)]. The coated plate was centrifuged with 0.5 mL virus supernatant (3000 x g, 90 min, 4 °C). Supernatant was removed and 2x105 58α-β- cells in 1 mL/well RPMI 1640 (Corning, Corning, NY, USA) 10 % FBS (Gemini, Sacramento, CA, USA), 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/mL penicillin, 100 µg/mL streptomycin (all purchased from Life Technologies, Carlsbad, CA, USA), 50 µM 2-mercaptoethanol (Thermo Fisher Scientific, Waltham, MA, USA), 50 µg/mL gentamicin (VWR, Radnor, PA, USA) were added. The plate was centrifuged at 800 x g for 30 min and 32 °C. On the subsequent day, 0.5 mL of corresponding virus supernatant containing 8 µg/mL protamine sulfate (Sigma-Aldrich, St. Louis, MO, USA) was added per well and cells were again spinoculated (800 x g, 90 min, 32 °C). Three days later, 58α-β- cells were stained for TCR β-chain and sorted using FACSAriaII (BD Bioscience, Franklin Lakes, NJ, USA). After cells recovered in vitro, the level of expression of the TCRs among the different TCR-engineered 58α-β- cells was determined by TCR β-chain staining using flow cytometry using NovoCyte Quanteon (Agilent, Santa Clara, CA, USA).
Cancer cell injection in mice
For generation of tumor bearing C3H/HeN wild type mice, 6132A-fragments were generated and injected s.c. as previously described (76). For treatment of established 6132A, 6132A-MCC-GFP, 4102-mL9-GFP or 4102-MCC-GFP tumors, 1x107 cancer cells were injected s.c. into the shaved back of C3H Rag2-/- mice.
Tumor preparation and isolation of CD11b+ and F4/80+ cells
6132A tumors, either grown in C3H/HeN mice for isolation of tetramer-binding CD4+ T cells or grown in C3H Rag2-/- mice for isolation of APCs, were removed and single cell suspensions were generated by enzymatic digestion (77). Tumors were minced, 2 mg/mL Collagenase D and 100 U/mL DNAse I (both Roche, Indianapolis, IN, USA) were added and suspension was incubated for 20 min at 37 °C in RPMI 1640 on a horizontal shaker. Following the addition of trypsin in Hanks’ Balance Salt Solution (HBSS, MP Biomedicals LLC, Solon, OH, USA) to a final concentration of 0.025%, cell suspension was incubated for additional 15 min at 37 °C on a horizontal shaker. Tumor cell suspension was filtered over a 40 µm cell strainer (Thermo Fisher Scientific, Waltham, MA, USA) and used subsequently. For the isolation of APCs, CD11b+ and F4/80+ cells were collected by magnetic cell sorting (Miltenyi, Bergisch Gladbach, Germany) following manufacturer’s protocol. Successful isolation was confirmed by FACS before both cell populations were used for T cell stimulation.
Tumor tissue analysis
At day six, seven and eight after ATT, tumors were isolated and about 100 mg were homogenized using Polytron (Kinematica, Lucern, Swiss) and spun down. Supernatants were used for determination of cytokines by flow cytometry using Legendplex according to manufacture protocol (Biolegend, San Diego, CA, USA). For endothelial cell analysis, single cell suspension from tumor tissue was generated as described under “Tumor preparation”. Tumor single cell suspensions were analyzed for dead CD31+ and CD146+ cell populations with Sytox Blue (Helix NP Blue, Biolegend, San Diego, CA, USA) by flow cytometry.
T cell stimulation and cytokine analysis
TCR-engineered T cells or TCR-engineered 58α-β- cells were cocultured for 24 h with APCs to determine specificity and sensitivity. In brief, 1x105 T cells were added to 1x105 stromal cells isolated from tumor. For TCR independent stimulation, 8 µg/mL αCD3 (University of Chicago, Frank W. Fitch Monoclonal Antibody Facility, Clone 145–2C11.1) and 2 µg/mL αCD28 (Clone 37.51, Biolegend, San Diego, CA, USA) was used. In addition, T cells were also cocultured with spleen cells isolated from C3H/HeN mice and 26mer mL9, wtL9 or MCC peptides at various concentrations indicated in the figure legends. After 24 h, supernatants were removed and tested for IFN-γ or IL-2 concentrations by enzyme-linked immunosorbent assay (ELISA, Ready-SET-Go!, eBioscience, San Diego, CA, USA), following the manufacturer’s protocol. Light absorbance at 450 nm was read with the microplate reader VERSAmax (Molecular Devices LLC, San Jose, CA, USA) respectively. Furthermore, supernatants were used for determination of various cytokines by flow cytometry using Legendplex according to manufacture protocol (Biolegend, San Diego, CA, USA).
