

Abstract
Androgen deprivation therapies, the key treatment options for prostate cancer, have shown resistance and disease progression in many patients receiving these treatments. Therefore, it is crucial to identify new targetable pathways. Epidermal growth factor receptor pathway substrate 8 (Eps8) is one such potential target. Although this pathway is associated with the progression of various cancers, studies on the role of Eps8 in prostate cancer remain limited. This study investigated the role of Eps8 in prostate cancer. The LNCaP cell line and enzalutamide-resistant LNCaP (LNCaP Enz-R) cell lines were utilized for the investigation. Overexpression of Eps8 was observed in the LNCaP Enz-R cells. Transfecting pCMV-EPS8 also increased the levels of epithelial-to-mesenchymal transition (EMT), cell proliferation, and cell viability in both cell lines. Conversely, knockdown of Eps8 expression decreased the levels of EMT, cell proliferation, and cell viability in both cell lines. Furthermore, EPS8-induced EMT activation could be reversed by suppressing the Ras/JAK/PI3K signaling pathway. In vivo animal study also confirmed the crucial role of Eps8 expression in prostate cancer progression. Therefore, we suggest that targeting Eps8 by knocking down its expression is promising as a therapeutic approach for prostate cancer treatment.
Keywords: Epidermal growth factor receptor pathway substrate 8, prostate cancer, castration-resistant, epithelial-to-mesenchymal transition, cancer progression
Introduction
Global Cancer Statistics 2022 report state prostate cancer to be the most frequently diagnosed cancer among men across 118 countries accounting for 1% of all cancer cases in men, and as the fifth leading cause of cancer-related deaths [1]. Approximately 50% of prostate cancers are diagnosed at metastatic stages. Prostate cancers have a 5-year survival rate of approximately 30% [2,3]. While metastatic prostate cancer can be initially controlled using androgen deprivation therapy, castration-resistant prostate cancer (CRPC) often develops occurs after a certain period of treatment. Novel antiandrogens such as abiraterone, enzalutamide, apalutamide, and darolutamide are currently promising in improving metastasis-free survival in CRPC patients [4-7]. These treatments can effectively inhibit prostate cancer cell growth and extend the life expectancy of patients. However, prostate cancer cells can overcome the restricting effects of these treatments, ultimately leading to mortality.
The mechanisms underlying prostate cancer development and progression have been extensively investigated. Androgen receptor (AR) expression and amplification play a pivotal role in cancer cell growth and differentiation, as well as in CRPC cell development [8,9]. Furthermore, neuroendocrine (NE) cells are involved in paracrine signaling that regulates the growth, differentiation, and secretion of normal prostate cells. However, NE cell-secreted paracrine factors, such as neurotensin, serotonin, calcitonin, and thyroid-stimulating hormone, are associated with tumor proliferation and metastasis [10]. NE differentiation is more commonly observed in CRPC than in hormone-sensitive prostate cancer and is associated with a poorer prognosis [11].
However, epithelial-to-mesenchymal transition (EMT) is a process through which compact epithelial cells are transformed into metastatic and invasive mesenchymal cells, thereby contributing to the aggressive behavior of the cancer and resistance to anticancer treatments [12]. Additionally, the epidermal growth factor receptor (EGFR) has been identified as an EMT regulator. In lung cancer, EMT reduces the sensitivity of cancer cells to EGFR inhibitors, thereby resulting in gefitinib resistance [12,13]. Furthermore, EGFR overexpression in cervical cancer is associated with poor disease prognosis and is linked to EMT regulation [14].
Initially, EGFR pathway substrate 8 (Eps8) was identified as a novel substrate of EGFR kinase. Eps8 is highly expressed in many human tumor types, including colorectal, pituitary, oral, esophageal, pancreatic, ovarian, lung, breast, thyroid, and cervical cancers [15]. Overexpression of Eps8 promotes mitosis and enhances malignant metastasis by activation of the EMT and EGFR-regulated downstream signaling pathways [15,16]. Conversely, progression of these cancers is inhibited by Eps8 knockdown [12,17]. Although the role of Eps8 in other cancers has been extensively investigated, its role in prostate cancer progression remains limited. Herein, we investigated Eps8 expression in enzalutamide-resistant prostate cancer cells and explored the impact of this expression on EMT and tumor cell activity in prostate cancer.
Materials and methods
Cell culture
LNCaP cells from the American Tissue Culture Collection (Rockville, Maryland, USA) were cultured in RPMI 1640 supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C under 5% CO2. The LNCaP Enz-R cell lines were generated from the LNCaP cells treated with 10 μM enzalutamide under 5% CO2 at 37°C for more than 6 months and maintained in the above mentioned media containing 5 μM enzalutamide. The LNCaP Enz-R cells were generated according to previous studies [18,19].
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
Cells (1 × 104 cells/dL) were inoculated in a 96-well plate and incubated for 24 h, followed by incubation with pCMV-EPS8 for 24 h, both at 37°C and 5% CO2. Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. After transfection for 24 h, the 50 μL of 5 mg/mL MTT solution was added to each well and incubated at 37°C for 3 h. Subsequently, 500 μL isopropyl alcohol was added to dissolve the reduced formazan product. The absorbance at 590 nm in each well was measured using a spectrophotometer (Sunrise-Basic Tecan, Tecan Austria GmbH Grödig, Austria) before cell viability was examined. Values represent the mean OD590 ± standard deviation (SD) from three or more independent reaction wells.