Analysis of TCR signaling by phosphorylation of ERK
For measuring strength of TCR signaling, 1x105 TCR-engineered 58α-β- T cell hybridomas were co-cultured with 1x105 LK35 cells. The LK35 cells were cultured prior overnight with 1 µM mutant L9 (mL9) or 1 µM wild type L9 (wtL9) peptide in 96-well U-bottom plates. TCR-engineered 58α-β- cells were life/dead stained with fixation resistant dye 510 (BD Bioscience, Franklin Lakes, NJ, USA) prior to addition to LK35 cells. Co-cultures were stopped at 0, 5, 10, 15, 20, 25 and 30 min after addition of TCR-engineered 58α-β- cells. Plates were centrifuged for 20 sec at 400 g after each T cell addition to initiate contact and placed in a 37 °C humidified incubator at 5% CO2. After the last time point, the plate was centrifuged at 400 g for 4 min, supernatants were discarded, and cells were immediately fixed with 100 µL/well ice-cold 10 % formalin solution (containing 4 % formaldehyde) for 15 min on ice. PBS (100 µL/well) was added, cells were centrifuged for 4 min at 400 g, and supernatants were discarded. Cells were then permeabilized with 100 µL/well ice-cold 90 % methanol for 15 min on ice. PBS (100 µL/well) was added, cells were centrifuged for 4 min at 400 g, and supernatants were discarded. Cells were then FcR-blocked (50 µL/well of anti-FcR clone 2G4 in PBS) for 10 min at 4 °C, washed and stained at a 1:50 dilution intracellularly for phosphorylated ERK1/2 and at a 1:100 dilution for I-Ek and TCR β-chain to distinguish LK35 cells from TCR-engineered 58α-β- cells for 30 min at room temperature, before resuspending in PBS and analyzing phosphorylated ERK1/2 by flow cytometry. The MFI of TCR-engineered 58α-β-cells stimulated by wtL9 peptide was averaged from all time points and considered background. The time point which showed the peak response to mL9 peptide was used and background was subtracted from both samples (stimulation with wtL9 or mL9).
Analysis of tumor-associated macrophages (TAMs)
6132A tumor tissue was harvested at day 0, 6 and 20 after transfer of T cells. Single cell suspensions were prepared as described under (Tumor preparation) and incubated with DAF-FM (Life Technologies, Carlsbad, CA, USA) following manufacturer protocol for detection of NO. The viability dye 780 (BD Bioscience, Franklin Lakes, NJ, USA) was used for detection of life/dead cells following the manufacture protocol. Afterwards, cells were fixed and permeabilized using cytofix/cytoperm solution (BD Bioscience, Franklin Lakes, NJ, USA) following the manufacture protocol followed by 1 µg Fc receptor block. At the end, intracellular stain was performed together with αCD11b and αF4/80 antibodies and TAMs were analyzed by flow cytometry using NovoCyte Quanteon (Agilent, Santa Clara, CA, USA).
BrdU injection and cleaved caspase 3
6132A-ECFP labeled cancer cells were used. Mice were injected i.p. twice a day with 100 µL BrdU (Sigma-Aldrich, Burlington, MA, USA) at a concentration of 10µg/µL for three consecutive days. Mice were sacrificed and tumor and spleen were taken out as described under tumor preparation and T cell cultures. BrdU stain was performed using the BD BrdU Flow kit (BD Bioscience, Franklin Lakes, NJ, USA) following manufacturer protocol. In addition, dye 780 (BD Bioscience, Franklin Lakes, NJ, USA) was used for detection of life/dead cells. The rabbit antibody clone 9661 (Cell Signaling Technology, Danvers, MA, USA) was used for detection of cleaved caspase 3 and anti-rabbit IgG clone 79408 [R-Phycoerythrin (PE), Cell Signaling Technology, Danvers, MA, USA] was used for detection by flow cytometry. Furthermore, αCD11b and αF4/80 antibodies were used to detect TAMs and αCD3, αCD4 antibodies together with mL9-tetramer was used to detect TILs.
Tumor infiltration and peripheral blood analysis
Blood was taken by buccal bleeding between day 45 and 75 as indicated in the figure legends with a 5 mm animal lancet (Medipoint Inc, Mineola, NY, USA). Blood (100 µL) was collected in tubes containing 50 µL heparin (80 U/mL, Pfizer, New York, NY, USA). Red blood cells were lysed and remaining peripheral blood cells were stained with Sytox Blue (Helix NP Blue, Biolegend, San Diego, CA, USA) for life/dead cells, and for CD3, CD4 and Vβ6 before being analyzed by flow cytometry with the NovoCyte Quanteon (Agilent, Santa Clara, CA, USA).