Transfection
To produce the pCMV-EPS8 plasmid, the open reading frame (ORF) sequence of Eps8 (accession no. NM_004447) was cloned into the human-tagged ORF clone plasmid (cat. no. RC205300; Origene Technologies, Inc.). The cells were cultured in six-well plates and treated with 2 μg/mL of the pCMV-EPS8 plasmid (2 μg/mL) (cat. no. RG205300; Origene Technologies, Inc.) or the pCMV-GFP plasmid (cat. no. PS100010; Origene Technologies, Inc.). The cells were transfected with the plasmids using the FuGENE HD Transfection Reagent (Roche Diagnostics, Inc.) and incubated at 37°C under 5% CO2 for 24 h as per manufacturer’s protocols. For Eps8 knockdown, ON-TARGETplus EPS8 shRNA SMARTpool (shRNA-EPS8; cat. no. TL313184; OriGene Technologies, Inc.) was used to silence the Eps8 gene. Eps8 shRNAs (5 μg) were transfected into the cells by using the TurboFectin Transfection Reagent (OriGene Technologies, Inc.) and incubated under 5% CO2 at 37°C for 24 h. The cells were subjected to reverse transcription-PCR (RT-PCR) and western blotting after 24 h of transfection.
Reverse transcription-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). 1 μg of the isolated RNA was reverse transcribed into cDNA using the SuperScript™ III First Strand Synthesis System for RT-PCR (Invitrogen; Thermo Fisher Scientific, Inc.). The cDNA was synthesized from 5 μg of RNA using oligo dT (50 μM) and 10 mM dNTP. For PCR, 1 μL of oligo dT primer and 1 μL of 10 mM dNTP mix were added to 8 μL of RNA, incubated at 65°C for 5 minutes, and then placed on ice for at least 1 minute. After that, 10 μL of cDNA synthesis mix (including 2 μL of 10× RT buffer, 4 μL of 25 mM MgCl2, 2 μL of 0.1 M DTT, 1 μL of RNase OUT™ [40 U/μL], and 1 μL of SuperScript™ III RT [200 U/μL]) was added to each RNA/primer mixture and incubated at 50°C for 50 minutes. The reaction was terminated by heating to 85°C for 5 minutes. For each PCR reaction, 10× PCR buffer (cat. no. 18067-017; Invitrogen; Thermo Fisher Scientific, Inc.), 50 mM MgCl2, 10 mM dNTP mix, cDNA, Taq DNA polymerase (5 U/μL), and primer pairs were added. The final product was stored at -20°C. The reaction mixtures, containing 5 μL of 10× PCR buffer, 1.5 μL of 50 mM MgCl2, 1 μL of 10 mM dNTP mix, 2 μg cDNA, 10 μM of each primer pair, 0.4 μL Taq DNA polymerase, and 38.1 μL DEPC-treated water, were incubated for an initial denaturation at 94°C for 2 minutes. The PCR was then carried out for 35 cycles, with an annealing temperature of 55°C, denaturation at 94°C, and extension at 72°C, each step lasting for 30 seconds.
RNA was used as a template for reverse transcription (Invitrogen; Thermo Fisher Scientific, Inc.), followed by PCR analysis using primers specific for Eps8 (forward, 5’-GATGGAGGAAGTGCAAGATG-3’ and reverse, 5’-GACTGTAACCACGTCTTCACA-3’) and GAPDH (forward, 5’-ATGTGTCCGTCGTGGATCTGAC-3’ and reverse, 5’-AGACAACCTGGTCCTCAGTGTAG-3’). The expression levels of total RNA were normalized to those of the GAPDH gene (assay ID, Hs03929097_g1; Thermo Fisher Scientific, Inc.). DNA (0.5 µg/lane) was visualized on a 2% agarose gel stained with SafeView™ Classic stains (Applied Biological Materials, Inc.).
Cell viability assay
Cells were inoculated into 96-well plates and grown to 60%-75% confluence. The cells were transfected with pCMV-control or EPS8/shRNA-control or EPS8 for 24 h. Cell viability was evaluated using the Cell Counting Kit-8 (CCK8) assay. Briefly, after exposure was terminated, the cells were aspirated, rinsed with PBS, and treated with 10 μL/well CCK-8 for 2 h at 37°C. Absorbance was measured at 450 nm spectrophotometrically (Bio-Rad, Hercules, CA, USA).
Western blotting
Cell lysates were produced using lysis buffer (20 μg/mL aprotinin, 10 mM Tris, 1% Triton X-100, 20 μg/mL leupeptin, 50 μg/mL phenylmethylsulfonyl fluoride, 1 m MEDTA, 1 mM dithiothreitol, and 1 mM Na3VO4). The lysates were subjected to 10% SDS-PAGE. Proteins were transferred to the PVDF membrane via an electrotransfer unit. The membranes were probed with primary antibodies overnight at 4°C. The primary antibodies included E-cadherin (ab53033, Abcam; 1:2000), α-SMA (ab5694, Abcam; 1:2000), AMPK (ab32047, Abcam; 1:2000), SIRT1 (ab110304, Abcam; 1:2000), anti-PGC1-α (ab54481, Abcam; 1:2000), anti-NRF1 (ab34682, Abcam; 1:2000), and β-actin (A5441, Sigma; 1:4000). Subsequently, the membranes were incubated at room temperature for 1 h with corresponding secondary antibodies. The immunoreactive bands were developed using a Western-Ready™ ECL Substrate Plus Kit (426316, BioLegend, San Diego, CA, USA) and detected using the MultiGel-21® image system. β-actin was used as an internal control.