Longitudinal confocal imaging
The method was described previously (63). Windows were implanted on the shaved back of C3H Rag-/- mice. 6132A-cerulean cancer cells were injected at 3 different sites in between the fascia and dermis of the rear skin layer. Mice were treated 15 days after window implantation with H6-engineered CD4+ T cells. For longitudinal in vivo imaging, mice were anesthetized and positioned on a custom-made stage adaptor. The three screws that are used to hold the window frame also fixed the mouse onto the stage adaptor. A motorized microscope XY scanning stage and Leica LAS-AF software allowed recording individual 3-dimensional positions per field-of-view and returning to them later with high precision (stated accuracy ± 3 μm; reproducibility < 1.0 μm). Blood vessels were used as “landmarks” and could be located within 50 μm on the same day and within 100 μm on the next day. Data were acquired using a Leica SP5 II TCS tandem scanner two-photon spectral confocal microscope (long-working distance 20x/NA 0.45 and 4x/NA 0.16 dry lenses, Olympus). Tumor blood flow was visualized by retro-orbital injection of 100 µL red blood cells labelled with DiD (1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine,4-chlorobenzenesulfonate salt, Thermo Fisher Scientific, Waltham, MA, USA). To determine the fraction of area occupied by vessels or cerulean fluorescent cancer cells, acquired images were analyzed using Fiji software (Laboratory for Optical and Computational Instrumentation; University of Wisconsin-Madison, WI, RRID:SCR_002285).
Flow cytometry and antibodies
1 µg Fc receptor block (anti-mouse 2.4G2) was added to samples and cells were incubated with 50 µL PBS containing 0.2 µg of indicated anti-mouse antibodies for 20 min at 4°C. Then samples were washed twice with PBS and acquired using NovoCyte Quanteon (Agilent, Santa Clara, CA, USA). Data analysis was performed using FlowJo software (TreeStar, Ashland, OR, USA, RRID:SCR_008520). Used fluorophores: Allophycocyanin (APC), Fluorescein isothiocyanate (FITC), Peridinin chlorophyll protein-Cyanine5.5 (PerCp/Cy5.5), Allophycocyanin-Cyanine7 (APC/Cy7), Brilliant Violet 421 (BV421), R-Phycoerythrin (PE) and Alexa Fluor 647 (AF647). Antibodies: arginase 1 [A1exF5, APC, eFluor 450, eBioscience, Hatfield, GB, RRID:AB_2734833)], anti-BrdU [3D4, FITC, RRID:AB_396304], CD3+ [145–2C11, FITC, PerCp/Cy5.5, RRID:AB_312671], CD4+ [GK1.5, APC, APC/Cy7, BV421, FITC, RRID:AB_312697], CD11b+ [M1/70, APC, APC/Cy7, BV421, PE, RRID:AB_312794], CD31+ [390, PE, RRID:AB_312902], CD40 [3/23, FITC, RRID:AB_1134090], CD146+ [ME-9F1, APC, RRID:AB_2563088], CD163 [S15049F, PE, RRID:AB_2860724], CD204 [1F8C33, APC, RRID:AB_2892311], CD206 [C068C2, BV421, RRID:AB_2562232], F4/80+ [BM8, FITC, PerCp/Cy5.5, RRID:AB_893502], IDO [mIDO-4B, PE, Invitrogen Carlsbad, CA, USA], I-Ek [14–4-4S, FITC, PerCp/Cy5.5, AF647, RRID:AB_313470], IL-10 [JES5–16E3, BV421, RRID:AB_2563240], IL-12 [C15.6, APC, RRID:AB_315369], pERK1/2 [4B11B69, AF647, RRID:AB_2571894], TGFβ (TW7–20B9, PE, PerCp/Cy5.5, RRID:AB_10720866], TCR β-chain [H57–597, PE, RRID:AB_313430], TNF [MP6-XT22, APC, PE, RRID:AB_315429], TCR Vb2 [B20.6, PE, RRID:AB_1227785], TCR Vb3 [KJ25, PE, BD Bioscience, Franklin Lakes, NJ, USA, RRID:AB_394709], TCR Vb6 [RR4–7, PE, RRID:AB_10643583], TCR Vb8.1,8.2 [KJ16–133.18, PE, RRID:AB_1134109], TCR Vb8.3 [1B3.3, PE, RRID:AB_2800699]. Unless indicated otherwise, antibodies were purchased from Biolegend (San Diego, CA, USA). Tetramers (I-Ek-mL9 and I-Ek-CLIP) were provided by the NIH Tetramer Core Facility. Samples were stained with 1.4 µg/mL tetramer for 1 h at 4 °C in RPMI 1640 (Corning, NY, USA) containing 10% FBS (Gemini, Sacramento, CA, USA). For life/dead distinction, Sytox Blue (Helix NP Blue, Biolegend, San Diego, CA, USA) or Fixation resistant dye 510 or 780 (BD Bioscience, Franklin Lakes, NJ, USA) were used. Gating strategy for Fig. 1B is included in Fig. S1. Gating strategies for Fig. 3, Fig. 4 and Fig. 5 and supplementary figures are explained in the first part of Supplementary Materials.