In vivo tumor growth and metastasis
The animal study protocol was approved by the Institutional Animal Care and Use Committee of E-Da Hospital (IACUC-EDAH-109002). NOD-SCID mice were purchased from BioLASCO Taiwan Co., Ltd. and maintained in microisolator cages. All mice were used in according to institutional guidelines, and the experiments were approved by the Use Committee for Animal Care. Different types of tumor cells including 1 million LNCap and control cells, and 1 million LNCap-EPS8 and control cells were resuspended in PBS with Matrigel medium and inoculated subcutaneously into 6-week-old NOD-SCID mice. The tumors were measured every 3 days after appearance, and the tumor volume was calculated as follows: length × width2/2. After subcutaneously injecting 1 × 106 cells per transfection with EPS8 or without EPS8, we monitored tumor development over 25 days. At 25 days after inoculation, all the mice were euthanized with CO2.
Hematoxylin and eosin staining
Tissues were fixed in 10% normal formalin (Sigma-Aldrich Co.), embedded in paraffin blocks, and cut using a sliding microtome (3-μm sections). After the sections were deparaffinized and rehydrated with xylene and ethanol, the slides were stained with hematoxylin and eosin (H&E) (Sigma-Aldrich Co.). The stained slides were hydrated and mounted using a hydrophobic mounting solution to prevent sample contamination. Tumor tissues morphology was observed using a BX51 light microscope (Olympus, Japan).
Immunohistochemistry
The paraffin-embedded sections were first deparaffinized and rehydrated through treatments with xylene, ethanol, and tap water to prepare them for histological analysis. Antigen retrieval was achieved by heating the slides in a microwave for 10 minutes in a chamber containing 0.01 M citrate buffer (pH 6.0). To inhibit endogenous peroxidase activity, the tissue slides were immersed in 0.3% methanol/hydrogen peroxide (Sigma-Aldrich Co.) for 30 minutes. To minimize non-specific binding of the primary antibodies, the slides were incubated with 10% normal goat serum (Vector Laboratories, Burlingame, CA, USA) for 1 hour. The slides were then incubated overnight at 4°C in a humidified chamber with primary antibodies, including E-cadherin (1:100, Abcam) or α-SMA (1:100, Abcam), diluted in 5% bovine serum albumin (Sigma-Aldrich Co.). The next day, the slides were rinsed three times with 1× PBS-T (pH 7.4) and incubated for 30 minutes at room temperature with the appropriate biotinylated secondary antibodies (1:500 dilution, Jackson ImmunoResearch). After a subsequent rinse with 1× PBS-T for 10 minutes, the Vectastain Universal Elite ABC kit reagent (Vector Laboratories) was applied for 30 minutes. Immunoreactive complexes were visualized using the DAB substrate (Sigma-Aldrich Co.) and counterstained with hematoxylin. Finally, the slides were mounted with coverslips using a mounting solution. The slides were examined under a BX51 light microscope, and images were captured.
Statistical analysis
Each experiment was performed at least thrice, and representative results were presented. Values in bar graphs are presented as mean ± SD. Statistical analyses were performed using SPSS software (version 22; SPSS, Chicago, IL, USA). Statistically significant differences were determined through one-way analysis of variance (ANOVA), followed by Tukey’s post hoc test or two-way ANOVA of repeated measures and the Bonferroni post hoc test. P < 0.05 was considered statistically significant.
Results
EPS8 expression is increased in LNCaP Enz-R cells
To determine the role of EPS8 in LNCaP and LNCaP Enz-R cells, RT-PCR was performed to first measure EPS8 expression levels. EPS8 expression was markedly higher in the LNCaP Enz-R cells, but was almost absent in the sensitive LNCaP cells (Figure 1A, 1B). EPS8 protein expression was evaluated through western blotting. Markedly higher EPS8 expression levels were noted in the LNCaP Enz-R cells than in the LNCaP cells (Figure 1C, 1D). Figure S1 presents the triplicates of western blot images. These observations suggest that EPS8 expression increased during enzalutamide resistance development.
Figure 1.
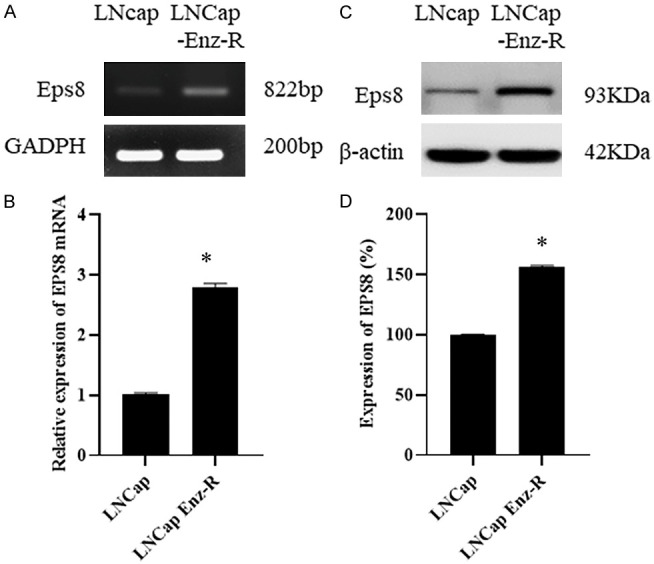
EPS8 expression in LNCaP cells and LNCaP Enz-R cells. A, B. RT-PCR revealed higher EPS8 expression in the LNCaP Enz-R cell line, which was negligible in the LNCaP cell line. C, D. Western blotting revealed higher expression of EPS8 protein in the LNCaP Enz-R cell line than that in LNCaP cell line. The triplicates of the western blotting images are shown in Figure S1. *P < 0.05 vs. LNCap. Enz-R, enzalutamide-resistant.