Histology and immunohistochemistry
Tumor bearing and/or moribund mice were sacrificed by cervical dislocation and were subjected to a full necropsy. Tissue samples were fixed for 24 h in 10 % buffered formalin (Sigma-Aldrich, Burlington, MA, USA) and then transferred to 70 % ethanol. Tissue processing and immunohistochemistry stainings were performed by the Human Tissue Resource Center at the University of Chicago. Tissues were processed, paraffin embedded and 5 µm sections mounted on glass slides were subsequently stained with hematoxylin and eosin (H&E). Histopathological analysis was performed blinded and independently by two experienced pathologists. Microscopic images were captured using an Olympus BX43 microscope equipped with a ProgRes Speed XT core5 camera (Jenoptik) or a Leitz Laborlux D (W.Nuhsbaum, Inc., Mc Henry, IL, USA) microscope with a Retiga 2000R (QImaging) camera and Adobe Photoshop 2014 2.2 (San Jose, CA) to compose images. Serial sections were stained for CD3 with rabbit monoclonal antibody SP162 (abcam ab135372). The slides were stained using Leica Bond RX automated stainer. After dewax and rehydration, tissue section was heat treated for 20 min with antigen retrieval solution (Leica Biosystems, AR9961). Anti-CD3 antibody (1:100) was applied on tissue sections for 60 min incubation at room temperature and the antigen-antibody binding was detected with Bond Polymer Refine Detection HRP detection system (Leica Biosystems, DS9800) without post primary antibody amplification. The peroxidase reaction was developed using liquid diaminobenzidine brown substrate chromogen provided in the kit. Sections were counterstained with hematoxylin, dehydrated in alcohol, cleared in Xylene and mounted in Tissue-Tek Glas Mounting Medium (Sakura Finetek Japan Co, Ltd., Tokyo, Japan) for microscopic evaluation.
Detection of DNA damage using TUNEL
6132A-ECFP labeled cancer cells were injected s.c. on the back of C3H Rag2-/- mice and around 40 days later treated with either H6- or αmL26-engineered CD4+ T cells. About 20 days after T cell transfer, tumors were isolated. For analysis of DNA damage using flow cytometry, tumor single cell suspensions were prepared (see “Tumor preparation”). Samples were stained with the viability dye 780 (BD Bioscience, Franklin Lakes, NJ, USA). TUNEL stain was performed using APO-BrdU™ TUNEL Assay Kit (Life Technologies/Invitrogen™, Carlsbad, CA, USA) according to manufacture protocol. Cells were fixed and permeabilized as described in TCR signaling. Detection of life, TUNEL-positive 6132A-ECFP cancer cells was done by flow cytometry using NovoCyte Quanteon (Agilent, Santa Clara, CA, USA). For detection of TUNEL positive cells by immunohistochemistry, FFPE slides were stained using ApopTag® plus Peroxidase in situ (Millipore, Burlington, MA, USA) and counterstained with hematoxylin, dehydrated in alcohol, cleared in Xylene and mounted in Tissue-Tek Glas Mounting Medium (Sakura Finetek Japan Co, Ltd., Tokyo, Japan) for microscopic evaluation using 40X magnification.
TCR sequencing analysis
The raw sequencing data were processed using the 10x Genomics Cell Ranger Software (v6.0.0, RRID:SCR_023221) with the command cellranger multi, the provided config csv files contain the information of mm10 reference genome, vdj GRCm38 reference and TotalSeq-C surface markers. The output from cellranger multi contains TCR diversity metric which includes clonotype frequency and barcode information.
Whole-exome and RNA sequencing of cancer cells
Both genomic DNA and total RNA were extracted from in vitro readapted 6132A cell lines, using AllPrep DNA/RNA mini kit (Qiagen, Venlo, The Netherlands). For whole-exome sequencing, 3 μg of genomic DNA was subjected to library construction, using SureSeletXT Mouse All Exon V1 (Agilent Technologies, Santa Clara, CA, USA). RNAseq libraries were prepared from 1 μg of total RNA using TruSeq Stranded Total RNA Library Prep kit (Illumina, San Diego, CA, USA). The prepared whole-exome and RNAseq libraries were quantified by 2200 Tape Station (Agilent Technologies, Santa Clara, CA, USA), and then sequenced by 150 bp paired-end reads on NextSeq 500 Sequencer (Illumina, San Diego, CA, USA).
Statistics
All statistical analyses, including survival data, were performed using GraphPad Prism software (GraphPad, San Diego, CA, USA, RRID:SCR_002798). Data points indicate either means of biological duplicates of a representative experiment or are experimental replicates summarized as mean ± standard deviation. An unpaired, two-tailed Students t-test was used to determine significance between TUNEL positive and negative samples. In all other experiments, the method used to present the statistical significance of the data is indicated in the figure legend. In all experiments, statistical significance was indicated as: n.s. – not significant with p > 0.5, *p ≤ 0.5, **p ≤ 0.01, and ***p ≤ 0.001.
Supplementary Material
Acknowledgments
We thank Vytas P. Bindokas, Thomas Krausz, Carl Nathan, Lisha Zhou, Markus Diehl, Doremi Feng, Esther Kim, Ching-En Lee and Tomohiro Tate for technical support and valuable suggestions. This work used the NIH tetramer core Facility (contract number 75N93020D00005) for tetramers. This work was supported by the Cytometry and Antibody Technology Core Facility, the Center for research informatics, the Integrated Light Microscopy Core and the Genomics Core at the University of Chicago. The core facilities are supported by the Cancer Center Support Grant (P30CA014599) at the University of Chicago.
Funding:
This work was supported by National Institutes of Health grants (R01-CA22677 to HS), the David and Etta Jonas Center for Cellular Therapy (to SPW, ML and HS), Harriet and Allan Wulfstat (to HS), the Gerald O. Mann Foundation (to HS), a gift of Nancy Carey (to HS) and a gift of Janet D. Rowley (to HS)
Footnotes
Supplementary Materials
Gating strategies of flow cytometric analyses
Fig. S1. Progressively growing 6132A tumors are heavily infiltrated by T cells.
Fig. S2. The spleen of the original 6132-tumor bearing mouse developed a preferentially selected TCR.
Fig S3. TCR sequences of single T cell clonotypes found in 6132A-tumor bearing mice.
Fig. S4. CD4TCRs therapeutic in the 6132A tumor model are also effective in a second UV-induced tumor model.
Fig. S5. The immune response against moth cytochrome c induces TCRs encoded by multiple or single T cell clonotypes.
Fig. S6. Comparing the therapeutic effects of MCC-specific TCRs encoded by multiple T cell clonotypes with MCC-specific TCRs from single T cell clonotypes.
Fig. S7. Destruction of tumor vessels but not of pre-existent vasculature after T cell transfer.
Fig. S8. Persistent detection of T cells in the original 6132A tumor as well as transplanted 6132A tumors.
Fig. S9. Non-proliferative, growth-arrested 6132A cancer cells can be recovered in vitro and form again treatable tumors in vivo.
Fig. S10. The TCR-engineered 58α-β- CD4+ T cell hybridomas express the different CD4TCRs similarly.
Fig. S11. Proportion of dead stromal macrophages is similar in growing and arrested tumors.
Fig. S12. Production of NO is selectively induced in TAMs following treatment with the therapeutically effective and preferentially selected TCR H6.
Table S1. Number of sorted mL9-tetramer+ CD4+ T cells and identified TCRs based on CDR3 amino acid sequences among tumor and spleen from tumor-bearing mice.
Table S2. Analysis of expressed nsSNVs in reisolated progressing or arrested 6132A tumors.