EPS8 overexpression increases LNCaP and LNCaP Enz-R cell viability
The effects of EPS8 on prostate cancer cells were determined on the basis of cell viability. MTT and CCK-8 assays were performed to measure cell viability (Figure 2). According to the results of the MTT assay, the pCMV-EPS8-transfected LNCaP cells exhibited the highest levels of viability compared with the cells in the other two control groups at 24 h after transfection (Figure 2A). A similar result was observed for the LNCaP Enz-R cells (Figure 2A). Next, the CCK-8 assay was performed to assess the viability of both LNCaP and LNCaP Enz-R cell lines. EPS8-overexpressing LNCaP and LNCaP Enz-R cells exhibited the highest levels of viability compared with the cells in the other two control groups at 24 h after transfection (Figure 2B). These results suggest that EPS8 overexpression can increase prostate cancer cell viability.
Figure 2.
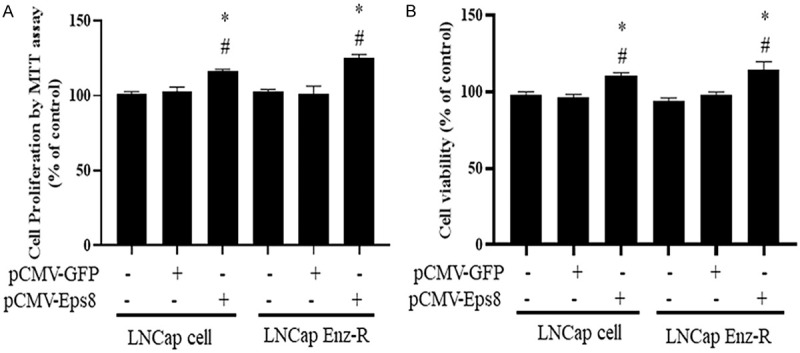
Effects of EPS8 overexpression on cell proliferation and viability. A. Cells transfected with pCMV-EPS8 exhibited higher levels of cell proliferation when compared with untreated cells and those transfected with the pCMV-GFP based on the MTT assay. B. Cells transfected with pCMV-EPS8 exhibited higher levels of cell viability when compared with untreated cells and those transfected with pCMV-GFP based on the CCK-8 assay. *P < 0.05 vs. Control and #P < 0.05 vs. pCMV-GFP. CCK-8, Cell Counting Kit-8; Enz-R, enzalutamide-resistant; GFP, green fluorescent protein; MMT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
EPS8 overexpression increases EMT in LNCaP and LNCaP Enz-R cells
To clarify the mechanism underlying the increase in cell migration after pCMV-EPS8 transfection, western blotting was performed to measure the expression of EMT markers, namely E-cadherin, α-SMA, Snail, and Slug. α-SMA, Snail, and Slug expression was positively correlated with EMT, whereas E-cadherin expression was negatively correlated. The expression patterns of these four markers in the untreated cells were similar to those in the empty vector-transfected cells. This possibly explains the similar cell migratory capacities of these two cell groups. In the pCMV-EPS8-transfected LNCaP and LNCaP Enz-R cells, E-cadherin was significantly downregulated, whereas the other three markers were significantly upregulated (Figure 3). The triplicates of western blotting images are presented in Figures S2 and S3. This suggests that EPS8 overexpression induces EMT in the LNCaP and LNCaP Enz-R cells, which may be why the pCMV-EPS8-transfected cells exhibited the highest cell migration levels. These results suggest that EPS8 overexpression can promote EMT to increase the migratory capacity of prostate cancer cells.
Figure 3.

Effects of EPS8 overexpression on the EMT. Western blotting revealed that LNCaP and LNCaP EnzaR cells transfected with pCMV-EPS8 exhibited lower levels of E-cadherin expression and higher levels of α-SMA, Snail, and Slug when compared with untreated cells and those transfected with pCMV-GFP. (A, C) LNCap cells and (B, D) LNCaP Enz-R cells. The triplicates of western blotting images are shown in Figures S2 and S3. *P < 0.05 vs. Control and #P < 0.05 vs. pCMV-GFP. α-SMA, α-smooth muscle actin; Enz-R, enzalutamide-resistant; GFP, green fluorescent protein.
EPS8 knockdown reduces LNCaP and LNCaP Enz-R cell viability
After EPS8 overexpression, the possible effects of EPS8 knockdown on prostate cancer cells were evaluated. MTT and CCK-8 assays were conducted to measure cell viability. shRNA-EPS8 transfection was performed to knockdown EPS8 expression. According to the MTT assay, the shRNA-EPS8-transfected LNCaP cells exhibited lowest levels of proliferation compared with the untreated cells and the shRNA-control-transfected cells (Figure 4A). In addition, shRNA-EPS8 transfection significantly reduced in the proliferation of LNCaP EnzaR cells (Figure 4A). Similar results were observed in the CCK-8 assay. Specifically, the shRNA-EPS8-transfected LNCaP and LNCaP Enz-R cells exhibited the lowest viability compared with the untreated cells and the shRNA-control-transfected cells (Figure 4B). These results suggest that EPS8 knockdown using shRNA reduces prostate cancer cell viability.
Figure 4.