Competing interest: SPW, ML, KS and HS have patent WO2023049733A3 pending. The authors declare no other competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials. Materials used to conduct this study will be made available upon request. Sequencing data are accessible at the Sequence Read Archive (SRA). Project number PRJNA1113628 includes original TCR-sequencing data and project number PRJNA1113704 includes original whole-exome-sequencing data for reisolated and readapted 6132A cancer cell lines referenced in Table S2.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW, Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monach PA, Meredith SC, Siegel CT, Schreiber H, A unique tumor antigen produced by a single amino acid substitution. Immunity 2, 45–59 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D, A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269, 1281–1284 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Tran E, Robbins PF, Rosenberg SA, 'Final common pathway' of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol 18, 255–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haslam A, Prasad V, Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2, e192535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Restifo NP, Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD, Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335, 723–727 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A, Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, Gartner JJ, Sindiri S, Malekzadeh P, Deniger DC, Lowery FJ, Parkhurst MR, Ngo LT, Ray S, Li YF, Hill V, Florentin M, Masi RV, Paria BC, Levin N, Bera A, Hedges EA, Choi A, Chatani PD, Parikh AY, Levi S, Seitter S, Lu YC, Zheng Z, Prickett TD, Jia L, Hernandez JM, Hoang CD, Robbins PF, Goff SL, Sherry RM, Yang JC, Rosenberg SA, Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor-Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol Res, OF1–OF15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, Leung A, Payne R, Sutcliffe K, Cramer J, Rosenberg SA, Fox BA, Urba WJ, Tran E, Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N Engl J Med 386, 2112–2119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leisegang M, Engels B, Schreiber K, Yew PY, Kiyotani K, Idel C, Arina A, Duraiswamy J, Weichselbaum RR, Uckert W, Nakamura Y, Schreiber H, Eradication of Large Solid Tumors by Gene Therapy with a T-Cell Receptor Targeting a Single Cancer-Specific Point Mutation. Clin Cancer Res 22, 2734–2743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leisegang M, Kammertoens T, Uckert W, Blankenstein T, Targeting human melanoma neoantigens by T cell receptor gene therapy. J Clin Invest 126, 854–858 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA, Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veatch JR, Lee SM, Fitzgibbon M, Chow IT, Jesernig B, Schmitt T, Kong YY, Kargl J, Houghton AM, Thompson JA, McIntosh M, Kwok WW, Riddell SR, Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest 128, 1563–1568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, Gartner J, Jia L, Trebska-McGowan K, Somerville RP, Robbins PF, Rosenberg SA, Goff SL, Feldman SA, Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24, 724–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolf SP, Anastasopoulou V, Drousch K, Diehl MI, Engels B, Yew PY, Kiyotani K, Nakamura Y, Schreiber K, Schreiber H, Leisegang M, One CD4+TCR and one CD8+TCR targeting autochthonous neoantigens are essential and sufficient for tumor eradication. Clin Cancer Res, (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward PL, Koeppen H, Hurteau T, Schreiber H, Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. J Exp Med 170, 217–232 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yassai MB, Naumov YN, Naumova EN, Gorski J, A clonotype nomenclature for T cell receptors. Immunogenetics 61, 493–502 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venturi V, Kedzierska K, Price DA, Doherty PC, Douek DC, Turner SJ, Davenport MP, Sharing of T cell receptors in antigen-specific responses is driven by convergent recombination. Proc Natl Acad Sci U S A 103, 18691–18696 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kedzierska K, Turner SJ, Doherty PC, Conserved T cell receptor usage in primary and recall responses to an immunodominant influenza virus nucleoprotein epitope. Proc Natl Acad Sci U S A 101, 4942–4947 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck-Engeser GB, Monach PA, Mumberg D, Yang F, Wanderling S, Schreiber K, Espinosa R 3rd, Le Beau MM, Meredith SC, Schreiber H, Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens. J Exp Med 194, 285–300 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McHeyzer-Williams LJ, Panus JF, Mikszta JA, McHeyzer-Williams MG, Evolution of antigen-specific T cell receptors in vivo: preimmune and antigen-driven selection of preferred complementarity-determining region 3 (CDR3) motifs. J Exp Med 189, 1823–1838 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schietinger A, Arina A, Liu RB, Wells S, Huang J, Engels B, Bindokas V, Bartkowiak T, Lee D, Herrmann A, Piston DW, Pittet MJ, Lin PC, Zal T, Schreiber H, Longitudinal confocal microscopy imaging of solid tumor destruction following adoptive T cell transfer. Oncoimmunology 2, e26677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, Li CY, Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell 58, 284–296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mumberg D, Monach PA, Wanderling S, Philip M, Toledano AY, Schreiber RD, Schreiber H, CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc Natl Acad Sci U S A 96, 8633–8638 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orentas RJ, Bircher LA, Roskopf S, Retroviral transfer of T-cell receptor genes produces cells with a broad range of lytic activity. Scand J Immunol 58, 33–42 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber KS, Donermeyer DL, Allen PM, Kranz DM, Class II-restricted T cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc Natl Acad Sci U S A 102, 19033–19038 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adachi K, Davis MM, T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc Natl Acad Sci U S A 108, 1549–1554 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biswas SK, Mantovani A, Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11, 889–896 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, Crawford JC, Clemens EB, Nguyen THO, Kedzierska K, La Gruta NL, Bradley P, Thomas PG, Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Looney TJ, Topacio-Hall D, Lowman G, Conroy J, Morrison C, Oh D, Fong L, Zhang L, TCR Convergence in Individuals Treated With Immune Checkpoint Inhibition for Cancer. Front Immunol 10, 2985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan M, Li B, T cell receptor convergence is an indicator of antigen-specific T cell response in cancer immunotherapies. Elife 11, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storkus WJ, Maurer D, Lin Y, Ding F, Bose A, Lowe D, Rose A, DeMark M, Karapetyan L, Taylor JL, Chelvanambi M, Fecek RJ, Filderman JN, Looney TJ, Miller L, Linch E, Lowman GM, Kalinski P, Butterfield LH, Tarhini A, Tawbi H, Kirkwood JM, Dendritic cell vaccines targeting tumor blood vessel antigens in combination with dasatinib induce therapeutic immune responses in patients with checkpoint-refractory advanced melanoma. J Immunother Cancer 9, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guedan S, Luu M, Ammar D, Barbao P, Bonini C, Bousso P, Buchholz CJ, Casucci M, De Angelis B, Donnadieu E, Espie D, Greco B, Groen R, Huppa JB, Kantari-Mimoun C, Laugel B, Mantock M, Markman JL, Morris E, Quintarelli C, Rade M, Reiche K, Rodriguez-Garcia A, Rodriguez-Madoz JR, Ruggiero E, Themeli M, Hudecek M, Marchiq I, Time 2EVOLVE: predicting efficacy of engineered T-cells - how far is the bench from the bedside? J Immunother Cancer 10, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brightman SE, Becker A, Thota RR, Naradikian MS, Chihab L, Zavala KS, Ramamoorthy Premlal AL, Griswold RQ, Dolina JS, Cohen EEW, Miller AM, Peters B, Schoenberger SP, Neoantigen-specific stem cell memory-like CD4(+) T cells mediate CD8(+) T cell-dependent immunotherapy of MHC class II-negative solid tumors. Nat Immunol 24, 1345–1357 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaettler MO, Desai R, Wang AZ, Livingstone AJ, Kobayashi DK, Coxon AT, Bowman-Kirigin JA, Liu CJ, Li M, Bender DE, White MJ, Kranz DM, Johanns TM, Dunn GP, TCR-engineered adoptive cell therapy effectively treats intracranial murine glioblastoma. J Immunother Cancer 11, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP, Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol 26, 111–117 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C, Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 358, 2698–2703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, Kerstann KW, Feigenbaum L, Chan CC, Restifo NP, Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112, 362–373 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP, Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 207, 637–650 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg PD, Kern DE, Cheever MA, Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2- T cells. Tumor eradication does not require participation of cytotoxic T cells. J Exp Med 161, 1122–1134 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zahavi DJ, Weiner LM, Tumor mechanisms of resistance to immune attack. Prog Mol Biol Transl Sci 164, 61–100 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Tveita AA, Schjesvold FH, Sundnes O, Haabeth OA, Haraldsen G, Bogen B, Indirect CD4+ T-cell-mediated elimination of MHC II(NEG) tumor cells is spatially restricted and fails to prevent escape of antigen-negative cells. Eur J Immunol 44, 2625–2637 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Nichols CA, Gibson WJ, Brown MS, Kosmicki JA, Busanovich JP, Wei H, Urbanski LM, Curimjee N, Berger AC, Gao GF, Cherniack AD, Dhe-Paganon S, Paolella BR, Beroukhim R, Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat Commun 11, 2517 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang MS, Mog BJ, Douglass J, Pearlman AH, Hsiue EH, Paul S, DiNapoli SR, Konig MF, Pardoll DM, Gabelli SB, Bettegowda C, Papadopoulos N, Vogelstein B, Zhou S, Kinzler KW, Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Sjoblom T, Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy. Pharmaceuticals (Basel) 14, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, Ivanov A, Szymborska A, Patone G, Kunz S, Sommermeyer D, Engels B, Leisegang M, Textor A, Fehling HJ, Fruttiger M, Lohoff M, Herrmann A, Yu H, Weichselbaum R, Uckert W, Hubner N, Gerhardt H, Beule D, Schreiber H, Blankenstein T, Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature 545, 98–102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teng MN, Park BH, Koeppen HK, Tracey KJ, Fendly BM, Schreiber H, Long-term inhibition of tumor growth by tumor necrosis factor in the absence of cachexia or T-cell immunity. Proc Natl Acad Sci U S A 88, 3535–3539 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV, Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 312, 724–729 (1984). [DOI] [PubMed] [Google Scholar]