Effect of EPS8 knockdown on cell proliferation and viability. A. Cells transfected with shRNA-EPS8 exhibited higher levels of cell proliferation when compared with untreated cells and those transfected with shRNA-control based on the MTT assay. B. Cells transfected with shRNA-EPS8 revealed lower levels of cell viability when compared with untreated cells and those transfected with shRNA-control based on the CCK-8 assays. *P < 0.05 vs. Control and #P < 0.05 vs. pCMV-GFP. CCK 8, Cell Counting Kit 8; Enz-R, enzalutamide resistant; MMT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; shRNA, short-hairpin RNA.
EPS8 knockdown inhibits EMT in LNCaP and LNCaP Enz-R cells
To assess the association between decreased EMT levels, the protein expression of E-cadherin, α-SMA, Snail and Slug in both LNCaP and LNCaP Enz-R cells was measured through western blotting. Consistent with this hypothesis, cells with EPS8 expression knockdown exhibited reduced EMT induction. Specifically, E-cadherin expression significantly increased, whereas α-SMA, Snail, and Slug expression significantly decreased in the shRNA-EPS8-transfected LNCaP and LNCaP Enz-R cells compared with the untreated cells and the shRNA-control-transfected cells (Figure 5). Figures S4 and S5 present the triplicates of western blotting images. These results suggest that EPS8 knockdown can inhibit EMT and EMT-related protein expression to suppress prostate cancer cell migration.
Figure 5.

Effects of EPS8 knockdown on the EMT. Western blotting revealed that LNCaP and LNCaP Enz-R cells transfected with shRNA-EPS8 exhibited increased E-cadherin expression and reduced α-SMA, Snail, and Slug expression when compared with untreated cells and those transfected with shRNA-control. A. Western blotting images of EMT markers in the LNCaP cells. B. Quantification of EMT markers expression in LNCaP cells. C. Western blotting images of EMT markers in the LNCaP Enz-R cells. D. Quantification of EMT markers expression in LNCaP Enz-R cells. The triplicates of western blotting images are shown in Figures S4 and S5. *P < 0.05 vs. Control and #P < 0.05 vs. pCMV-GFP. α-SMA, α-smooth muscle actin; EMT, epithelial-to-mesenchymal transition; Enz-R, enzalutamide-resistant; GFP, green fluorescent protein; shRNA, short-hairpin RNA.
Suppression of Ras/p53/JAK/PI3K signaling reverses Eps8 overexpression-induced upregulation of EMT
Subsequently, we evaluated whether the regulation of Ras/p53/JAK/PI3K signaling participates in EPS8-induced EMT. We showed that Ras (salirasib) inhibition reverts EPS8-induced reduction in E-cadherin expression and induction of α-SMA, Snail, and Slug expression (Figure 6). Similarly, when administered, the JAK inhibitor (baricitinib) and PI3K inhibitor (buparlisib) upregulated E-cadherin expression levels and downregulated α-SMA, Snail, and Slug expression levels in prostate cancer cells under EPS8 stimulation in the pCMV-treated group (Figure 6). The triplicates of western blotting images are displayed in Figures S6 and S7. These findings demonstrate that blockade of the Ras/JAK/PI3K pathway has the potential to improve prostate cancer migration by downregulating EMT.
Figure 6.

Regulation of EMT by administration of Ras/p53/JAK/PI3K signaling inhibitors. The administration of Ras inhibitor (salirasib), JAK inhibitor (Bacrivitinib), and PI3K inhibitor (Buparlisib) upregulated E-cadherin expression and downregulated α-SMA, Snail, and Slug expression in prostate cancer cells transfected with pCMV-EPS8 when compared with prostate cancer cells treated with pCMV-GFP. A. Western blotting images of EMT markers in the LNCaP cells. B. Quantification of EMT markers expression in LNCaP cells. C. Western blotting images of EMT markers in the LNCaP Enz-R cells. D. Quantification of EMT markers expression in LNCaP Enz-R cells. The triplicates of western blotting images are shown in Figures S6 and S7. *P < 0.05 vs. Control and #P < 0.05 vs. pCMV-GFP. α-SMA, α-smooth muscle actin; EMT, epithelial-to-mesenchymal transition; Enz-R, enzalutamide-resistant; GFP, green fluorescent protein.
EPS8 induces EMT in xenograft tumor sections
Changes in xenograft tumor sections were observed through H&E staining after EPS8 overexpression (Figure 7). The pCMV-EPS8 plasmid was used to investigate a possible correlation between EMT and enhanced tumorigenesis in prostate cancer cells. After subcutaneously injecting 2.5 × 106 cells per transfection with EPS8 or without EPS8, tumor development was monitored over 25 days. The median tumor weight was greater in the pcDNA-EPS8 and shRNA-control groups compared with the pcDNA-control and shRNA-EPS8 groups (Table S1). The mice were sacrificed and no tumor metastases were detected during autopsy.
Figure 7.

EPS8 transfection in xenograft tumor sections. H&E staining for xenograft tumor sections after 25 days of transfection. Aggressive infiltration was detected in pCMV-EPS8-transfected tumors whereas fewer nuclei and infiltration were detected in shRNA-EPS8-transfected tumors. shRNA, short-hairpin RNA.
Effect of inhibiting EPS8 on EMT and metastasis in vivo
To examine the inhibitory effect of EPS8 on EMT and metastasis in vivo, the expression levels of EMT marker proteins in xenograft tumors were determined through immunochemistry (IHC). As shown in Figure 7, EPS8 possibly induces EMT of prostate cancer cells. To further confirm these observations, EMT marker proteins were assessed. We further confirmed this change in the EMT marker expression pattern in xenograft tumor sections through immunochemical staining. Moreover, EPS8 expression in LNCaP cells transfected with EPS8-overexpressing tumor cells of the xenograft mouse model was observed through immunochemical staining (Figure 8A). EPS8-overexpressing tumor cells exhibited downregulation of the epithelial cell marker (E-cadherin) but upregulation of the mesenchymal cell marker (α-SMA), as observed through immunochemical staining (Figure 8B and 8C). By contrast, EPS8 downregulation resulted in E-cadherin upregulation and α-SMA downregulation (Figure 8D). These results collectively indicate that EPS8 promotes EMT of prostate cancer cells.