- 50.Shen J, Xiao Z, Zhao Q, Li M, Wu X, Zhang L, Hu W, Cho CH, Anti-cancer therapy with TNFalpha and IFNgamma: A comprehensive review. Cell Prolif 51, e12441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothstein JL, Schreiber H, Synergy between tumor necrosis factor and bacterial products causes hemorrhagic necrosis and lethal shock in normal mice. Proc Natl Acad Sci U S A 85, 607–611 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seung LP, Rowley DA, Dubey P, Schreiber H, Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A 92, 6254–6258 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF, Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singer K, Gottfried E, Kreutz M, Mackensen A, Suppression of T-cell responses by tumor metabolites. Cancer Immunol Immunother 60, 425–431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu SJ, Ma C, Heinrich B, Brown ZJ, Sandhu M, Zhang Q, Fu Q, Agdashian D, Rosato U, Korangy F, Greten TF, Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J Hepatol 70, 449–457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li S, Zhuang S, Heit A, Koo SL, Tan AC, Chow IT, Kwok WW, Tan IB, Tan DSW, Simoni Y, Newell EW, Bystander CD4(+) T cells infiltrate human tumors and are phenotypically distinct. Oncoimmunology 11, 2012961 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meier SL, Satpathy AT, Wells DK, Bystander T cells in cancer immunology and therapy. Nat Cancer 3, 143–155 (2022). [DOI] [PubMed] [Google Scholar]
- 58.Lee HG, Cho MJ, Choi JM, Bystander CD4(+) T cells: crossroads between innate and adaptive immunity. Exp Mol Med 52, 1255–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stuehr DJ, Nathan CF, Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med 169, 1543–1555 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fauskanger M, Haabeth OAW, Skjeldal FM, Bogen B, Tveita AA, Tumor Killing by CD4(+) T Cells Is Mediated via Induction of Inducible Nitric Oxide Synthase-Dependent Macrophage Cytotoxicity. Front Immunol 9, 1684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bogen B, Fauskanger M, Haabeth OA, Tveita A, CD4(+) T cells indirectly kill tumor cells via induction of cytotoxic macrophages in mouse models. Cancer Immunol Immunother 68, 1865–1873 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, Yu P, Fu YX, Weichselbaum RR, Rowley DA, Kranz DM, Schreiber H, Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med 204, 49–55 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang B, Zhang Y, Bowerman NA, Schietinger A, Fu YX, Kranz DM, Rowley DA, Schreiber H, Equilibrium between host and cancer caused by effector T cells killing tumor stroma. Cancer Res. 68, 1563–1571 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, Ranta F, Ullrich S, Mocikat R, Braungart K, Mehra T, Fehrenbacher B, Berdel J, Niessner H, Meier F, van den Broek M, Haring HU, Handgretinger R, Quintanilla-Martinez L, Fend F, Pesic M, Bauer J, Zender L, Schaller M, Schulze-Osthoff K, Rocken M, T-helper-1-cell cytokines drive cancer into senescence. Nature 494, 361–365 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, Wong CS, Wu HY, Law HT, Liu K, Talbot CC Jr., Lau WK, Montell DJ, Fung MC, Cell survival DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell 23, 2240–2252 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, Dimmeler S, Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J Biol Chem 274, 6823–6826 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Sun G, Guzman E, Balasanyan V, Conner CM, Wong K, Zhou HR, Kosik KS, Montell DJ, A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J Cell Biol 216, 3355–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gong YN, Crawford JC, Heckmann BL, Green DR, To the edge of cell death and back. FEBS J 286, 430–440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Preston BD, Albertson TM, Herr AJ, DNA replication fidelity and cancer. Semin Cancer Biol 20, 281–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diehl MI, Wolf SP, Bindokas VP, Schreiber H, Automated cell cluster analysis provides insight into multi-cell-type interactions between immune cells and their targets. Exp Cell Res, 112014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kappler J, White J, Wegmann D, Mustain E, Marrack P, Antigen presentation by Ia+ B cell hybridomas to H-2-restricted T cell hybridomas. Proc Natl Acad Sci U S A 79, 3604–3607 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morita S, Kojima T, Kitamura T, Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7, 1063–1066 (2000). [DOI] [PubMed] [Google Scholar]
- 73.Engels B, Chervin AS, Sant AJ, Kranz DM, Schreiber H, Long-term persistence of CD4(+) but rapid disappearance of CD8(+) T cells expressing an MHC class I-restricted TCR of nanomolar affinity. Mol Ther 20, 652–660 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, Kranz DM, Meredith SC, Rowley DA, Schreiber H, Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 23, 516–526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, Uckert W, Song JY, Haanen JB, Schumacher TN, Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med 16, 565–570, 561p following 570 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Ward PL, Koeppen HK, Hurteau T, Rowley DA, Schreiber H, Major histocompatibility complex class I and unique antigen expression by murine tumors that escaped from CD8+ T-cell-dependent surveillance. Cancer Res 50, 3851–3858 (1990). [PubMed] [Google Scholar]
- 77.Spiotto MT, Rowley DA, Schreiber H, Bystander elimination of antigen loss variants in established tumors. Nat Med 10, 294–298 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. Materials used to conduct this study will be made available upon request. Sequencing data are accessible at the Sequence Read Archive (SRA). Project number PRJNA1113628 includes original TCR-sequencing data and project number PRJNA1113704 includes original whole-exome-sequencing data for reisolated and readapted 6132A cancer cell lines referenced in Table S2.