Figure 8.

Immunohistochemical evaluation of EMT marker expression in xenograft tumors. LNCaP xenograft tumors were analyzed for the expression of (A) EPS8, (B) α-SMA, and (C) E-cadherin by IHC after 25 days of transfection. (D) Higher EPS8, higher α-SMA, and lower E-cadherin levels were detected in the pCMV-EPS8-transfected xenograft tumor cells. Conversely, lower EPS8, lower α-SMA, and higher E-cadherin levels were detected in the shRNA-EPS8-transfected xenograft tumor cells. #P < 0.05 vs. Control, *P < 0.05 vs. pCMV-GFP, + P < 0.05 vs. shRNA-control. α-SMA, α smooth muscle actin; EMT, epithelial-to-mesenchymal transition; GFP, green fluorescent protein; IHC, immunohistochemistry; shRNA, short-hairpin RNA.
Discussion
In our study, Eps8 expression was higher in LNCaP Enz-R cells than in LNCaP cells. Furthermore, Eps8 activation increases in the levels of EMT, cell proliferation, and cell viability in both LNCaP and LNCaP Enz-R cells. Conversely, after Eps8 knockdown, the levels of EMT, cell proliferation, and cell viability decreased in both LNCaP and LNCaP Enz-R cells. These findings strongly suggest that Eps8 plays a critical role in prostate cancer progression and can be a treatment target for CRPC.
Eps8 is a skeletal protein involved in EGFR kinase activity and is primarily expressed in various tissues, including fat, brain, bowel, gall bladder, endometrium, placenta, ovary, kidney, and bladder. The mechanisms underlying the effects of EPS8 on EMT and the biological behavior of cancer cells have been extensively studied. Eps8 activates mTOR, FAK, STAT3, and Src, thereby promoting protein synthesis, oncogenesis, and tumor proliferation [15]. These pathways are also related to the development of drug-resistant diseases [14,15]. Increased mTOR activation enhances protein synthesis and cellular growth. Increased activation of FAK and STAT3 signaling pathways amplifies oncogenic signaling. The activation of FAK and STAT3 further boosts Src activity, thereby promoting tyrosine phosphorylation and Eps8 synthesis and reinforcing signaling cascades.
The Ras/Raf/MEK pathway is a pathway frequently upregulated in cancer. Eps8 can influence the regulation of this pathway [20]. Ras binds to related Raf proteins such as c-Raf1, B-Raf, and A-Raf, which causes Ras to translocate to the plasma membrane and activate MEK. Activated MEK subsequently activates ERK. Activation of the ERK pathway enables tumor cells to progress through the cell cycle [21,22]. Targeting this pathway has been challenging and considered an “undruggable” target because effective agents inhibiting Ras signaling were lacking [23]. However, recent advancements in therapeutic agents have shown the potential to inhibit Ras in clinical trials [24].
EPS8 has been also implicated in regulating the PI3K/AKT/mTOR pathway. PI3K enzymes phosphorylate inositol lipids on the cell membrane, thereby facilitating cellular signal transduction. PI3K activation induces the generation of phosphatidylinositol (3-5)-trisphosphate from phosphatidylinositol 4,5-bisphosphate, thereby subsequently activating AKT/PKB kinases. These kinases then phosphorylate downstream targets that are crucial for cell survival, proliferation, cell cycle progression, growth, migration, and angiogenesis [25,26]. The tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) negatively regulates AKT activation [26]. PTEN loss or PI3K/AKT pathway activation results in increased cancer cell proliferation, survival, migration, and even castration-resistant growth [27]. Inhibiting the PI3K pathway can promote AR activity, and the combined inhibition of PI3K and AR pathways can cause significant tumor regressions [28,29].
Although several Eps8-associated pathways have been extensively studied, its specific activation and impact in prostate cancer have received limited attention. No clinical data is available on the impact of EPS8 expression on prostate cancer patient survival. Only a few relevant preclinical studies involving Eps8 expression in prostate cancer are present. According to Kretschmer et al., Eps8 can be activated by clusterin, YB-1, and AR variant proteins. This activation is a critical mediator in the formation of tunneling nanotubes (TNTs). TNTs are actin-based membranous structures connecting distant cells, enabling intercellular communication under AR antagonism-induced stress. Silencing Eps8 expression reduces TNT formation as well as intercellular communication and cell survival mechanisms in prostate cancer cells under AR antagonism-induced stress [30]. Similarly, in our study, Eps8 expression was higher in the LNCaP Enz-R cells than in the LNCaP cells. Eps8 expression knockdown could reduce EMT, cell proliferation, and cell viability in both LNCaP Enz-R and LNCaP cells. These findings indicate that Eps8 has a great potential in prostate cancer treatment.
Mithramycin (MTM), an antibiotic obtained from Streptomyces, can reduce EPS8 levels, leading to decreased cancer cell growth and migration [31]. MTM can suppress prostate cancer cell proliferation and inhibit the expression and transcriptional activity of AR and AR variants [32-35]. Additionally, MTM impairs DNA damage repair and enhances the effectiveness of ionizing radiation or bleomycin, a radiomimetic agent, in prostate cancer [35]. However, use of MTM in clinics has been limited because of its adverse effects, including gastrointestinal, hepatic, renal, and bone marrow toxicities [31]. Fortunately, a novel MTM analog exhibiting reduced toxicity has sparked renewed interest. Further in vivo studies are warranted to confirm the efficacy of this new analog in prostate cancer treatment [36].
To the best of our knowledge, this study was the first to investigate and report the effect of Eps8 on prostate cancer cell proliferation. The findings provide novel perspective on potential treatment strategies for prostate cancer. However, there are several limitations. First, our analysis was limited to the LNCaP cell line and its subline, LNCaP Enz-R. Eps8 overexpression and knockdown may have different effects on EMT or cell behavior in different cell lines. The most widely used human prostate cancer cell lines are DU145, PC3, and LNCaP. Of these, only LNCaP expresses the AR and is responsive to androgen. Therefore, many studies use LNCaP cells to establish various castration-resistant cell lines for research [37]. The current study focuses on enzalutamide-resistant prostate cancer cells and LNCaP is a most suitable cell line for this purpose; however, the results of the present study should be compared with other cell lines with caution. Second, our CRPC-related investigation was focused solely on the LNCaP Enz-R cell line. Further studies must be conducted using other CRPC cell lines and androgen antagonists to corroborate our results in the CRPC context. Third, subcutaneous inoculation of tumor cells was performed in our in vivo study, which may have a different biological environment compared with prostate cancer. This discrepancy should be considered while interpreting and extrapolating the results to the prostate setting. Moreover, the mechanisms through which EPS8 regulates EMT and induces drug resistance are still entirely understood and further studies are warranted.
Conclusion
The study highlights the significance of Eps8 expression in prostate cancer progression and prostate cancer cell viability. Eps8 activation promotes EMT, which is linked to the aggressive behavior of cancer and its resistance to drugs. A reduction in EMT and cancer cell viability was observed on silencing Eps8. These findings indicate the key role of Eps8 in prostate cancer progression and drug resistance. Eps8 might be a promising target for developing innovative treatments.
Acknowledgements
We are thankful to consultants and specialist registrars of the E-Da Hospital, I-Shou University, Chang Gung Memorial Hospital, Chang Gung University, and Chung-Hua University of Medical Technology for their contribution in database construction and study consultation. We are also grateful to medical staff associated with our project for all their support. This work was supported by E-Da Hospital Research Grants (EDPJ109065, EDPJ109035, EDPJ110028, and EDPJ110070) and National Science Council Grants, Taiwan (MOST 109 2314 B 650 013 MY2).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–263. doi: 10.3322/caac.21834. [DOI] [PubMed] [Google Scholar]
- 2.Desai MM, Cacciamani GE, Gill K, Zhang J, Liu L, Abreu A, Gill IS. Trends in incidence of metastatic prostate cancer in the US. JAMA Netw Open. 2022;5:e222246. doi: 10.1001/jamanetworkopen.2022.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel DA, O’Neil ME, Richards TB, Dowling NF, Weir HK. Prostate cancer incidence and survival, by stage and race/ethnicity - United States, 2001-2017. MMWR Morb Mortal Wkly Rep. 2020;69:1473–1480. doi: 10.15585/mmwr.mm6941a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller K, Carles J, Gschwend JE, Van Poppel H, Diels J, Brookman-May SD. The phase 3 COU-AA-302 study of abiraterone acetate plus prednisone in men with chemotherapy-naïve metastatic castration-resistant prostate cancer: stratified analysis based on pain, prostate-specific antigen, and gleason score. Eur Urol. 2018;74:17–23. doi: 10.1016/j.eururo.2017.08.035. [DOI] [PubMed] [Google Scholar]
- 5.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, Davis ID, de Bono JS, Evans CP, Fizazi K, Joshua AM, Kim CS, Kimura G, Mainwaring P, Mansbach H, Miller K, Noonberg SB, Perabo F, Phung D, Saad F, Scher HI, Taplin ME, Venner PM, Tombal B PREVAIL Investigators. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, Olmos D, Mainwaring PN, Lee JY, Uemura H, Lopez-Gitlitz A, Trudel GC, Espina BM, Shu Y, Park YC, Rackoff WR, Yu MK, Small EJ SPARTAN Investigators. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408–1418. doi: 10.1056/NEJMoa1715546. [DOI] [PubMed] [Google Scholar]
- 7.Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, Jievaltas M, Luz M, Alekseev B, Kuss I, Kappeler C, Snapir A, Sarapohja T, Smith MR ARAMIS Investigators. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2019;380:1235–1246. doi: 10.1056/NEJMoa1815671. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt KT, Huitema ADR, Chau CH, Figg WD. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat Rev Urol. 2021;18:209–226. doi: 10.1038/s41585-021-00438-4. [DOI] [PubMed] [Google Scholar]
- 9.Feng Q, He B. Androgen receptor signaling in the development of castration-resistant prostate cancer. Front Oncol. 2019;9:858. doi: 10.3389/fonc.2019.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Y, Ning S, Hu J. Molecular mechanisms of neuroendocrine differentiation in prostate cancer progression. J Cancer Res Clin Oncol. 2022;148:1813–1823. doi: 10.1007/s00432-022-04061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler W, Huang J. Neuroendocrine cells of the prostate: histology, biological functions, and molecular mechanisms. Precis Clin Med. 2021;4:25–34. doi: 10.1093/pcmedi/pbab003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen Q, Jiao X, Kuang F, Hou B, Zhu Y, Guo W, Sun G, Ba Y, Yu D, Wang D, Zhang F, Qiao HC, Wang S, Tang S, Qiao H. FoxO3a inhibiting expression of EPS8 to prevent progression of NSCLC: a new negative loop of EGFR signaling. EBioMedicine. 2019;40:198–209. doi: 10.1016/j.ebiom.2019.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 14.Chen YJ, Shen MR, Chen YJ, Maa MC, Leu TH. Eps8 decreases chemosensitivity and affects survival of cervical cancer patients. Mol Cancer Ther. 2008;7:1376–1385. doi: 10.1158/1535-7163.MCT-07-2388. [DOI] [PubMed] [Google Scholar]
- 15.Luo K, Zhang L, Liao Y, Zhou H, Yang H, Luo M, Qing C. Effects and mechanisms of Eps8 on the biological behaviour of malignant tumours (Review) Oncol Rep. 2021;45:824–834. doi: 10.3892/or.2021.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li YH, Xue TY, He YZ, Du JW. Novel oncoprotein EPS8: a new target for anticancer therapy. Future Oncol. 2013;9:1587–1594. doi: 10.2217/fon.13.104. [DOI] [PubMed] [Google Scholar]
- 17.Chen YJ, Shen MR, Chen YJ, Maa MC, Leu TH. Eps8 decreases chemosensitivity and affects survival of cervical cancer patients. Mol Cancer Ther. 2008;7:1376–1385. doi: 10.1158/1535-7163.MCT-07-2388. [DOI] [PubMed] [Google Scholar]
- 18.Lee GT, Rosenfeld JA, Kim WT, Kwon YS, Palapattu G, Mehra R, Kim WJ, Kim IY. TCF4 induces enzalutamide resistance via neuroendocrine differentiation in prostate cancer. PLoS One. 2019;14:e0213488. doi: 10.1371/journal.pone.0213488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, Fessenden TB, Isikbay M, Paner GP, Szmulewitz RZ, Vander Griend DJ. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget. 2016;7:26259–26274. doi: 10.18632/oncotarget.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Logue JS, Cartagena-Rivera AX, Baird MA, Davidson MW, Chadwick RS, Waterman CM. Erk regulation of actin capping and bundling by Eps8 promotes cortex tension and leader bleb-based migration. Elife. 2015;4:e08314. doi: 10.7554/eLife.08314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 22.Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, Kato T, Curioni-Fontecedro A, Sacher A, Spira A, Ramalingam SS, Takahashi T, Besse B, Anderson A, Ang A, Tran Q, Mather O, Henary H, Ngarmchamnanrith G, Friberg G, Velcheti V, Govindan R. Sotorasib for lung cancers with KRAS p. G12C mutation. N Engl J Med. 2021;384:2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 26.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 27.Choudhury AD. PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate. 2022;82(Suppl 1):S60–S72. doi: 10.1002/pros.24372. [DOI] [PubMed] [Google Scholar]
- 28.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 2020;21:4507. doi: 10.3390/ijms21124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kretschmer A, Zhang F, Somasekharan SP, Tse C, Leachman L, Gleave A, Li B, Asmaro I, Huang T, Kotula L, Sorensen PH, Gleave ME. Stress-induced tunneling nanotubes support treatment adaptation in prostate cancer. Sci Rep. 2019;9:7826. doi: 10.1038/s41598-019-44346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang TP, Chiou HL, Maa MC, Wang CJ. Mithramycin inhibits human epithelial carcinoma cell proliferation and migration involving downregulation of Eps8 expression. Chem Biol Interact. 2010;183:181–186. doi: 10.1016/j.cbi.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 32.Wang LG, Ferrari AC. Mithramycin targets sp1 and the androgen receptor transcription level-potential therapeutic role in advanced prostate cancer. Transl Oncogenomics. 2006;1:19–31. [PMC free article] [PubMed] [Google Scholar]
- 33.Choi ES, Chung T, Kim JS, Lee H, Kwon KH, Cho NP, Cho SD. Mithramycin A induces apoptosis by regulating the mTOR/Mcl-1/tBid pathway in androgen-independent prostate cancer cells. J Clin Biochem Nutr. 2013;53:89–93. doi: 10.3164/jcbn.13-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malek A, Núñez LE, Magistri M, Brambilla L, Jovic S, Carbone GM, Morís F, Catapano CV. Modulation of the activity of Sp transcription factors by mithramycin analogues as a new strategy for treatment of metastatic prostate cancer. PLoS One. 2012;7:e35130. doi: 10.1371/journal.pone.0035130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Gilbreath C, Kollipara RK, Sonavane R, Huo X, Yenerall P, Das A, Ma S, Raj GV, Kittler R. Mithramycin suppresses DNA damage repair via targeting androgen receptor in prostate cancer. Cancer Lett. 2020;488:40–49. doi: 10.1016/j.canlet.2020.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Núñez LE, Nybo SE, González-Sabín J, Pérez M, Menéndez N, Braña AF, Shaaban KA, He M, Morís F, Salas JA, Rohr J, Méndez C. A novel mithramycin analogue with high antitumor activity and less toxicity generated by combinatorial biosynthesis. J Med Chem. 2012;55:5813–5825. doi: 10.1021/jm300234t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abate-Shen C, Nunes de Almeida F. Establishment of the LNCaP cell line - the dawn of an era for prostate cancer research. Cancer Res. 2022;82:1689–1691. doi: 10.1158/0008-5472.CAN-22-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.